
Supramolecular Hydrogels for Protein Delivery in Tissue Engineering


Abstract
1. Introduction
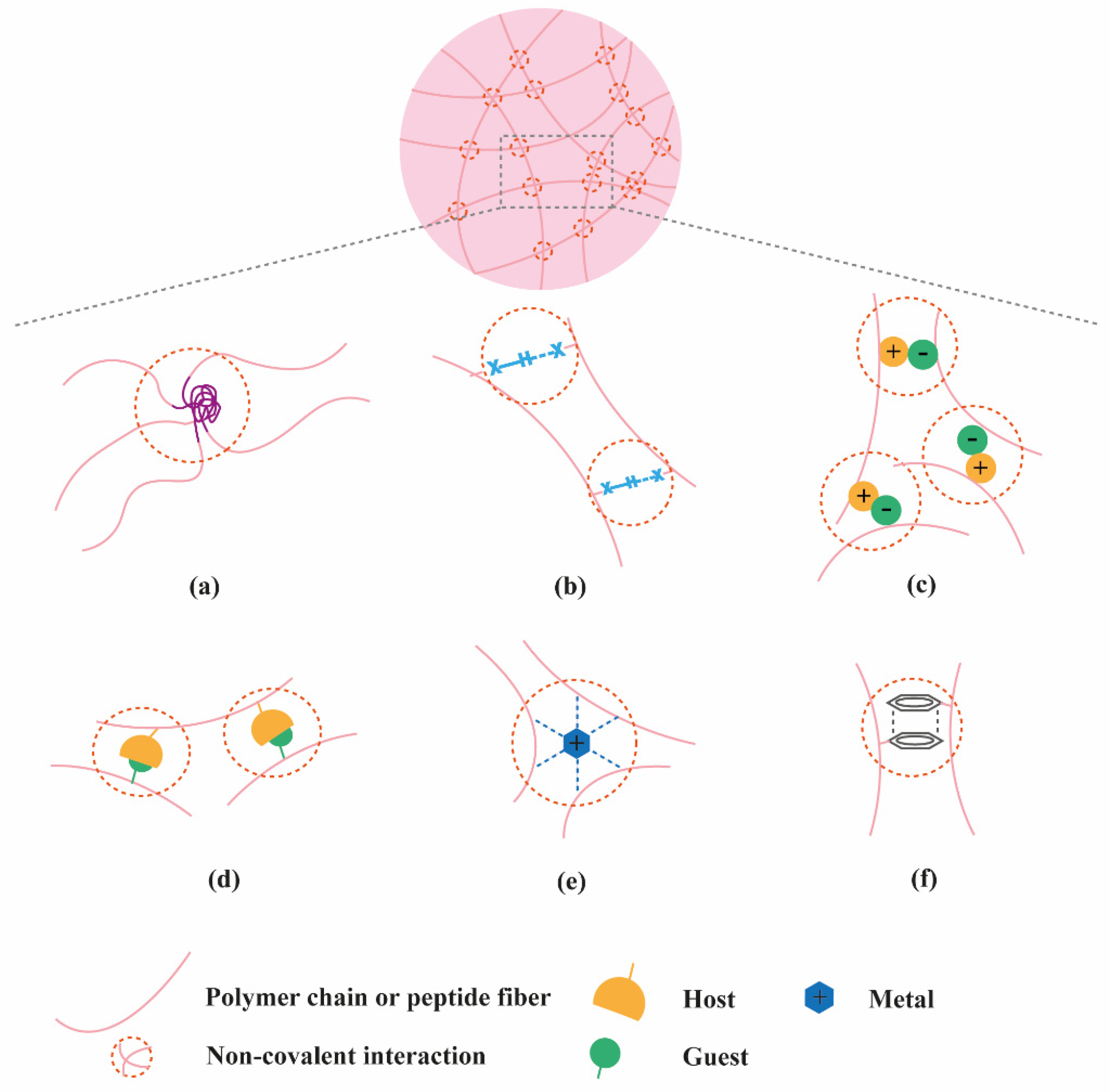
2. Classification of Supramolecular Hydrogels Based on Their Composition
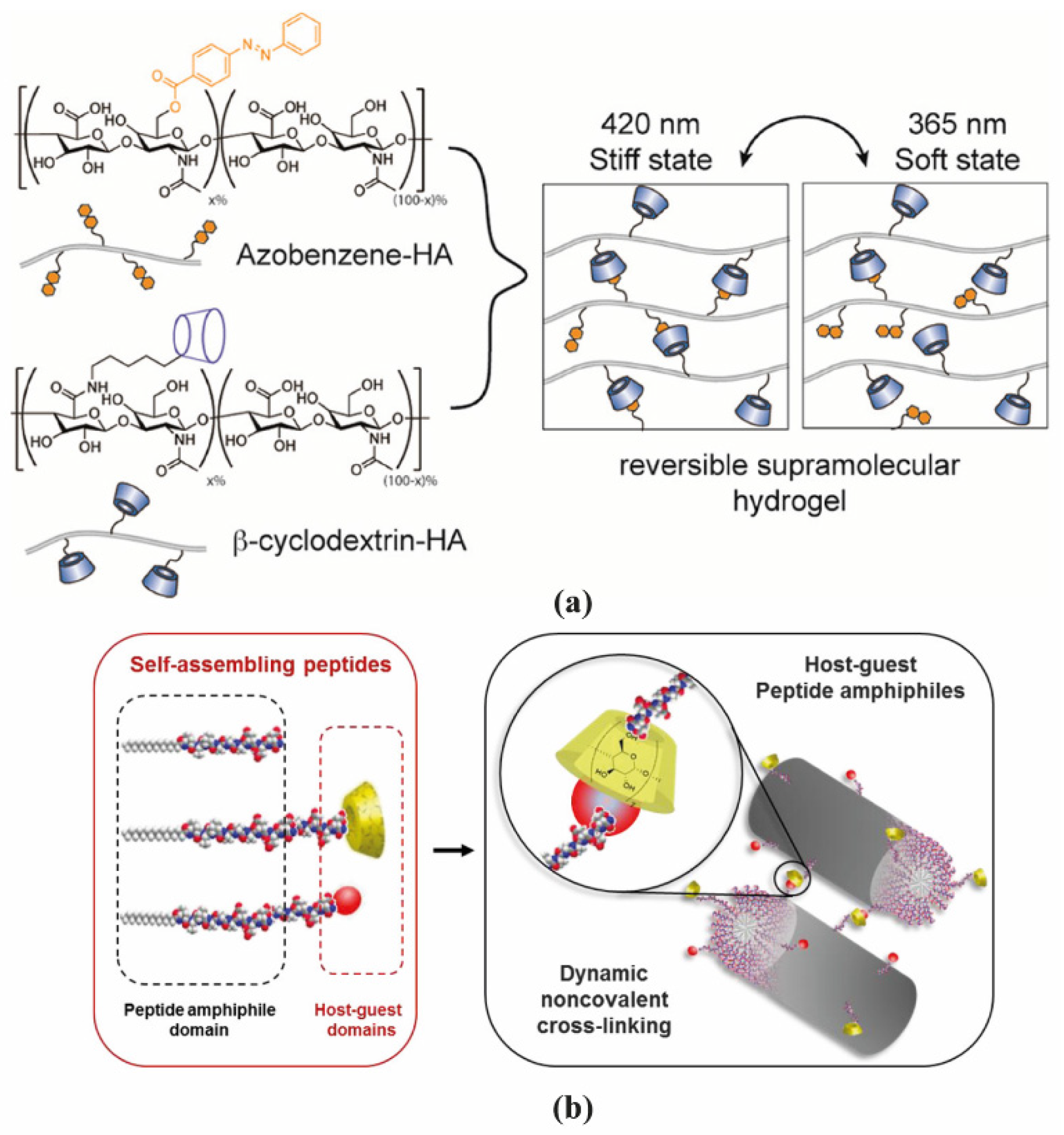
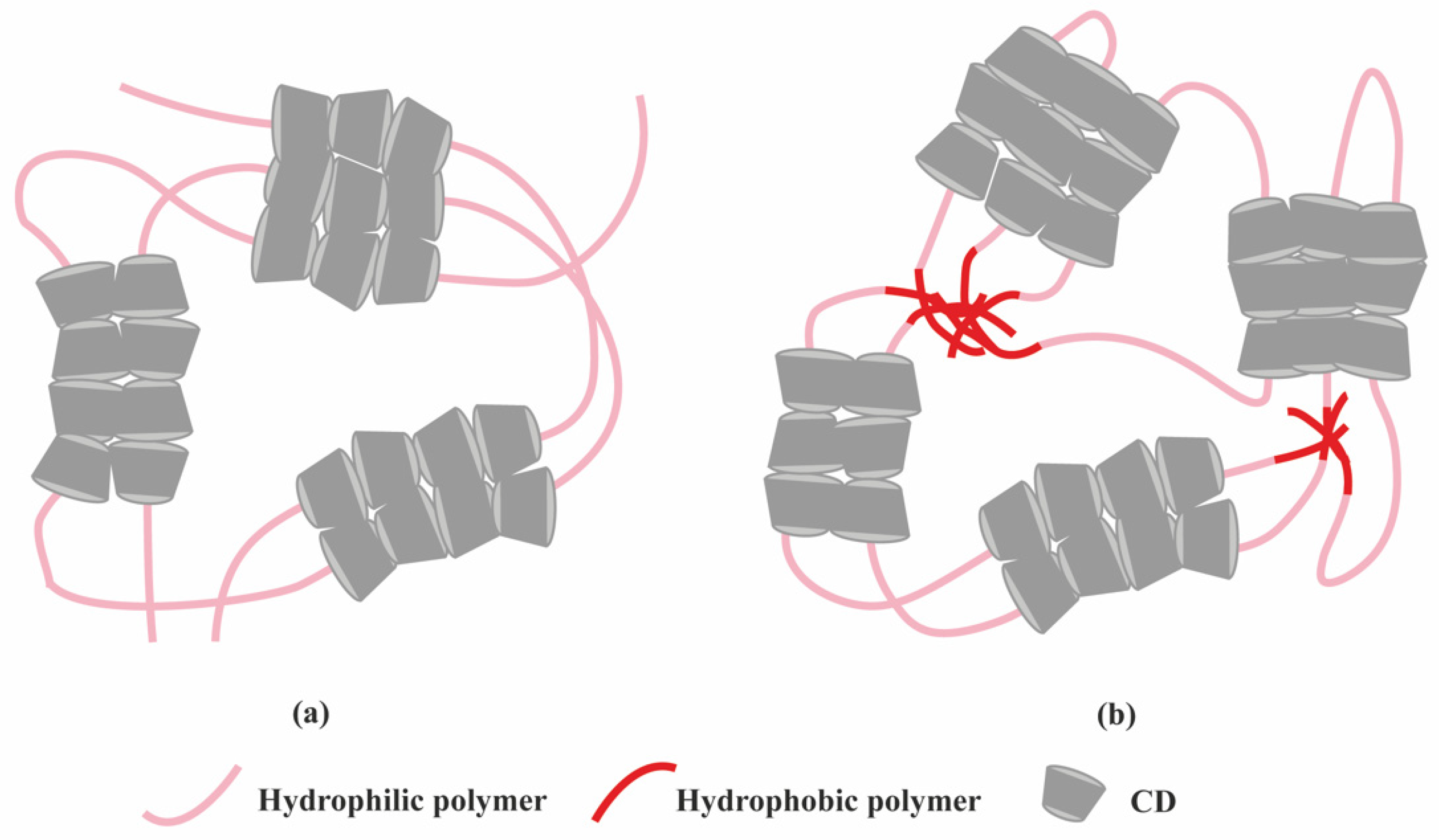
2.1. Polymer-Based Hydrogels
2.2. Peptide-Based Hydrogels
2.3. Nucleic Acid-Based Hydrogels
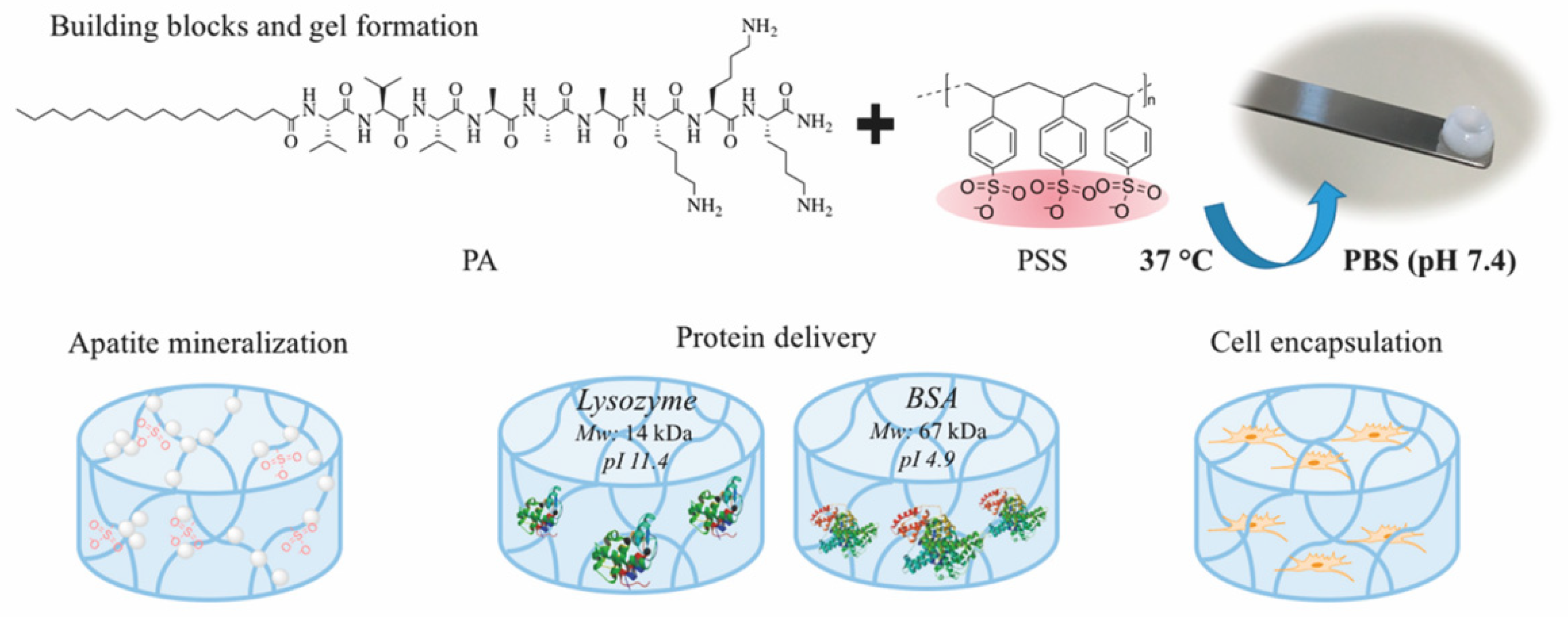
2.4. Multi-Component Hydrogels
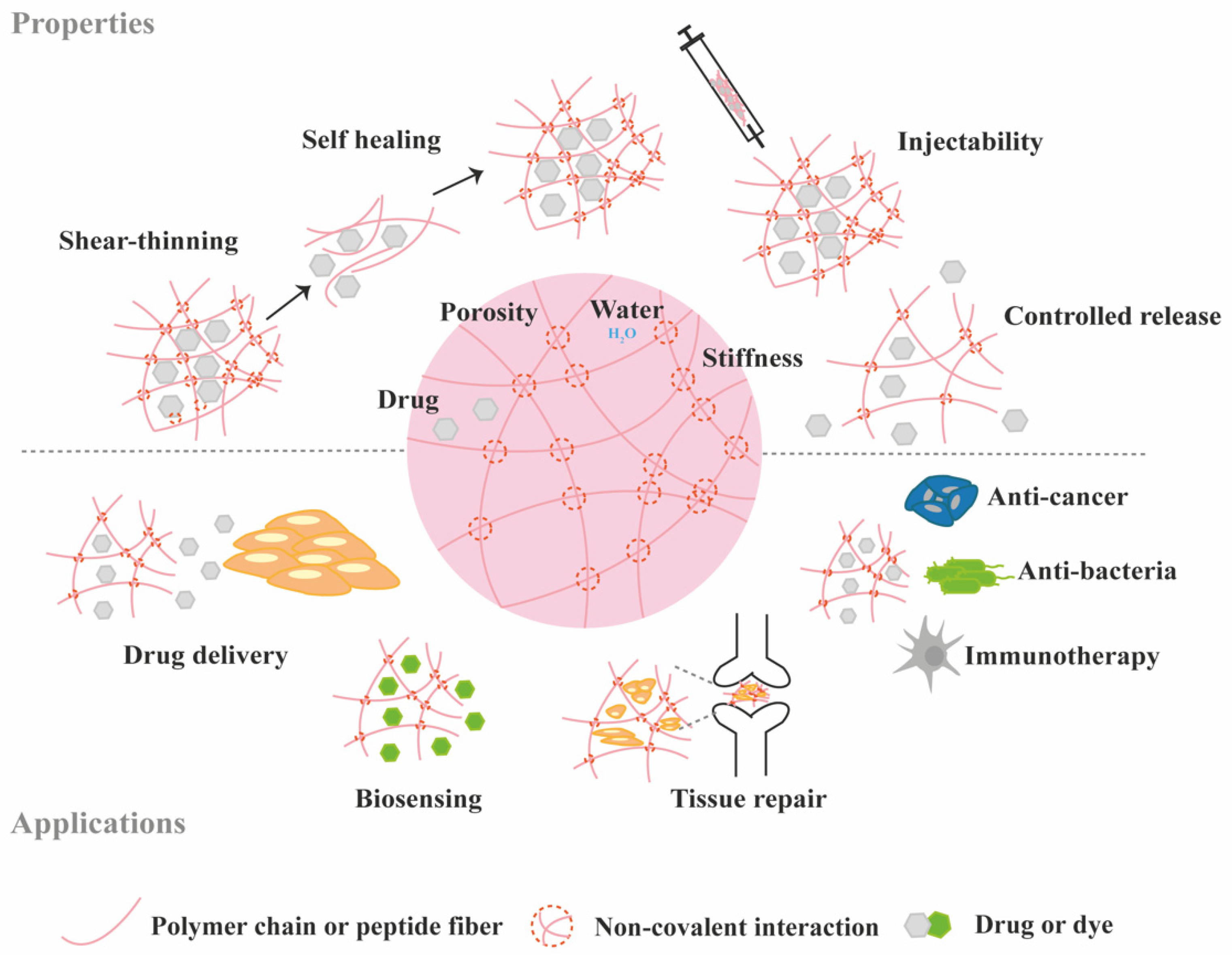
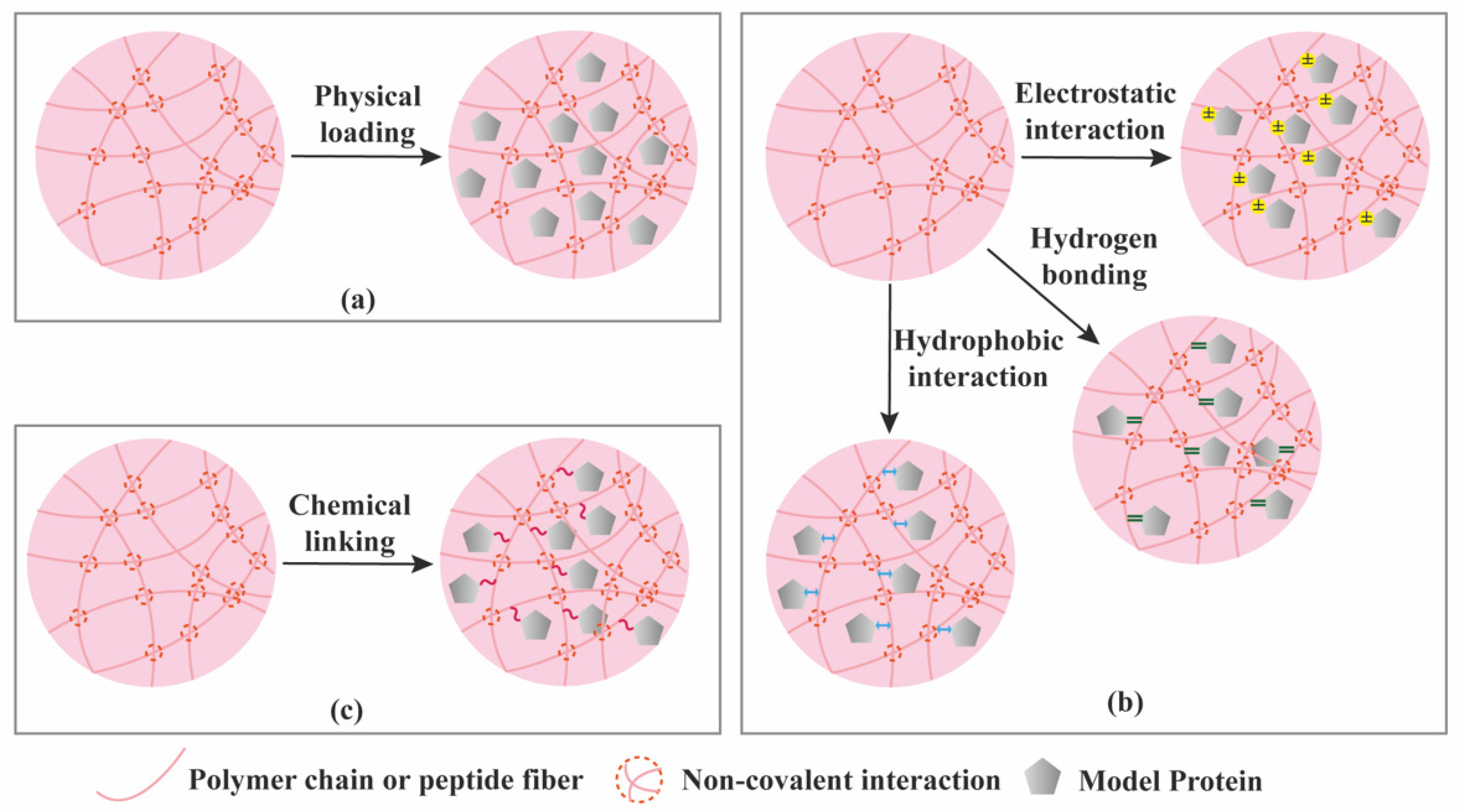
3. Protein Loading and Release from Supramolecular Hydrogels
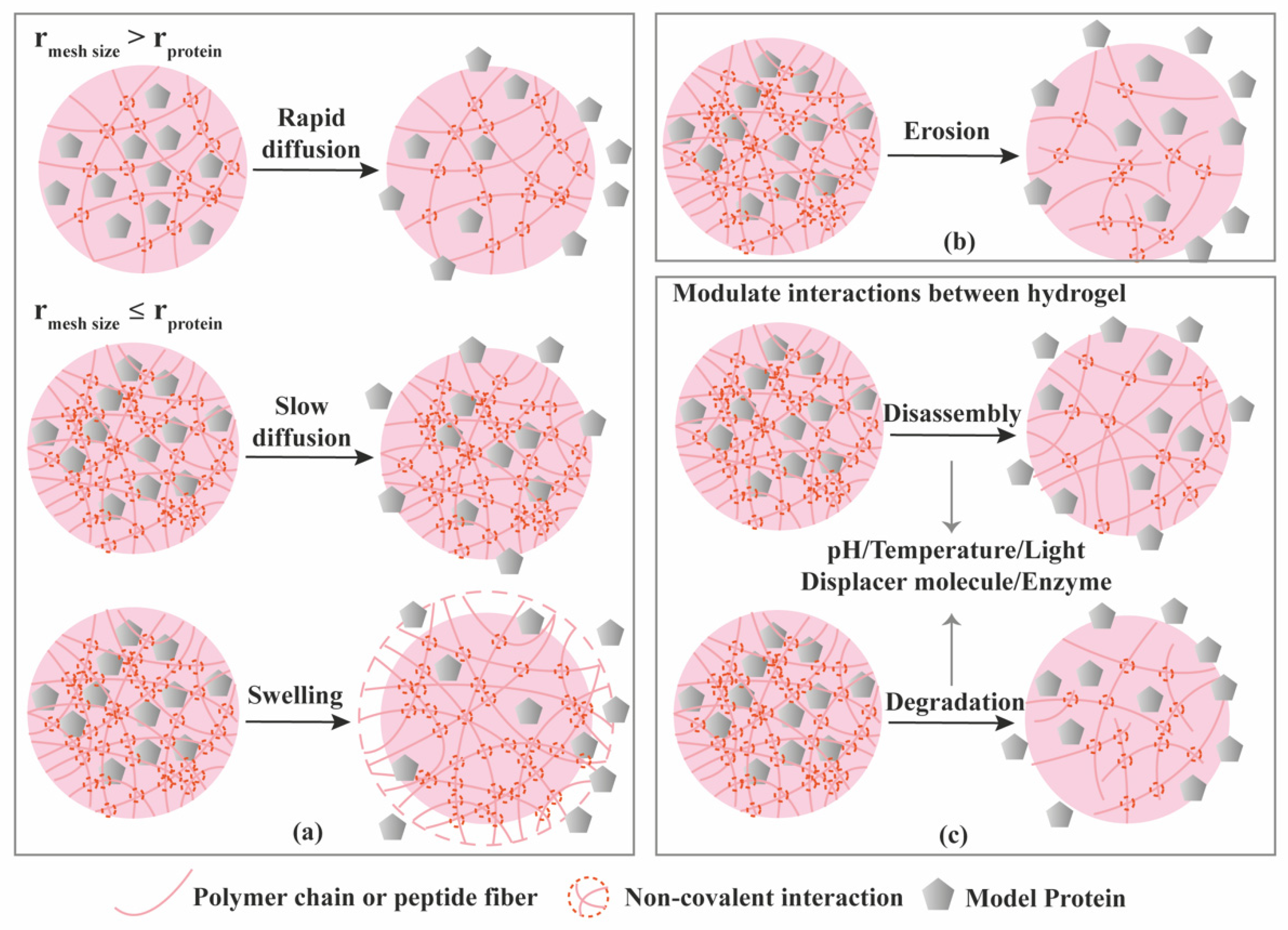
3.1. Diffusion-Controlled Release
3.2. Erosion-Controlled Release
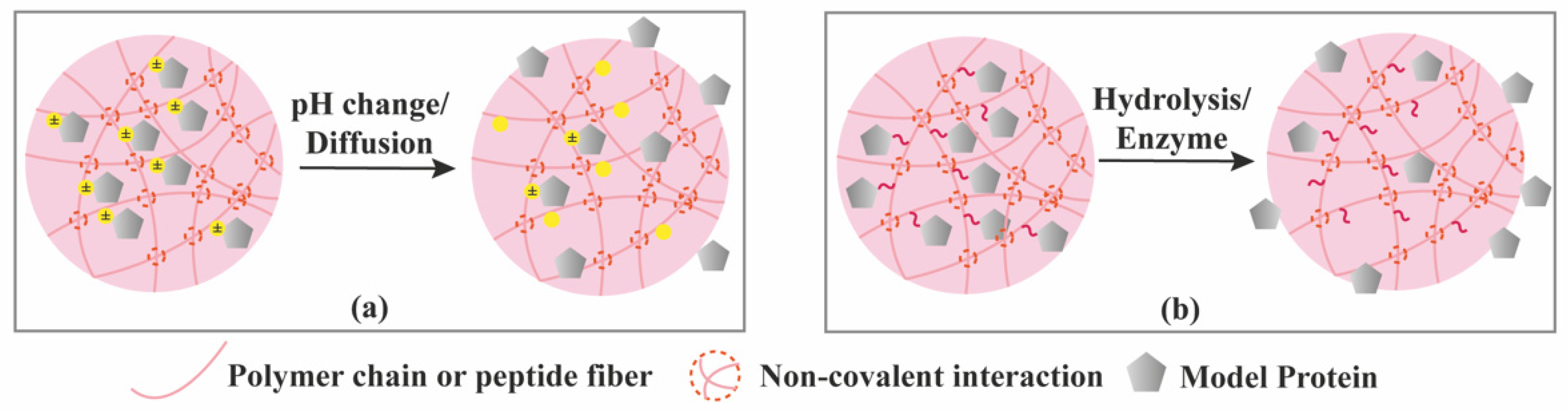
3.3. Stimuli-Controlled Release
3.4. Chemical Interactions-Mediated Release
4. Supramolecular Hydrogels for the Delivery of Bioactive Proteins for TE Applications
4.1. Vascular Tissues
4.2. Bone
4.3. Cartilage
4.4. Skin
4.5. Others
5. Challenges in the Design of Supramolecular Hydrogels
6. Clinical Challenges of Supramolecular Hydrogels
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Langer, R.; Vacanti, J.P. Tissue engineering. Science 1993, 260, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Caballero Aguilar, L.M.; Silva, S.M.; Moulton, S.E. Growth factor delivery: Defining the next generation platforms for tissue engineering. J. Control. Release Off. J. Control. Release Soc. 2019, 306, 40–58. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Silva, E.A.; Mooney, D.J. Growth factor delivery-based tissue engineering: General approaches and a review of recent developments. J. R. Soc. Interface 2011, 8, 153–170. [Google Scholar] [CrossRef]
- Rode, M.P.; Batti Angulski, A.B.; Gomes, F.A.; da Silva, M.M.; Jeremias, T.D.S.; de Carvalho, R.G.; Calloni, G.W. Carrageenan hydrogel as a scaffold for skin-derived multipotent stromal cells delivery. J. Biomater. Appl. 2018, 33, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Miranda, D.G.; Malmonge, S.M.; Campos, D.M.; Attik, N.G.; Grosgogeat, B.; Gritsch, K. A chitosan-hyaluronic acid hydrogel scaffold for periodontal tissue engineering. J. Biomed. Mater. Res. Part. B Appl. Biomater. 2016, 104, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Zhao, X.; Guo, B.; Ma, P.X. Self-Healing Conductive Injectable Hydrogels with Antibacterial Activity as Cell Delivery Carrier for Cardiac Cell Therapy. ACS Appl. Mater. Interfaces 2016, 8, 17138–17150. [Google Scholar] [CrossRef]
- Wang, H.; Heilshorn, S.C. Adaptable hydrogel networks with reversible linkages for tissue engineering. Adv. Mater. 2015, 27, 3717–3736. [Google Scholar] [CrossRef]
- Rosales, A.M.; Anseth, K.S. The design of reversible hydrogels to capture extracellular matrix dynamics. Nat. Rev. Mater. 2016, 1, 15012. [Google Scholar] [CrossRef]
- Chen, J.X.; Cao, L.J.; Shi, Y.; Wang, P.; Chen, J.H. In situ supramolecular hydrogel based on hyaluronic acid and dextran derivatives as cell scaffold. J. Biomed. Mater. Res. Part A 2016, 104, 2263–2270. [Google Scholar] [CrossRef]
- Dang, I.A.; Kousar, A.; Liu, J.; Mukwaya, V.; Zhao, C.; Wang, F.; Feng, C.L. Mechanically Stable C2-Phenylalanine Hybrid Hydrogels for Manipulating Cell Adhesion. ACS Appl. Mater. Interfaces 2019, 11, 28657–28664. [Google Scholar] [CrossRef]
- Bhattarai, N.; Gunn, J.; Zhang, M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 83–99. [Google Scholar] [CrossRef]
- Wahid, F.; Zhou, Y.N.; Wang, H.S.; Wan, T.; Zhong, C.; Chu, L.Q. Injectable self-healing carboxymethyl chitosan-zinc supramolecular hydrogels and their antibacterial activity. Int. J. Biol. Macromol. 2018, 114, 1233–1239. [Google Scholar] [CrossRef]
- Chandramohan, Y.; Jeganathan, K.; Sivanesan, S.; Koka, P.; Amritha, T.M.S.; Vimalraj, S.; Dhanasekaran, A. Assessment of human ovarian follicular fluid derived mesenchymal stem cells in chitosan/PCL/Zn scaffold for bone tissue regeneration. Life Sci. 2021, 264, 118502. [Google Scholar] [CrossRef]
- O’Connor, J.P.; Kanjilal, D.; Teitelbaum, M.; Lin, S.S.; Cottrell, J.A. Zinc as a Therapeutic Agent in Bone Regeneration. Materials 2020, 13, 2211. [Google Scholar] [CrossRef]
- Rodell, C.B.; Kaminski, A.L.; Burdick, J.A. Rational design of network properties in guest-host assembled and shear-thinning hyaluronic acid hydrogels. Biomacromolecules 2013, 14, 4125–4134. [Google Scholar] [CrossRef] [PubMed]
