
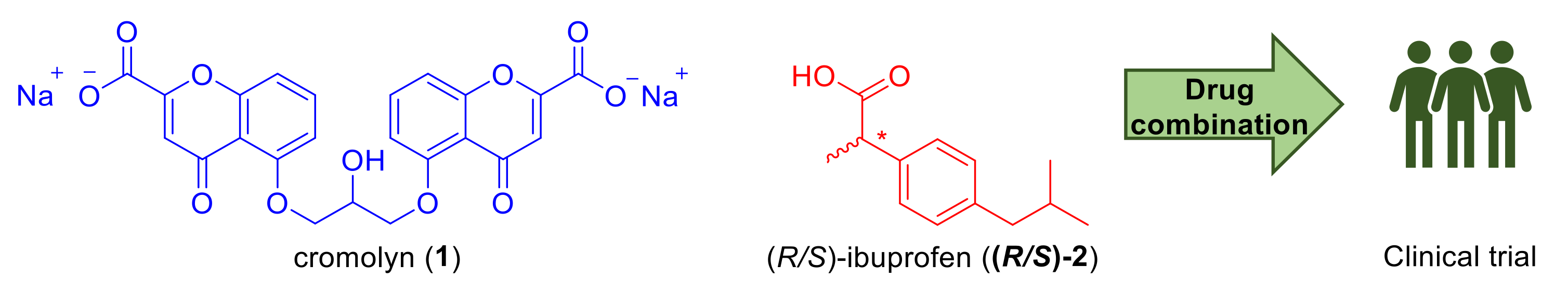
From Combinations to Single-Molecule Polypharmacology—Cromolyn-Ibuprofen Conjugates for Alzheimer’s Disease

 , , ,
, , ,  ,
,  , and
, and
Abstract
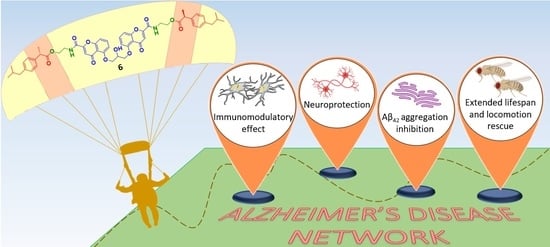
1. Introduction
2. Results
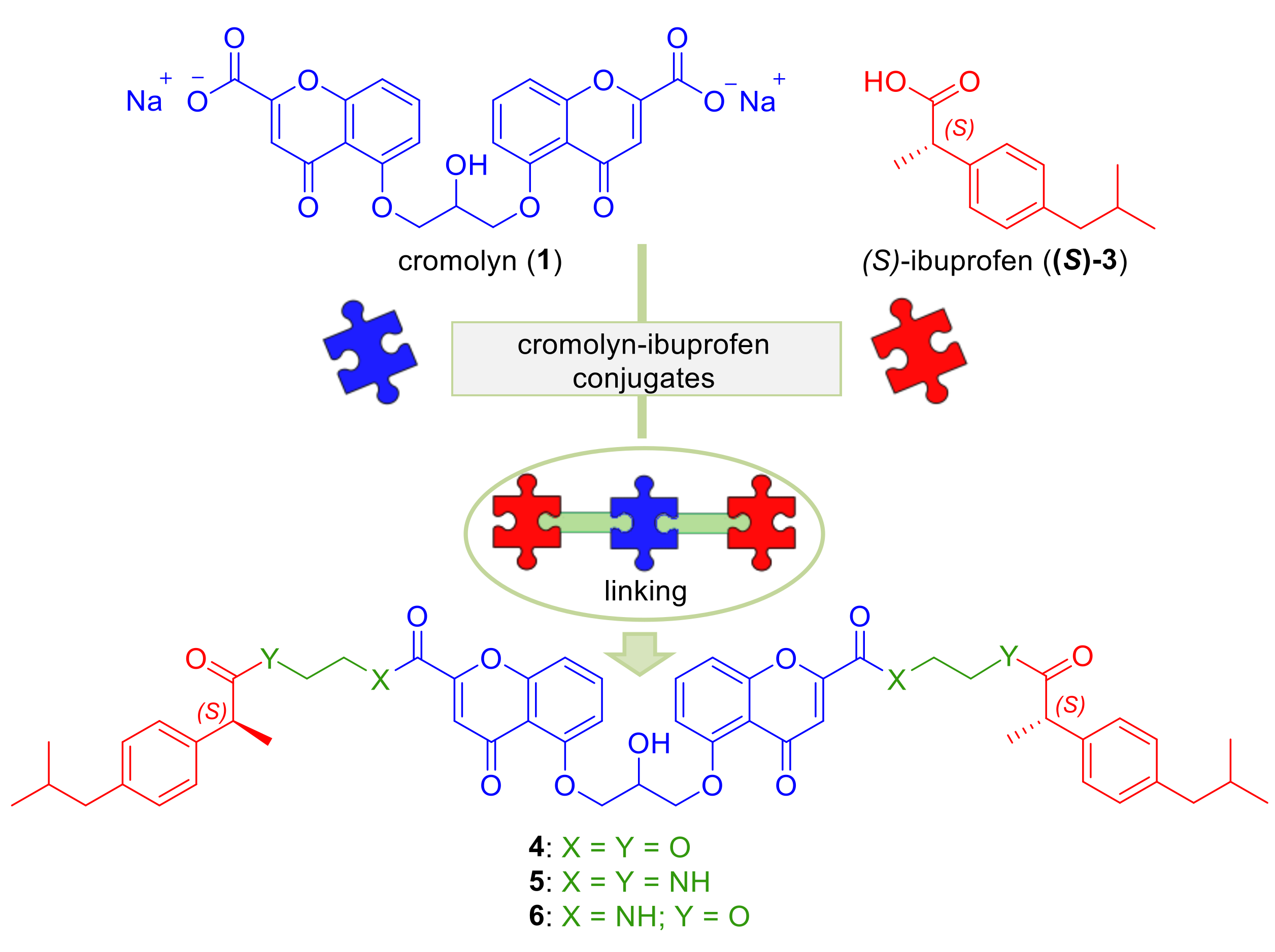
2.1. Design
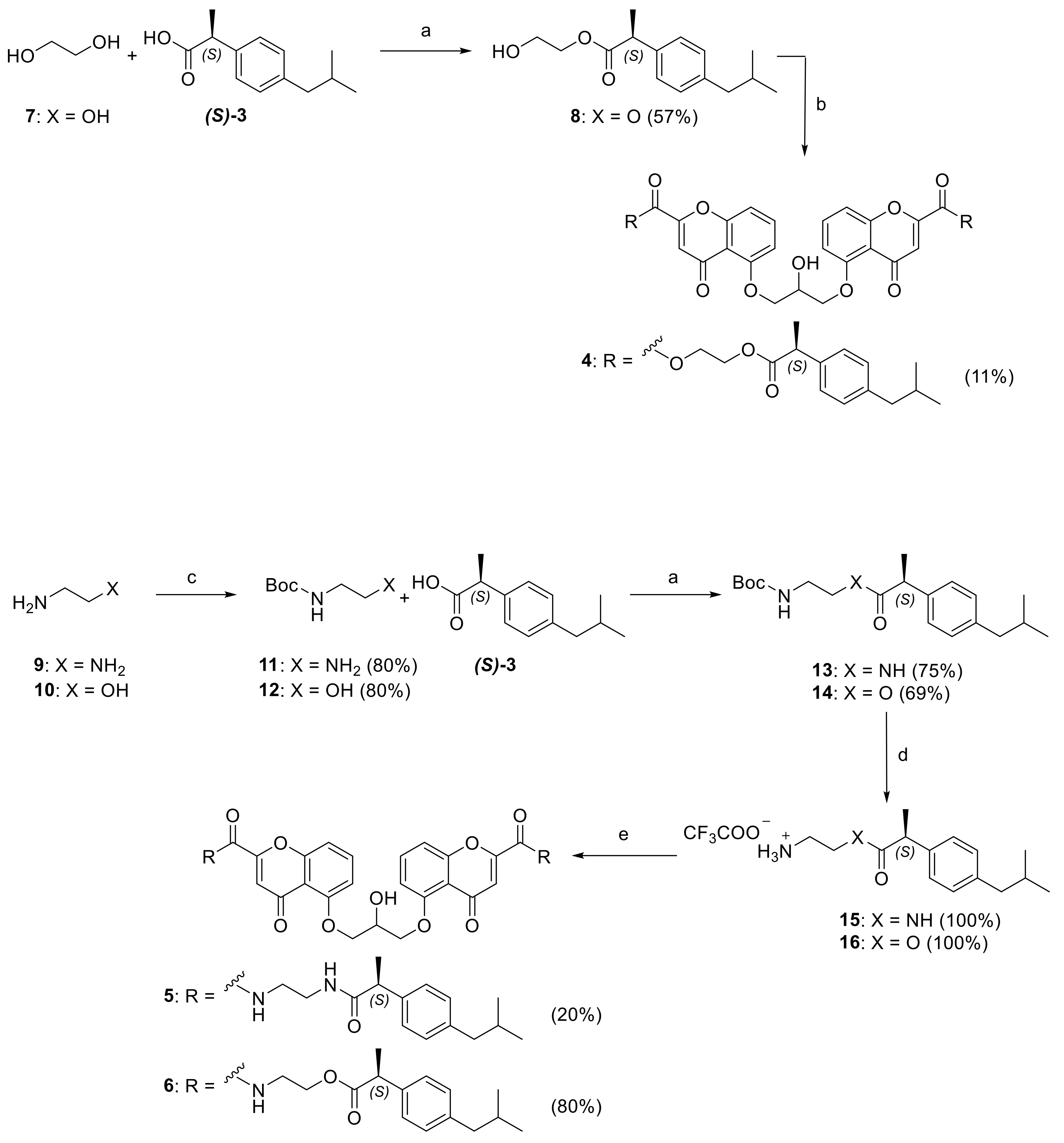
2.2. Synthesis
2.3. Biologic Evaluation
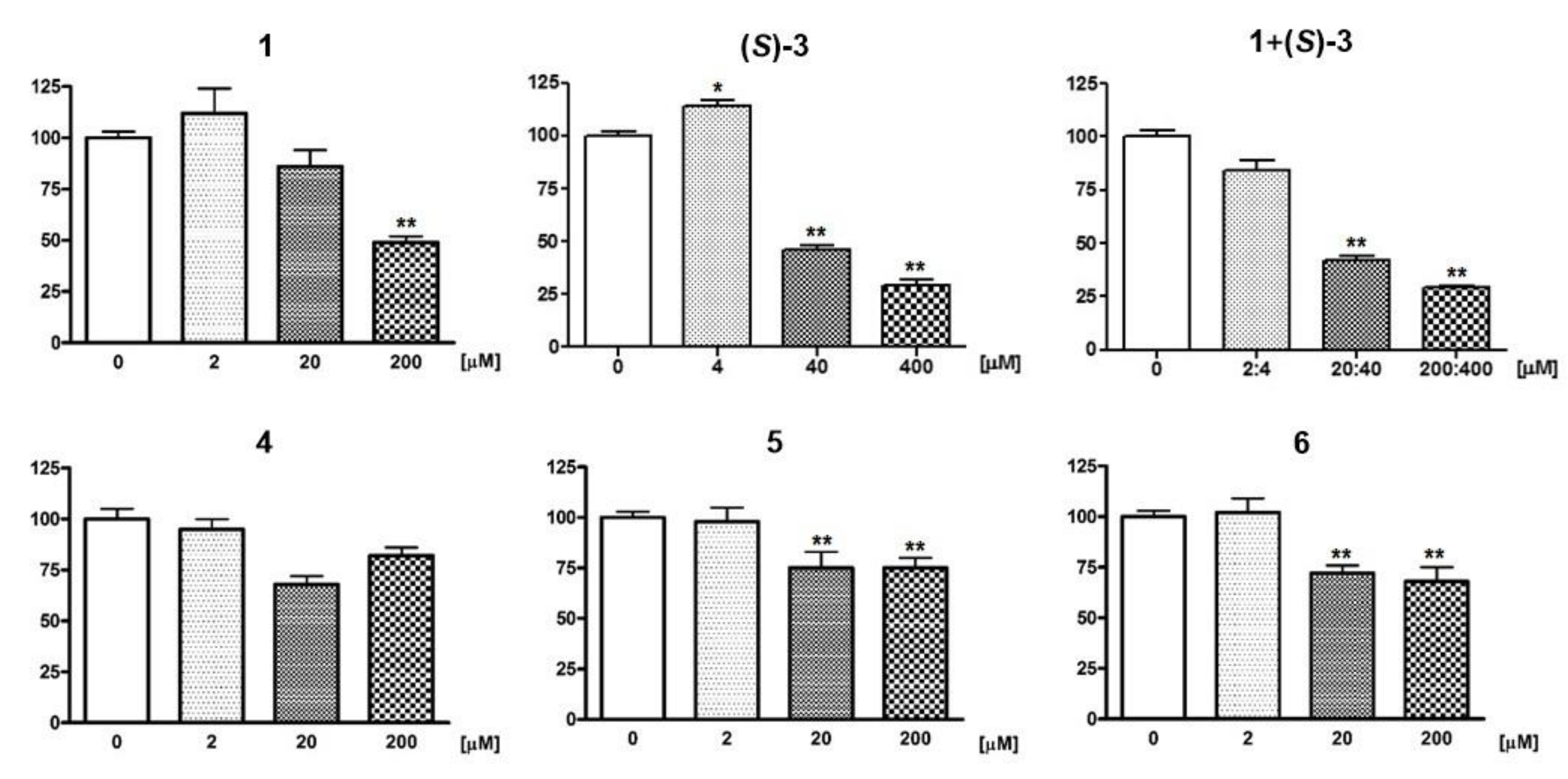
2.3.1. Neurotoxicity on Primary Cerebellar Granule Neurons (CGNs)
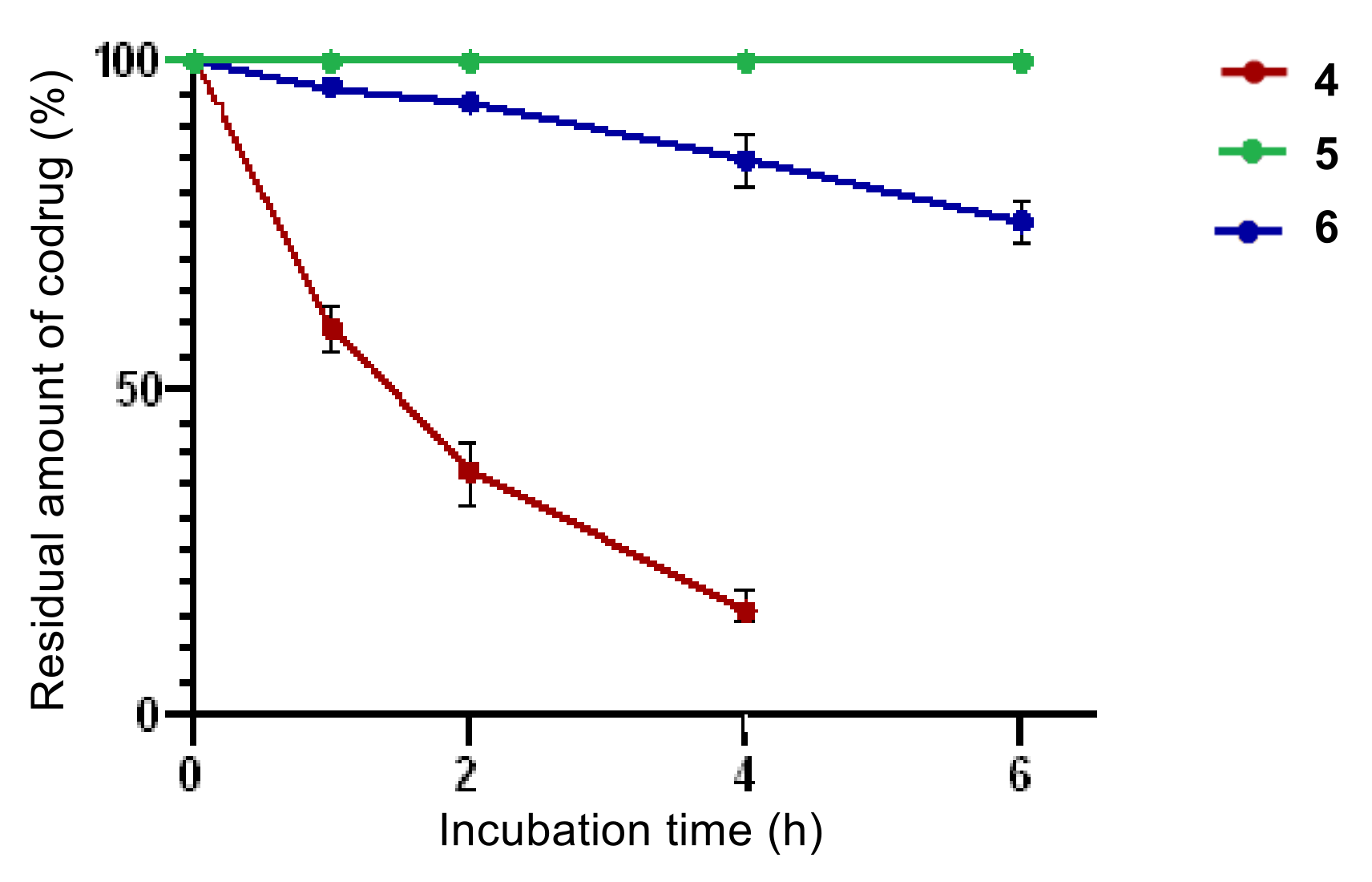
2.3.2. Plasma Stability
2.3.3. Determination of Percentages of Compound 5 and 6 Internalization in Microglial Cells
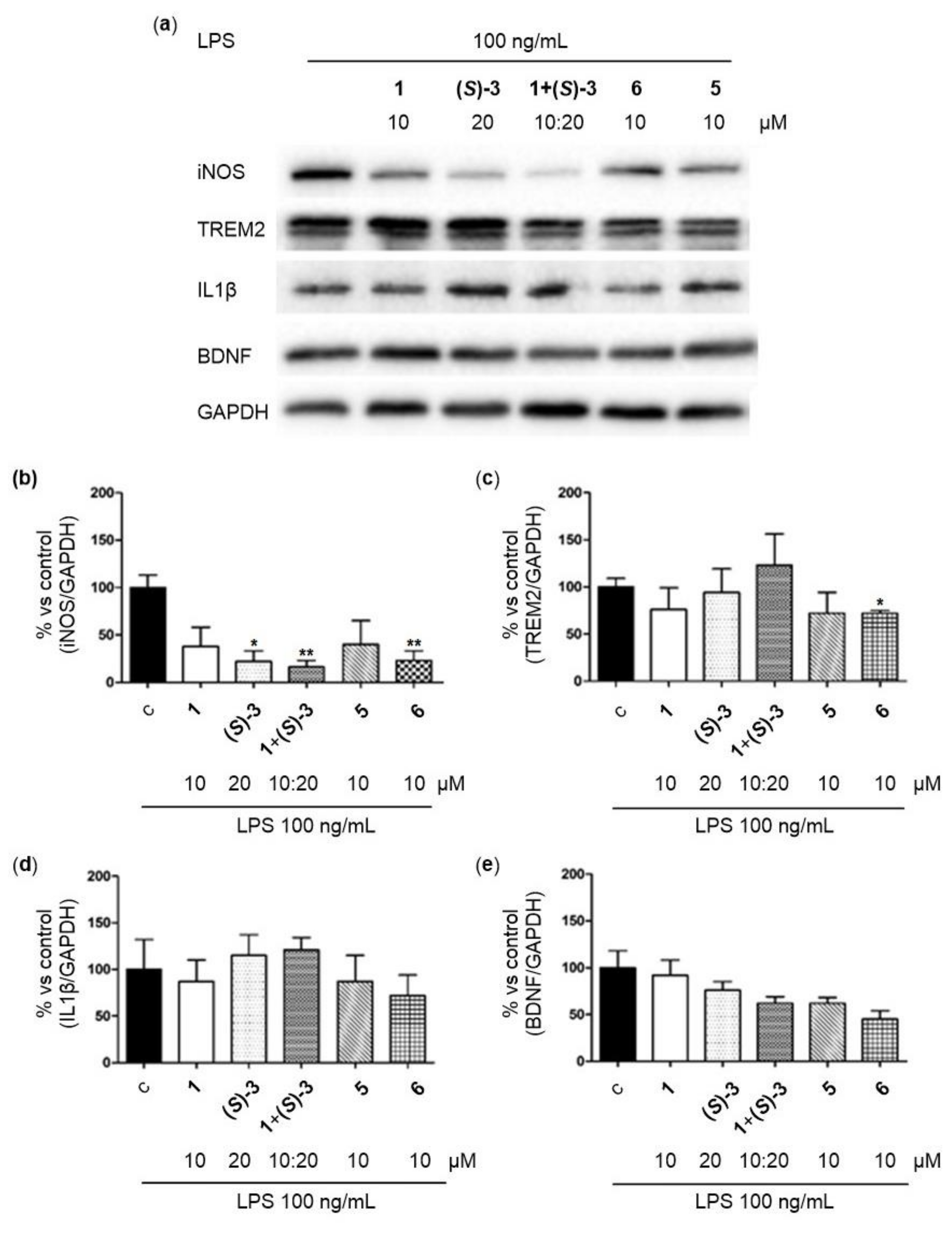
