
The Role of the Inflammasome in Neurodegenerative Diseases





Abstract
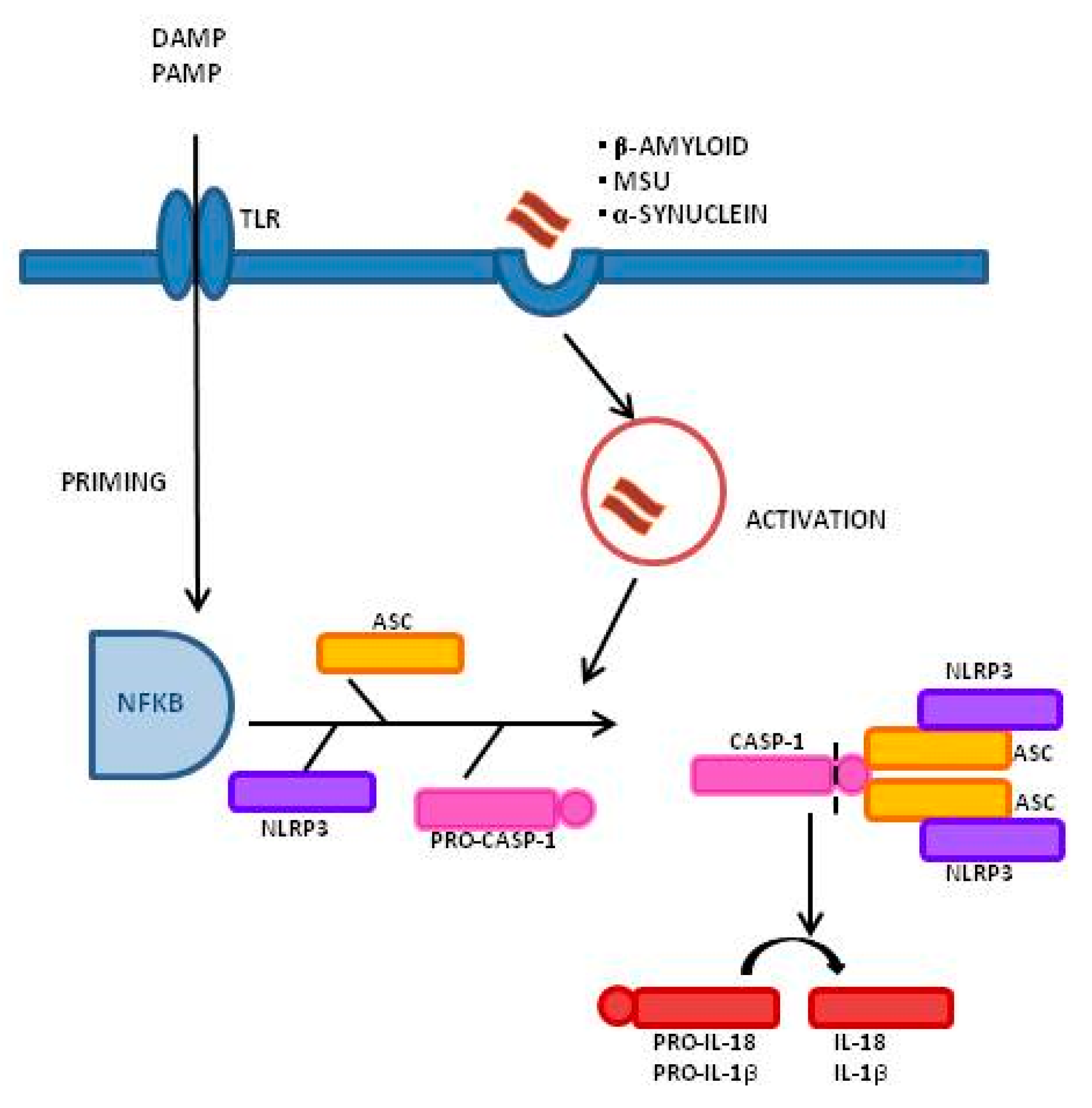
1. Introduction
2. Alzheimer’s Disease
3. Multiple Sclerosis
4. Parkinson’s Disease
5. Amyotrophic Lateral Sclerosis
6. Pharmacological Modulation of the Inflammasome
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schain, M.; Kreisl, W.C. Neuroinflammation in neurodegenerative disorders-a review. Curr. Neurol. Neurosci. Rep. 2017, 17, 25. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases. Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cheng, X.; Zhong, S.; Liu, C.; Liu, F.; Zhao, C. Peripheral and Central Nervous System Immune Response Crosstalk in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 575. [Google Scholar] [CrossRef]
- Ciccocioppo, F.; Bologna, G.; Ercolino, E.; Pierdomenico, L.; Simeone, P.; Lanuti, P.; Pieragostino, D.; Del Boccio, P.; Marchisio, M.; Miscia, S. Neurodegenerative diseases as proteinopathies-driven immune disorders. Neural. Regen. Res. 2020, 15, 850–856. [Google Scholar] [CrossRef]
- Bayer, T.A. Proteinopathies, a core concept for understanding and ultimately treating degenerative disorders? Eur. Neuropsychopharmacol. 2015, 25, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Sami, N.; Rahman, S.; Kumar, V.; Zaidi, S.; Islam, A.; Ali, S.; Ahmad, F.; Hassan, M.I. Protein aggregation, misfolding and consequential human neurodegenerative diseases. Int. J. Neurosci. 2017, 127, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Ann. Rev. Cell. Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Z.; Xiao, T.S. Post-translational regulation of inflammasomes. Cell. Mol. Immunol. 2017, 14, 65–79. [Google Scholar] [CrossRef]
- Shi, J.Q.; Zhang, C.C.; Sun, X.L.; Cheng, X.X.; Wang, J.B.; Zhang, Y.D.; Xu, J.; Zou, H.Q. Antimalarial drug artemisinin extenuates amyloidogenesis and neuroinflammation in APPswe/PS1dE9 transgenic mice via inhibition of nuclear factor-kappaB and NLRP3 inflammasome activation. CNS. Neurosci. Ther. 2013, 19, 262–268. [Google Scholar] [CrossRef]
- Allan, S.M. Pragmatic target discovery from novel gene to functionally defined drug target: The interleukin-1 story. Methods Mol. Med. 2005, 104, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Alboni, S.; Cervia, D.; Sugama, S.; Conti, B. Interleukin 18 in the CNS. J. Neuroinflamm. 2010, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Awad, F.; Assrawi, E.; Jumeau, C.; Georgin-Lavialle, S.; Cobret, L.; Duquesnoy, P.; Piterboth, W.; Thomas, L.; Stankovic-Stojanovic, K.; Louvrier, C.; et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS ONE 2017, 12, e0175336. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, F.; Saresella, M.; Marventano, I.; Piancone, F.; Ripamonti, E.; Al-Daghri, N.; Bazzini, C.; Zoia, C.P.; Conti, E.; Ferrarese, C.; et al. Stavudine Reduces NLRP3 Inflammasome Activation and Modulates Amyloid-β Autophagy. J. Alzheimers Dis. 2019, 72, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 1–14. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Heneka, M.T. Inflammasome activation and innate immunity in Alzheimer’s disease. Brain Pathol. 2017, 27, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, B.; Sonobe, Y.; Horiuchi, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Oligomeric amyloid β induces IL-1β processing via production of ROS: Implication in Alzheimer’s disease. Cell Death Dis. 2013, 4, e975. [Google Scholar] [CrossRef] [PubMed]
