
Easy-To-Access Quinolone Derivatives Exhibiting Antibacterial and Anti-Parasitic Activities

 , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussions
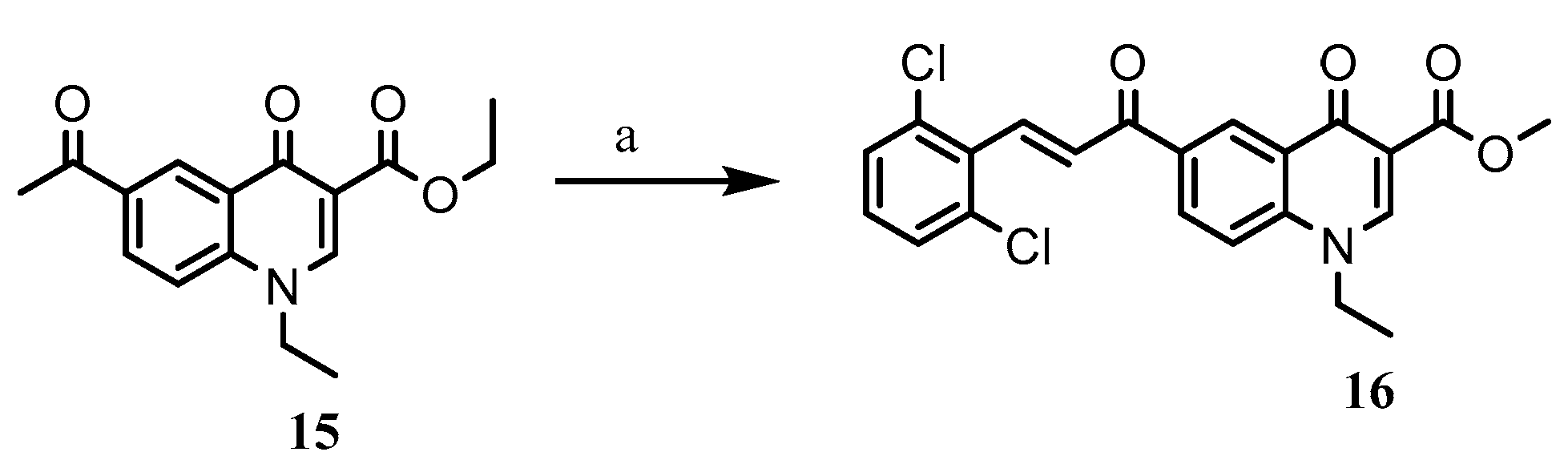
2.1. Chemistry
2.2. In Vitro Biological Evaluation
2.2.1. Anti-Mtb Activity
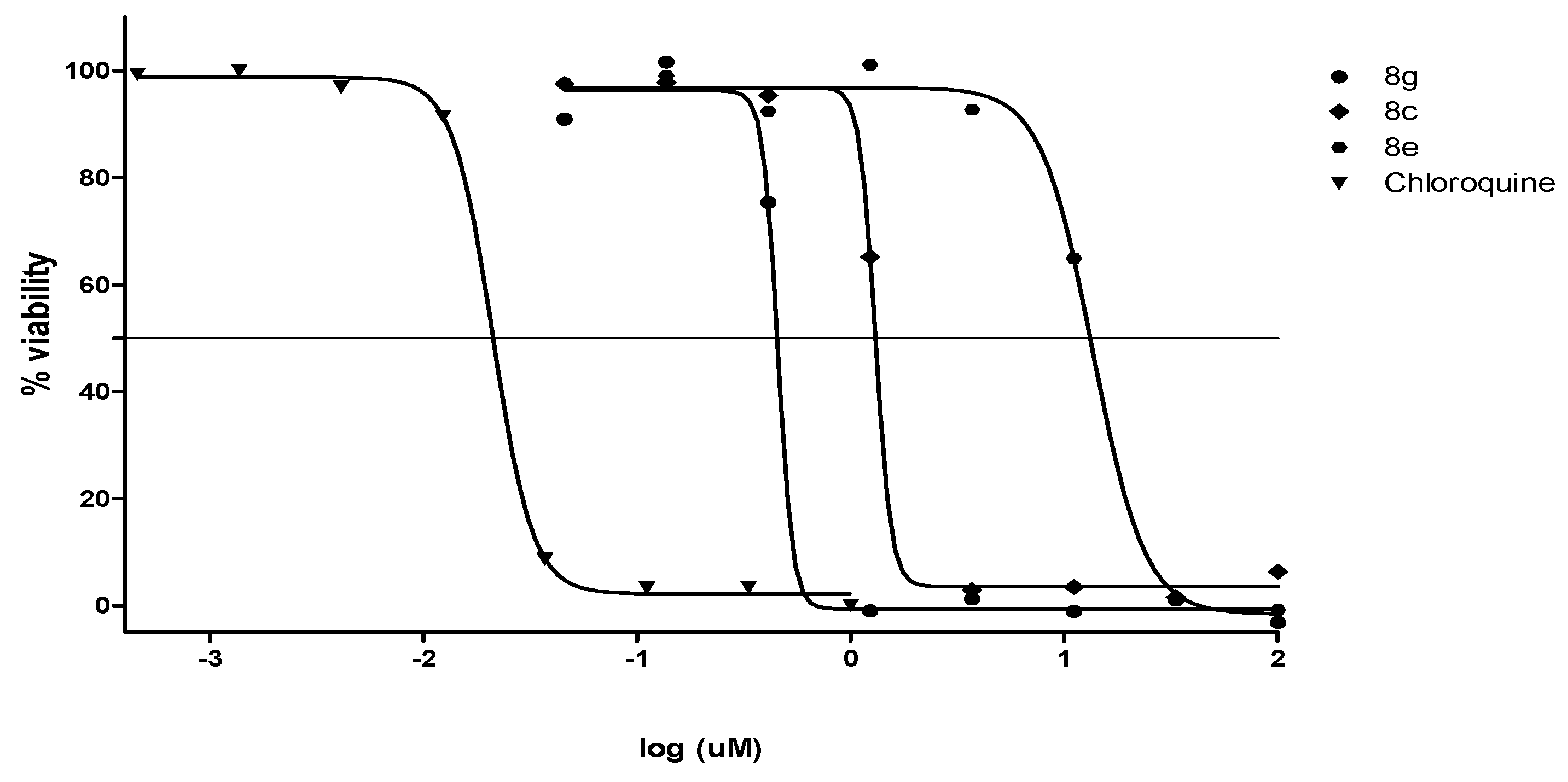
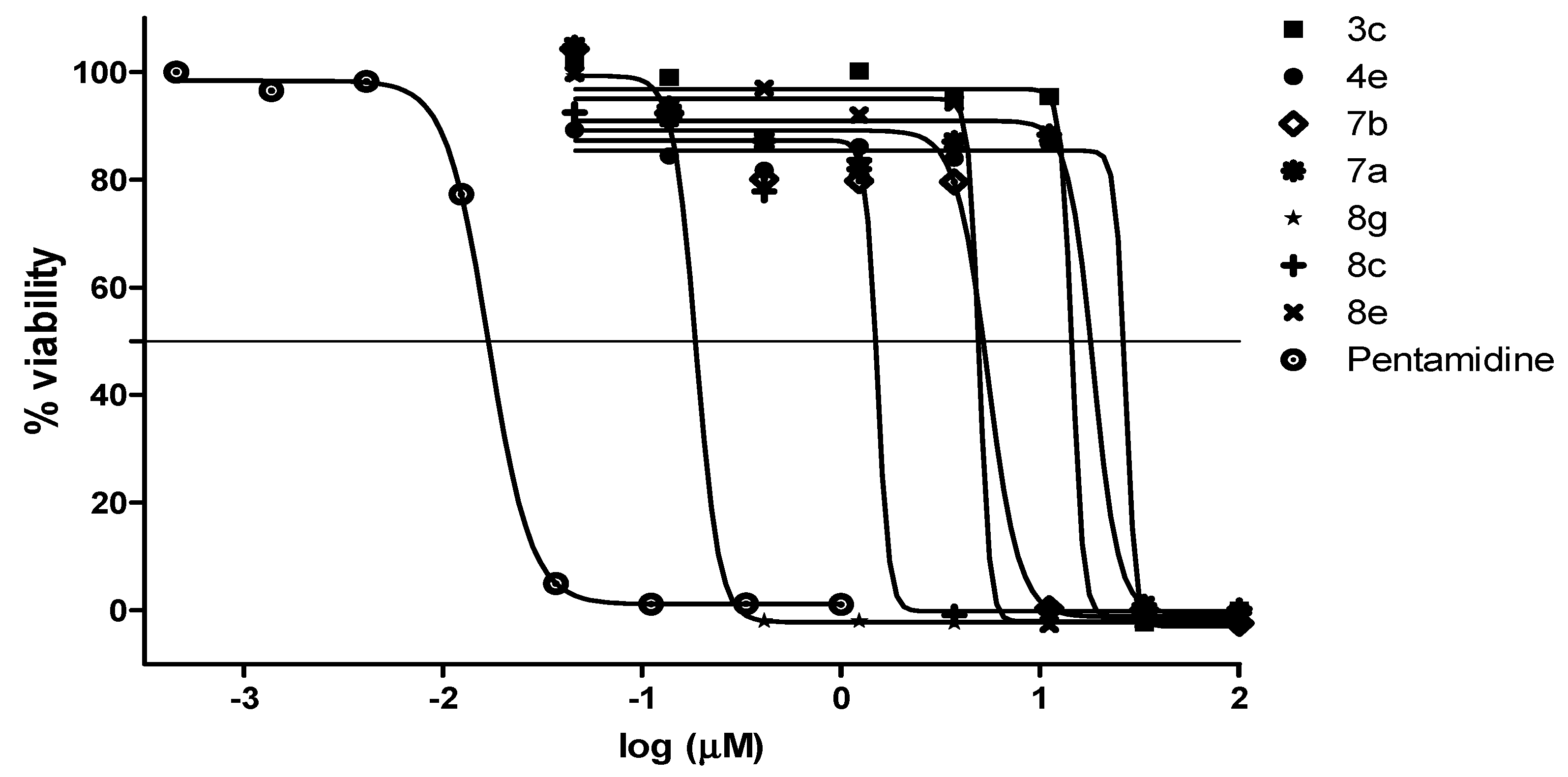
2.2.2. Antiplasmodial and Anti-Trypanosomal Activities
2.2.3. Antimicrobial Activities and Cytotoxicity Evaluation
2.3. Lipophilicity
3. Materials and Methods
3.1. Chemistry: Materials and Instrumentation
3.2. Synthesis of Target Compounds
3.2.1. N-alkylation
3.2.2. Ester Aminolysis
3.2.3. Imine Formation
3.3. In Vitro Antimycobacterial Assay
3.4. In Vitro Antiplasmodial Assay
3.5. In Vitro Cytotoxicity Assay
3.6. In Vitro Anti-Trypanosomal Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Brites, D.; Gagneux, S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol. Rev. 2015, 264, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Cambau, E.; Drancourt, D. Steps towards the discovery of Mycobacterium tuberculosis by Robert Koch, 1882. Clin. Microbiol. Infect. 2014, 20, 196–201. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Tuberculosis Report 2019; WHO: Geneva, Switzerland, 2019; Available online: https://apps.who.int/iris/bitstream/handle/10665/329368/9789241565714-eng.pdf (accessed on 1 May 2020).