2.3.4. Immunomodulation Assays
2.3.5. Anti-Aggregating Activity towards β-Amyloid Self-Aggregation
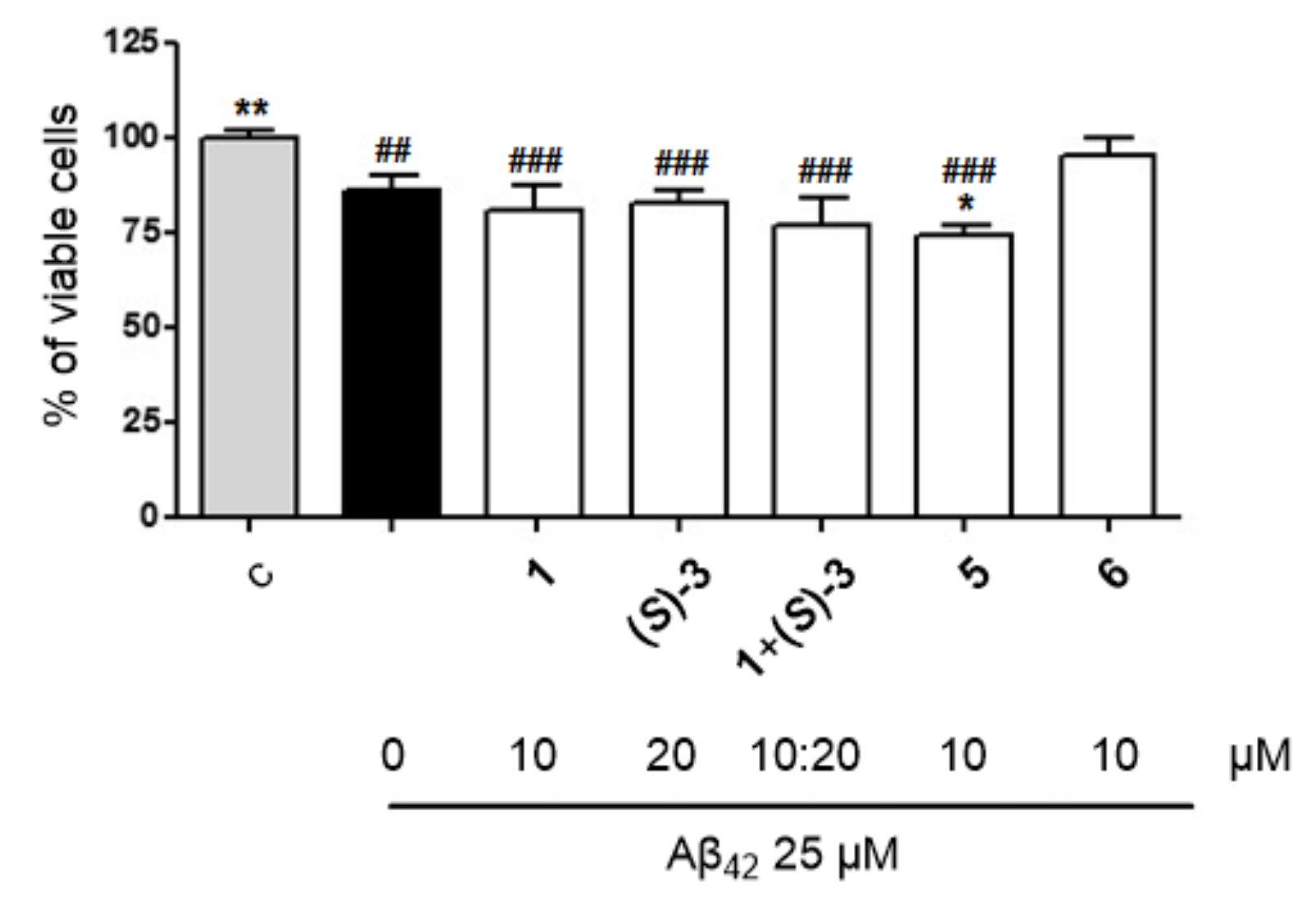
2.3.6. Neuroprotective Effect on Aβ42-Induced Neurotoxicity in CGNs
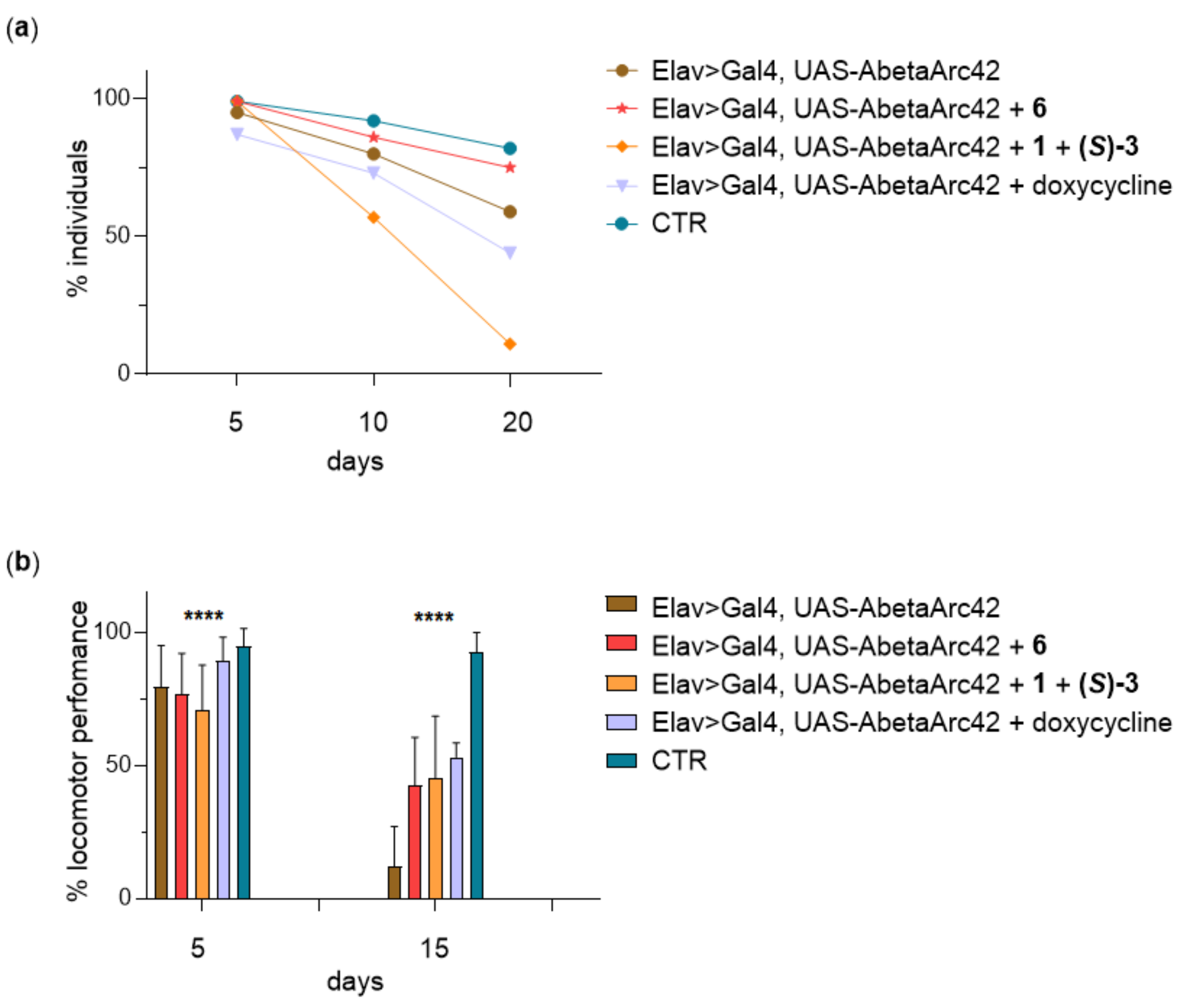
2.3.7. Lifespan and Climbing Assay in Aβ42-Expressing Flies
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis
4.1.1. bis(2-(((S)-2-(4-isobutylphenyl)propanoyl)oxy)ethyl) 5,5′-((2-hydroxypropane-1,3-diyl)bis(oxy)) bis(4-oxo-4H-chromene-2-carboxylate) 4
4.1.2. 5,5′-((2-hydroxypropane-1,3-diyl)bis(oxy))bis(N-(2-((S)-2-(4-isobutylphenyl)propanamido)ethyl)-4-oxo-4H-chromene-2-carboxamide) 5