- Pontillo, A.; Catamo, E.; Arosio, B.; Mari, D.; Crovella, S. NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2012, 26, 277–281. [Google Scholar] [CrossRef]
- Tan, M.S.; Tan, L.; Jiang, T.; Zhu, X.C.; Wang, H.F.; Jia, C.D.; Yu, J.T. Amyloid-beta induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1382. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.H.; LeBlanc, A.C. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686. [Google Scholar] [CrossRef]
- Rezai-Zadeh, K.; Gate, D.; Gowing, G.; Town, T. How to get from here to there: Macrophage recruitment in Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 156–163. [Google Scholar] [CrossRef]
- Nascimento, C.M.; Pereira, J.R.; de Andrade, L.P.; Garuffi, M.; Talib, L.L.; Forlenza, O.V.; Cancela, J.M.; Cominetti, M.R.; Stella, F. Physical exercise in MCI elderly promotes reduction of pro-inflammatory cytokines and improvements on cognition and BDNF peripheral levels. Curr. Alzheimer Res. 2014, 11, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Dionisio-Santos, D.A.; Olschowka, J.A.; O’Banion, M.K. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 74. [Google Scholar] [CrossRef]
- Town, T.; Tan, J.; Mullan, M. CD40 signaling and Alzheimer’s disease pathogenesis. Neurochem. Int. 2001, 39, 371–380. [Google Scholar] [CrossRef]
- Town, T.; Nikolic, V.; Tan, J. The microglial “activation” continuum: From innate to adaptive responses. J. Neuroinflamm. 2005, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Town, T.; Laouar, Y.; Pittenger, C.; Mori, T.; Szekely, C.A.; Tan, J.; Duman, R.S.; Flavell, R.A. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 2008, 14, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; McLaurin, J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.S.; Yu, J.T.; Jiang, T.; Zhu, X.C.; Tan, L. The NLRP3 inflammasome in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 875–882. [Google Scholar] [CrossRef]
- Townsend, K.P.; Town, T.; Mori, T.; Lue, L.F.; Shytle, D.; Sanberg, P.R.; Morgan, D.; Fernandez, F.; Flavell, R.A.; Tan, J. CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur. J. Immunol. 2005, 35, 901–910. [Google Scholar] [CrossRef]
- Feng, Y.; Li, L.; Sun, X.H. Monocytes and Alzheimer’s disease. Neurosci. Bull. 2011, 2, 115–122. [Google Scholar] [CrossRef]
- Fiala, M.; Lin, J.; Ringman, J.; Kermani-Arab, V.; Tsao, G.; Patel, A.; Lossinsky, A.S.; Graves, M.C.; Gustavson, A.; Sayre, J.; et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J. Alzheimers Dis. 2005, 3, 221–232. [Google Scholar] [CrossRef]
- Simard, A.R.; Rivest, S. Neuroprotective properties of the innate immune system and bone marrow stem cells in Alzheimer’s disease. Mol. Psychiatry 2006, 11, 327–335. [Google Scholar] [CrossRef]