- Rosales, A.M.; Rodell, C.B.; Chen, M.H.; Morrow, M.G.; Anseth, K.S.; Burdick, J.A. Reversible Control of Network Properties in Azobenzene-Containing Hyaluronic Acid-Based Hydrogels. Bioconj. Chem. 2018, 29, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Montanari, E.; Zoratto, N.; Mosca, L.; Cervoni, L.; Lallana, E.; Angelini, R.; Matricardi, P. Halting hyaluronidase activity with hyaluronan-based nanohydrogels: Development of versatile injectable formulations. Carbohydr. Polym. 2019, 221, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Montanari, E.; Di Meo, C.; Coviello, T.; Gueguen, V.; Pavon-Djavid, G. Intracellular Delivery of Natural Antioxidants via Hyaluronan Nanohydrogels. Pharmaceutics 2019, 11, 532. [Google Scholar] [CrossRef]
- Lee, F.; Chung, J.E.; Kurisawa, M. An injectable hyaluronic acid-tyramine hydrogel system for protein delivery. J. Control. Release Off. J. Control. Release Soc. 2009, 134, 186–193. [Google Scholar] [CrossRef]
- Redondo-Gómez, C.; Abdouni, Y.; Becer, C.R.; Mata, A. Self-Assembling Hydrogels Based on a Complementary Host-Guest Peptide Amphiphile Pair. Biomacromolecules 2019, 20, 2276–2285. [Google Scholar] [CrossRef]
- Van de Manakker, F.; van der Pot, M.; Vermonden, T.; van Nostrum, C.F.; Hennink, W.E. Self-Assembling Hydrogels Based on β-Cyclodextrin/Cholesterol Inclusion Complexes. Macromolecules 2008, 41, 1766–1773. [Google Scholar] [CrossRef]
- Van de Manakker, F.; Braeckmans, K.; Morabit, N.E.; De Smedt, S.C.; van Nostrum, C.F.; Hennink, W.E. Protein-Release Behavior of Self-Assembled PEG–b-Cyclodextrin/PEG–Cholesterol Hydrogels. Adv. Funct. Mater. 2009, 19, 2992–3001. [Google Scholar] [CrossRef]
- Li, J. Self-assembled supramolecular hydrogels based on polymer–cyclodextrin inclusion complexes for drug delivery. NPG Asia Mater. 2010, 2, 112–118. [Google Scholar] [CrossRef]
- Harada, A.; Li, J.; Kamachi, M. The molecular necklace: A rotaxane containing many threaded α-cyclodextrins. Nature 1992, 356, 325–327. [Google Scholar] [CrossRef]
- Khodaverdi, E.; Heidari, Z.; Tabassi, S.A.; Tafaghodi, M.; Alibolandi, M.; Tekie, F.S.; Hadizadeh, F. Injectable supramolecular hydrogel from insulin-loaded triblock PCL-PEG-PCL copolymer and γ-cyclodextrin with sustained-release property. AAPS Pharmscitech 2015, 16, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Song, L.; Chen, L.; Zhang, W.; Chen, Y.; Zang, F.; Zhang, Y. Injectable magnetic supramolecular hydrogel with magnetocaloric liquid-conformal property prevents post-operative recurrence in a breast cancer model. Acta Biomater. 2018, 74, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Qin, M.; Wu, J.; Luo, D.; Jiang, Q.; Li, Y.; Wang, W. Electroresponsive Supramolecular Graphene Oxide Hydrogels for Active Bacteria Adsorption and Removal. ACS Appl. Mater. Interfaces 2016, 8, 15120–15127. [Google Scholar] [CrossRef]
- Vermonden, T.; Censi, R.; Hennink, W.E. Hydrogels for protein delivery. Chem. Rev. 2012, 112, 2853–2888. [Google Scholar] [CrossRef]
- He, C.; Kim, S.W.; Lee, D.S. In situ gelling stimuli-sensitive block copolymer hydrogels for drug delivery. J. Control. Release Off. J. Control. Release Soc. 2008, 127, 189–207. [Google Scholar] [CrossRef]
- Sanna, D.; Alzari, V.; Nuvoli, D.; Nuvoli, L.; Rassu, M.; Sanna, V.; Mariani, A. β-Cyclodextrin-based supramolecular poly(N-isopropylacrylamide) hydrogels prepared by frontal polymerization. Carbohydr. Polym. 2017, 166, 249–255. [Google Scholar] [CrossRef]
- Song, X.; Zhang, Z.; Zhu, J. Thermoresponsive Hydrogel Induced by Dual Supramolecular Assemblies and Its Controlled Release Property for Enhanced Anticancer Drug Delivery. Biomacromolecules 2020, 21, 1516–1527. [Google Scholar] [CrossRef]
- Zou, H.; Guo, W.; Yuan, W. Supramolecular hydrogels from inclusion complexation of alpha-cyclodextrin with densely grafted chains in micelles for controlled drug and protein release. J. Mater. Chem. B 2013, 1, 6235–6244. [Google Scholar] [CrossRef]
- Bastings, M.M.; Koudstaal, S.; Kieltyka, R.E.; Nakano, Y.; Pape, A.C.; Feyen, D.A.; Dankers, P.Y. A fast pH-switchable and self-healing supramolecular hydrogel carrier for guided, local catheter injection in the infarcted myocardium. Adv. Healthc. Mater. 2014, 3, 70–78. [Google Scholar] [CrossRef]
- Zhang, S.; Bellinger, A.M.; Glettig, D.L.; Barman, R.; Lee, Y.A.; Zhu, J.; Traverso, G. A pH-responsive supramolecular polymer gel as an enteric elastomer for use in gastric devices. Nat. Mater. 2015, 14, 1065–1071. [Google Scholar] [CrossRef]
