
Novel Inhibitors of Nicotinamide-N-Methyltransferase for the Treatment of Metabolic Disorders

 , and
, and
Abstract
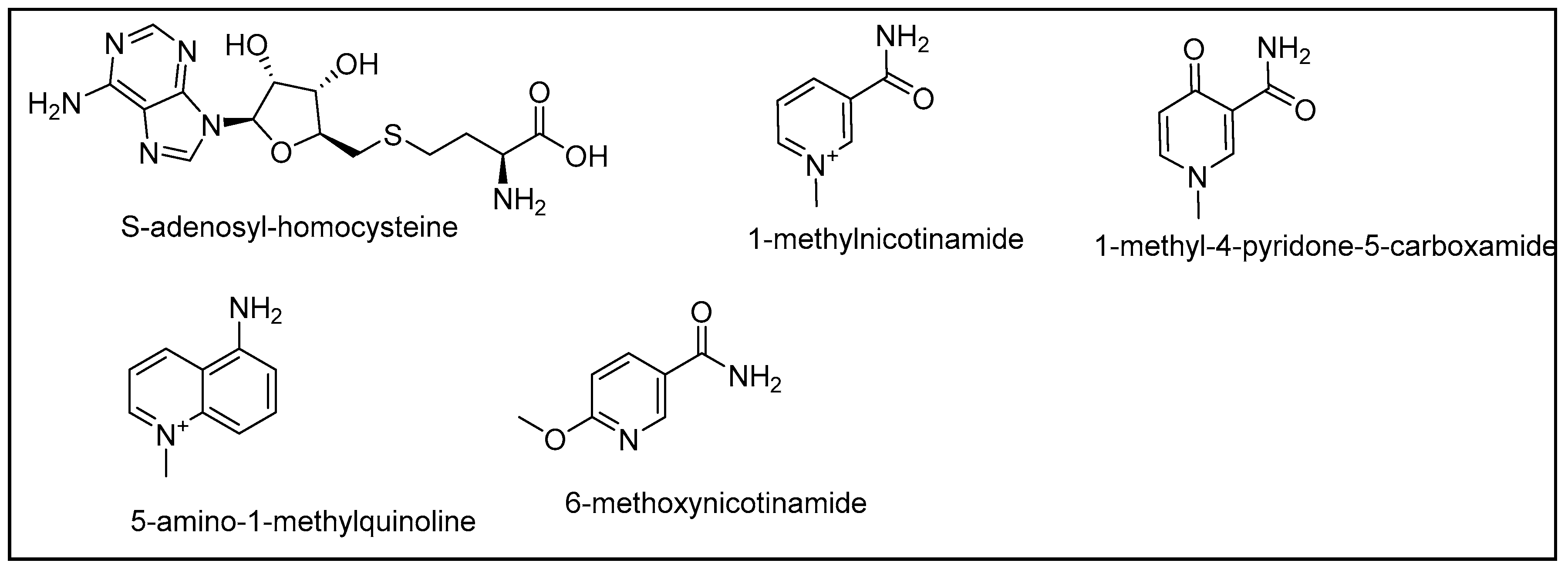
1. Introduction
2. Results and Discussion
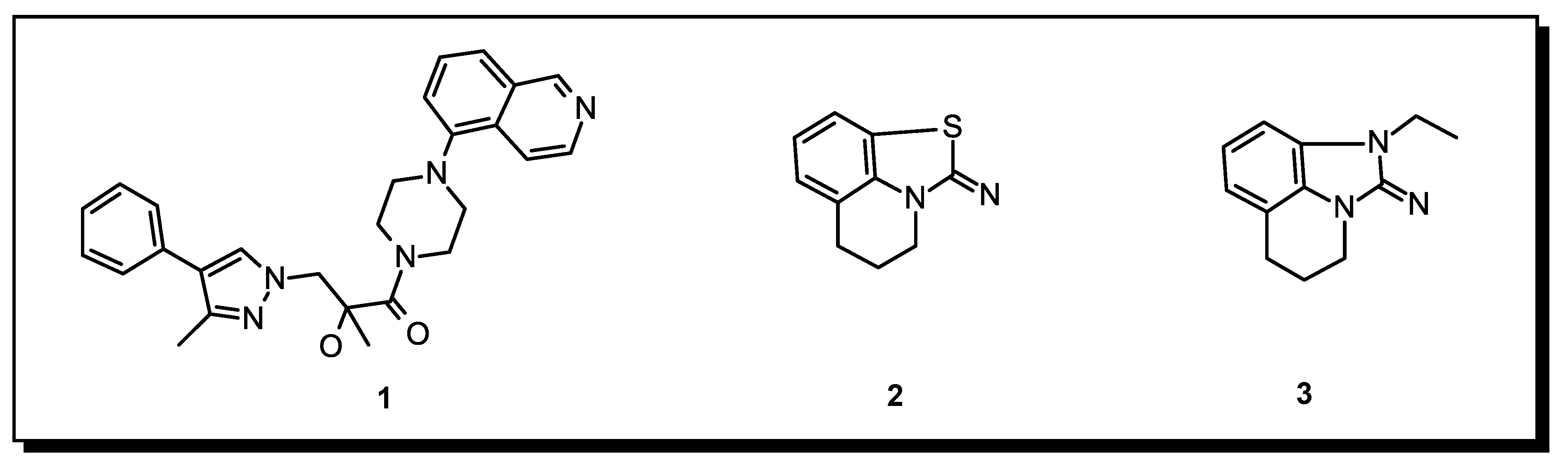
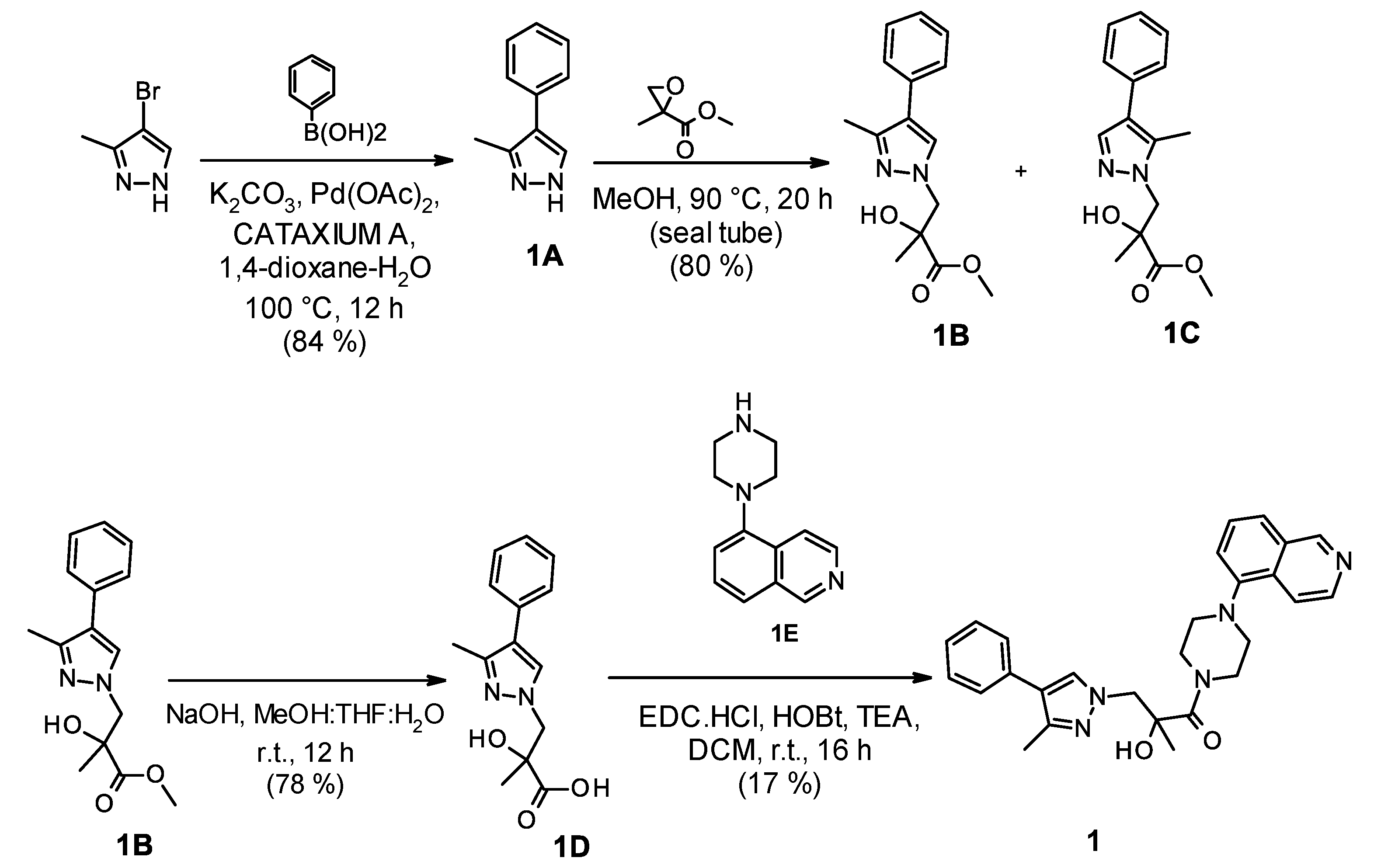
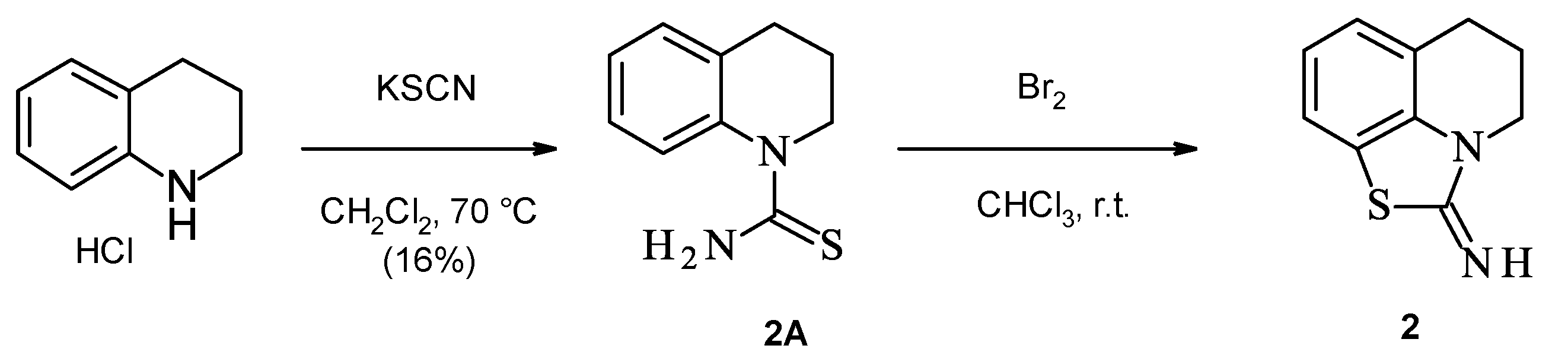
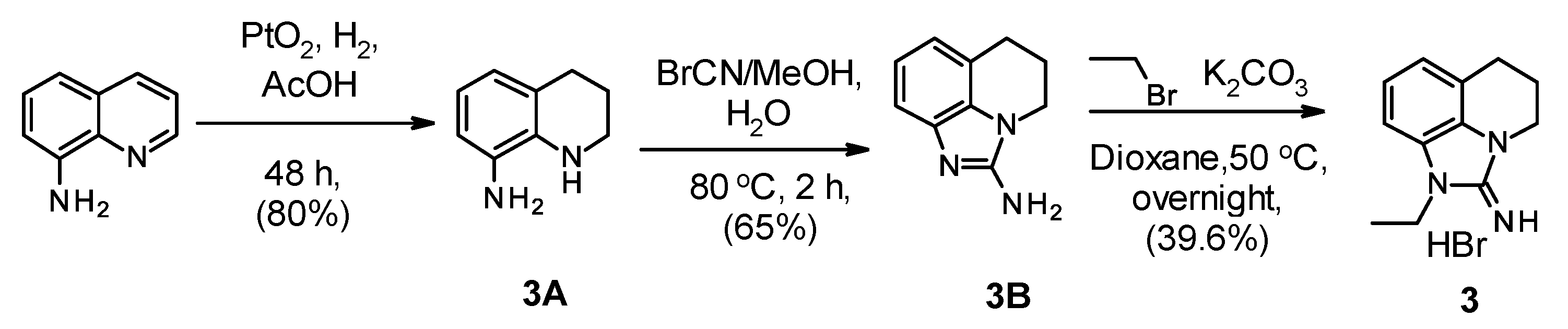
2.1. Chemistry
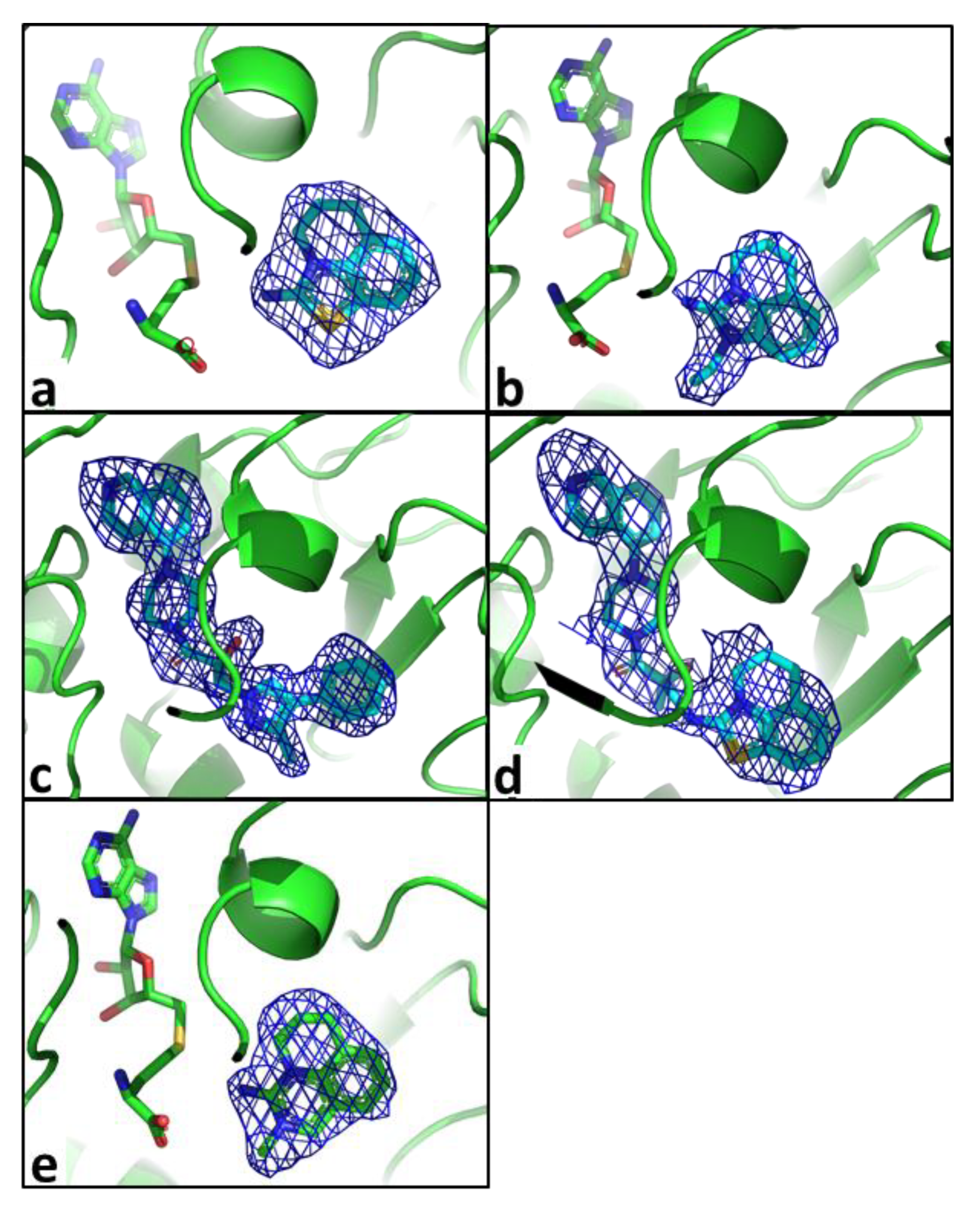
2.2. Structural Biology
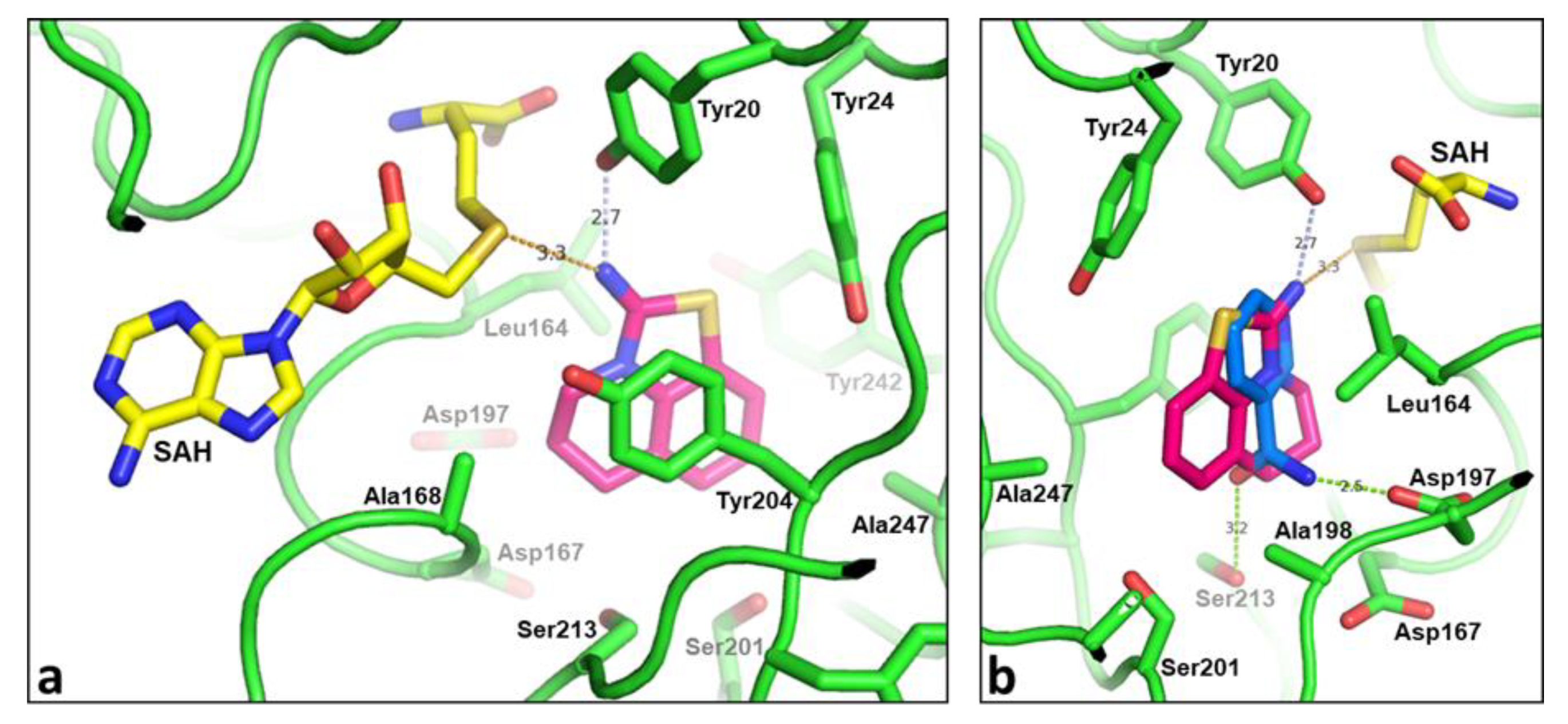
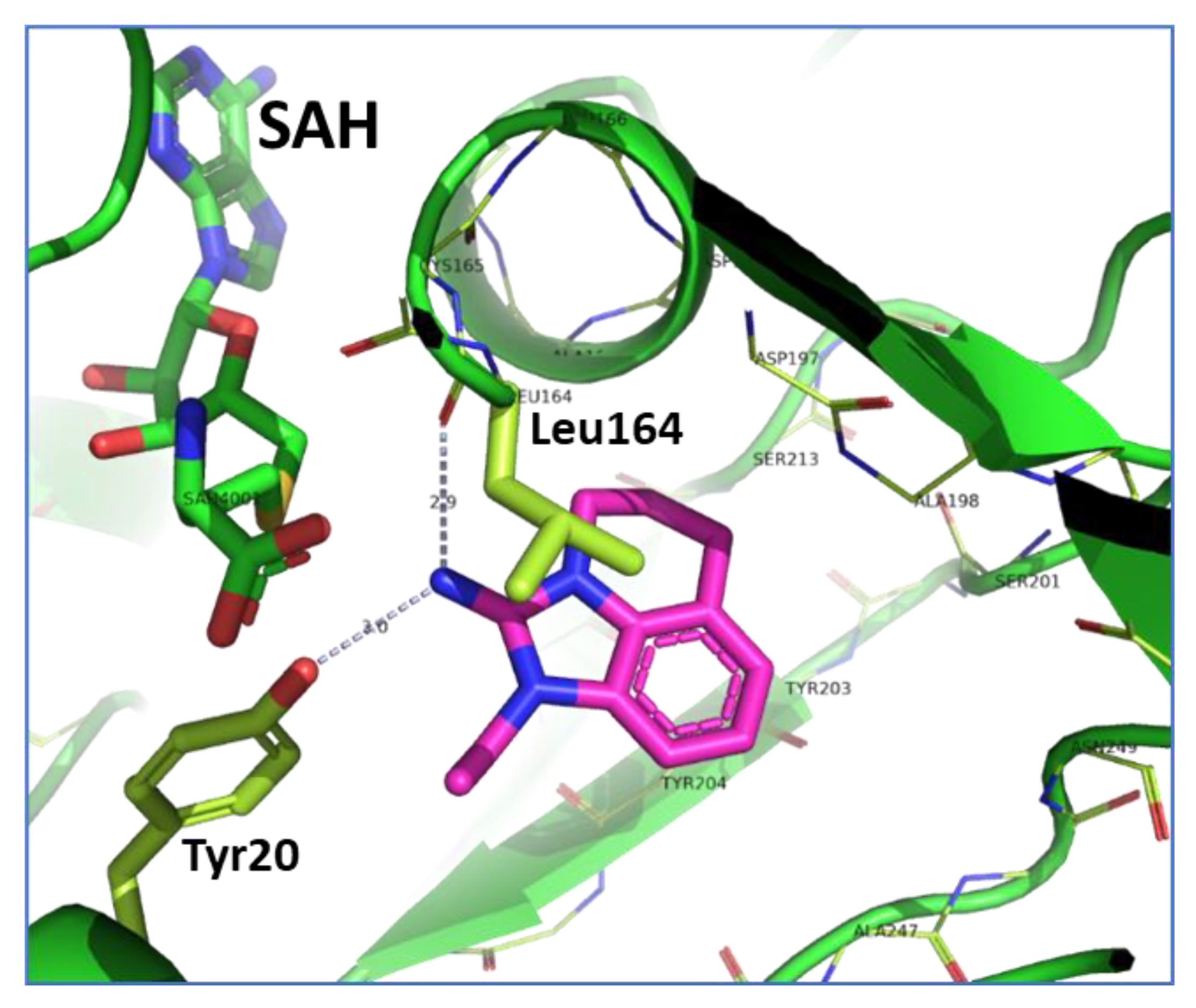
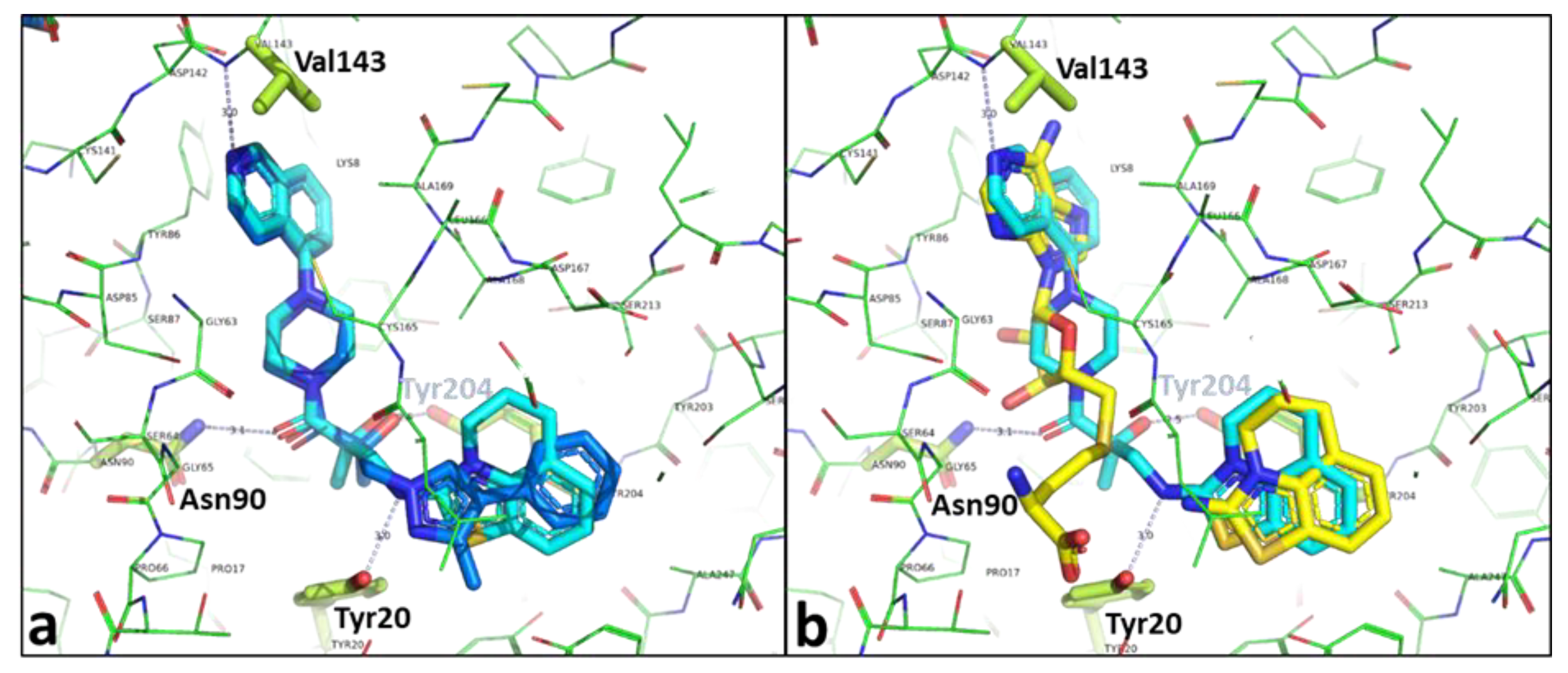
2.3. Binding Mode of the High Throughput Screening Hits
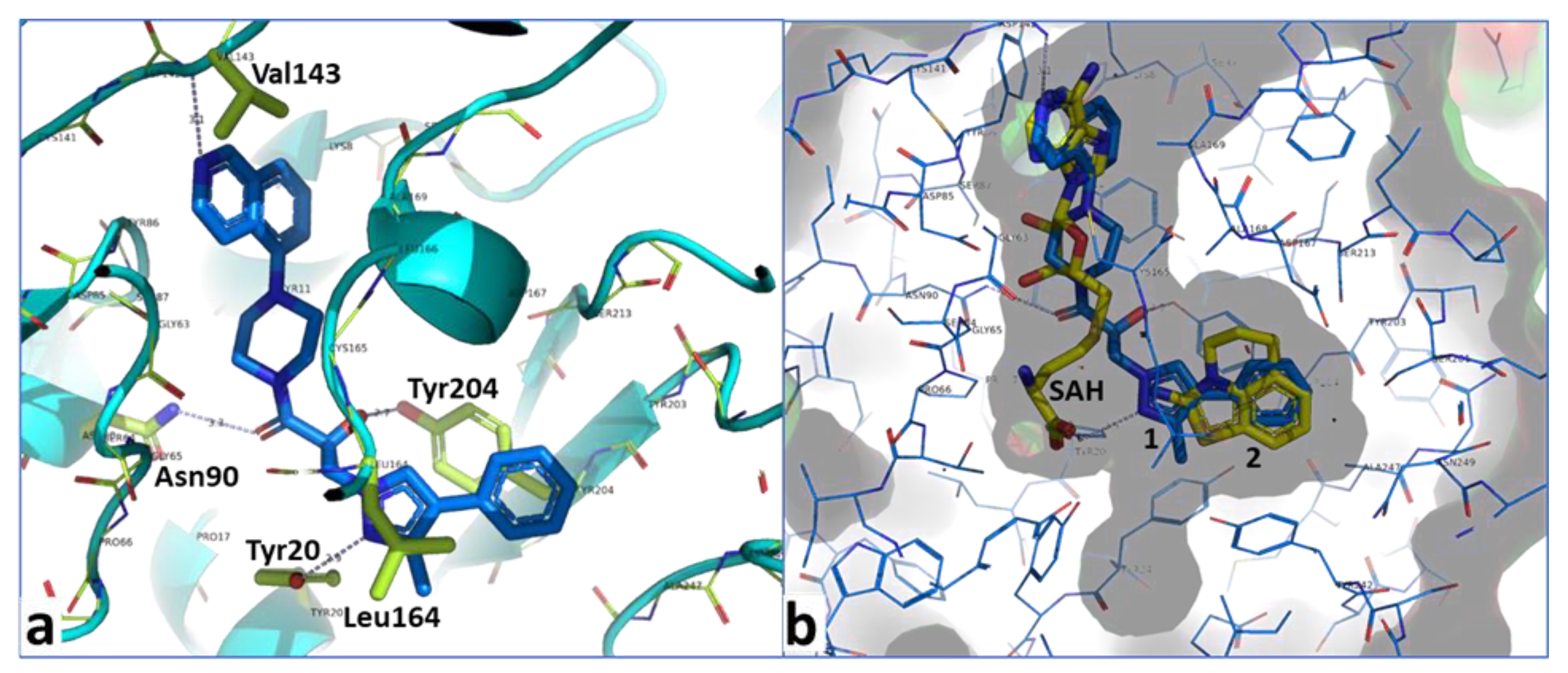
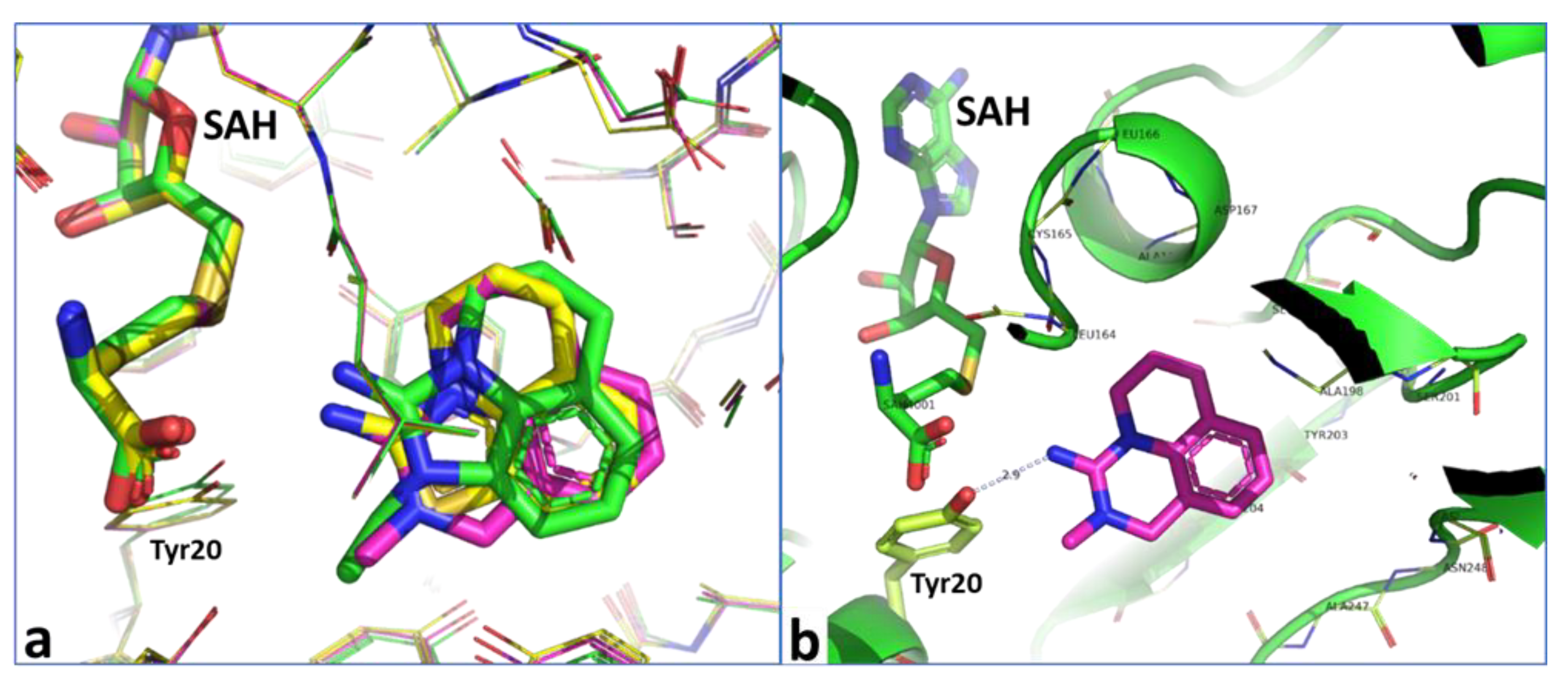
2.4. Optimization of a Bisubstrate-Like Inhibitor Series Based on Compound 1
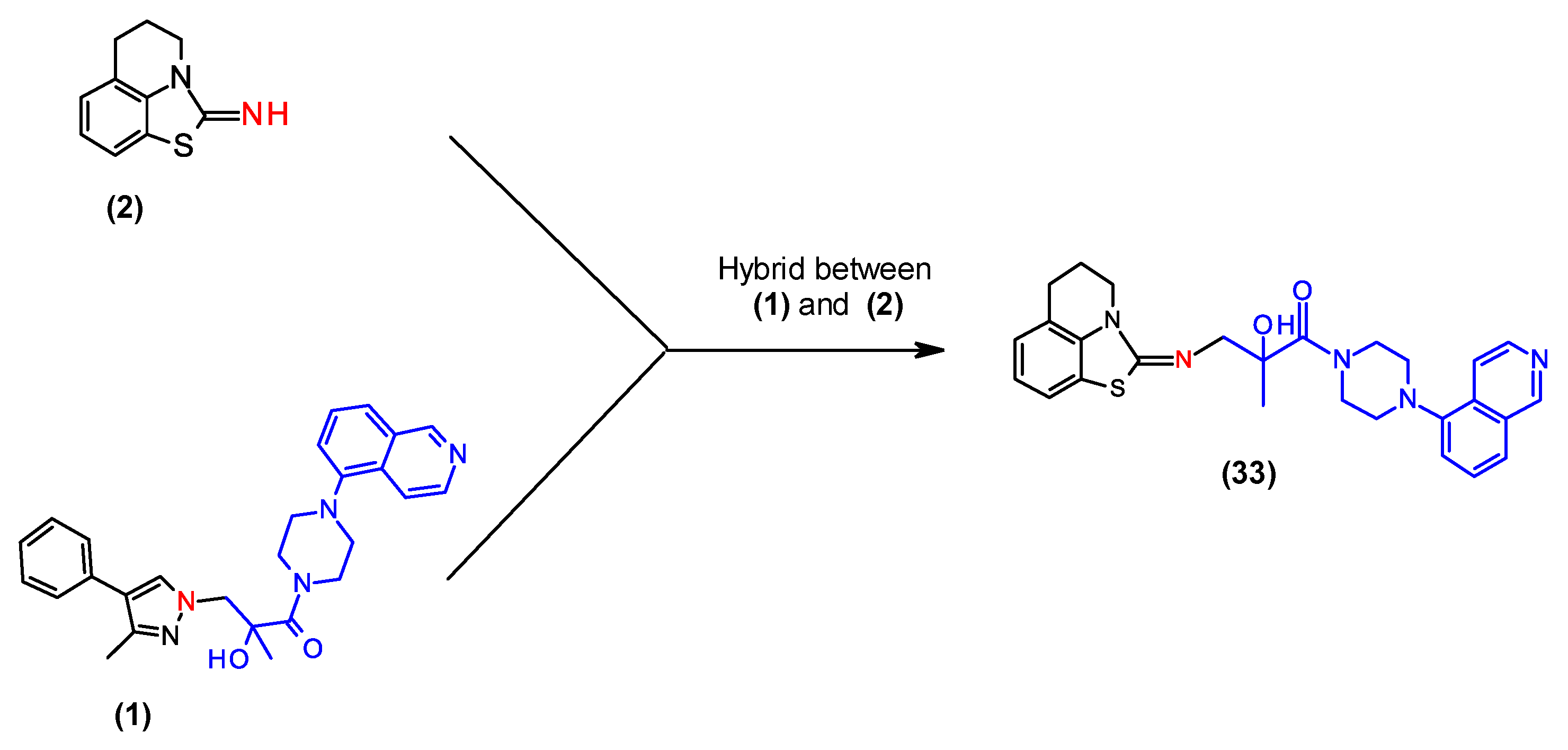
2.5. Combination of Our Bisubstrate-Like Inhibitor Series with the HTS Leads 2 and 3
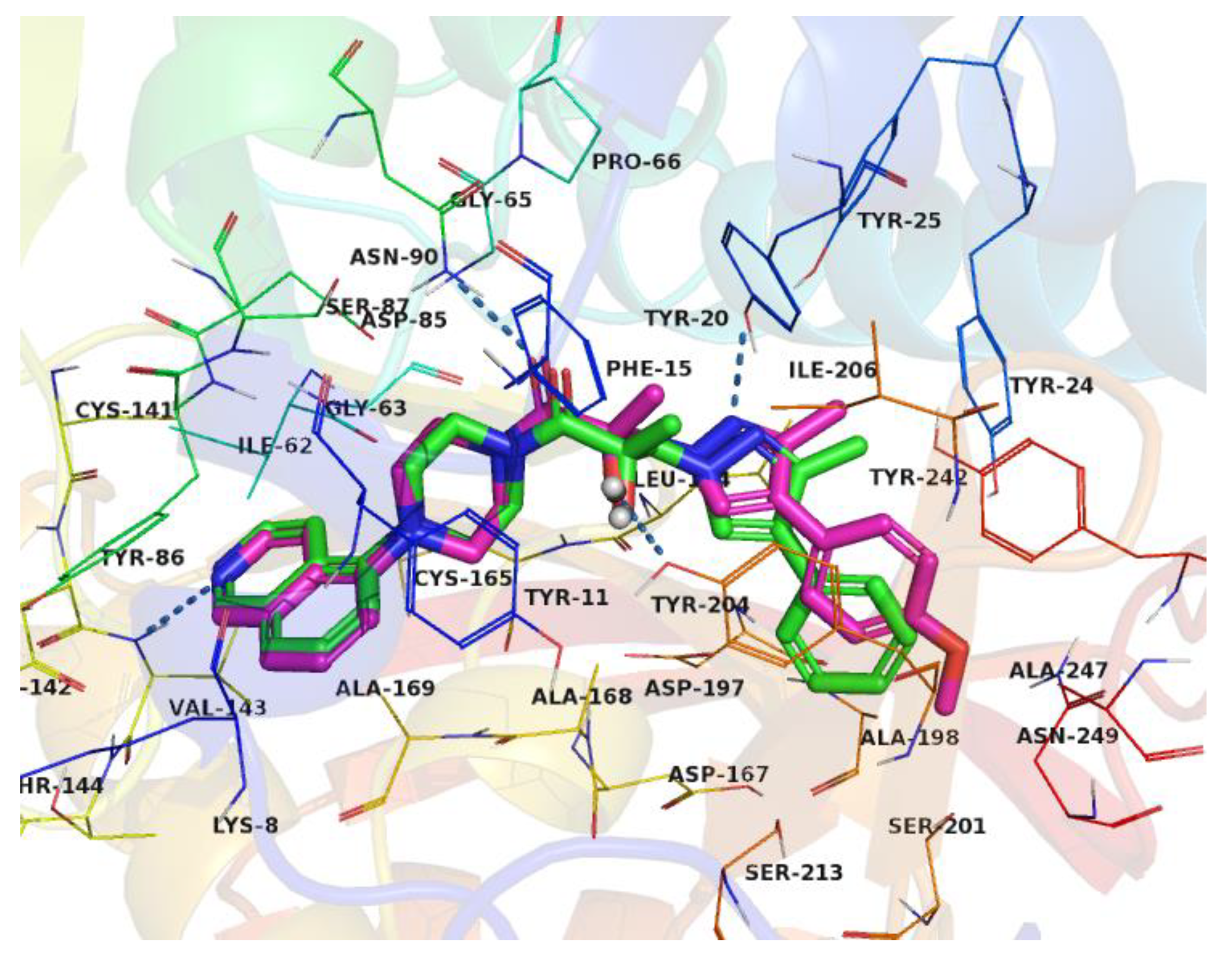
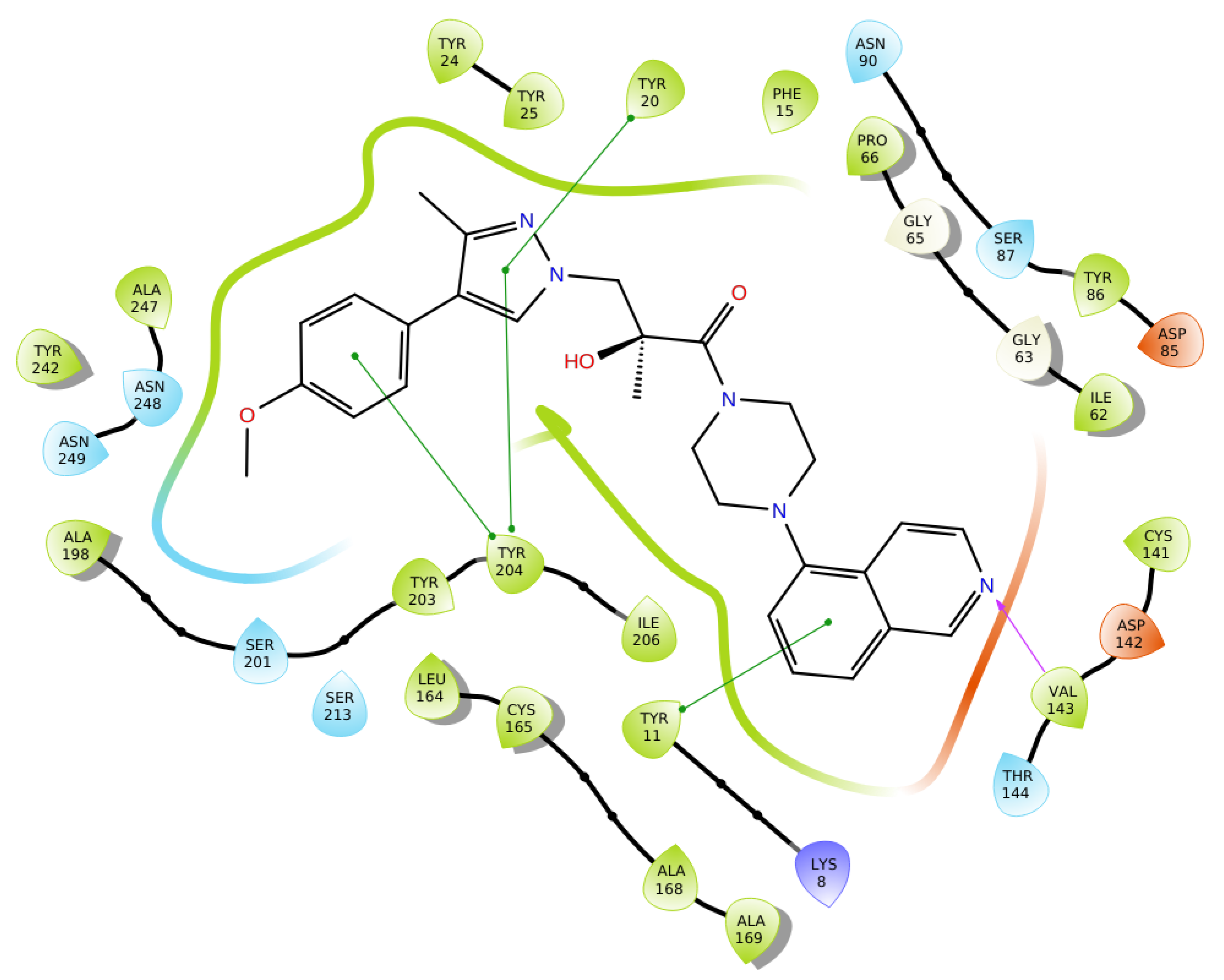
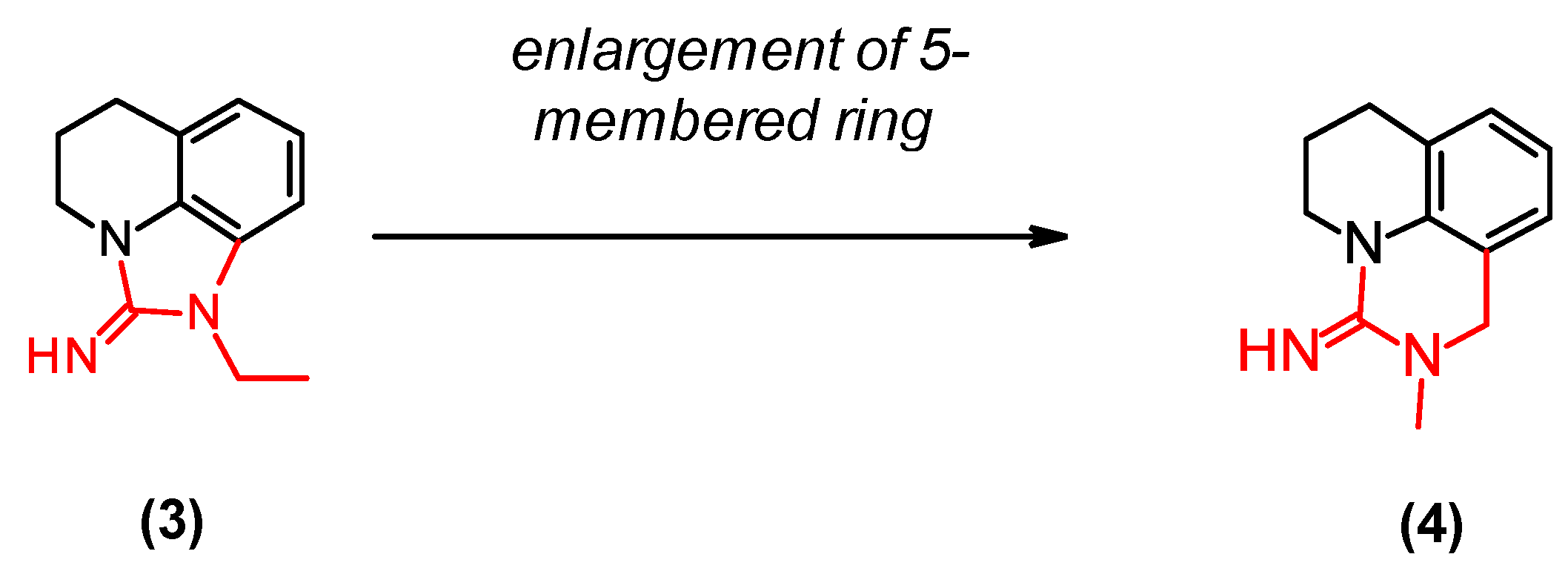
2.6. Optimization of a Series of Tricyclic Compounds Binding to the Nam Pocket Only
3. Materials and Methods
Crystallographic Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pissios, P. Nicotinamide N-Methyltransferase: More Than a Vitamin B3 Clearance Enzyme. Trends. Endocrinol. Metab. 2017, 28, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human liver nicotinamide N-methyltransferase. cDNA cloning, expression, and biochemical characterization. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar] [CrossRef]