- Saresella, M.; Marventano, I.; Calabrese, E.; Piancone, F.; Rainone, V.; Gatti, A.; Alberoni, M.; Nemni, R.; Clerici, M. A complex proinflammatory role for peripheral monocytes in Alzheimer’s disease. J. Alzheimers Dis. 2014, 38, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging. 2017, 49, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Basilico, N.; Marventano, I.; Perego, F.; La Rosa, F.; Piancone, F.; Taramelli, D.; Banks, H.; Clerici, M. Leishmania infantum infection reduces the amyloid β42-stimulated NLRP3 inflammasome activation. Brain Behav. Immun. 2020, 88, 597–605. [Google Scholar] [CrossRef]
- Trumble, B.C.; Stieglitz, J.; Blackwell, A.D.; Allayee, H.; Beheim, B.; Finch, C.E.; Gurven, M.; Kaplan, H. Apolipoprotein E4 is associated with improved cognitive function in Amazonian forager-horticulturalists with a high parasite burden. FASEB J. 2017, 31, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.W. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef] [PubMed]
- Gris, D.; Ye, Z.; Iocca, H.A.; Wen, H.; Craven, R.R.; Gris, P.; Huang, M.; Schneider, M.; Miller, S.D.; Ting, J.P. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J. Immunol. 2010, 185, 974–981. [Google Scholar] [CrossRef]
- Ransohoff, R.M. Animal models of multiple sclerosis: The good, the bad and the bottom line. Nat. Neurosci. 2012, 15, 1074–1077. [Google Scholar] [CrossRef]
- Peelen, E.; Damoiseaux, J.; Muris, A.H.; Knippenberg, S.; Smolders, J.; Hupperts, R.; Thewissen, M. Increased inflammasome related gene expression profile in PBMC may facilitate T helper 17 cell induction in multiple sclerosis. Mol. Immunol. 2015, 63, 521–529. [Google Scholar] [CrossRef]
- Inoue, M.; Shinohara, M.L. NLRP3 Inflammasome and MS/EAE. Autoimmune Dis. 2013, 2013, 859145. [Google Scholar] [CrossRef]
- Mamik, M.K.; Power, C. Inflammasomes in neurological diseases: Emerging pathogenic and therapeutic concepts. Brain 2017, 140, 2273–2285. [Google Scholar] [CrossRef]
- Furlan, R.; Filippi, M.; Bergami, A.; Rocca, M.A.; Martinelli, V.; Poliani, P.L.; Grimaldi, L.M.; Desina, G.; Comi, G.; Martino, G. Peripheral levels of caspase-1 mRNA correlate with disease activity in patients with multiple sclerosis; a preliminary study. J. Neurol. Neurosurg. Psychiatry 1999, 67, 785–788. [Google Scholar] [CrossRef]
- Ming, X.; Li, W.; Maeda, Y.; Blumberg, B.; Raval, S.; Cook, S.D.; Dowling, P.C. Caspase-1 expression in multiple sclerosis plaques and cultured glial cells. J. Neurol. Sci. 2002, 197, 9–18. [Google Scholar] [CrossRef]
- Keane, R.W.; Dietrich, W.D.; de Rivero Vaccari, J.P. Inflammasome Proteins As Biomarkers of Multiple Sclerosis. Front. Neurol. 2018, 9, 135. [Google Scholar] [CrossRef]
- Losy, J.; Niezgoda, A. IL-18 in patients with multiple sclerosis. Acta Neurol. Scand. 2001, 104, 171–173. [Google Scholar] [CrossRef]
- Nicoletti, F.; Di Marco, R.; Mangano, K.; Patti, F.; Reggio, E.; Nicoletti, A.; Bendtzen, K.; Reggio, A. Increased serum levels of interleukin-18 in patients with multiple sclerosis. Neurology 2001, 57, 342–344. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chen, S.D.; Miao, L.; Liu, Z.G.; Li, W.; Zhao, Z.X.; Sun, X.J.; Jiang, G.X.; Cheng, Q. Serum levels of interleukin (IL)-18, IL-23 and IL-17 in Chinese patients with multiple sclerosis. J. Neuroimmunol. 2012, 243, 56–60. [Google Scholar] [CrossRef] [PubMed]
