
Exploring the Interplay between Drug Release and Targeting of Lipid-Like Polymer Nanoparticles Loaded with Doxorubicin

 , , and
, , and
Abstract

1. Introduction
2. Results and Discussion
2.1. Physicochemical Characterization of Nanoparticles
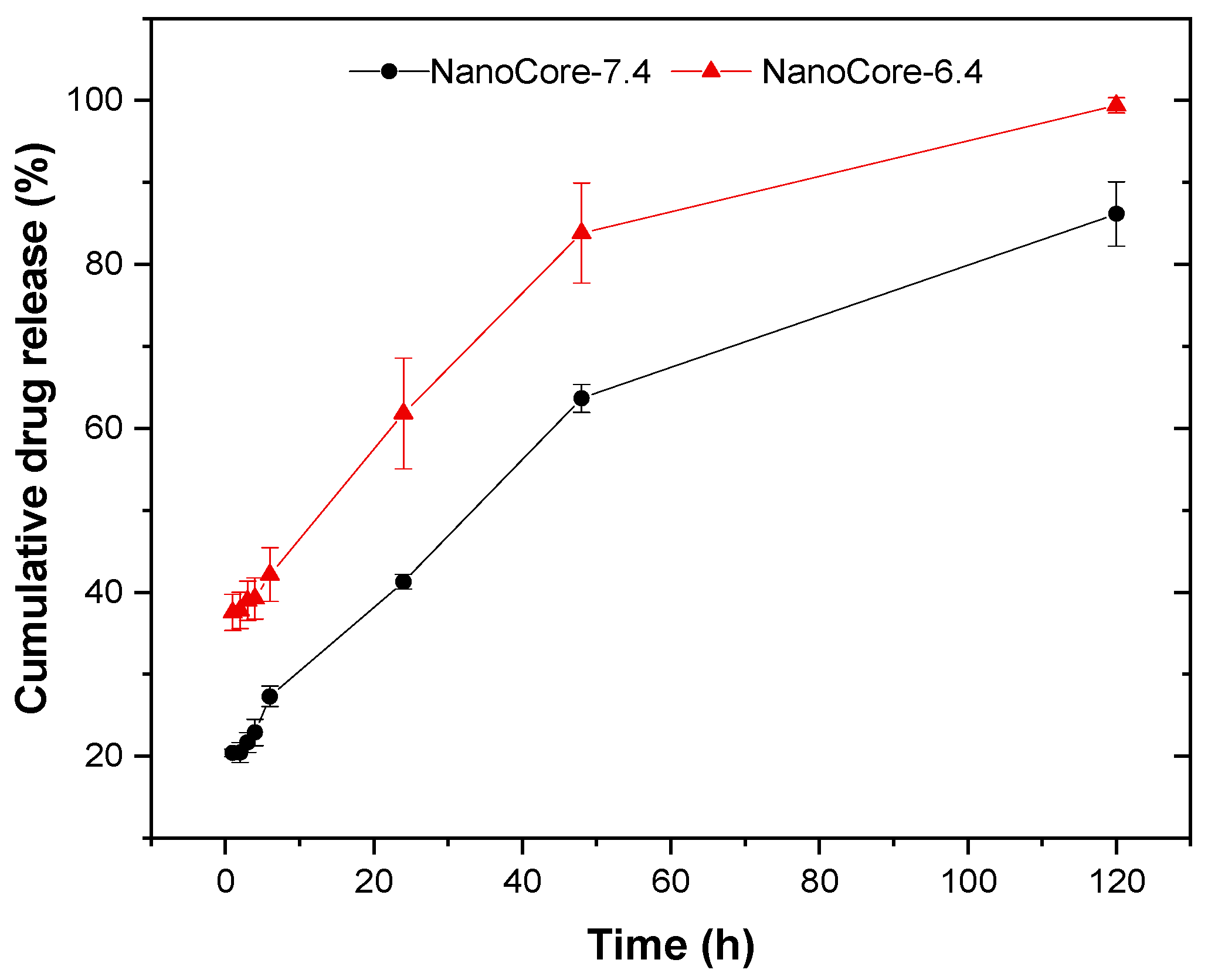
2.2. Evaluation of the In Vitro Release Kinetics of Doxorubicin
2.3. Evaluation of In Vitro Release Kinetics in Human Blood Plasma
2.4. Interaction of Doxorubicin and Nanoparticles with Red Blood Cells
2.5. Analysis of Pharmacokinetics
2.5.1. Non-Compartmental Analysis
2.5.2. Physiologically-Based Nanocarrier Biopharmaceutics Model
3. Materials and Methods
3.1. Materials
3.2. Preparation of NanoCore-7.4 and NanoCore-6.4
3.3. Characterization of Formulation Prototypes
3.3.1. Measurement of Nanoparticle Size, Size Distribution, and Zeta-Potential
3.3.2. Evaluation of Drug Content and Encapsulation Efficiency
3.3.3. Evaluation of PLGA Content
3.3.4. Evaluation of Poloxamer 188 Content
3.4. In Vitro Release Studies Using Simplified Release Media
3.5. In Vitro Release Studies Using Human Plasma
3.6. Quantification of Doxorubicin in Human Plasma
3.7. Analysis of In Vitro Drug Release Using Different Mathematical Models
3.8. Interaction of Nanoparticles with Human Red Blood Cells
3.9. Pharmacokinetic Study in Rats
3.9.1. Animal Experiments
3.9.2. Sample Preparation Technique Used to Determine Free and Total Doxorubicin in Rat Plasma
3.9.3. Quantification of Doxorubicin
3.10. Analysis of Plasma Pharmacokinetics Using Non-Compartmental Modeling
3.11. Analyzing Plasma Pharmacokinetics Using the Physiologically-Based Nanocarrier Biopharmaceutics Model
3.12. Data Analysis and Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Storm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38. [Google Scholar] [CrossRef]
- Siddique, S.; Chow, J.C.L. Application of Nanomaterials in Biomedical Imaging and Cancer Therapy. Nanomaterials 2020, 10, 1700. [Google Scholar] [CrossRef]
- Wacker, M. Nanocarriers for intravenous injection--the long hard road to the market. Int. J. Pharm. 2013, 457, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Wacker, M.G. Frontiers in pharmaceutical nanotechnology. Beilstein. J. Nanotechnol. 2019, 10, 2538–2540. [Google Scholar] [CrossRef]
- Cheo, S.T.T.; Lim, G.H.; Lim, K.H.C. Glioblastoma multiforme outcomes of 107 patients treated in two Singapore institutions. Singap. Med. J. 2017, 58, 41–45. [Google Scholar] [CrossRef]
- Feczko, T.; Piiper, A.; Ansar, S.; Blixt, F.W.; Ashtikar, M.; Schiffmann, S.; Ulshofer, T.; Parnham, M.J.; Harel, Y.; Israel, L.L.; et al. Stimulating brain recovery after stroke using theranostic albumin nanocarriers loaded with nerve growth factor in combination therapy. J. Control. Release 2019, 293, 63–72. [Google Scholar] [CrossRef]