- GTEx Consortium. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef]
- Kannt, A.; Rajagopal, S.; Kadnur, S.V.; Suresh, J.; Bhamidipati, R.K.; Swaminathan, S.; Hallur, M.S.; Kristam, R.; Elvert, R.; Czech, J.; et al. A small molecule inhibitor of Nicotinamide N-methyltransferase for the treatment of metabolic disorders. Sci. Rep. 2018, 8, 3660. [Google Scholar] [CrossRef] [PubMed]
- Brachs, S.; Polack, J.; Brachs, M.; Jahn-Hofmann, K.; Elvert, R.; Pfenninger, A.; Bärenz, F.; Margerie, D.; Mai, K.; Spranger, J.; et al. Genetic Nicotinamide N-Methyltransferase (Nnmt) Deficiency in Male Mice Improves Insulin Sensitivity in Diet-Induced Obesity but Does Not Affect Glucose Tolerance. Diabetes 2019, 68, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Matsubara, K.; Okada, K.; Fukushima, S.; Shimizu, K.; Yamaguchi, S.; Uezono, T.; Satomi, M.; Hayase, N.; Ohta, S.; et al. N-methylation ability for azaheterocyclic amines is higher in Parkinson’s disease: Nicotinamide loading test. J. Neural. Transm. 2000, 107, 985–995. [Google Scholar] [CrossRef]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Nair, S.; Rousseau, E.; Allison, D.B.; Page, G.P.; Tataranni, P.A.; Bogardus, C.; Permana, P.A. Microarray profiling of isolated abdominal subcutaneous adipocytes from obese vs