- De Jong, B.A.; Huizinga, T.W.; Bollen, E.L.; Uitdehaag, B.M.; Bosma, G.P.; van Buchem, M.A.; Remarque, E.J.; Burgmans, A.C.; Kalkers, N.F.; Polman, C.H.; et al. Production of IL-1beta and IL-1Ra as risk factors for susceptibility and progression of relapse-onset multiple sclerosis. J. Neuroimmunol. 2002, 126, 172–179. [Google Scholar] [CrossRef]
- Burger, D.; Molnarfi, N.; Weber, M.S.; Brandt, K.J.; Benkhoucha, M.; Gruaz, L.; Chofflon, M.; Zamvil, S.S.; Lalive, P.H. Glatiramer acetate increases IL-1 receptor antagonist but decreases T cell-induced IL-1β in human monocytes and multiple sclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 4355–4359. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Patti, F.; DiMarco, R.; Zaccone, P.; Nicoletti, A.; Meroni, P.; Reggio, A. Circulating serum levels of IL-1ra in patients with relapsing remitting multiple sclerosis are normal during remission phases but significantly increased either during exacerbations or in response to IFN-β treatment. Cytokine 1996, 8, 395–400. [Google Scholar] [CrossRef]
- Martin, B.N.; Wang, C.; Zhang, C.J.; Kang, Z.; Gulen, M.F.; Zepp, J.A.; Zhao, J.; Bian, G.; Do, J.S.; Min, B.; et al. T cell-intrinsic ASC critically promotes T(H)17-mediated experimental autoimmune encephalomyelitis. Nat. Immunol. 2016, 17, 583–592. [Google Scholar] [CrossRef]
- Pierini, R.; Perret, M.; Djebali, S.; Juruj, C.; Michallet, M.C.; Förster, I.; Marvel, J.; Walzer, T.; Henry, T. ASC controls IFN-γ levels in an IL-18-dependent manner in caspase-1-deficient mice infected with Francisella novicida. J. Immunol. 2013, 191, 3847–3857. [Google Scholar] [CrossRef]
- Amorini, A.M.; Petzold, A.; Tavazzi, B.; Eikelenboom, J.; Keir, G.; Belli, A.; Giovannoni, G.; Di Pietro, V.; Polman, C.; D’Urso, S.; et al. Increase of uric acid and purine compounds in biological fluids of multiple sclerosis patients. Clin. Biochem. 2009, 42, 1001–1006. [Google Scholar] [CrossRef]
- Liu, B.; Shen, Y.; Xiao, K.; Tang, Y.; Cen, L.; Wei, J. Serum uric acid levels in patients with multiple sclerosis: A meta-analysis. Neurol. Res. 2012, 34, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Piancone, F.; Saresella, M.; Marventano, I.; La Rosa, F.; Santangelo, M.A.; Caputo, D.; Mendozzi, L.; Rovaris, M.; Clerici, M. Monosodium Urate Crystals Activate the Inflammasome in Primary Progressive Multiple Sclerosis. Front. Immunol. 2018, 9, 983. [Google Scholar] [CrossRef] [PubMed]
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Matute, C. Interaction between glutamate signalling and immune attack in damaging oligodendrocytes. Neuron. Glia. Biol. 2007, 3, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Oyanguren-Desez, O.; Rodriguez-Antiguedad, A.; Villoslada, P.; Domercq, M.; Alberdi, E.; Matute, C. Gain-of-function of P2X7 receptor gene variants in multiple sclerosis. Cell Calcium. 2011, 50, 468–472. [Google Scholar] [CrossRef]
- Caragnano, M.; Tortorella, P.; Bergami, A.; Ruggieri, M.; Livrea, P.; Specchio, L.M.; Martino, G.; Trojano, M.; Furlan, R.; Avolio, C. Monocytes P2X7 purinergic receptor is modulated byglatiramer acetate in multiple sclerosis. J. Neuroimmunol. 2012, 245, 93–97. [Google Scholar] [CrossRef]
- Petrucci, S.; Consoli, F.; Valente, E.M. Parkinson Disease Genetics: A “Continuum” from Mendelian to Multifactorial Inheritance. Curr. Mol. Med. 2014, 14, 1079–1088. [Google Scholar] [CrossRef]