- Salvalaio, M.; Rigon, L.; Belletti, D.; D’Avanzo, F.; Pederzoli, F.; Ruozi, B.; Marin, O.; Vandelli, M.A.; Forni, F.; Scarpa, M.; et al. Targeted Polymeric Nanoparticles for Brain Delivery of High Molecular Weight Molecules in Lysosomal Storage Disorders. PLoS ONE 2016, 11, e0156452. [Google Scholar] [CrossRef]
- Petri, B.; Bootz, A.; Khalansky, A.; Hekmatara, T.; Muller, R.; Uhl, R.; Kreuter, J.; Gelperina, S. Chemotherapy of brain tumour using doxorubicin bound to surfactant-coated poly(butyl cyanoacrylate) nanoparticles: Revisiting the role of surfactants. J. Control. Release 2007, 117, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Wohlfart, S.; Khalansky, A.S.; Gelperina, S.; Maksimenko, O.; Bernreuther, C.; Glatzel, M.; Kreuter, J. Efficient chemotherapy of rat glioblastoma using doxorubicin-loaded PLGA nanoparticles with different stabilizers. PLoS ONE 2011, 6, e19121. [Google Scholar] [CrossRef] [PubMed]
- Ganipineni, L.P.; Danhier, F.; Preat, V. Drug delivery challenges and future of chemotherapeutic nanomedicine for glioblastoma treatment. J. Control Release 2018, 281, 42–57. [Google Scholar] [CrossRef]
- Kim, S.S.; Harford, J.B.; Pirollo, K.F.; Chang, E.H. Effective treatment of glioblastoma requires crossing the blood-brain barrier and targeting tumors including cancer stem cells: The promise of nanomedicine. Biochem. Biophys. Res. Commun. 2015, 468, 485–489. [Google Scholar] [CrossRef]
- Zhao, M.; van Straten, D.; Broekman, M.L.D.; Preat, V.; Schiffelers, R.M. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics 2020, 10, 1355–1372. [Google Scholar] [CrossRef]
- Kreuter, J.; Ramge, P.; Petrov, V.; Hamm, S.; Gelperina, S.E.; Engelhardt, B.; Alyautdin, R.; von Briesen, H.; Begley, D.J. Direct evidence that polysorbate-80-coated poly(butylcyanoacrylate) nanoparticles deliver drugs to the CNS via specific mechanisms requiring prior binding of drug to the nanoparticles. Pharm. Res. 2003, 20, 409–416. [Google Scholar] [CrossRef]
- Kreuter, J.; Shamenkov, D.; Petrov, V.; Ramge, P.; Cychutek, K.; Koch-Brandt, C.; Alyautdin, R. Apolipoprotein-mediated transport of nanoparticle-bound drugs across the blood-brain barrier. J. Drug Target 2002, 10, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Steiniger, S.C.; Kreuter, J.; Khalansky, A.S.; Skidan, I.N.; Bobruskin, A.I.; Smirnova, Z.S.; Severin, S.E.; Uhl, R.; Kock, M.; Geiger, K.D.; et al. Chemotherapy of glioblastoma in rats using doxorubicin-loaded nanoparticles. Int. J. Cancer 2004, 109, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Zensi, A.; Begley, D.; Pontikis, C.; Legros, C.; Mihoreanu, L.; Wagner, S.; Buchel, C.; von Briesen, H.; Kreuter, J. Albumin nanoparticles targeted with Apo E enter the CNS by transcytosis and are delivered to neurones. J. Control. Release 2009, 137, 78–86. [Google Scholar] [CrossRef]