non-obese Pima Indians: Increased expression of inflammation-related genes. Diabetologia 2005, 48, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Kannt, A.; Pfenninger, A.; Teichert, L.; Tönjes, A.; Dietrich, A.; Schön, M.R.; Klöting, N.; Blüher, M. Association of nicotinamide-N-methyltransferase mRNA expression in human adipose tissue and the plasma concentration of its product, 1-methylnicotinamide, with insulin resistance. Diabetologia 2015, 58, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, L.; Chu, J.; Zhu, B.; Zhang, Q.; Yin, X.; Jiang, W.; Dai, G.; Ju, W.; Wang, Z.; et al. Serum N(1)-Methylnicotinamide Is Associated With Obesity and Diabetes in Chinese. J. Clin. Endocrinol. Metab. 2015, 100, 3112–3117. [Google Scholar] [CrossRef]
- Salek, R.M.; Maguire, M.L.; Bentley, E.; Rubtsov, D.V.; Hough, T.; Cheeseman, M.; Nunez, D.; Sweatman, B.C.; Haselden, J.N.; Cox, R.D.; et al. A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiol. Genomics. 2007, 29, 99–108. [Google Scholar] [CrossRef]
- Bañales-Luna, M.; Figueroa-Vega, N.; Marín-Aragón, C.I.; Perez-Luque, E.; Ibarra-Reynoso, L.; Gallardo-Blanco, H.L.; López-Aguilar, I.; Malacara, J.M. Associations of nicotinamide-N-methyltransferase, FTO and IRX3 genetic variants with body mass index and resting energy expenditure in Mexican subjects. Sci. Rep. 2020, 10, 11478. [Google Scholar] [CrossRef] [PubMed]