4.1.3. ((5,5′-((2-hydroxypropane-1,3-diyl)bis(oxy))bis(4-oxo-4H-chromene-5,2-diyl-2-carbonyl))bis(azanediyl))bis(ethane-2,1-diyl)(2S,2’S)-bis(2-(4-isobutylphenyl)propanoate) 6
4.1.4. 2-hydroxyethyl (S)-2-(4-isobutylphenyl)propanoate 8
4.1.5. tert-butyl (2-aminoethyl)carbamate 11
4.1.6. tert-butyl (2-hydroxyethyl)carbamate 12
4.1.7. tert-butyl (S)-(2-(2-(4-isobutylphenyl)propanamido)ethyl)carbamate 13
4.1.8. 2-((tert-butoxycarbonyl)amino)ethyl (S)-2-(4-isobutylphenyl)propanoate 14
4.1.9. (S)-N-(2-aminoethyl)-2-(4-isobutylphenyl)propenamide 15
4.1.10. 2-aminoethyl (S)-2-(4-isobutylphenyl)propanoate 16
4.2. Determination of 4–6 Purity
4.3. Cytotoxicity Assay in Primary Neurons
4.4. Plasma Stability Evaluation
4.5. Determination of the Percentage of Conjugate Internalization in N9 Cells
4.5.1. Sample Preparation
4.5.2. LC-UV Analysis
4.6. Immunomodulation
4.7. Western Blot Analysis
4.8. Statistical Analysis
4.9. Neuroprotection Assay
4.10. Inhibition of Aβ42 Self-Aggregation
4.11. Flies Used and Treatment
4.12. Climbing Assay and Survival Rate
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 15 November 2020).
- Vos, T.; Abajobir, A.A.; Abate, K.H.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abdulkader, R.S.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef]
- Cummings, J.; Feldman, H.H.; Scheltens, P. The “rights” of precision drug development for Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Carreiras, M.C.; Mendes, E.; Perry, M.J.; Francisco, A.P.; Marco-Contelles, J. The multifactorial nature of Alzheimer’s disease for developing potential therapeutics. Curr. Top. Med. Chem. 2013, 13, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- Oset-Gasque, M.J.; Marco-Contelles, J. Alzheimer’s disease, the “one-molecule, one-target” paradigm, and the multitarget directed ligand approach. ACS Chem. Neurosci. 2018, 9, 401–403. [Google Scholar] [CrossRef]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef]
- Peters, J.U. Polypharmacology—Foe or friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Bolognesi, M.L. Polypharmacology in a single drug: Multitarget drugs. Curr. Med. Chem. 2013, 20, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Albertini, C.; Salerno, A.; de Sena Murteira Pinheiro, P.; Bolognesi, M.L. From combinations to multitarget-directed ligands: A continuum in Alzheimer’s disease polypharmacology. Med. Res. Rev. 2020. [Google Scholar] [CrossRef]
- Sun, W.; Sanderson, P.E.; Zheng, W. Drug combination therapy increases successful drug repositioning. Drug Discov. Today 2016, 21, 1189–1195. [Google Scholar] [CrossRef]
- ClinicalTrials. Available online: https://clinicaltrials.gov/ct2/show/NCT04570644?term=cromolyn&cond=Alzheimer+Disease&draw=2&rank=1 (accessed on 10 January 2021).
