
Mesoporous Carbon Fibers with Tunable Mesoporosity for Electrode Materials in Energy Devices

 ,
,
Abstract
1. Introduction
2. Results and Discussion
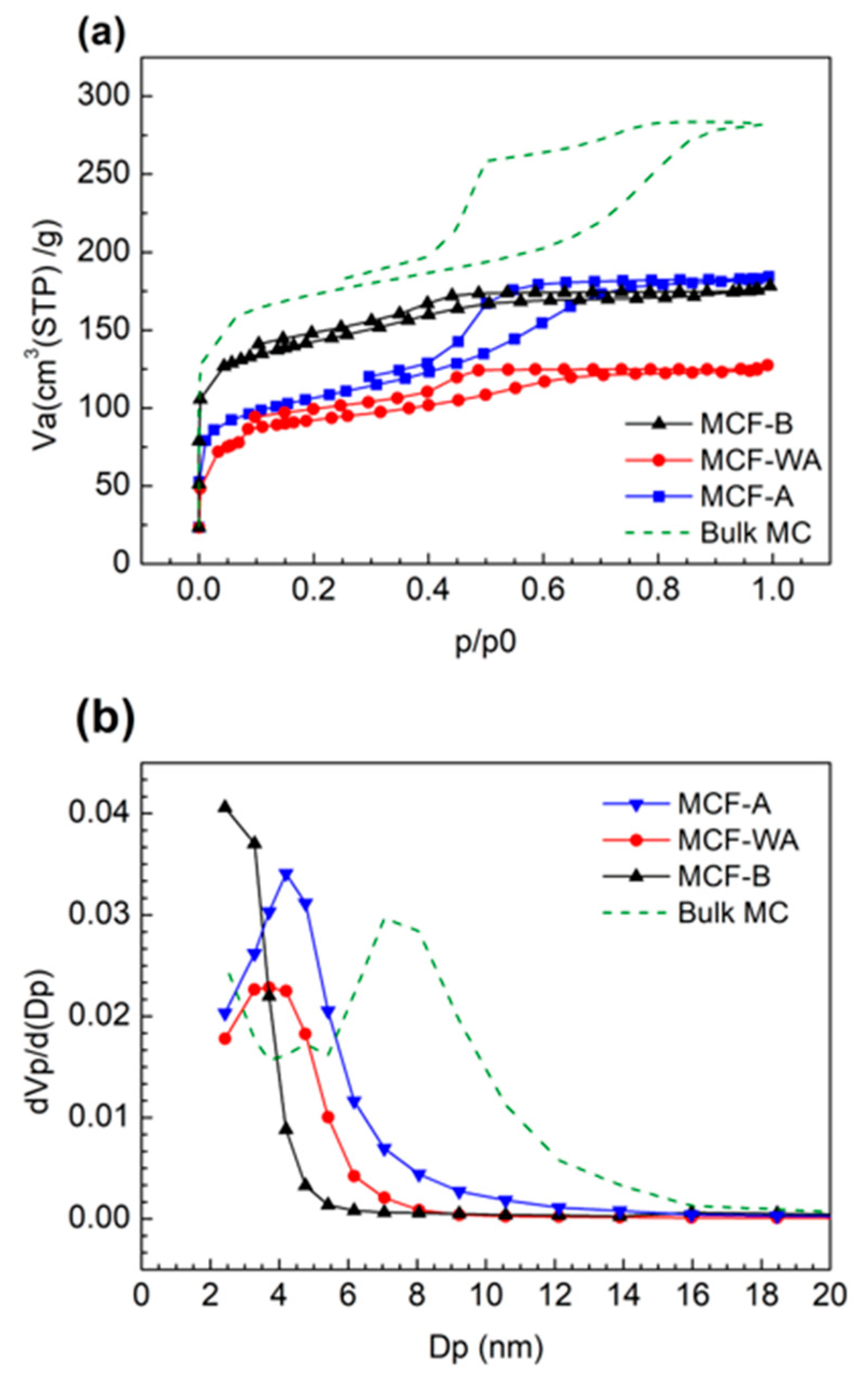
2.1. Characterization of the Porous Structure of MCFs
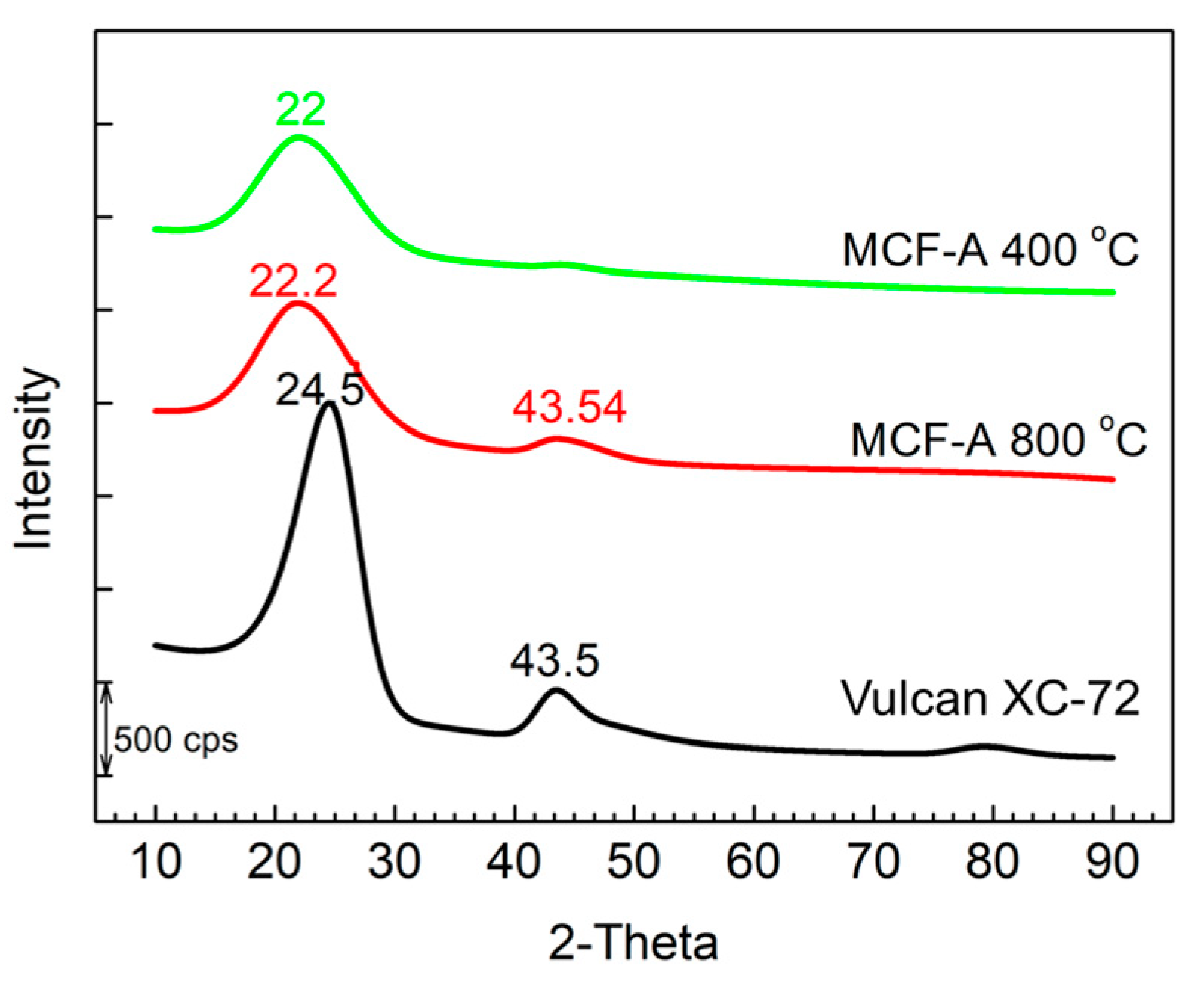
