
Aqueous Solubility of Organic Compounds for Flow Battery Applications: Symmetry and Counter Ion Design to Avoid Low-Solubility Polymorphs



Abstract
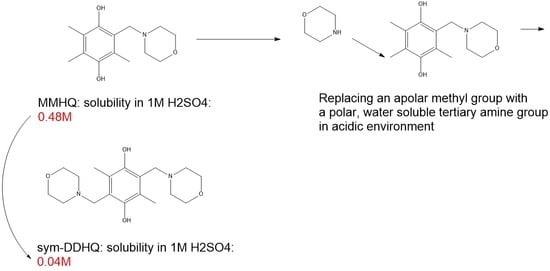
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Nuclear Magnetic Resonance Spectroscopy (NMR)
4.2. Liquid Chromatography-Mass Spectroscopy (LC-MS)
4.3. X-ray Crystallography
4.4. Synthesis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Alotto, P.; Guarnieri, M.; Moro, F. Redox flow batteries for the storage of renewable energy: A review. Renew. and Sustain. Energy Rev. 2014, 29, 325–335. [Google Scholar] [CrossRef]
- Soloveichik, G.L. Flow Batteries: Current Status and Trends. Chem. Rev. 2015, 115, 11533–11558. [Google Scholar] [CrossRef] [PubMed]
- Huskinson, B.; Marshak, M.P.; Suh, C.; Er, S.; Gerhardt, M.R.; Galvin, C.J.; Chen, X.; Aspuru-Guzik, A.; Gordon, R.G.; Aziz, M.J. A metal-free organic-inorganic aqueous flow battery. Nature 2014, 505, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Hoober-Burkhardt, L.; Wang, F.; Surya Prakash, G.K.; Narayanan, S.R. An Inexpensive Aqueous Flow Battery for Large-Scale Electrical Energy Storage Based on Water-Soluble Organic Redox Couples. J. Electrochem. Soc. 2014, 161, A1371–A1380. [Google Scholar] [CrossRef]
- Winsberg, J.; Hagemann, T.; Janoschka, T.; Hager, M.D.; Schubert, U.S. Redox-Flow Batteries: From Metals to Organic Redox-Active Materials. Angew. Chem. Int. Ed. 2017, 56, 686–711. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Pan, W.; Duan, W.; Hollas, A.; Yang, Z.; Li, B.; Nie, Z.; Liu, J.; Reed, D.; Wang, W.; et al. Materials and Systems for Organic Redox Flow Batteries: Status and Challenges. ACS Energy Lett. 2017, 2, 2187–2204. [Google Scholar] [CrossRef]
- Wang, W.; Sprenkle, V. Energy storage: Redox flow batteries go organic. Nat. Chem. 2016, 8, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Comer, J.; Tam, K. Lipophilicity Profiles: Theory and Measurement. In Pharmacokinetic Optimization in Drug Research: Biological, Physicochemical, and Computational Strategies; Testa, B., van de Waterbeemd, H., Folkers, G., Guy, R., Eds.; Helvetica Chimica Acta: Zurich, Switzerland, 2001; pp. 275–304. [Google Scholar]
- Chemburkar, S.R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W.; et al. Dealing with the Impact of Ritonavir Polymorphs on the Late Stages of Bulk Drug Process Development. Org. Process. Res. Dev. 2000, 4, 413–417. [Google Scholar] [CrossRef]