- Zensi, A.; Begley, D.; Pontikis, C.; Legros, C.; Mihoreanu, L.; Buchel, C.; Kreuter, J. Human serum albumin nanoparticles modified with apolipoprotein A-I cross the blood-brain barrier and enter the rodent brain. J. Drug Target 2010, 18, 842–848. [Google Scholar] [CrossRef]
- Pereverzeva, E.; Treschalin, I.; Bodyagin, D.; Maksimenko, O.; Kreuter, J.; Gelperina, S. Intravenous tolerance of a nanoparticle-based formulation of doxorubicin in healthy rats. Toxicol. Lett. 2008, 178, 9–19. [Google Scholar] [CrossRef]
- Sulheim, E.; Iversen, T.-G.; To Nakstad, V.; Klinkenberg, G.; Sletta, H.; Schmid, R.; Hatletveit, A.R.; Wågbø, A.M.; Sundan, A.; Skotland, T.; et al. Cytotoxicity of Poly(Alkyl Cyanoacrylate) Nanoparticles. Int. J. Mol. Sci. 2017, 18, 2454. [Google Scholar] [CrossRef]
- Khalin, I.; Severi, C.; Heimburger, D.; Wehn, A.; Hellal, F.; Reisch, A.; Klymchenko, A.S.; Plesnila, N. Highly fluorescent biodegradable PLGA nano-carriers allow real-time tracking of individual particles in vivo. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wohlfart, S.; Khalansky, A.S.; Gelperina, S.; Begley, D.; Kreuter, J. Kinetics of transport of doxorubicin bound to nanoparticles across the blood-brain barrier. J. Control. Release 2011, 154, 103–107. [Google Scholar] [CrossRef]
- Gelperina, S.; Maksimenko, O.; Khalansky, A.; Vanchugova, L.; Shipulo, E.; Abbasova, K.; Berdiev, R.; Wohlfart, S.; Chepurnova, N.; Kreuter, J. Drug delivery to the brain using surfactant-coated poly(lactide-co-glycolide) nanoparticles: Influence of the formulation parameters. Eur. J. Pharm. Biopharm. 2010, 74, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Filon, O.; Krivorotko, P.; Kobyakov, G.; Razjivina, V.; Maximenko, O.; Gelperina, S.; Kreuter, J. A phase I study of safety and pharmacokinetics of NanoBB-1-Dox in patients with advanced solid tumors. J. Clin. Oncol. 2017, 35, e13537. [Google Scholar] [CrossRef]
- Rodrigues de Azevedo, C.; von Stosch, M.; Costa, M.S.; Ramos, A.M.; Cardoso, M.M.; Danhier, F.; Préat, V.; Oliveira, R. Modeling of the burst release from PLGA micro- and nanoparticles as function of physicochemical parameters and formulation characteristics. Int. J. Pharm. 2017, 532, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Janas, C.; Mast, M.P.; Kirsamer, L.; Angioni, C.; Gao, F.; Mantele, W.; Dressman, J.; Wacker, M.G. The dispersion releaser technology is an effective method for testing drug release from nanosized drug carriers. Eur. J. Pharm. Biopharm. 2017, 115, 73–83. [Google Scholar] [CrossRef]