- Pescarini, J.M.; Rodrigues, L.C.; Gabriela, M.; Gomes, M.; Waldman, E.A. Migration to middle-income countries and tuberculosis—Global policies for global economies. Glob. Health 2017, 13, 15. [Google Scholar] [CrossRef]
- Millet, J.-P.; Moreno, A.; Fina, L.; del Baño, L.; Orcau, A.; García de Olalla, P.; Caylà, J.A. Factors that influence current tuberculosis epidemiology. Eur. Spine J. 2013, 22, 539–548. [Google Scholar] [CrossRef]
- Kwan, C.K.; Ernst, J.D. HIV and tuberculosis: A deadly human syndemic. Clin. Microbiol. Rev. 2011, 24, 351–376. [Google Scholar] [CrossRef]
- Marriner, G.A.; Nayyar, A.; Uh, E.; Wong, S.Y.; Mukherjee, T.; Via, L.E.; Carroll, M.; Edwards, R.L.; Gruber, T.D.; Choi, I.; et al. The medicinal chemistry of tuberculosis chemotherapy. Top. Med. Chem. 2011, 7, 47–124. [Google Scholar]
- Tiberi, S.; Muñoz-Torrico, M.; Duarted, R.; Dalcolmo, M.; D’Ambrosio, L.; Migliori, G.-B. New drugs and perspectives for new anti-tuberculosis regimens. Pulmonology 2018, 24, 86–98. [Google Scholar] [CrossRef]
- Nguyen, L. Antibiotic resistance mechanisms in M. tuberculosis: An update. Arch. Toxicol. 2016, 90, 1585–1604. [Google Scholar] [CrossRef] [PubMed]
- Genestet, C.; Ader, F.; Pichat, C.; Lina, G.; Dumitrescu, O.; Goutelle, S. Assessing the combined antibacterial effect of isoniazid and rifampin on four Mycobacterium tuberculosis strains using in vitro experiments and response-surface modeling. Antimicrob. Agents Chemother. 2018, 62, e01413-17. [Google Scholar] [CrossRef]
- Wirth, D.; Dass, R.; Hettle, R. Cost-effectiveness of adding novel or group 5 interventions to a background regimen for the treatment of multidrug-resistant tuberculosis in Germany. BMC Health Serv. Res. 2017, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.J. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 2003, 83, 91–97. [Google Scholar] [CrossRef]
- Bhat, Z.S.; Rather, M.A.; Maqbool, M.; UL Lah, H.; Yousuf, S.K.; Ahmad, Z. Cell wall: A versatile fountain of drug targets in Mycobacterium tuberculosis. Biomed. Pharmacother. 2017, 95, 1520–1534. [Google Scholar] [CrossRef] [PubMed]
- Favrot, L.; Ronning, D.R. Targeting the mycobacterial envelope for tuberculosis drug development. Expert Rev. Anti-Infect. Ther. 2012, 10, 1023–1036. [Google Scholar] [CrossRef]
- Chen, H.; Nyantakyi, S.A.; Li, M.; Gopal, P.; Aziz, D.B.; Yang, T.; Moreira, W.; Gengenbacher, M.; Dick, T.; Go, M.L. The mycobacterial membrane: A novel target space for anti-tubercular drugs. Front. Microbiol. 2018, 9, 1627. [Google Scholar] [CrossRef] [PubMed]
- SilvaMiranda, M.; Breiman, A.; Allain, S.; Deknuydt, F.; Altare, F. The tuberculous granuloma: An unsuccessful host defence mechanism providing a safety shelter for the bacteria? Clin. Dev. Immunol. 2012, 139127. [Google Scholar] [CrossRef]
- Yuan, T.; Sampson, N.S. Hit Generation in TB Drug Discovery: From Genome to Granuloma. Chem. Rev. 2018, 118, 1887–1916. [Google Scholar] [CrossRef]
- Ehlers, S.; Schaible, U.E. The Granuloma in Tuberculosis: Dynamics of a Host–Pathogen Collusion. Front. Immunol. 2013, 3, 411. [Google Scholar] [CrossRef]