- Souto, J.C.; Blanco-Vaca, F.; Soria, J.M.; Buil, A.; Almasy, L.; Ordoñez-Llanos, J.; Martín-Campos, J.M.; Lathrop, M.; Stone, W.; Blangero, J.; et al. A genomewide exploration suggests a new candidate gene at chromosome 11q23 as the major determinant of plasma homocysteine levels: Results from the GAIT project. Am. J. Hum. Genet. 2005, 76, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Sazci, A.; Ozel, M.D.; Ergul, E.; Aygun, C. Association of nicotinamide-N-methyltransferase gene rs694539 variant with patients with nonalcoholic steatohepatitis. Genet. Test Mol. Biomark. 2013, 17, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.J.; Lin, Y.J.; Chen, W.; Wang, Y.H.; Qiu, L.Q.; Cai, C.X.; Xiong, Q.; Chen, F.; Chen, L.H.; Zhou, Q.; et al. Physiological Study on Association between Nicotinamide N-Methyltransferase Gene Polymorphisms and Hyperlipidemia. Biomed. Res. Int. 2016, 45, 50. [Google Scholar] [CrossRef] [PubMed]
- Neelakantan, H.; Vance, V.; Wetzel, M.D.; Wang, H.L.; McHardy, S.F.; Finnerty, C.C.; Hommel, J.D.; Watowich, S.J. Selective and membrane-permeable small molecule inhibitors of nicotinamide N-methyltransferase reverse high fat diet-induced obesity in mice. Biochem. Pharmacol. 2018, 147, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Ruf, S.; Hallur, M.S.; Anchan, N.K.; Swamy, I.N.; Murugesan, K.R.; Sarkar, S.; Narasimhulu, L.K.; Putta, V.P.R.K.; Shaik, S.; Chandrasekar, D.V.; et al. Novel nicotinamide analog as inhibitor of nicotinamide N-methyltransferase. Bioorg. Med. Chem. Lett. 2018, 28, 92–925. [Google Scholar] [CrossRef]