- Abramov, Y.A.; Pencheva, K. Thermodynamics and Relative Solubility Prediction of Polymorphic Systems. In Chemical Engineering in the Pharmaceutical Industry: R&D to Manufacturing, 2nd ed.; J. am Ende, D., T. am Ende, M., Eds.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2019; pp. 505–518. [Google Scholar]
- Llinas, A.; Glen, R.C.; Goodman, J.M. Solubility challenge: Can you predict solubilities of 32 molecules using a database of 100 reliable measurements? J. Chem. Inf. Model. 2008, 48, 1289–1303. [Google Scholar] [CrossRef]
- Hopfinger, A.J.; Esposito, E.X.; Llinas, A.; Glen, R.C.; Goodman, J.M. Findings of the challenge to predict aqueous solubility. J. Chem. Inf. Model. 2009, 49, 1–5. [Google Scholar] [CrossRef]
- Morissette, S.L.; Soukasene, S.; Levinson, D.; Cima, M.J.; Almarsson, O. Elucidation of crystal form diversity of the HIV protease inhibitor ritonavir by high-throughput crystallization. Proc. Natl. Acad. Sci. USA 2003, 100, 2180–2184. [Google Scholar] [CrossRef] [PubMed]
- Threlfall, T. Structural and Thermodynamic Explanations of Ostwald’s Rule. Org. Process. Res. Dev. 2003, 7, 1017–1027. [Google Scholar] [CrossRef]
- Hoober-Burkhardt, L.; Krishnamoorthy, S.; Yang, B.; Murali, A.; Nirmalchandar, A.; Prakash, G.K.S.; Narayanan, S.R. A New Michael-Reaction-Resistant Benzoquinone for Aqueous Organic Redox Flow Batteries. J. Electrochem. Soc. 2017, 164, A600–A607. [Google Scholar] [CrossRef]
- Fields, D.L.; Miller, J.B.; Reynolds, D.D. Preparation of Acetoxybenzyl Bromides. J. Org. Chem. 1964, 9, 2640–2647. [Google Scholar] [CrossRef]
- Mao, J.; Ruan, W.; Chen, Q. Understanding the Aqueous Solubility of Anthraquinone Sulfonate Salts: The Quest for High Capacity Electrolytes of Redox Flow Batteries. J. Electrochem. Soc. 2020, 167, 070522. [Google Scholar] [CrossRef]
- Avdeef, A.; Fuguet, E.; Llinàs, A.; Ràfols, C.; Bosch, E.; Völgyi, G.; Verbić, T.; Boldyreva, E.; Takács-Novák, K. Equilibrium solubility measurement of ionizable drugs—Consensus recommendations for improving data quality. ADMET DMPK 2016, 4, 117. [Google Scholar] [CrossRef]







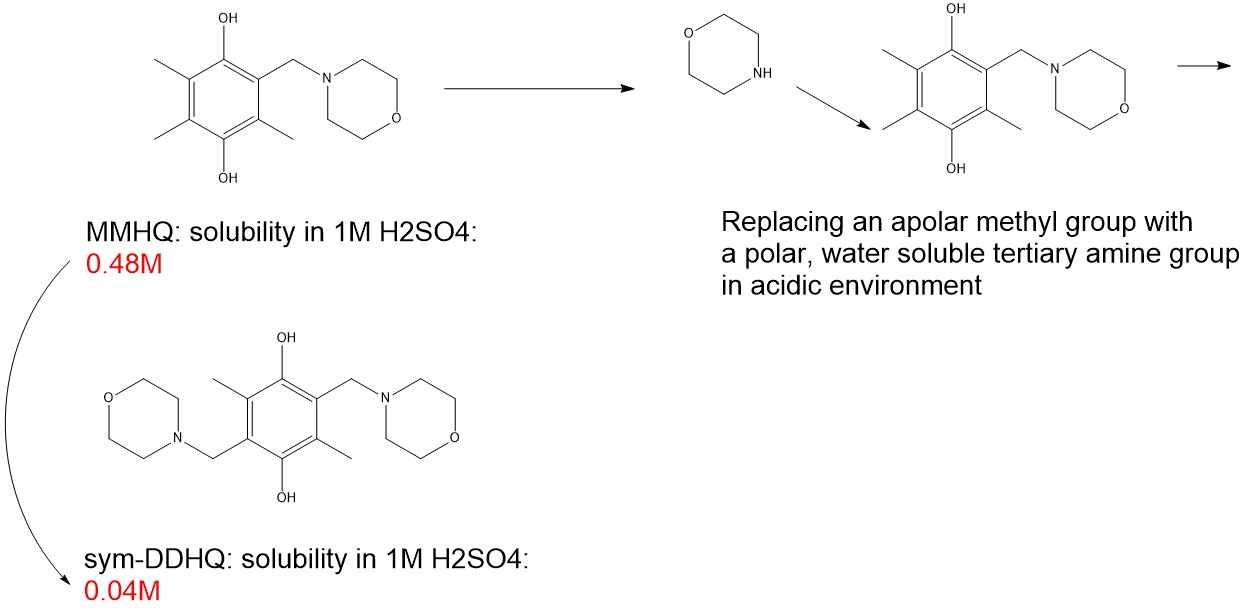{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | DMDQ.2HCl |
|---|---|
| Empirical formula | C16 H30 Cl2 N2 O6 |
| Formula weight | 417.32 |
| Temperature | 140(2) |
| Radiation and wavelength | Cu-Kα, λ = 1.54187 Å |
| Crystal system | monoclinic |