- Ndlovu, H.; Marakalala, M.J. Granulomas and inflammation: Host-Directed Therapies for Tuberculosis. Front. Immunol. 2016, 7, 434. [Google Scholar] [CrossRef] [PubMed]
- Machado, D.; Girardini, M.; Viveiros, M.; Pieroni, M. Challenging the drug-likeness dogma for new drug discovery in tuberculosis. Front. Microbiol. 2018, 9, 1367. [Google Scholar] [CrossRef]
- Goldman, R.C. Why are membrane targets discovered by phenotypic screens and genome sequencing in Mycobacterium tuberculosis? Tuberculosis 2013, 93, 569–588. [Google Scholar] [CrossRef] [PubMed]
- Cholo, M.C.; Mothiba, M.T.; Fourie, B.; Anderson, R. Mechanisms of action and therapeutic efficacies of the lipophilic antimycobacterial agents clofazimine and bedaquiline. J. Antimicrob. Chemother. 2017, 72, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, D.T.; Liu, J.; Lee, R.B.; Lee, R.E. New agents for the treatment of drug-resistant Mycobacterium tuberculosis. Adv. Drug Deliver. Rev. 2016, 102, 55–72. [Google Scholar] [CrossRef]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; Arena, A.; Arrieta, F.; Rebolledo, J.C.J.; Smith, D.K. Nitrotriazole- and imidazole-based amides and sulfonamides as antitubercular agents. Antimicrob. Agents Chemother. 2014, 58, 6828–6836. [Google Scholar] [CrossRef]
- Vitoria, M.; Granich, R.; Gilks, C.F.; Gunneberg, C.; Hosseini, M.; Were, W.; Raviglione, M.; De Cock, K.M. The Global fight against HIV/AIDS, tuberculosis, and malaria. Am. J. Clin. Pathol. 2009, 131, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Piperaki, E.T.; Daikos, G.L. Malaria in Europe: Emerging threat or minor nuisance? Clin. Microbiol. Infect. 2016, 22, 487–493. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Malaria Report 2019; WHO: Geneva, Switzerland, 2019; Available online: https://www.who.int/publications-detail/world-malaria-report-2019 (accessed on 9 April 2020).
- Schlitzer, M. Antimalarial drugs–what is in use and what is in the pipeline. Arch. Pharm. Chem. Life Sci. 2008, 341, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Dini, S.; Zaloumis, S.; Cao, P.; Price, R.N.; Fowkes, F.J.I.; van der Pluijm, R.W.; McCaw, J.M.; Simpson, J.A. Investigating the efficacy of triple artemisinin-based combination therapies for treating Plasmodium falciparum malaria patients using mathematical modeling. Antimicrob. Agents Chemother. 2018, 62, e0106818. [Google Scholar] [CrossRef]
- Okomboa, J.; Chibale, K. Recent updates in the discovery and development of novel antimalarial drug candidates. Med. Chem. Commun. 2018, 9, 437–453. [Google Scholar] [CrossRef]
- Thu, A.M.; Phyo, A.P.; Landier, J.; Parker, D.M.; Nosten, F.H. Combating multidrug-resistant Plasmodium falciparum malaria. FEBS J. 2017, 284, 2569–2578. [Google Scholar] [CrossRef]
- Brun, R.; Schumacher, R.; Schmid, C.; Kunz, C.; Burri, C. The phenomenon of treatment failures in human African trypanosomiasis. Trop. Med. Int. Health. 2001, 6, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Global Health Observatory Data. Available online: https://apps.who.int/gho/data/node.main.A1635?lang=en (accessed on 9 April 2020).