- Tamesue, S.; Takashima, Y.; Yamaguchi, H.; Shinkai, S.; Harada, A. Photoswitchable Supramolecular Hydrogels Formed by Cyclodextrins and Azobenzene Polymers. Angew. Chem. Int. Ed. 2010, 49, 7461–7464. [Google Scholar] [CrossRef] [PubMed]
- Mandl, G.A.; Rojas-Gutierrez, P.A.; Capobianco, J.A. A NIR-responsive azobenzene-based supramolecular hydrogel using upconverting nanoparticles. Chem. Commun. 2018, 54, 5847–5850. [Google Scholar] [CrossRef]
- Mehrban, N.; Abelardo, E.; Wasmuth, A.; Hudson, K.L.; Mullen, L.M.; Thomson, A.R.; Woolfson, D.N. Assessing cellular response to functionalized α-helical peptide hydrogels. Adv. Healthc. Mater. 2014, 3, 1387–1391. [Google Scholar] [CrossRef]
- Zhang, S.; Holmes, T.; Lockshin, C.; Rich, A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc. Natl. Acad. Sci. USA 1993, 90, 3334–3338. [Google Scholar] [CrossRef] [PubMed]
- Koutsopoulos, S.; Zhang, S. Two-layered injectable self-assembling peptide scaffold hydrogels for long-term sustained release of human antibodies. J. Control. Release Off. J. Control. Release Soc. 2012, 160, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.E.; Parmenter, C.D.J.; Scherman, O.A. Tunable Pentapeptide Self-Assembled β-Sheet Hydrogels. Angew. Chem. Int. Ed. 2018, 57, 7709–7713. [Google Scholar] [CrossRef]
- Dasgupta, A.; Das, D. Designer Peptide Amphiphiles: Self-Assembly to Applications. Langmuir ACS J. Surf. Colloids 2019, 35, 10704–10724. [Google Scholar] [CrossRef]
- Ghosh, G.; Barman, R.; Sarkar, J.; Ghosh, S. pH-Responsive Biocompatible Supramolecular Peptide Hydrogel. J. Phys. Chem. B 2019, 123, 5909–5915. [Google Scholar] [CrossRef]
- Dehsorkhi, A.; Castelletto, V.; Hamley, I.W. Self-assembling amphiphilic peptides. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2014, 20, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Webber, M.J.; Stupp, S.I. Self-assembly of peptide amphiphiles: From molecules to nanostructures to biomaterials. Biopolymers 2010, 94, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, X.; Zhang, P.; Liu, Y.; Ran, W.; Cai, Y.; Li, Y. Injectable peptide hydrogel as intraperitoneal triptolide depot for the treatment of orthotopic hepatocellular carcinoma. Acta Pharm. Sin. B 2019, 9, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Freeman, R.; Han, M.; Álvarez, Z.; Lewis, J.A.; Wester, J.R.; Stephanopoulos, N.; Stupp, S.I. Reversible self-assembly of superstructured networks. Science 2018, 362, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Gačanin, J.; Synatschke, C.V.; Weil, T. Biomedical Applications of DNA-based hydrogels. Adv. Funct. Mater. 2020, 30, 1906253. [Google Scholar] [CrossRef]
- Shao, Y.; Jia, H.; Cao, T.; Liu, D. Supramolecular Hydrogels Based on DNA Self-Assembly. Acc. Chem Res. 2017, 50, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kuang, Y.; Lin, H.C.; Gao, Y.; Shi, J.; Xu, B. Supramolecular nanofibers and hydrogels of nucleopeptides. Angewandte Chemie 2011, 50, 9365–9369. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.; Noblett, A.D.; Ren, P.; Suggs, L.J. Design and Characterization of Nucleopeptides for Hydrogel Self Assembly. ACS Appl. Mater. Interfaces 2019, 2, 2812–2821. [Google Scholar] [CrossRef]
- Daly, M.L.; Gao, Y.; Freeman, R. Encoding Reversible Hierarchical Structures with Supramolecular Peptide-DNA Materials. Bioconj. Chem. 2019, 30, 1864–1869. [Google Scholar] [CrossRef]
- Boback, K.; Bacchi, K.; O’Neill, S.; Brown, S.; Dorsainvil, J. Impact of C-Terminal Chemistry on Self-Assembled Morphology of Guanosine Containing Nucleopeptides. Molecules 2020, 25, 5493. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kuang, Y.; Shi, J.; Gao, Y.; Lin, H.C.; Xu, B. Multifunctional, biocompatible supramolecular hydrogelators consist only of nucleobase, amino acid, and glycoside. J. Am. Chem. Soc. 2011, 133, 17513–17518. [Google Scholar] [CrossRef]
- Radvar, E.; Azevedo, H.S. Supramolecular Peptide/Polymer Hybrid Hydrogels for Biomedical Applications. Macromol. Biosci. 2019, 19, e1800221. [Google Scholar] [CrossRef] [PubMed]
- Xing, R.; Li, S.; Zhang, N.; Shen, G.; Möhwald, H.; Yan, X. Self-Assembled Injectable Peptide Hydrogels Capable of Triggering Antitumor Immune Response. Biomacromolecules 2017, 18, 3514–3523. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Zou, Q.; Xing, R.; Jiao, T.; Yan, X. An injectable dipeptide-fullerene supramolecular hydrogel for photodynamic antibacterial therapy. J. Mater. Chem. B 2018, 6, 7335–7342. [Google Scholar] [CrossRef]