- Nagpal, S.; Braner, S.; Modh, H.; Tan, A.X.X.; Mast, M.P.; Chichakly, K.; Albrecht, V.; Wacker, M.G. A physiologically-based nanocarrier biopharmaceutics model to reverse-engineer the in vivo drug release. Eur. J. Pharm. Biopharm. 2020, 153, 257–272. [Google Scholar] [CrossRef]
- Fulop, Z.; Gref, R.; Loftsson, T. A permeation method for detection of self-aggregation of doxorubicin in aqueous environment. Int. J. Pharm. 2013, 454, 559–561. [Google Scholar] [CrossRef]
- Maksimenko, O.; Malinovskaya, J.; Shipulo, E.; Osipova, N.; Razzhivina, V.; Arantseva, D.; Yarovaya, O.; Mostovaya, U.; Khalansky, A.; Fedoseeva, V.; et al. Doxorubicin-loaded PLGA nanoparticles for the chemotherapy of glioblastoma: Towards the pharmaceutical development. Int. J. Pharm. 2019, 572, 118733. [Google Scholar] [CrossRef]
- Kreuter, J. Drug delivery to the central nervous system by polymeric nanoparticles: What do we know? Adv. Drug Deliv. Rev. 2014, 71, 2–14. [Google Scholar] [CrossRef]
- Pereverzeva, E.; Treschalin, I.; Treschalin, M.; Arantseva, D.; Ermolenko, Y.; Kumskova, N.; Maksimenko, O.; Balabanyan, V.; Kreuter, J.; Gelperina, S. Toxicological study of doxorubicin-loaded PLGA nanoparticles for the treatment of glioblastoma. Int. J. Pharm. 2019, 554, 161–178. [Google Scholar] [CrossRef]
- Fugit, K.D.; Xiang, T.X.; Choi du, H.; Kangarlou, S.; Csuhai, E.; Bummer, P.M.; Anderson, B.D. Mechanistic model and analysis of doxorubicin release from liposomal formulations. J. Control. Release 2015, 217, 82–91. [Google Scholar] [CrossRef]
- Nothnagel, L.; Wacker, M.G. How to measure release from nanosized carriers? Eur. J. Pharm. Sci. 2018, 120, 199–211. [Google Scholar] [CrossRef]
- Feczko, T.; Piiper, A.; Pleli, T.; Schmithals, C.; Denk, D.; Hehlgans, S.; Rodel, F.; Vogl, T.J.; Wacker, M.G. Theranostic Sorafenib-Loaded Polymeric Nanocarriers Manufactured by Enhanced Gadolinium Conjugation Techniques. Pharmaceutics 2019, 11, 489. [Google Scholar] [CrossRef] [PubMed]
- Hines, D.J.; Kaplan, D.L. Poly(lactic-co-glycolic) acid-controlled-release systems: Experimental and modeling insights. Crit. Rev. Ther. Drug Carr. Syst. 2013, 30, 257–276. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Mohammadi, G.; Barzegar-Jalali, M.; Valizadeh, H.; Nazemiyeh, H.; Barzegar-Jalali, A.; Siahi Shadbad, M.R.; Adibkia, K.; Zare, M. Reciprocal powered time model for release kinetic analysis of ibuprofen solid dispersions in oleaster powder, microcrystalline cellulose and crospovidone. J. Pharm. Pharm. Sci. 2010, 13, 152–161. [Google Scholar] [CrossRef]
- Wallenwein, C.M.; Nova, M.V.; Janas, C.; Jablonka, L.; Gao, G.F.; Thurn, M.; Albrecht, V.; Wiehe, A.; Wacker, M.G. A dialysis-based in vitro drug release assay to study dynamics of the drug-protein transfer of temoporfin liposomes. Eur. J. Pharm. Biopharm. 2019, 143, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Laubrock, N.; Hempel, G.; Schulze-Westhoff, P.; Würthwein, G.; Flege, S.; Boos, J. The stability of doxorubicin and ldarubicin in plasma and whole blood. Chromatographia 2000, 52, 9–13. [Google Scholar] [CrossRef]