- Simarro, P.; Cecchi, G.; Franco, J.; Paone, M.; Diarra, A.; Ruiz-Postigo, J.; Fèvre, E.; Mattioli, R.; Jannin, G. Estimating and mapping the population at risk of sleeping sickness. PLoS. Negl. Trop. Dis. 2012, 6, e1859. [Google Scholar] [CrossRef]
- Pohlig, G.; Bernhard, S.; Blum, J.; Burri, C.; Mpanya, A.; Lubaki, J.P.; Mpoto, A.M.; N’tombe, P.M.; Deo, G.K.; Mutantu, P.N.; et al. Efficacy and safety of pafuramidine versus pentamidine maleate for treatment of first stage sleeping sickness in a randomized, comparator-controlled, international phase 3 clinical trial. PLoS Negl. Trop. Dis. 2016, 10, e0004363. [Google Scholar] [CrossRef]
- Burri, C. Chemotherapy against human African trypanosomiasis: Is there a road to success? Parasitology 2010, 137, 1987–1994. [Google Scholar] [CrossRef]
- Chappuis, F.; Udayraj, N.; Stietenroth, K.; Meussen, A.; Bovier, P.A. Eflornithine is safer than melarsoprol for the treatment of second-stage Trypanosoma brucei gambiense human African trypanosomiasis. Clin. Inf. Dis. 2005, 41, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Berninger, M.; Schmidt, I.; Ponte-Sucre, A.; Holzgrabe, U. Novel lead compounds in pre-clinical development against African sleeping sickness. Med. Chem. Commun. 2017, 8, 1872–1890. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Fexinidazole: First global approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef]
- Beteck, R.M.; Seldon, R.; Jordaan, A.; Warner, D.F.; Hoppe, H.C.; Laming, D.; Legoabe, L.J.; Khanye, S.D. Quinolone-isoniazid hybrids: Synthesis and preliminary in vitro cytotoxicity and anti-tuberculosis evaluation. Med. Chem. Commun. 2019, 10, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Beteck, R.M.; Isaacs, M.; Hoppe, H.C.; Khanye, S.D. Synthesis, in vitro cytotoxicity and trypanocidal evaluation of novel 1,3,6-substituted non-fluoroquinolones. S. Afr. J. Chem. 2018, 71, 188–195. [Google Scholar] [CrossRef]
- Dhiman, P.; Arora, N.; Thanikachalam, P.V.; Monga, V. Recent advances in the synthetic and medicinal perspective of quinolones: A review. Bioorg. Chem. 2019, 92, 103291. [Google Scholar] [CrossRef]
- Alemu, A.; Kebede, A.; Dagne, B.; Amare, M.; Diriba, G.; Yenew, B.; Tesfaye, E.; Tadesse, M.; Sinshaw, W.; Challa, D.; et al. Intestinal parasites co-infection and associated factors among active pulmonary tuberculosis patients in selected health centers, Addis Ababa, Ethiopia: Unmatched case control study. BMC Infect. Dis. 2019, 19, 407. [Google Scholar] [CrossRef]
- Abate, E.; Belayneh, M.; Gelaw, A.; Idh, J.; Getachew, A.; Alemu, S.; Diro, E.; Fikre, N.; Britton, S.; Elias, D.; et al. The impact of asymptomatic Helminth co-infection in patients with newly diagnosed tuberculosis in North-West Ethiopia. PLoS ONE 2012, 7, e42901. [Google Scholar] [CrossRef]
- Li, X.-X.; Zhou, X.-N. Co-infection of tuberculosis and parasitic diseases in humans: A systemic review. Parasit. Vectors 2014, 6, 79. [Google Scholar] [CrossRef]
- Hotez, P.J.; Molyneux, D.H.; Fenwick, A.; Ottesen, E.; Sachs, S.E.; Sachs, J.D. Incorporating a rapid-impact package for neglected tropical diseases with programs for HIV/AIDS, tuberculosis, and malaria. PLoS Med. 2006, 3, e102. [Google Scholar] [CrossRef]
- Daly, J.W.; Padgett, W.L.; Shamim, M.T. Analogs of caffeine and theophylline: Effect of structural alterations on affinity at adenosine receptors. J. Med. Chem. 1986, 29, 1305–1308. [Google Scholar] [CrossRef]
