
Impact of the Metal Center and Leaving Group on the Anticancer Activity of Organometallic Complexes of Pyridine-2-carbothioamide †

 , , ,
, , ,  and
and
Abstract
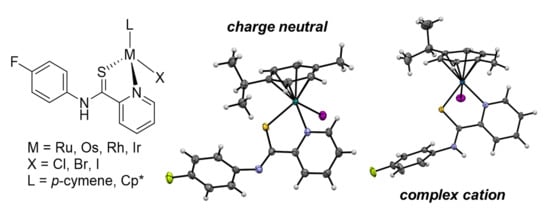
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Physical Measurements
3.2. Syntheses
3.3. Sulforhodamine B Cytotoxicity Assay
3.4. Stability and Amino Acid Reactivity Studies
3.5. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Haas, K.L.; Franz, K.J. Application of Metal Coordination Chemistry To Explore and Manipulate Cell Biology. Chem. Rev. 2009, 109, 4921–4960. [Google Scholar] [CrossRef]
- Leung, C.-H.; He, H.-Z.; Liu, L.-J.; Wang, M.; Chan, D.S.-H.; Ma, D.-L. Metal complexes as inhibitors of transcription factor activity. Coord. Chem. Rev. 2013, 257, 3139–3151. [Google Scholar] [CrossRef]
- Medici, S.; Peana, M.; Nurchi, V.M.; Lachowicz, J.I.; Crisponi, G.; Zoroddu, M.A. Noble metals in medicine: Latest advances. Coord. Chem. Rev. 2015, 284, 329–350. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The next generation of platinum drugs: Targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Hanif, M.; Hartinger, C.G. Anticancer metallodrugs: Where is the next cisplatin? Future Med. Chem. 2018, 10, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Guo, Z. Metal-based anticancer chemotherapeutic agents. Curr. Opin. Chem. Biol. 2014, 19, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Allardyce, C.S.; Dyson, P.J. Ruthenium in medicine: Current clinical uses and future prospects. Platin. Met. Rev. 2001, 45, 62–69. [Google Scholar]
- Hartinger, C.G.; Zorbas-Seifried, S.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From bench to bedside–preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef]
- Sava, G.; Bergamo, A.; Dyson, P.J. Metal-based antitumour drugs in the post-genomic era: What comes next? Dalton Trans. 2011, 40, 9069–9075. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; van den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H. A phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef]
- Yan, Y.K.; Melchart, M.; Habtemariam, A.; Sadler, P.J. Organometallic chemistry, biology and medicine: Ruthenium arene anticancer complexes. Chem. Commun. 2005, 38, 4764–4776. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, C.G.; Dyson, P.J. Bioorganometallic chemistry—From teaching paradigms to medicinal applications. Chem. Soc. Rev. 2009, 38, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic anticancer compounds. J. Med. Chem. 2010, 54, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Gasser, G.; Metzler-Nolte, N. The potential of organometallic complexes in medicinal chemistry. Curr. Opin. Chem. Biol. 2012, 16, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Metzler-Nolte, N.; Dyson, P.J. Challenges and Opportunities in the Development of Organometallic Anticancer Drugs. Organometallics 2012, 31, 5677–5685. [Google Scholar] [CrossRef]
- Agonigi, G.; Riedel, T.; Zacchini, S.; Păunescu, E.; Pampaloni, G.; Bartalucci, N.; Dyson, P.J.; Marchetti, F. Synthesis and Antiproliferative Activity of New Ruthenium Complexes with Ethacrynic-Acid-Modified Pyridine and Triphenylphosphine Ligands. Inorg. Chem. 2015, 54, 6504–6512. [Google Scholar] [CrossRef]