- Rajangam, K.; Behanna, H.A.; Hui, M.J.; Han, X.; Hulvat, J.F.; Lomasney, J.W.; Stupp, S.I. Heparin binding nanostructures to promote growth of blood vessels. Nano Lett. 2006, 6, 2086–2090. [Google Scholar] [CrossRef] [PubMed]
- Borges, J.; Sousa, M.P.; Cinar, G.; Caridade, S.G.; Guler, M.O.; Mano, J.F. Nanoengineering Hybrid Supramolecular Multilayered Biomaterials Using Polysaccharides and Self-Assembling Peptide Amphiphiles. Adv. Funct. Mater. 2017, 27, 1605122. [Google Scholar] [CrossRef]
- Damanik, F.F.R.; Brunelli, M.; Pastorino, L.; Ruggiero, C.; van Blitterswijk, C.; Rotmans, J.; Moroni, L. Sustained delivery of growth factors with high loading efficiency in a layer by layer assembly. Biomater. Sci. 2019, 8, 174–188. [Google Scholar] [CrossRef]
- Radvar, E.; Azevedo, H.S. Supramolecular Nanofibrous Peptide/Polymer Hydrogels for the Multiplexing of Bioactive Signals. ACS Biomater. Sci. Eng. 2019, 5, 4646–4656. [Google Scholar] [CrossRef]
- Du, X.; Zhou, J.; Li, X.; Xu, B. Self-assembly of nucleopeptides to interact with DNAs. Interface Focus 2017, 7, 20160116. [Google Scholar] [CrossRef]
- Li, Y.; Wang, F.; Cui, H. Peptide-Based Supramolecular Hydrogels for Delivery of Biologics. Bioeng. Transl. Med. 2016, 1, 306–322. [Google Scholar] [CrossRef]
- Uzunalli, G.; Guler, M.O. Peptide gels for controlled release of proteins. Ther. Deliv. 2020, 11, 193–211. [Google Scholar] [CrossRef]
- Baek, K.; Noblett, A.D.; Ren, P.; Suggs, L.J. Self-assembled nucleo-tripeptide hydrogels provide local and sustained doxorubicin release. Biomater. Sci. 2020, 8, 3130–3137. [Google Scholar] [CrossRef]
- Censi, R.; Di Martino, P.; Vermonden, T.; Hennink, W.E. Hydrogels for protein delivery in tissue engineering. J. Control. Release Off. J. Control. Release Soc. 2012, 161, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Koutsopoulos, S.; Unsworth, L.D.; Nagai, Y.; Zhang, S. Controlled release of functional proteins through designer self-assembling peptide nanofiber hydrogel scaffold. Proc. Natl. Acad. Sci. USA 2009, 106, 4623–4628. [Google Scholar] [CrossRef] [PubMed]
- Van Tomme, S.R.; De Geest, B.G.; Braeckmans, K.; De Smedt, S.C.; Siepmann, F.; Siepmann, J.; Hennink, W.E. Mobility of model proteins in hydrogels composed of oppositely charged dextran microspheres studied by protein release and fluorescence recovery after photobleaching. J. Control. Release Off. J. Control. Release Soc. 2005, 110, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Appel, E.A.; Loh, X.J.; Jones, S.T.; Dreiss, C.A.; Scherman, O.A. Sustained release of proteins from high water content supramolecular polymer hydrogels. Biomaterials 2012, 33, 4646–4652. [Google Scholar] [CrossRef]
- Dankers, P.Y.; Hermans, T.M.; Baughman, T.W.; Kamikawa, Y.; Kieltyka, R.E.; Bastings, M.M.; Meijer, E.W. Hierarchical formation of supramolecular transient networks in water: A modular injectable delivery system. Adv. Mater 2012, 24, 2703–2709. [Google Scholar] [CrossRef]
- Kaplan, J.A.; Barthélémy, P.; Grinstaff, M.W. Self-Assembled Nanofiber Hydrogels for Mechanoresponsive Therapeutic Anti-TNFα Antibody Delivery. Chem. Commun. 2016, 52, 5860–5863. [Google Scholar]
- Branco, M.C.; Pochan, D.J.; Wagner, N.J.; Schneider, J.P. Macromolecular diffusion and release from self-assembled beta-hairpin peptide hydrogels. Biomaterials 2009, 30, 1339–1347. [Google Scholar] [CrossRef]
- Branco, M.C.; Pochan, D.J.; Wagner, N.J.; Schneider, J.P. The effect of protein structure on their controlled release from an injectable peptide hydrogel. Biomaterials 2010, 31, 9527–9534. [Google Scholar] [CrossRef]
- Nagy-Smith, K.; Yamada, Y.; Schneider, J.P. Protein release from highly charged peptide hydrogel networks. J. Mater. Chem. B 2016, 4, 1999–2007. [Google Scholar] [CrossRef]
- Oyen, E.; Martin, C.; Caveliers, V.; Madder, A. In Vivo Imaging of the Stability and Sustained Cargo Release of an Injectable Amphipathic Peptide-Based Hydrogel. Biomacromolecules 2017, 18, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, X.Y.; Zhao, Y.; Yang, Y. pH-Switchable Antimicrobial Nanofiber Networks of Hydrogel Eradicate Biofilm and Rescue Stalled Healing in Chronic Wounds. ACS Nano 2019, 13, 11686–11697. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, Y.; Hou, Z.; Cui, X.; Zhao, Y.; Xu, H. Rational Design of Short Peptide-Based Hydrogels with MMP-2 Responsiveness for Controlled Anticancer Peptide Delivery. Biomacromolecules 2017, 18, 3563–3571. [Google Scholar] [CrossRef] [PubMed]