- Beteck, R.M.; Seldon, R.; Coertzen, D.; van der Watt, M.E.; Reader, J.; Mackenzie, J.S.; Lamprecht, D.A.; Abraham, M.; Eribez, K.; Müller, J.; et al. Accessible and distinct decoquinate derivatives active against Mycobacterium tuberculosis and apicomplexan parasites. Comm. Chem. 2018, 1, 62. [Google Scholar] [CrossRef]
- Zewge, D.; Chen, C.-Y.; Deer, C.; Dormer, P.G.; Hughes, D.L. A mild and efficient synthesis of 4-quinolones and quinolone heterocycles. J. Org. Chem. 2007, 72, 4276–4279. [Google Scholar] [CrossRef] [PubMed]
- Yempalla, K.R.; Munagala, G.; Singh, S.; Magotra, A.; Kumar, S.; Singh, V.; Rajput; Bharate, S.S.; Tikoo, M.; Singh, G.D.; et al. Nitrofuranyl methyl piperazines as new anti-TB agents: Identification, validation, medicinal chemistry and PK studies. ACS Med. Chem. Lett. 2015, 6, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J. Lipophilicity in drug discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.W.; Gallego, R.A.; Edwards, M.P. Lipophilic efficiency as an important metric in drug design. J. Med. Chem. 2018, 61, 6401–6420. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Piccaro, G.; Poce, G.; Biava, M.; Giannoni, F.; Fattorini, L. Activity of lipophilic and hydrophilic drugs against dormant and replicating Mycobacterium tuberculosis. J. Antibiot. 2015, 68, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Marvadi, S.K.; Krishna, V.S.; Sriram, D.; Kantevari, S. Synthesis of novel morpholine, thiomorpholine and N-substituted piperazine coupled 2-(thiophen-2-yl)dihydroquinolines as potent inhibitors of Mycobacterium tuberculosis. Eur. J. Med. Chem. 2019, 164, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Takács-Novák, K.; Avdeef, A. Inter-laboratory study of log P determination by shake-flask and potentiometric methods. J. Pharm. Biomed. Anal. 1996, 14, 1405–1413. [Google Scholar] [CrossRef]
- Bha, S.K. logP—Making Sense of the Value, Advanced Chemistry Development Inc. (ACD/Labs). 2018. Available online: https://www.acdlabs.com/products/percepta/predictors/logp/index.php (accessed on 16 February 2021).
- Kapustikova, I.; Bak, A.; Gonec, T.; Kos, J.; Kozik, V.; Jampilek, J. Investigation of hydro-lipophilic properties of N-alkoxyphenylhydroxynaphthalenecarboxamides. Molecules 2018, 23, 1635. [Google Scholar] [CrossRef]
- Nycz–Empel, A.; Bober, K.; Wyszomirski, M.; Kisiel, E.; Zieba, A. The application of CA and PCA to the evaluation of lipophilicity and physicochemical properties of tetracyclic diazaphenothiazine derivatives. J. Anal. Methods Chem. 2019, 2019, 8131235. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, G.L.; Kumar, A.; Savvi, S.; Hung, A.W.; Wen, S.; Abell, C.; Barry III, C.E.; Sherman, D.R.; Boshoff, H.I.; Mizrahi, V. Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem. Biol. 2012, 19, 844–854. [Google Scholar] [CrossRef]
- Collins, L.; Franzblau, S.G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997, 41, 1004–1009. [Google Scholar] [CrossRef]
- Collins, L.A.; Torrero, M.N.; Franzblau, S.G. Green fluorescent protein reporter microplate assay for high-throughput screening of compounds against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1998, 42, 344–347. [Google Scholar] [CrossRef]
- Ollinger, J.; Bailey, M.A.; Moraski, G.C.; Casey, A.; Florio, S.; Alling, T.; Miller, M.J.; Parish, T. A dual read-out assay to evaluate the potency of compounds active against Mycobacterium tuberculosis. PLoS ONE 2013, 8, e60531. [Google Scholar] [CrossRef]