- Van Haren, M.J.; Taig, R.; Kuppens, J.; Toraño, J.S.; Moret, E.E.; Parsons, R.B.; Sartini, D.; Emanuelli, M.; Martin, N.I. Inhibitors of nicotinamide N-methyltransferase designed to mimic the methylation reaction transition state. Org. Biomol. Chem. 2017, 15, 6656–6667. [Google Scholar] [CrossRef]
- Babault, N.; Allali-Hassani, A.; Li, F.; Fan, J.; Yue, A.; Ju, K.; Liu, F.; Vedadi, M.; Liu, J.; Jin, J. Discovery of Bisubstrate Inhibitors of Nicotinamide N-Methyltransferase (NNMT). J. Med. Chem. 2018, 61, 1541–1551. [Google Scholar] [CrossRef]
- Policarpo, R.L.; Decultot, L.; May, E.; Kuzmič, P.; Carlson, S.; Huang, D.; Chu, V.; Wright, B.A.; Dhakshinamoorthy, S.; Kannt, A.; et al. High-Affinity Alkynyl Bisubstrate Inhibitors of Nicotinamide N-Methyltransferase (NNMT). J. Med. Chem. 2019, 62, 9837–9873. [Google Scholar] [CrossRef]
- Gao, Y.; van Haren, M.J.; Moret, E.E.; Rood, J.J.M.; Sartini, D.; Salvucci, A.; Emanuelli, M.; Craveur, P.; Babault, N.; Jin, J.; et al. Bisubstrate Inhibitors of Nicotinamide N-Methyltransferase (NNMT) with Enhanced Activity. J. Med. Chem. 2019, 62, 6597–6614. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Li, L.; Diaz, K.; Iyamu, I.D.; Yadav, R.; Noinaj, N.; Huang, R. Novel Propargyl-Linked Bisubstrate Analogues as Tight-Binding Inhibitors for Nicotinamide N-Methyltransferase. J. Med. Chem. 2019, 62, 10783–10797. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Suciu, R.M.; Horning, B.D.; Vinogradova, E.V.; Ulanovskaya, O.A.; Cravatt, B.F. Covalent inhibitors of nicotinamide N-methyltransferase (NNMT) provide evidence for target engagement challenges in situ. Bioorg. Med. Chem. Lett. 2018, 28, 2682–2687. [Google Scholar] [CrossRef]
- Peng, Y.; Sartini, D.; Pozzi, V.; Wilk, D.; Emanuelli, M.; Yee, V.C. Structural Basis of Substrate Recognition in Human Nicotinamide N-Methyltransferase. Biochemistry 2011, 50, 7800–7808. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Bolleddula, J.; DeMent, K.; Driscoll, J.P.; Worboys, P.; Brassil, P.J.; Bourdet, D.L. Biotransformation and bioactivation reactions of alicyclic amines in drug molecules. Drug Metab. Rev. 2014, 46, 379–419. [Google Scholar] [CrossRef]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Bricogne, G.; Blanc, E.; Brandl, M.; Flensburg, C.; Keller, P.; Paciorek, W.; Roversi, P.; Sharff, A.; Smart, O.S.; Vonrhein, C.; et al. Buster; Global Phasing Ltd.: Cambridge, UK, 2011. [Google Scholar]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Number | R1 | R2 | R3 | R4 | R5 | hNNMT IC50 [µM] | mNNMT IC50 [µM] | HLM/RLM/MLM % Remaining at 1 h |
|---|---|---|---|---|---|---|---|---|
| 1 |  | OH | Me | H | H | 0.26 | 0.84 | NA |
| 6 |  | H | H | H | H | 1.67 | 2.2 | NA |
| 7 |  | OH | Me | F | H | 0.30 | 0.44 | 30/<5/<5 |