- Piazza, E.; Broggini, M.; Trabattoni, A.; Natale, N.; Libretti, A.; Donelli, M.G. Adriamycin distribution in plasma and blood cells of cancer patients with altered hematocrit. Eur. J. Cancer Clin. Oncol. 1981, 17, 1089–1096. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Gupta, V.; Zern, B.J.; Pan, D.; Zakrewsky, M.; Muzykantov, V.; Mitragotri, S. Delivering nanoparticles to lungs while avoiding liver and spleen through adsorption on red blood cells. ACS Nano 2013, 7, 11129–11137. [Google Scholar] [CrossRef] [PubMed]
- Colombo, T.; Broggini, M.; Garattini, S.; Donelli, M.G. Differential adriamycin distribution to blood components. Eur. J. Drug Metab. Pharm. 1981, 6, 115–122. [Google Scholar] [CrossRef]
- Yu, S.; Li, S.; Yang, H.; Lee, F.; Wu, J.T.; Qian, M.G. A novel liquid chromatography/tandem mass spectrometry based depletion method for measuring red blood cell partitioning of pharmaceutical compounds in drug discovery. Rapid Commun. Mass Spectrom. 2005, 19, 250–254. [Google Scholar] [CrossRef]
- Kalamaridis, D.; DiLoreto, K. Drug Partition in Red Blood Cells. In Optimization in Drug Discovery: In Vitro Methods; Caldwell, G.W., Yan, Z., Eds.; Humana Press: Totowa, NJ, USA, 2014; pp. 39–47. [Google Scholar] [CrossRef]
- Rahman, A.; Carmichael, D.; Harris, M.; Roh, J.K. Comparative pharmacokinetics of free doxorubicin and doxorubicin entrapped in cardiolipin liposomes. Cancer Res. 1986, 46, 2295–2299. [Google Scholar]
- Le Ray, A.M.; Vert, M.; Gautier, J.C.; Benoît, J.P. Fate of [14C]poly(DL-lactide-co-glycolide) nanoparticles after intravenous and oral administration to mice. Int. J. Pharm. 1994, 106, 201–211. [Google Scholar] [CrossRef]
- Rafiei, P.; Haddadi, A. Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: Pharmacokinetics and biodistribution profile. Int. J. Nanomed. 2017, 12, 935–947. [Google Scholar] [CrossRef]
- Ishihara, T.; Maeda, T.; Sakamoto, H.; Takasaki, N.; Shigyo, M.; Ishida, T.; Kiwada, H.; Mizushima, Y.; Mizushima, T. Evasion of the accelerated blood clearance phenomenon by coating of nanoparticles with various hydrophilic polymers. Biomacromolecules 2010, 11, 2700–2706. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.B.; Blaufox, M.D. Blood volume in the rat. J. Nucl. Med. 1985, 26, 72–76. [Google Scholar]
- Yuan, D.; He, H.; Wu, Y.; Fan, J.; Cao, Y. Physiologically Based Pharmacokinetic Modeling of Nanoparticles. J. Pharm. Sci. 2019, 108, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Nothnagel, L.; Jung, F.; Rossmanith, T.; Thurn, M.; Ashtikar, M.; Geisslinger, G.; Parnham, M.J.; Wacker, M.G. Predictive PBPK modeling as a tool in the formulation of the drug candidate TMP-001. Eur. J. Pharm. Biopharm. 2019, 134, 144–152. [Google Scholar] [CrossRef]