| Space group | P 21/c |
| Unit cell dimensions | a = 9.2629(2) Å |
| b = 15.4919(3) Å | |
| c = 7.4246(2) Å | |
| α = 90° | |
| β = 110.973(8)° | |
| γ = 90° | |
| Volume | 994.84(6) Å3 |
| Z | 2 |
| Density (calculated) | 1.393 Mg/m3 |
| Absorption coefficient, μ | 3.238 mm−1 |
| F(000) | 444 |
| Crystal color | colorless |
| Crystal description | prism |
| Crystal size | 0.72 × 0.48 × 0.38 (mm) |
| Absorption correction | numerical |
| Max. and min. transmission | 0.5390.766 |
| θ−range for data collection | 5.114 ≤ θ ≤ 68.172° |
| Index ranges | −11 ≤ h ≤ 10; −18 ≤ k ≤ 18; −8 ≤ l ≤ 7 |
| Reflections collected | 17,903 |
| Completeness to 2θ | 0.972 |
| Independent reflections | 1763 [R(int) = 0.0332] |
| Reflections I > 2σ(I) | 1733 |
| Refinement method | full-matrix least-squares on F2 |
| Data/restraints/parameters | 1763/0/134 |
| Goodness-of-fit on F2 | 1.184 |
| Final R indices [I > 2σ(I)] | R1 = 0.0308, wR2 = 0.0787 |
| R indices (all data) | R1 = 0.0317, wR2 = 0.0793 |
| Max. and mean shift/esd | 0.000; 0.000 |
| Largest diff. peak and hole | 0.249; −0.244 e.Å−3 |
| Compound | Cl− | ClO4− | SO42− | PO33− |
|---|---|---|---|---|
| MMHQ | 0.07 1 | 0.02 | 0.48 | 0.48 |
| DMHQ | 0.03 1 | 0.01 | 0.08 2 | 0.53 |
| sym-DDHQ | - | - | 0.06 3 | 0.40 |
| asym-DDHQ | - | - | 0.44 | 0.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, S.N.; Yang, X.; Bereczki, L.; Kónya, D. Aqueous Solubility of Organic Compounds for Flow Battery Applications: Symmetry and Counter Ion Design to Avoid Low-Solubility Polymorphs. Molecules 2021, 26, 1203. https://doi.org/10.3390/molecules26051203
Garcia SN, Yang X, Bereczki L, Kónya D. Aqueous Solubility of Organic Compounds for Flow Battery Applications: Symmetry and Counter Ion Design to Avoid Low-Solubility Polymorphs. Molecules. 2021; 26(5):1203. https://doi.org/10.3390/molecules26051203
Chicago/Turabian StyleGarcia, Sergio Navarro, Xian Yang, Laura Bereczki, and Dénes Kónya. 2021. "Aqueous Solubility of Organic Compounds for Flow Battery Applications: Symmetry and Counter Ion Design to Avoid Low-Solubility Polymorphs" Molecules 26, no. 5: 1203. https://doi.org/10.3390/molecules26051203
APA StyleGarcia, S. N., Yang, X., Bereczki, L., & Kónya, D. (2021). Aqueous Solubility of Organic Compounds for Flow Battery Applications: Symmetry and Counter Ion Design to Avoid Low-Solubility Polymorphs. Molecules, 26(5), 1203. https://doi.org/10.3390/molecules26051203