| 8 |  | OH | Me | F | H | >10 | NA | <5/<5/<5 |
| 9 |  | OH | H | F | H | 0.88 | 2.43 | NA |
| 10 |  | OH | Me | F | F | 0.29 | >3 | 7/NA/<5 |
| 11 |  | OH | Me | F | H | 0.2 | 1.87 | <5/NA/<5 |
| 12 |  | OH | Me | H | OMe | 0.008 | 0.0008 | NA |
| 13 |  | OH | Me | F | H | >3 | NA | 73/22/22 |
| Compound Number | R1 | R2 | R3 | R4 | R5 | hNNMT IC50 [µM] | mNNMT IC50 [µM] | HLM/RLM/MLM % Remaining at 1 h |
|---|---|---|---|---|---|---|---|---|
| 14 |  | OH | Me | F | H | 0.49 | 3.48 | <5/NA/<5 |
| 15 |  | OH | Me | F | H | 0.79 | 5.1 | <5/<5/<5 |
| 16 |  | OH | Me | F | H | 1.1 | 1.63 | <5/<5/<5 |
| 17 |  | OH | Me | F | H | >3 | NA | 27/<5/8 |
| 18 |  | OH | Me | F | H | >30 | NA | 35/<5/30 |
| 19 |  | OH | Me | F | H | >10 | NA | NA |
| 20 |  | OH | Me | F | H | >3 | NA | 18/NA/<5 |
| 21 |  | OH | Me | F | H | 0.25 | 1.64 | 20/<5/<5 |
| 22 |  | OH | Me | F | H | >30 | NA | 16/31/<5 |
| 23 |  | OH | Me | F | H | 0.15 | 1.0 | <5/NA/<5 |
| 24 |  | OH | Me | F | H | >10 | NA | <5/NA/<5 |
| Compound Number | Structure | hNNMT IC50 [uM] | mNNMT IC50 [uM] | HLM/MLM |
|---|---|---|---|---|
| 25 |  | 0.75 | >3 | 17/<5 |
| 26 |  | >10 | NA | 12/<5 |
| 27 |  | >30 | NA | NA |
| 28 |  | >3 | >30 | NA |
| 29 |  | >30 | NA | <5/<5 |
| 30 |  | >10 | NA | <5/<5 |
| 31 |  | 0.11 | 0.56 | 23/<5 |
| 32 |  | >30 | NA | 72/32 |
| Compound | hNNMT IC50 [µM] | mNNMT IC50 [µM] |
|---|---|---|
| (2) | 1.3 | 2.3 |