- Malinovskaya, Y.; Melnikov, P.; Baklaushev, V.; Gabashvili, A.; Osipova, N.; Mantrov, S.; Ermolenko, Y.; Maksimenko, O.; Gorshkova, M.; Balabanyan, V.; et al. Delivery of doxorubicin-loaded PLGA nanoparticles into U87 human glioblastoma cells. Int. J. Pharm. 2017, 524, 77–90. [Google Scholar] [CrossRef]
- Heron, M.W.; Paton, B.C. A method for measuring a nonionic surface-active agent (Pluronic F-68) in biological fluids. Anal. Biochem. 1968, 24, 491–495. [Google Scholar] [CrossRef]
- Bian, X.; Liang, S.; John, J.; Hsiao, C.-H.; Wei, X.; Liang, D.; Xie, H. Development of PLGA-based itraconazole injectable nanospheres for sustained release. Int. J. Nanomed. 2013, 8, 4521–4531. [Google Scholar] [CrossRef][Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | pH of External Aqueous Phase | Encapsulation Efficiency (%) | Drug Load (%) | Size Distribution | Zeta Potential (mV) | |||
|---|---|---|---|---|---|---|---|---|
| Mean Particle Diameter (nm) | PDI | Intensity (nm) | Volume (nm) | |||||
| NanoCore-7.4 | 7.4 | 91 ± 1 | 9.1 ± 0.3 | 105 ± 12 | 0.144 ± 0.011 | 118 (100%) | 94 (100%) | −10.46 ± 1.33 |
| NanoCore-6.4 | 6.4 | 80 ± 1 | 7.8 ± 0.1 | 137 ± 7 | 0.301 ± 0.069 | 157 (94%) | 119 (91%) | −6.40 ± 2.27 |
| Formulation | pH of External Aqueous Phase | Size Distribution | |||
|---|---|---|---|---|---|
| Mean Particle Diameter (nm) | PDI | Intensity (nm) | Volume (nm) | ||
| NanoCore-7.4 | 7.4 | 93 ± 7 | 0.092 ± 0.012 | 106 (100%) | 85 (100%) |
| NanoCore-6.4 | 6.4 | 89 ± 12 | 0.110 ± 0.003 | 99.8 (100%) | 80 (100%) |
| Formulation | Time, h | K | n | R2 |
|---|---|---|---|---|
| NanoCore-7.4 | 1–6 | 0.1792 | 0.1745 | 0.9199 |
| 1–48 | 0.1629 | 0.3152 | 0.9030 | |
| 1–120 | 0.1581 | 0.3115 | 0.9584 | |
| NanoCore-6.4 | 1–6 | 0.3614 | 0.0521 | 0.8603 |
| 1–48 | 0.3424 | 0.1865 | 0.8726 | |
| 1–120 | 0.3009 | 0.2459 | 0.9293 |
| Time | Doxorubicin Concentration (μg/mL) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 10 | 50 | 100 | |||||||
| Free Doxorubicin | Nano- Core-7.4 | Nano- Core-6.4 | Free Doxorubicin | Nano- Core-7.4 | Nano- Core-6.4 | Free Doxorubicin | Nano- Core-7.4 | Nano- Core-6.4 | |
| 5 min | 35 | 33 | 36 | 33 | 32 | 33 | 33 | 31 | 32 |
| 15 min | 59 | 49 | 57 | 62 | 46 | 58 | 63 | 49 | 58 |
| 30 min | 60 | 50 | 55 | 61 | 47 | 50 | 64 | 47 | 57 |
| Formulation | Parameter | Mean | Min | Max | SD | CV |
|---|---|---|---|---|---|---|
| NanoCore-7.4total (5 mg/kg) | Half-life (h) | 0.374 | 0.375 | 0.372 | 0.001 | 0.003 |
| m | 0.468 | 0.468 | 0.466 | 0.000 | 0.001 | |
| b | 0.832 | 0.840 | 0.826 | 0.005 | 0.006 | |
| c | 9.052 | 9.058 | 9.046 | 0.004 | 0.000 | |