- De Voss, J.J.; Rutter, K.; Schroeder, B.G.; Su, H.; Zhu, Y.; Barry III, C.E. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. USA 2000, 97, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Mbaba, M.; Mabhula, A.N.; Boel, N.; Edkins, A.L.; Isaacs, M.; Hoppe, H.C.; Khanye, S.D. Ferrocenyl and organic novobiocin derivatives: Synthesis and their in vitro biological activity. J. Inorg. Biochem. 2017, 172, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Oderinlo, O.O.; Tukulula, M.; Isaacs, M.; Hoppe, H.C.; Taylor, D.; Smith, V.J.; Khanye, S.D. New thiazolidine-2,4-dione derivatives combined with organometallic ferrocene: Synthesis, structure and antiparasitic activity. Appl. Organomet. Chem. 2018, 32, e4385. [Google Scholar] [CrossRef]
- Hirumi, H.; Hirumi, K. Contiuous cultvation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989, 75, 985–989. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | a MIC90 (µM) | IC50 (µM) | bClogP | ||
|---|---|---|---|---|---|
| Pf 3D7 | T. b. brucei | HEK293 | |||
| 3a | >125 | >20 | >20 | >32 | 1.2 |
| 3b | >125 | >20 | >20 | >32 | 2.2 |
| 3c | >125 | >20 | 14.5 | >32 | 2.5 |
| 3d | 15.6 | 13.6 | 21.6 | >32 | 3.3 |
| 4a | >125 | >20 | >20 | >32 | 0.9 |
| 4b | >125 | >20 | >20 | >32 | 0.7 |
| 4c | >125 | >20 | >20 | >32 | 0.3 |
| 4d | 7.9 | >20 | >20 | >32 | 0.4 |
| 4e | >125 | >20 | 26.8 | >32 | 0.6 |
| 4f | >125 | >20 | >20 | >32 | 1.3 |
| 4g | 7.9 | 4.1 | >20 | >32 | 1.2 |
| 4h | >125 | 0.68 | >20 | >32 | 1.3 |
| 7a | >125 | 2.3 | 18.5 | >32 | 4.0 |
| 7b | >125 | >20 | 5.4 | >32 | 5.9 |
| 8a | 7.8 | 6.8 | 14.1 | >32 | 2.0 |
| 8b | >125 | 2.4 | >20 | >32 | 3.1 |
| 8c | 1.9 | 1.3 | 1.5 | >32 | 2.4 |
| 8d | >125 | >20 | >20 | >32 | 2.6 |
| 8e | 15.6 | 13.6 | 4.9 | >32 | 3.7 |
| 8f | >125 | >20 | >20 | >32 | 4.3 |
| 8g | 0.9 | 0.46 | 0.18 | >32 | 3.7 |
| 8h | 16 | >20 | >20 | >32 | 3.9 |
| 11 | 31.3 | >20 | >20 | >32 | 1.6 |
| 12 | >125 | 10 | >20 | >32 | 1.9 |
| 13 | 8.0 | 6.5 | 5.2 | >32 | 2.3 |
| 14 | >125 | >20 | >20 | >32 | 0.03 |
| 16 | 0.9 | 11.7 | 1.5 | >32 | 3.8 |
| RIF | 0.001 | - | - | - | 1.0 |
| CQ | - | 0.021 | - | - | - |
| PE | - | - | 0.017 | - | - |
| TMF | - | - | - | 9 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beteck, R.M.; Jordaan, A.; Seldon, R.; Laming, D.; Hoppe, H.C.; Warner, D.F.; Khanye, S.D. Easy-To-Access Quinolone Derivatives Exhibiting Antibacterial and Anti-Parasitic Activities. Molecules 2021, 26, 1141. https://doi.org/10.3390/molecules26041141
Beteck RM, Jordaan A, Seldon R, Laming D, Hoppe HC, Warner DF, Khanye SD. Easy-To-Access Quinolone Derivatives Exhibiting Antibacterial and Anti-Parasitic Activities. Molecules. 2021; 26(4):1141. https://doi.org/10.3390/molecules26041141
Chicago/Turabian StyleBeteck, Richard M., Audrey Jordaan, Ronnett Seldon, Dustin Laming, Heinrich C. Hoppe, Digby F. Warner, and Setshaba D. Khanye. 2021. "Easy-To-Access Quinolone Derivatives Exhibiting Antibacterial and Anti-Parasitic Activities" Molecules 26, no. 4: 1141. https://doi.org/10.3390/molecules26041141
APA StyleBeteck, R. M., Jordaan, A., Seldon, R., Laming, D., Hoppe, H. C., Warner, D. F., & Khanye, S. D. (2021). Easy-To-Access Quinolone Derivatives Exhibiting Antibacterial and Anti-Parasitic Activities. Molecules, 26(4), 1141. https://doi.org/10.3390/molecules26041141