- Faidra Angelerou, M.G.; Markus, R.; Paraskevopoulou, V.; Foralosso, R.; Clarke, P.; Alvarez, C.V.; Chenlo, M. Mechanistic investigations into the encapsulation and release of small molecules and proteins from a supramolecular nucleoside gel in vitro and in vivo. J. Control. Release Off. J. Control. Release Soc. 2020, 317, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Shigemitsu, H.; Kubota, R.; Nakamura, K.; Matsuzaki, T.; Minami, S.; Aoyama, T.; Hamachi, I. Protein-responsive protein release of supramolecular/polymer hydrogel composite integrating enzyme activation systems. Nat. Commun. 2020, 11, 3859. [Google Scholar] [CrossRef]
- Wu, Y.; Li, C.; Boldt, F.; Wang, Y.; Kuan, S.L.; Tran, T.T.; Weil, T. Programmable protein-DNA hybrid hydrogels for the immobilization and release of functional proteins. Chem. Commun. 2014, 50, 14620–14622. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Cheng, I.; Luo, K.Q.; Mi, Y. Capture and release of protein by a reversible DNA-induced sol-gel transition system. Angew. Chem. 2008, 47, 331–333. [Google Scholar] [CrossRef]
- Hoque, J.; Sangaj, N.; Varghese, S. Stimuli-Responsive Supramolecular Hydrogels and Their Applications in Regenerative Medicine. Macromol. Biosci. 2019, 19, e1800259. [Google Scholar] [CrossRef]
- Zelzer, M.; Todd, S.J.; Hirst, A.R.; McDonald, T.O.; Ulijn, R.V. Enzyme responsive materials: Design strategies and future developments. Biomater. Sci. 2013, 1, 11–39. [Google Scholar] [CrossRef]
- Liang, H.P.H.; Xu, J.; Xue, M.; Jackson, C. Matrix metalloproteinases in bone development and pathology: Current knowledge and potential clinical utility. Met. Med. 2016, 3, 93–102. [Google Scholar] [CrossRef]
- Tada, S.; Kitajima, T.; Ito, Y. Design and synthesis of binding growth factors. Int. J. Mol. Sci. 2012, 13, 6053–6072. [Google Scholar] [CrossRef]
- Hajimiri, M.; Shahverdi, S.; Kamalinia, G.; Dinarvand, R. Growth factor conjugation: Strategies and applications. J. Biomed. Mater. Res. Part A 2015, 103, 819–838. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, X.; Pan, Y.; Cui, Y.; Zhou, X.; Kong, D.; Zhao, Q. Enhanced vascularization in hybrid PCL/gelatin fibrous scaffolds with sustained release of VEGF. BioMed Res. Int. 2015, 2015, 865076. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Muiños, T.; Recha-Sancho, L.; López-Chicón, P.; Castells-Sala, C.; Mata, A.; Semino, C.E. Bimolecular based heparin and self-assembling hydrogel for tissue engineering applications. Acta Biomater. 2015, 16, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Yan, Y.; Qi, J.; Deng, L.; Shao, Z.W.; Zhang, K.Q. Cooperative Assembly of a Peptide Gelator and Silk Fibroin Afford an Injectable Hydrogel for Tissue Engineering. ACS Appl. Mater. Interfaces 2018, 10, 12474–12484. [Google Scholar] [CrossRef]
- Tan, J.; Zhang, M.; Hai, Z.; Wu, C.; Lin, J.; Kuang, W.; Liang, G. Sustained Release of Two Bioactive Factors from Supramolecular Hydrogel Promotes Periodontal Bone Regeneration. ACS Nano 2019, 13, 5616–5622. [Google Scholar] [CrossRef]
- Lu, H.H.; Subramony, S.D.; Boushell, M.K.; Zhang, X. Tissue engineering strategies for the regeneration of orthopedic interfaces. Ann. Biomed. Eng. 2010, 38, 2142–2154. [Google Scholar] [CrossRef]
- Hou, S.; Wang, X.; Park, S.; Jin, X.; Ma, P.X. Rapid Self-Integrating, Injectable Hydrogel for Tissue Complex Regeneration. Adv. Healthc. Mater. 2015, 4, 1491–1495. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Hsu, E.L.; Mendoza, M.; Ghodasra, J.; Nickoli, M.S.; Ashtekar, A.; Stupp, S.I. Gel scaffolds of BMP-2-binding peptide amphiphile nanofibers for spinal arthrodesis. Adv. Healthc. Mater. 2015, 4, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Feng, Q.; Lin, S.; Yuan, W.; Li, R.; Li, J.; Bian, L. Injectable stem cell-laden supramolecular hydrogels enhance in situ osteochondral regeneration via the sustained co-delivery of hydrophilic and hydrophobic chondrogenic molecules. Biomaterials 2019, 210, 51–61. [Google Scholar] [CrossRef]
- Alvarez-Lorenzo, C.; García-González, C.A.; Concheiro, A. Cyclodextrins as versatile building blocks for regenerative medicine. J. Control. Release Off. J. Control. Release Soc. 2017, 268, 269–281. [Google Scholar] [CrossRef]
- Wei, K.; Zhu, M.; Sun, Y.; Xu, J.; Feng, Q.; Lin, S.; Bian, L. Robust Biopolymeric Supramolecular “Host−Guest Macromer”Hydrogels Reinforced by in Situ Formed Multivalent Nanoclusters for Cartilage Regeneration. Macromolecules 2016, 49, 866–875. [Google Scholar] [CrossRef]