- Nazarov, A.A.; Meier, S.M.; Zava, O.; Nosova, Y.N.; Milaeva, E.R.; Hartinger, C.G.; Dyson, P.J. Protein ruthenation and DNA alkylation: Chlorambucil-functionalized RAPTA complexes and their anticancer activity. Dalton Trans. 2015, 44, 3614–3623. [Google Scholar] [CrossRef]
- Kubanik, M.; Holtkamp, H.; Söhnel, T.; Jamieson, S.M.F.; Hartinger, C.G. Impact of the Halogen Substitution Pattern on the Biological Activity of Organoruthenium 8-Hydroxyquinoline Anticancer Agents. Organometallics 2015, 34, 5658–5668. [Google Scholar] [CrossRef]
- Movassaghi, S.; Hanif, M.; Holtkamp, H.U.; Sohnel, T.; Jamieson, S.M.F.; Hartinger, C.G. Making organoruthenium complexes of 8-hydroxyquinolines more hydrophilic: Impact of a novel L-phenylalanine-derived arene ligand on the biological activity. Dalton Trans. 2018, 47, 2192–2201. [Google Scholar] [CrossRef] [PubMed]
- Tremlett, W.D.J.; Tong, K.K.H.; Steel, T.R.; Movassaghi, S.; Hanif, M.; Jamieson, S.M.F.; Sohnel, T.; Hartinger, C.G. Hydroxyquinoline-derived anticancer organometallics: Introduction of amphiphilic PTA as an ancillary ligand increases their aqueous solubility. J. Inorg. Biochem. 2019, 199, 110768. [Google Scholar] [CrossRef] [PubMed]
- Kljun, J.; Bytzek, A.K.; Kandioller, W.; Bartel, C.; Jakupec, M.A.; Hartinger, C.G.; Keppler, B.K.; Turel, I. Physicochemical studies and anticancer potency of ruthenium η6-p-cymene complexes containing antibacterial quinolones. Organometallics 2011, 30, 2506–2512. [Google Scholar] [CrossRef] [PubMed]
- Kandioller, W.; Balsano, E.; Meier, S.M.; Jungwirth, U.; Göschl, S.; Roller, A.; Jakupec, M.A.; Berger, W.; Keppler, B.K.; Hartinger, C.G. Organometallic anticancer complexes of lapachol: Metal centre-dependent formation of reactive oxygen species and correlation with cytotoxicity. Chem. Commun. 2013, 49, 3348–3350. [Google Scholar] [CrossRef]
- Kurzwernhart, A.; Kandioller, W.; Bächler, S.; Bartel, C.; Martic, S.; Buczkowska, M.; Mühlgassner, G.; Jakupec, M.A.; Kraatz, H.-B.; Bednarski, P.J.; et al. Structure–Activity Relationships of Targeted RuII(η6-p-Cymene) Anticancer Complexes with Flavonol-Derived Ligands. J. Med. Chem. 2012, 55, 10512–10522. [Google Scholar] [CrossRef]
- Movassaghi, S.; Leung, E.; Hanif, M.; Lee, B.Y.T.; Holtkamp, H.U.; Tu, J.K.Y.; Sohnel, T.; Jamieson, S.M.F.; Hartinger, C.G. A Bioactive L-Phenylalanine-Derived Arene in Multitargeted Organoruthenium Compounds: Impact on the Antiproliferative Activity and Mode of Action. Inorg. Chem. 2018, 57, 8521–8529. [Google Scholar] [CrossRef]
- Aman, F.; Hanif, M.; Siddiqui, W.A.; Ashraf, A.; Filak, L.K.; Reynisson, J.; Söhnel, T.; Jamieson, S.M.F.; Hartinger, C.G. Anticancer Ruthenium(η6-p-cymene) Complexes of Nonsteroidal Anti-inflammatory Drug Derivatives. Organometallics 2014, 33, 5546–5553. [Google Scholar] [CrossRef]
- Aman, F.; Hanif, M.; Kubanik, M.; Ashraf, A.; Söhnel, T.; Jamieson, S.M.F.; Siddiqui, W.A.; Hartinger, C.G. Anti-Inflammatory Oxicams as Multi-donor Ligand Systems: pH- and Solvent-Dependent Coordination Modes of Meloxicam and Piroxicam to Ru and Os. Chem. Eur. J. 2017, 23, 4893–4902. [Google Scholar] [CrossRef]
- Ashraf, A.; Aman, F.; Movassaghi, S.; Zafar, A.; Kubanik, M.; Siddiqui, W.A.; Reynisson, J.; Söhnel, T.; Jamieson, S.M.F.; Hanif, M.; et al. Structural Modifications of the Antiinflammatory Oxicam Scaffold and Preparation of Anticancer Organometallic Compounds. Organometallics 2019, 38, 361–374. [Google Scholar] [CrossRef]