| (33) | 2.5 | 3.0 |
| Compound | hNNMT IC50 [µM] | mNNMT IC50 [µM] |
|---|---|---|
| (3) | 0.2 | 0.5 |
| (4) | 0.07 | 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kannt, A.; Rajagopal, S.; Hallur, M.S.; Swamy, I.; Kristam, R.; Dhakshinamoorthy, S.; Czech, J.; Zech, G.; Schreuder, H.; Ruf, S. Novel Inhibitors of Nicotinamide-N-Methyltransferase for the Treatment of Metabolic Disorders. Molecules 2021, 26, 991. https://doi.org/10.3390/molecules26040991
Kannt A, Rajagopal S, Hallur MS, Swamy I, Kristam R, Dhakshinamoorthy S, Czech J, Zech G, Schreuder H, Ruf S. Novel Inhibitors of Nicotinamide-N-Methyltransferase for the Treatment of Metabolic Disorders. Molecules. 2021; 26(4):991. https://doi.org/10.3390/molecules26040991
Chicago/Turabian StyleKannt, Aimo, Sridharan Rajagopal, Mahanandeesha S. Hallur, Indu Swamy, Rajendra Kristam, Saravanakumar Dhakshinamoorthy, Joerg Czech, Gernot Zech, Herman Schreuder, and Sven Ruf. 2021. "Novel Inhibitors of Nicotinamide-N-Methyltransferase for the Treatment of Metabolic Disorders" Molecules 26, no. 4: 991. https://doi.org/10.3390/molecules26040991
APA StyleKannt, A., Rajagopal, S., Hallur, M. S., Swamy, I., Kristam, R., Dhakshinamoorthy, S., Czech, J., Zech, G., Schreuder, H., & Ruf, S. (2021). Novel Inhibitors of Nicotinamide-N-Methyltransferase for the Treatment of Metabolic Disorders. Molecules, 26(4), 991. https://doi.org/10.3390/molecules26040991