- Jung, H.; Park, J.S.; Yeom, J.; Selvapalam, N.; Park, K.M.; Oh, K.; Kim, K. 3D tissue engineered supramolecular hydrogels for controlled chondrogenesis of human mesenchymal stem cells. Biomacromolecules 2014, 15, 707–714. [Google Scholar] [CrossRef]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- Zhao, W.; Li, Y.; Zhang, X.; Zhang, R.; Hu, Y.; Boyer, C.; Xu, F.J. Photo-responsive supramolecular hyaluronic acid hydrogels for accelerated wound healing. J. Control. Release Off. J. Control. Release Soc. 2020, 323, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Xuan, X.; Zhou, Y.; Chen, A.; Zheng, S.; An, Y.; He, H.; Wu, J. Silver crosslinked injectable bFGF-eluting supramolecular hydrogels speed up infected wound healing. J. Mater. Chem. B 2020, 8, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jiang, X.J.; Lin, T.; Ren, S.; Li, X.Y.; Zhang, X.Z.; Tang, Q.Z. The inhibition of postinfarct ventricle remodeling without polycythaemia following local sustained intramyocardial delivery of erythropoietin within a supramolecular hydrogel. Biomaterials 2009, 30, 4161–4167. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.Q.; Wang, T.; Lu, B.; Xu, X.D.; Cheng, S.X.; Jiang, X.J.; Zhuo, R.X. Fabrication of supramolecular hydrogels for drug delivery and stem cell encapsulation. Langmuir ACS J. Surf. Colloids 2008, 24, 10306–10312. [Google Scholar] [CrossRef] [PubMed]
- Rodell, C.B.; Rai, R.; Faubel, S.; Burdick, J.A.; Soranno, D.E. Local immunotherapy via delivery of interleukin-10 and transforming growth factor beta antagonist for treatment of chronic kidney disease. J. Control. Release Off. J. Control. Release Soc. 2015, 206, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Pang, Z.; He, H.; Lee, S.; Qin, J.; Wu, J.; Yang, V.C. Drug depot-anchoring hydrogel: A self-assembling scaffold for localized drug release and enhanced stem cell differentiation. J. Control. Release Off. J. Control. Release Soc. 2017, 261, 234–245. [Google Scholar] [CrossRef]
- Segredo-Morales, E.; Reyes, R.; Arnau, M.R.; Delgado, A.; Evora, C. In situ gel-forming system for dual BMP-2 and 17beta-estradiol controlled release for bone regeneration in osteoporotic rats. Drug Deliv. Transl. Res. 2018, 8, 1103–1113. [Google Scholar] [CrossRef]
- Shah, R.N.; Shah, N.A.; Del Rosario Lim, M.M.; Hsieh, C.; Nuber, G.; Stupp, S.I. Supramolecular design of self-assembling nanofibers for cartilage regeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 3293–3298. [Google Scholar] [CrossRef]
- Govender, S.; Csimma, C.; Genant, H.K.; Valentin-Opran, A.; Amit, Y.; Arbel, R.; Aro, H. Recombinant human bone morphogenetic protein-2 for treatment of open tibial fractures: A prospective, controlled, randomized study of four hundred and fifty patients. J. Bone Jt. Surg. Am. Vol. 2002, 84, 2123–2134. [Google Scholar] [CrossRef]
- Vaccaro, A.R.; Lawrence, J.P.; Patel, T.; Katz, L.D.; Anderson, D.G.; Albert, T.J.; Fischgrund, J.S. The safety and efficacy of OP-1 (rhBMP-7) as a replacement for iliac crest autograft in posterolateral lumbar arthrodesis: A long-term (>4 years) pivotal study. Spine 2008, 33, 2850–2862. [Google Scholar] [CrossRef]
- Vojtova, L.; Michlovska, L.; Valova, K.; Zboncak, M.; Trunec, M.; Castkova, K.; Montufar, E.B. The Effect of the Thermosensitive Biodegradable PLGA-PEG-PLGA Copolymer on the Rheological, Structural and Mechanical Properties of Thixotropic Self-Hardening Tricalcium Phosphate Cement. Int J. Mol. Sci. 2019, 20, 391. [Google Scholar] [CrossRef]
- Choi, S.; Baudys, M.; Kim, S.W. Control of blood glucose by novel GLP-1 delivery using biodegradable triblock copolymer of PLGA-PEG-PLGA in type 2 diabetic rats. Pharm. Res. 2004, 21, 827–831. [Google Scholar] [CrossRef]
- Kim, Y.J.; Choi, S.; Koh, J.J.; Lee, M.; Ko, K.S.; Kim, S.W. Controlled release of insulin from injectable biodegradable triblock copolymer. Pharm. Res. 2001, 18, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Santoveña, A.; Monzón, C.; Alvarez-Lorenzo, C.; Del Rosario, C.; Delgado, A.; Evora, C.; Fariña, J.B. Structure-Performance Relationships of Temperature-Responsive PLGA-PEG-PLGA Gels for Sustained Release of Bone Morphogenetic Protein-2. J. Pharm. Sci. 2017, 106, 3353–3362. [Google Scholar] [CrossRef]
- Qiao, M.; Chen, D.; Hao, T.; Zhao, X.; Hu, H.; Ma, X. Injectable thermosensitive PLGA-PEG-PLGA triblock copolymers-based hydrogels as carriers for interleukin-2. Die Pharmazie 2008, 63, 27–30. [Google Scholar]