- Dorcier, A.; Dyson, P.J.; Gossens, C.; Rothlisberger, U.; Scopelliti, R.; Tavernelli, I. Binding of organometallic ruthenium(II) and osmium(II) complexes to an oligonucleotide: A combined mass spectrometric and theoretical study. Organometallics 2005, 24, 2114–2123. [Google Scholar] [CrossRef]
- Pizarro, A.M.; Habtemariam, A.; Sadler, P.J. Activation mechanisms for organometallic anticancer complexes. In Medicinal Organometallic Chemistry; Springer: Berlin/Heidelberg, Germany, 2010; pp. 21–56. [Google Scholar]
- Noffke, A.L.; Habtemariam, A.; Pizarro, A.M.; Sadler, P.J. Designing organometallic compounds for catalysis and therapy. Chem. Commun. 2012, 48, 5219–5246. [Google Scholar] [CrossRef] [PubMed]
- Hanif, M.; Arshad, J.; Astin, J.W.; Rana, Z.; Zafar, A.; Movassaghi, S.; Leung, E.; Patel, K.; Sohnel, T.; Reynisson, J.; et al. A Multitargeted Approach: Organorhodium Anticancer Agent Based on Vorinostat as a Potent Histone Deacetylase Inhibitor. Angew. Chem. Int. Ed. Engl. 2020, 59, 14609–14614. [Google Scholar] [CrossRef]
- Meier, S.M.; Hanif, M.; Adhireksan, Z.; Pichler, V.; Novak, M.; Jirkovsky, E.; Jakupec, M.A.; Arion, V.B.; Davey, C.A.; Keppler, B.K.; et al. Novel metal(II) arene 2-pyridinecarbothioamides: A rationale to orally active organometallic anticancer agents. Chem. Sci. 2013, 4, 1837–1846. [Google Scholar] [CrossRef]
- Arshad, J.; Hanif, M.; Movassaghi, S.; Kubanik, M.; Waseem, A.; Söhnel, T.; Jamieson, S.M.; Hartinger, C.G. Anticancer Ru(η6-p-cymene) complexes of 2-pyridinecarbothioamides: A structure–activity relationship study. J. Inorg. Biochem. 2017, 177, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.M.; Kreutz, D.; Winter, L.; Klose, M.H.M.; Cseh, K.; Weiss, T.; Bileck, A.; Alte, B.; Mader, J.C.; Jana, S.; et al. An Organoruthenium Anticancer Agent Shows Unexpected Target Selectivity For Plectin. Angew. Chem. Int. Ed. Engl. 2017, 56, 8267–8271. [Google Scholar] [CrossRef]
- Hanif, M.; Moon, S.; Sullivan, M.P.; Movassaghi, S.; Kubanik, M.; Goldstone, D.C.; Söhnel, T.; Jamieson, S.M.; Hartinger, C.G. Anticancer activity of Ru-and Os (arene) compounds of a maleimide-functionalized bioactive pyridinecarbothioamide ligand. J. Inorg. Biochem. 2016, 165, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Arshad, J.; Hanif, M.; Zafar, A.; Movassaghi, S.; Tong, K.; Reynisson, J.; Kubanik, M.; Waseem, A.; Söhnel, T.; Jamieson, S. Organoruthenium and-osmium complexes of 2-pyridinecarbothioamides functionalized with a sulfonamide motif: Synthesis, cytotoxicity and biomolecule interaction. Chem. Plus. Chem. 2018, 83, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Kinney, W.A.; Lee, N.E.; Blank, R.M.; Demerson, C.A.; Sarnella, C.S.; Scherer, N.T.; Mir, G.N.; Borella, L.E.; DiJoseph, J.F.; Wells, C. N-Phenyl-2-pyridinecarbothioamides as gastric mucosal protectants. J. Med. Chem. 1990, 33, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Meier-Menches, S.M.; Zappe, K.; Bileck, A.; Kreutz, D.; Tahir, A.; Cichna-Markl, M.; Gerner, C. Time-dependent shotgun proteomics revealed distinct effects of an organoruthenium prodrug and its activation product on colon carcinoma cells. Metallomics 2018, 11, 118–127. [Google Scholar] [CrossRef]
- Bennett, M.A.; Smith, A.K. Arene ruthenium(II) complexes formed by dehydrogenation of cyclohexadienes with ruthenium(III) trichloride. J. Chem. Soc. Dalton Trans. 1974, 233–241. [Google Scholar] [CrossRef]