- Brazier, D.; Perry, R.; Keane, J.; Barrett, K.; Elmaleh, D.R. Pharmacokinetics of Cromolyn and Ibuprofen in Healthy Elderly Volunteers. Clin. Drug Investig. 2017, 37, 1025–1034. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement. 2020, 6, e12050. [Google Scholar] [CrossRef]
- ClinicalTrials. Available online: https://clinicaltrials.gov/ct2/show/NCT02547818?term=cromolyn&cond=Alzheimer+Disease&draw=2&rank=2 (accessed on 15 November 2020).
- Alzheimer Drug Discovery Foundation. Available online: https://www.alzdiscovery.org/research-and-grants/priorities (accessed on 15 November 2020).
- Ismail, R.; Parbo, P.; Madsen, L.S.; Hansen, A.K.; Hansen, K.V.; Schaldemose, J.L.; Kjeldsen, P.L.; Stokholm, M.G.; Gottrup, H.; Eskildsen, S.F.; et al. The relationships between neuroinflammation, beta-amyloid and tau deposition in Alzheimer’s disease: A longitudinal PET study. J. Neuroinflamm. 2020, 17, 151. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Zhang, C.; Griciuc, A.; Hudry, E.; Wan, Y.; Quinti, L.; Ward, J.; Forte, A.M.; Shen, X.; Ran, C.; Elmaleh, D.R. Cromolyn reduces levels of the Alzheimer’s disease-associated amyloid β-protein by promoting microglial phagocytosis. Sci. Rep. 2018, 8, 1144. [Google Scholar] [CrossRef]
- Hori, Y.; Takeda, S.; Cho, H.; Wegmann, S.; Shoup, T.M.; Takahashi, K.; Irimia, D.; Elmaleh, D.R.; Hyman, B.T.; Hudry, E. A Food and Drug Administration-approved asthma therapeutic agent impacts amyloid β in the brain in a transgenic model of Alzheimer disease. J. Biol. Chem. 2015, 290, 1966–1978. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Das, N.; Dhanawat, M.; Dash, B.; Nagarwal, R.C.; Shrivastava, S.K. Codrug: An efficient approach for drug optimization. Eur. J. Pharm. Sci. 2010, 41, 571–588. [Google Scholar] [CrossRef]
- Mori, T.; Nishimura, K.; Tamaki, S.; Nakamura, S.; Tsuda, H.; Kakeya, N. Pro-drugs for the oral delivery of disodium cromoglycate. Chem. Pharm. Bull. 1988, 36, 338–344. [Google Scholar] [CrossRef]
- Ozkan, S.; Adapinar, D.O.; Elmaci, N.T.; Arslantas, D. Apraxia for differentiating Alzheimer’s disease from subcortical vascular dementia and mild cognitive impairment. Neuropsychiatr. Dis. Treat. 2013, 9, 947. [Google Scholar]
- Rakesh, R.; Anoop, K.R. Formulation and optimization of nano-sized ethosomes for enhanced transdermal delivery of cromolyn sodium. J. Pharm. Bioallied Sci. 2012, 4, 333–340. [Google Scholar]
- Li, Y.; Zhou, Y.; Jiang, J.; Wang, X.; Fu, Y.; Gong, T.; Sun, X.; Zhang, Z. Mechanism of brain targeting by dexibuprofen prodrugs modified with ethanolamine-related structures. J. Cereb. Blood Flow Metab. 2015, 35, 1985–1994. [Google Scholar] [CrossRef]
- Reichel, C.; Brugger, R.; Bang, H.; Geisslinger, G.; Brune, K. Molecular cloning and expression of a 2-arylpropionyl-coenzyme A epimerase: A key enzyme in the inversion metabolism of ibuprofen. Mol. Pharmacol. 1997, 51, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Placzek, A.T.; Ferrara, S.J.; Hartley, M.D.; Sanford-Crane, H.S.; Meinig, J.M.; Scanlan, T.S. Sobetirome prodrug esters with enhanced blood-brain barrier permeability. Bioorganic Med. Chem. 2016, 24, 5842–5854. [Google Scholar] [CrossRef] [PubMed]
- Levine, H., III. Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef]
- Bolus, H.; Crocker, K.; Boekhoff-Falk, G.; Chtarbanova, S. Modeling Neurodegenerative Disorders in Drosophila melanogaster. Int. J. Mol. Sci. 2020, 21, 3055. [Google Scholar] [CrossRef]
- Fernandez-Funez, P.; de Mena, L.; Rincon-Limas, D.E. Modeling the complex pathology of Alzheimer’s disease in Drosophila. Exp. Neurol. 2015, 274, 58–71. [Google Scholar] [CrossRef]
- Crowther, D.C.; Kinghorn, K.J.; Miranda, E.; Page, R.; Curry, J.A.; Duthie, F.A.; Gubb, D.C.; Lomas, D.A. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience 2005, 132, 123–135. [Google Scholar] [CrossRef]
- Costa, R.; Speretta, E.; Crowther, D.C.; Cardoso, I. Testing the therapeutic potential of doxycycline in a Drosophila melanogaster model of Alzheimer disease. J. Biol. Chem. 2011, 286, 41647–41655. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef]
- Sozio, P.; D’Aurizio, E.; Iannitelli, A.; Cataldi, A.; Zara, S.; Cantalamessa, F.; Nasuti, C.; Di Stefano, A. Ibuprofen and lipoic acid diamides as potential codrugs with neuroprotective activity. Arch. Pharm. 2010, 343, 133–142. [Google Scholar] [CrossRef]