| VDC (mL) | 11.935 | 12.018 | 11.862 | 0.050 | 0.004 | |
| Ftarget (%) | 7.415 | 7.615 | 7.310 | 0.110 | 0.015 | |
| NanoCore-6.4total (5 mg/kg) | Half-life (h) | 0.395 | 0.401 | 0.391 | 0.004 | 0.009 |
| m | 0.460 | 0.470 | 0.449 | 0.008 | 0.017 | |
| b | 0.880 | 0.883 | 0.877 | 0.002 | 0.002 | |
| c | 11.227 | 11.256 | 11.173 | 0.031 | 0.003 | |
| VDC (mL) | 10.521 | 10.595 | 10.433 | 0.051 | 0.005 | |
| Ftarget (%) | 5.899 | 6.057 | 5.740 | 0.106 | 0.018 | |
| NanoCore-6.4total (2 mg/kg) | Half-life (h) | 0.388 | 0.392 | 0.384 | 0.002 | 0.006 |
| m | 0.413 | 0.430 | 0.398 | 0.012 | 0.029 | |
| b | 0.870 | 0.874 | 0.866 | 0.002 | 0.003 | |
| c | 11.364 | 11.405 | 11.301 | 0.035 | 0.003 | |
| VDC (mL) | 10.795 | 10.871 | 10.723 | 0.045 | 0.004 | |
| Ftarget (%) | 5.143 | 5.426 | 4.885 | 0.183 | 0.036 |
| Zero-order model | Higuchi model | ||
| First-order model | Hickson-Crowell model | ||
| Korsmeyer-Peppas model | 3-parametric reciprocal powered time model |
| Parameter | Range |
|---|---|
| VDC (L) [48] | 8.325 ± 4.03 (ω: 0.5) |
| t½ (h) a [46,53] | 0.5 ± 0.71 (ω: 0.5) |
| t½ (h) b | 0.364 ± 0.017 (ω: 0.5) |
| kF (h−1) | 1.074 ± 0.46 (ω: 0.5) |
| k12 (h−1) | 13.77 ± 2.62 (ω: 0.5) |
| k21 (h−1) | 0.68 ± 0.21 (ω: 0.5) |
| VDF (mL) | 787.69 ± 293.13 (ω: 0.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovshova, T.; Osipova, N.; Alekseeva, A.; Malinovskaya, J.; Belov, A.; Budko, A.; Pavlova, G.; Maksimenko, O.; Nagpal, S.; Braner, S.; et al. Exploring the Interplay between Drug Release and Targeting of Lipid-Like Polymer Nanoparticles Loaded with Doxorubicin. Molecules 2021, 26, 831. https://doi.org/10.3390/molecules26040831
Kovshova T, Osipova N, Alekseeva A, Malinovskaya J, Belov A, Budko A, Pavlova G, Maksimenko O, Nagpal S, Braner S, et al. Exploring the Interplay between Drug Release and Targeting of Lipid-Like Polymer Nanoparticles Loaded with Doxorubicin. Molecules. 2021; 26(4):831. https://doi.org/10.3390/molecules26040831
Chicago/Turabian StyleKovshova, Tatyana, Nadezhda Osipova, Anna Alekseeva, Julia Malinovskaya, Alexey Belov, Andrey Budko, Galina Pavlova, Olga Maksimenko, Shakti Nagpal, Svenja Braner, and et al. 2021. "Exploring the Interplay between Drug Release and Targeting of Lipid-Like Polymer Nanoparticles Loaded with Doxorubicin" Molecules 26, no. 4: 831. https://doi.org/10.3390/molecules26040831
APA StyleKovshova, T., Osipova, N., Alekseeva, A., Malinovskaya, J., Belov, A., Budko, A., Pavlova, G., Maksimenko, O., Nagpal, S., Braner, S., Modh, H., Balabanyan, V., Wacker, M. G., & Gelperina, S. (2021). Exploring the Interplay between Drug Release and Targeting of Lipid-Like Polymer Nanoparticles Loaded with Doxorubicin. Molecules, 26(4), 831. https://doi.org/10.3390/molecules26040831