- Kanjickal, D.; Lopina, S.; Evancho-Chapman, M.M.; Schmidt, S.; Donovan, D. Effects of sterilization on poly(ethylene glycol) hydrogels. J. Biomed. Mater. Res. Part A 2008, 87, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Galante, R.; Pinto, T.J.A.; Colaço, R.; Serro, A.P. Sterilization of hydrogels for biomedical applications: A review. J. Biomed. Mater. Res. Part B Appl. Biomater. 2018, 106, 2472–2492. [Google Scholar] [CrossRef]
- Ilochonwu, B.C.; Urtti, A.; Hennink, W.E.; Vermonden, T. Intravitreal hydrogels for sustained release of therapeutic proteins. J. Control. Release Off. J. Control. Release Soc. 2020, 326, 419–441. [Google Scholar] [CrossRef]
- Azevedo, H.S.; Pashkuleva, I. Biomimetic supramolecular designs for the controlled release of growth factors in bone regeneration. Adv. Drug Deliv. Rev. 2015, 94, 63–76. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criterion | Covalently (Permanent Bonds) Crosslinked Hydrogels | Supramolecular Hydrogels |
|---|---|---|
| Processability | More complex; covalent bonds form during hydrogelation requiring additional reagents or inputs (e.g., light source). | Simple; it occurs spontaneously upon mixing hydrogel components or when in contact with electrolytes present in body fluids or culture medium. |
| Injectability | Difficult, unless if polymerizable in situ (e.g., under UV light). | Common in most hydrogels due to their shear thinning properties. |
| Self-healing ability | Absent, except if made of dynamic covalent bonds (e.g., disulfide). | Observed in most systems. |
| Protein loading | Mainly physical entrapment. Creating affinity interactions typically requires additional chemical modification of the hydrogel components. | Various possibilities (physical entrapment + affinity interactions) not requiring additional chemical modification. |
| Protein compatibility | Risk of protein deactivation during hydrogel formation if subjected to denaturing agents (e.g., catalysts, UV light). | Mostly compatible, unless there are strong electrostatic interactions between the protein and the hydrogel. |
| Stability | Stable at physiological conditions. | Less stable at physiological conditions (e.g., can be affected by differences in ionic strength). |
| Type of Hydrogels | Pros | Cons |
|---|---|---|
| Polymer-based |
|
|
| Peptide-based |
|
|
| Hydrogel Components | Hydrogel Type | Driving Force in Hydrogel Formation | Protein Loading Methods | Model Macromolecules | Release Period | Driving Force in Protein Release | Reference |
|---|---|---|---|---|---|---|---|
| HA-β-CD; HA-Ad | polymer | host-guest interaction | physical entrapment | BSA | 60 days | erosion/ diffusion | [16] |
| HA-β-CD; HA-Azo | polymer | host-guest interaction | physical entrapment | BSA | 8 days | stimuli (light)/ diffusion | [17] |
| PEG8- Cholesterol; PEG8-β-CD | polymer | hydrophobic and van der Waals interactions | physical entrapment | lysozyme; BSA | 250 h | erosion/ diffusion | [23] |
| γ-CD; PCL-PEG-PCL | polymer | hydrophobic interactions | physical entrapment | insulin | 37 days | diffusion/ erosion | [26] |
| α-CD; Py-PCL-b-POEGMA | polymer | host-guest interaction | physical entrapment | DOX; BSA | 64 h | stimuli (temperature) | [33] |
| dex-HEMA-MAA; dex-HEMA-DMAEMA | polymer | electrostatic interactions | physical entrapment | IgG, BSA, lysozyme | 60 days | diffusion | [68] |
| PVA-MV, HEC-Np, CB[8] | polymer | host-guest interaction | physical entrapment | BSA, lysozyme | 20–160 days | diffusion | [69] |
| UPy-X-PEG-Zk (X = (CH2)n; Z = molecular weight of PEG) | polymer | hydrogen bonding | physical entrapment | CFP | 4000 min | erosion | [70] |
| oleoylamide glycosyl-nucleoside-lipid | polymer (nucleoside-lipid) | hydrogen bonding; hydrophobic interactions; π-π stacking | physical entrapment | dextrans; IgG | - | stimuli (shear-mediated) | [71] |
| Ac-(RADA)4-NH2; Ac-(KLDL)3-NH2 | peptide | electrostatic interactions | physical entrapment | IgG | 100 days | diffusion | [40] |
| Ac-(RADA)4-NH2 | peptide | electrostatic interactions | physical entrapment | IgG, BSA, lysozyme trypsin inhibitor | 30–50 h | diffusion | [67] |
| MAX1/MAX8 | peptide | electrostatic interactions | physical entrapment | dextran, lactoferrin | 30 days | diffusion | [72] |
| MAX8 | peptide | electrostatic interactions | physical entrapment | lysozyme, IgG, lactoferrin, α-lactalbumin, myoglobin, BSA | 30 days | diffusion | [73] |