- Vu, C.B.; Bemis, J.E.; Benson, E.; Bista, P.; Carney, D.; Fahrner, R.; Lee, D.; Liu, F.; Lonkar, P.; Milne, J.C.; et al. Synthesis and Characterization of Fatty Acid Conjugates of Niacin and Salicylic Acid. J. Med. Chem. 2016, 59, 1217–1231. [Google Scholar] [CrossRef]
- Rossi, M.; Petralla, S.; Protti, M.; Baiula, M.; Kobrlova, T.; Soukup, O.; Spampinato, S.M.; Mercolini, L.; Monti, B.; Bolognesi, M.L. α-Linolenic Acid–Valproic Acid Conjugates: Toward Single-Molecule Polypharmacology for Multiple Sclerosis. ACS Med. Chem. Lett. 2020, 11, 2406–2413. [Google Scholar] [CrossRef]
- DeGoey, D.A.; Chen, H.-J.; Cox, P.B.; Wendt, M.D. Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection: Miniperspective. J. Med. Chem. 2017, 61, 2636–2651. [Google Scholar] [CrossRef]
- Uliassi, E.; Peña-Altamira, L.E.; Morales, A.V.; Massenzio, F.; Petralla, S.; Rossi, M.; Roberti, M.; Martinez Gonzalez, L.; Martinez, A.; Monti, B.; et al. A Focused Library of Psychotropic Analogues with Neuroprotective and Neuroregenerative Potential. ACS Chem. Neurosci. 2018. [Google Scholar] [CrossRef]
- Giorgetti, S.; Greco, C.; Tortora, P.; Aprile, F.A. Targeting amyloid aggregation: An overview of strategies and mechanisms. Int. J. Mol. Sci. 2018, 19, 2677. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Forloni, G. Doxycycline for Alzheimer’s disease: Fighting β-Amyloid oligomers and neuroinflammation. Front. Pharmacol. 2019, 10, 738. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, M.; Bertucci, C.; Bolognesi, M.L.; Cavalli, A.; Melchiorre, C.; Andrisano, V. Insight into the kinetic of amyloid beta (1-42) peptide self-aggregation: Elucidation of inhibitors’ mechanism of action. ChemBioChem 2007, 8, 2152–2161. [Google Scholar] [CrossRef] [PubMed]
- Naiki, H.; Higuchi, K.; Hosokawa, M.; Takeda, T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal. Biochem. 1989, 177, 244–249. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Type | Sample | Number of Cells (×106) | % Compound Internalized/Cell ± SD (n = 3) | % Compound Internalized/Cell mean ± SD 1 |
|---|---|---|---|---|---|
| 5 | N9_Ctrl | N9 Ctrl-1 | 2.60 | 9.03 ± 1.10 | 17.64 ± 12.81 |
| N9 Ctrl-2 | 1.48 | 32.36 ± 2.49 | |||
| N9 Ctrl-3 | 2.80 | 11.54 ± 0.38 | |||
| N9_LPS | N9 LPS-1 | 2.13 | 7.40 ± 0.59 | 16.15 ± 10.73 | |
| N9 LPS-2 | 1.48 | 28.13 ± 0.35 | |||
| N9 LPS-3 | 2.40 | 12.92 ± 0.40 | |||
| 6 | N9_Ctrl | N9 Ctrl-1 | 2.18 | 0.77 ± 0.05 | 1.35 ± 0.96 |
| N9 Ctrl-2 | 1.80 | 0.82 ± 0.13 | |||
| N9 Ctrl-3 | 1.90 | 2.46 ± 0.03 | |||
| N9_LPS | N9 LPS-1 | 1.45 | 1.76 ± 0.05 | 2.47 ± 1.56 | |
| N9 LPS-2 | 1.65 | 1.39 ± 0.03 | |||
| N9 LPS-3 | 2.30 | 4.25 ± 0.11 |
| Compound |  | Inhibition of Amyloid-Self Aggregation (%) [I] = 50 µM |
|---|---|---|
| R | ||
| 5 |  | Not soluble in the assay conditions |
| 6 |  | 72.0 ± 0.3 |
| 17 |  | < 5 |
| 1 (cromolyn) | 69.7 ± 5.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albertini, C.; Naldi, M.; Petralla, S.; Strocchi, S.; Grifoni, D.; Monti, B.; Bartolini, M.; Bolognesi, M.L. From Combinations to Single-Molecule Polypharmacology—Cromolyn-Ibuprofen Conjugates for Alzheimer’s Disease. Molecules 2021, 26, 1112. https://doi.org/10.3390/molecules26041112
Albertini C, Naldi M, Petralla S, Strocchi S, Grifoni D, Monti B, Bartolini M, Bolognesi ML. From Combinations to Single-Molecule Polypharmacology—Cromolyn-Ibuprofen Conjugates for Alzheimer’s Disease. Molecules. 2021; 26(4):1112. https://doi.org/10.3390/molecules26041112
Chicago/Turabian StyleAlbertini, Claudia, Marina Naldi, Sabrina Petralla, Silvia Strocchi, Daniela Grifoni, Barbara Monti, Manuela Bartolini, and Maria Laura Bolognesi. 2021. "From Combinations to Single-Molecule Polypharmacology—Cromolyn-Ibuprofen Conjugates for Alzheimer’s Disease" Molecules 26, no. 4: 1112. https://doi.org/10.3390/molecules26041112
APA StyleAlbertini, C., Naldi, M., Petralla, S., Strocchi, S., Grifoni, D., Monti, B., Bartolini, M., & Bolognesi, M. L. (2021). From Combinations to Single-Molecule Polypharmacology—Cromolyn-Ibuprofen Conjugates for Alzheimer’s Disease. Molecules, 26(4), 1112. https://doi.org/10.3390/molecules26041112