- Kiel, W.A.; Ball, R.G.; Graham, W.A. Carbonyl-η-hexamethylbenzene complexes of osmium. Carbon-hydrogen activation by (η-C6Me6)Os(CO)(H)2. J. Organomet. Chem. 1990, 383, 481–496. [Google Scholar] [CrossRef]
- Booth, B.; Haszeldine, R.; Hill, M. Organic reactions involving transition metals. Part I. Formation of pentamethylcyclopentadienylrhodium complexes by reaction of hexamethylbicyclo[2,2,0]hexa-2,5-diene with rhodium trichloride. J. Chem. Soc. A 1969, 1299–1303. [Google Scholar] [CrossRef]
- Ball, R.G.; Graham, W.A.G.; Heinekey, D.M.; Hoyano, J.K.; McMaster, A.D.; Mattson, B.M.; Michel, S.T. Synthesis and structure of [(η-C5Me5)Ir(CO)]2. Inorg. Chem. 1990, 29, 2023–2025. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment–Olex2 dissected. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tong, K.K.H.; Hanif, M.; Lovett, J.H.; Hummitzsch, K.; Harris, H.H.; Söhnel, T.; Jamieson, S.M.F.; Hartinger, C.G. Thiourea-Derived Chelating Ligands and Their Organometallic Compounds: Investigations into Their Anticancer Activity. Molecules 2020, 25, 3661. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | 3·EtAc | 4 | 6 |
|---|---|---|---|
| Bond length/Å | |||
| M–S | 2.3430(11) | 2.3671(10) | 2.3519(9) |
| C6–S | 1.689(4) | 1.731(4) | 1.682(4) |
| C6–N2 | 1.326(5) | 1.285(5) | 1.330(4) |
| M–N1 | 2.103(3) | 2.096(3) | 2.099(3) |
| M–X1 | 2.5465(6) | 2.7331(4) | 2.7165(3) |
| M–cymcentroid | 1.701 | 1.706 | 1.690 |
| Bond angle/° | |||
| S–M–N1 | 81.54(10) | 81.55(9) | 80.50(8) |
| N1–M–X1 | 82.36(10) | 84.14(9) | 83.56(8) |
| S–M–X | 89.50(3) | 89.75(3) | 89.74(2) |
| Compound | IC50 Values (μM) | |||
|---|---|---|---|---|
| HCT116 | NCI-H460 | SiHa | SW480 | |
| 1a | 5.7 ± 0.7 | 7.8 ± 1.8 | 16 ± 6 | 9.9 ± 0.7 |
| 2a | 6.5 ± 0.3 | 10 ± 2 | 8.3 ± 0.7 | 4.3 ± 1.2 |
| 3 | 7.7 ± 0.5 | 6.8 ± 1.0 | 17 ± 2 | 7.6 ± 0.7 |
| 4 | 7.5 ± 0.3 | 7.1 ± 0.9 | 17 ± 1 | 7.5 ± 0.8 |
| 5 | 18 ± 1 | 24 ± 2 | 21 ± 3 | 10 ± 2 |
| 6 | 19 ± 1 | 18 ± 1 | 31 ± 2 | 24 ± 1 |
| 7 | 11 ± 2 | 12 ± 2 | 22 ± 5 | 8.9 ± 1.1 |
| 8 | 15 ± 2 | 18 ± 4 | 46 ± 6 | 24 ± 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arshad, J.; Tong, K.K.H.; Movassaghi, S.; Söhnel, T.; Jamieson, S.M.F.; Hanif, M.; Hartinger, C.G. Impact of the Metal Center and Leaving Group on the Anticancer Activity of Organometallic Complexes of Pyridine-2-carbothioamide. Molecules 2021, 26, 833. https://doi.org/10.3390/molecules26040833
Arshad J, Tong KKH, Movassaghi S, Söhnel T, Jamieson SMF, Hanif M, Hartinger CG. Impact of the Metal Center and Leaving Group on the Anticancer Activity of Organometallic Complexes of Pyridine-2-carbothioamide. Molecules. 2021; 26(4):833. https://doi.org/10.3390/molecules26040833
Chicago/Turabian StyleArshad, Jahanzaib, Kelvin K. H. Tong, Sanam Movassaghi, Tilo Söhnel, Stephen M. F. Jamieson, Muhammad Hanif, and Christian G. Hartinger. 2021. "Impact of the Metal Center and Leaving Group on the Anticancer Activity of Organometallic Complexes of Pyridine-2-carbothioamide" Molecules 26, no. 4: 833. https://doi.org/10.3390/molecules26040833
APA StyleArshad, J., Tong, K. K. H., Movassaghi, S., Söhnel, T., Jamieson, S. M. F., Hanif, M., & Hartinger, C. G. (2021). Impact of the Metal Center and Leaving Group on the Anticancer Activity of Organometallic Complexes of Pyridine-2-carbothioamide. Molecules, 26(4), 833. https://doi.org/10.3390/molecules26040833