- Lee, S.J. Origins and effects of extracellular alpha-synuclein: Implications in Parkinson’s disease. J. Mol. Neurosci. 2008, 34, 17–22. [Google Scholar] [CrossRef]
- Beraud, D.; Maguire-Zeiss, K.A. Misfolded alpha-synuclein and Toll-like receptors: Therapeutic targets for Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. 1), S17–S20. [Google Scholar] [CrossRef]
- Harms, A.S.; Delic, V.; Thome, A.D.; Bryant, N.; Liu, Z.; Chandra, S.; Jurkuvenaite, A.; West, A.B. α-Synuclein fibrils recruit peripheral immune cells in the rat brain prior to neurodegeneration. Acta Neuropathol. Commun. 2017, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef]
- Lee, E.; Hwang, I.; Park, S.; Hong, S.; Hwang, B.; Cho, Y.; Son, J.; Yu, J.W. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019, 26, 213–228. [Google Scholar] [CrossRef]
- Daniels, M.J.D.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Shao, X.Y.; Qi, G.J.; Chen, Q.; Bu, L.L.; Chen, L.J.; Shi, J.; Ming, J.; Tian, B. Cdk5-Dependent Activation of Neuronal Inflammasomes in Parkinson’s Disease. Mov. Disord. 2016, 31, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Pan, Y.T.; Zhang, Z.Y.; Yang, H.; Yu, S.Y.; Zheng, Y.; Ma, J.H.; Wang, X.M. Systemic activation of NLRP3 inflammasome and plasma α-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J. Neuroinflamm. 2020, 17, 11. [Google Scholar] [CrossRef]
- McCombe, P.A.; Henderson, R.D. The Role of immune and inflammatory mechanisms in ALS. Curr. Mol. Med. 2011, 11, 246–254. [Google Scholar] [CrossRef]
- Lall, D.; Baloh, R.H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Investig. 2017, 127, 3250–3258. [Google Scholar] [CrossRef]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Mutant superoxide dismutase 1-induced IL-1β accelerates ALS pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 13046–13050. [Google Scholar] [CrossRef]
- Debye, B.; Schmülling, L.; Zhou, L.; Rune, G.; Beyer, C.; Johann, S. Neurodegeneration and NLRP3 inflammasome expression in the anterior thalamus of SOD1(G93A) ALS mice. Brain Pathol. 2018, 28, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Johann, S.; Heitzer, M.; Kanagaratnam, M.; Goswami, A.; Rizo, T.; Weis, J.; Troost, D.; Beyer, C. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 2015, 63, 2260–2273. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Carlesi, C.; Giungato, P.; Puxeddu, I.; Borroni, B.; Bossu, P.; Migliorini, P.; Siciliano, G.; Boraschi, D. Evaluating the levels of interleukin-1 family cytokines in sporadic amyotrophic lateral sclerosis. J. Neuroinflamm. 2014, 11, 94. [Google Scholar] [CrossRef]
- Kadhim, H.; Deltenre, P.; Martin, J.J.; Sebire, G. In-situ expression of Interleukin-18 and associated mediators in the human brain of sALS patients: Hypothesis for a role for immune-inflammatory mechanisms. Med. Hypotheses 2016, 86, 14–17. [Google Scholar] [CrossRef]
- Maier, N.K.; Leppla, S.H.; Moayeri, M. The cyclopentenone prostaglandin 15d-PGJ2 inhibits the NLRP1 and NLRP3 inflammasomes. J. Immunol. 2015, 194, 2776–2785. [Google Scholar] [CrossRef]