| HLT2 or VEQ3 | peptide | electrostatic interactions | electrostatic interactions | α-lactalbumin, myoglobin, lactoferrin | 4–28 days | diffusion | [74] |
| H-FEFQFK-NH2 | peptide | hydrophobic interaction | physical entrapment | 4-amino-2-cyclohexylmethyl-indolo[3,4-c]azepin-2-on (small molecule); 15 residue peptide; cAbVCAM1-5 Nb (protein) | 12 h | diffusion (small molecule)/erosion (peptide, protein) | [75] |
| Ac-IKFQFHFD-NH2 | peptide | π-π stacking; electrostatic interactions | physical entrapment (cypate); possible interactions with peptide due to structural similarities (proline) | cypate; proline | 24 h; 75 h | stimuli (pH) | [76] |
| Ac-I3SLKG-NH2 | peptide | hydrophobic interactions; hydrogen bonding | physical entrapment | peptide G3 | 16 days | stimuli (enzyme) | [77] |
| N4-octanoyl-2′-deoxycytidine | nucleoside | hydrogen bonding | non-covalent protein-gel association | β-lactoglobulin, BSA, lysozyme, insulin | 24 h | erosion | [78] |
| PA/PSS | multi-component (peptide/ polymer) | electrostatic interactions | electrostatic interactions | BSA, lysozyme | 30 days | diffusion | [61] |
| APmoc-F(CF3)F-OH/agarose | multi-component (peptide/ polymer) | hydrophobic interaction | physical entrapment | myoglobin | 3 h | stimuli (enzyme) | [79] |
| HSA/DNA | multi-component (protein/ DNA) | multi-arm DNA crosslinking | ssDNA hybridization | GFP; YFP | 30 min (trypsin) 15 min (DNase I) | enzyme (DNase I & trypsin) -mediated degradation | [80] |
| PAM/DNA | multi-component (polymer/ DNA) | DNA hybridization | thrombin-binding aptamer | human α-thrombin | - | reversible sol–gel transition by DNA strand displacement | [81] |
| Therapeutic Protein(s) | Hydrogel | Release Period | Application | In Vivo Model | Reference |
|---|---|---|---|---|---|
| VEGF/FGF-2 | PA-heparin | 10 days | angiogenesis | rat cornea angiogenesis | [58] |
| VEGF165/TGF-β1/FGFβ | RAD16-I/heparin | 36 h | angiogenesis | - | [88] |
| VEGF | SF/NapFF-RGD | 21 days | angiogenesis | mice model | [89] |
| VEGF | RADA16/RADA16-PEG-PLGA | >30 days | angiogenesis | - | [104] |
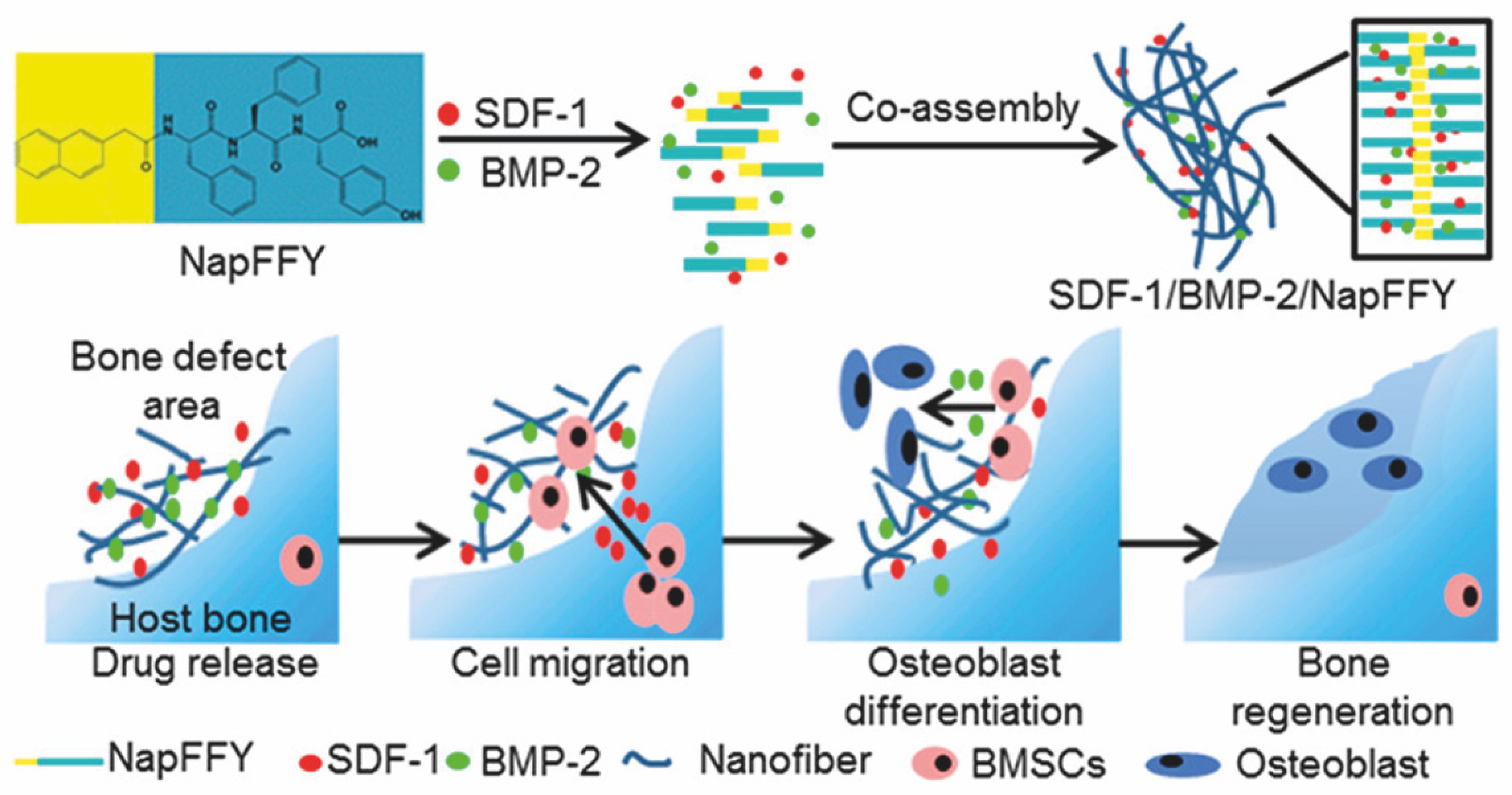
| BMP-2 | NapFFY nanofiber | 35 days | bone regeneration | critical-sized periodontal bone defect models of maxillae in rats | [90] |
| BMP-2 | BMP-2-binding PA nanofibers | 28 days | bone regeneration | posterolateral lumbar intertransverse spinal fusion model in rats | [93] |
| BMP-2 | Pluronic®127/ Tetronic®1307/α-CD | >7 weeks in vitro; 2 weeks in vivo | bone regeneration | osteoporosis model in rats | [105] |
| BMP-2 | DEX-UPy | >1 month | bone-cartilage complex | subcutaneous implantation model in nude mice | [92] |
| TGF-β1 | Ac-β-CDs/gelatin | >28 days | cartilage regeneration | knee osteochondral defects in rats | [94] |
| TGF-β1 | Ac-β-CDs/HA-Ad | > 21 days | cartilage regeneration | knee osteochondral defects in rats | [96] |
| TGF-β3 | monoCB[6]/DAH-HA | >3 weeks | cartilage regeneration | mice model | [97] |
| TGF-β1 | TGF-β binding PA nanofibers | >72 h | cartilage regeneration | chondral defect microfracture model in rabbits | [106] |
| EGF | HA-β-CD/HA-Azo | depending on the provided light stimuli | skin healing | excisional full-thickness wound model in rats | [99] |
| bFGF | CS-Ag | >11 days | skin healing | infected wound model in mice | [100] |
| HGF/IGF-1 | UPy-PEG | 7 days | cardiac repair | porcine model of chronic ischemia | [34] |
| EPO | α-CD/MPEG-PCL-MPEG | >7 days | cardiac repair | myocardial infarction model in rats | [101] |
| anti-TGF-β/IL-10 | HA-β-CD/HA-Ad | 13 days | kidney | unilateral ureteral obstruction model in mice | [103] |
| BMP-7 | UPy-X-PEG-Zk (X = (CH2)n; Z = molecular weight of PEG) | 1 week | kidney | rats | [70] |
| Challenges | Solutions |
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, Y.; Azevedo, H.S. Supramolecular Hydrogels for Protein Delivery in Tissue Engineering. Molecules 2021, 26, 873. https://doi.org/10.3390/molecules26040873
Lyu Y, Azevedo HS. Supramolecular Hydrogels for Protein Delivery in Tissue Engineering. Molecules. 2021; 26(4):873. https://doi.org/10.3390/molecules26040873
Chicago/Turabian StyleLyu, Yaqi, and Helena S. Azevedo. 2021. "Supramolecular Hydrogels for Protein Delivery in Tissue Engineering" Molecules 26, no. 4: 873. https://doi.org/10.3390/molecules26040873
APA StyleLyu, Y., & Azevedo, H. S. (2021). Supramolecular Hydrogels for Protein Delivery in Tissue Engineering. Molecules, 26(4), 873. https://doi.org/10.3390/molecules26040873