- Heitzer, M.; Kaiser, S.; Kanagaratnam, M.; Zendedel, A.; Hartmann, P.; Beyer, C.; Johann, S. Administration of 17beta-Estradiol Improves Motoneuron Survival and Down-regulates Inflammasome Activation in Male SOD1 (G93A) ALS Mice. Mol. Neurobiol. 2017, 54, 8429–8443. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; van de Veerdonk, F.L.; van der Meer, J.W.; Dinarello, C.A.; Joosten, L.A. Inflammasome-independent regulation of IL-1-family cytokines. Annu. Rev. Immunol. 2015, 33, 49–77. [Google Scholar] [CrossRef]
- Duan, Y.; Kelley, N.; He, Y. Role of the NLRP3 inflammasome in neurodegenerative diseases and therapeutic implications. Neural Regen Res. 2020, 15, 1249–1250. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef]
- Ahn, H.; Kim, J.; Jeung, E.B.; Lee, G.S. Dimethyl sulfoxide inhibits NLRP3 inflammasome activation. Immunobiology 2014, 219, 315–322. [Google Scholar] [CrossRef]
- Inoue, M.; Williams, K.L.; Oliver, T.; Vandenabeele, P.; Rajan, J.V.; Miao, E.A.; Shinohara, M.L. Interferon-β therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci. Signal. 2012, 5, ra38. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [PubMed]
- Fowler, B.J.; Gelfand, B.D.; Kim, Y.; Kerur, N.; Tarallo, V.; Hiran, Y.; Amarnath, S.; Fowler, D.H.; Radwan, M.; Young, M.T. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014, 346, 1000–1003. [Google Scholar] [CrossRef]


{kind=link}
| Disease | System | Inflammasome Component/Effector | References |
|---|---|---|---|
| Alzheimer’s | Human | ↑ IL-1β, ↑ IL-18 ↑ NLRP1, ↑ ASC, ↑ caspase-1 ↑ NLRP3, ↑ IL-1β | Awad et al., 2017 [16] Heneka et al., 2013 [17] La Rosa et al., 2017 [18] Saresella et al., 2017 [19] Kaushal et al., 2015 [26] Cattaneo et al., 2016 [40] |
| Multiple Sclerosis | Human | ↑ NLRP3, ↑ IL-1β, ↑caspase-1 ↑ caspase-1, ↑ IL-1β ↑ caspase-1 ↑ IL-18 ↑ IL-1β ↑ NLRP3, ↑ ASC, ↑ IL-18, ↑ caspase-8 | Peleen et al., 2015 [46] Inoue & Shinohara, 2013 [47] Mamik&Power, 2017 [48] Furlan R et al., 1999 [49] Ming X et al., 2002 [50] Losy J et al., 2001 [52] Nicoletti F et al., 2001 [53] Chen YC et al., 2012 [54] Gris D et al., 2010 [44] de Jong et al., 2002 [55] Piancone et al., 2018 [62] |
| Parkinson’s Disease | Human | ↑ IL-1β ↑ IL-1β, ↑ IL-18 ↑ IL-1β, ↑ caspase-1 ↑ NLRP3, ↑ IL-1β, ↑ caspase-1 | Codolo et al., 2013 [71] Zhang et al., 2016 [77] Zhou Y et al., 2015 [74] Fan et al., 2020 [78] |
| Amyotrophic Lateral Sclerosis | Human | ↑ IL-18 ↑ NLRP3, ↑ ASC, ↑ IL-18, ↑ caspase-1 ↑ NLRP3, ↑ caspase-1 | Italiani et al., 2014 [85] Johann et al., 2015 [84] Kadhim et al., 2016 [86] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 2021, 26, 953. https://doi.org/10.3390/molecules26040953
Piancone F, La Rosa F, Marventano I, Saresella M, Clerici M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules. 2021; 26(4):953. https://doi.org/10.3390/molecules26040953
Chicago/Turabian StylePiancone, Federica, Francesca La Rosa, Ivana Marventano, Marina Saresella, and Mario Clerici. 2021. "The Role of the Inflammasome in Neurodegenerative Diseases" Molecules 26, no. 4: 953. https://doi.org/10.3390/molecules26040953
APA StylePiancone, F., La Rosa, F., Marventano, I., Saresella, M., & Clerici, M. (2021). The Role of the Inflammasome in Neurodegenerative Diseases. Molecules, 26(4), 953. https://doi.org/10.3390/molecules26040953