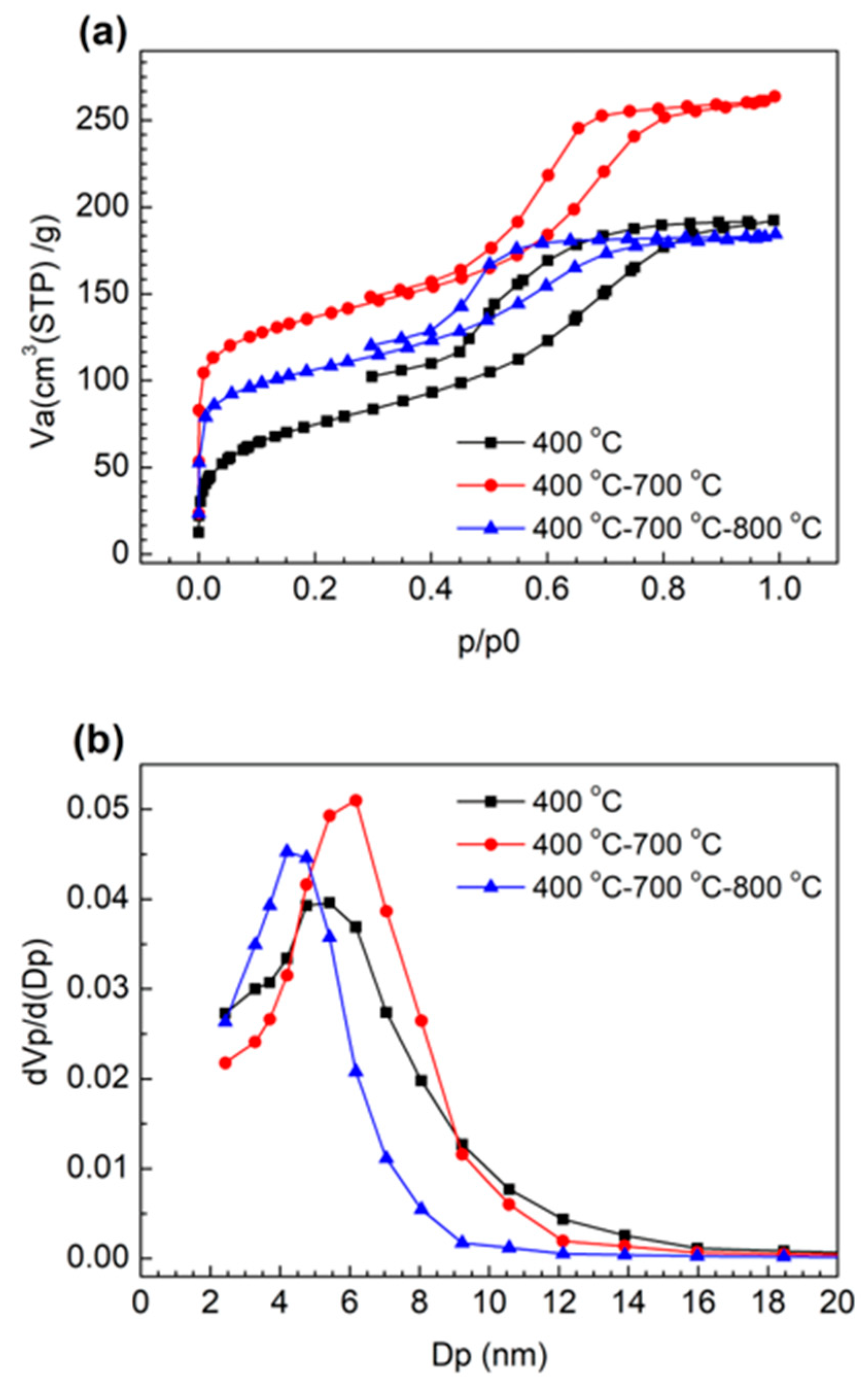
2.2. Optimization of the Heat Treatment Condition of MCF-A
2.3. Characterization of Morphology of MCF-A
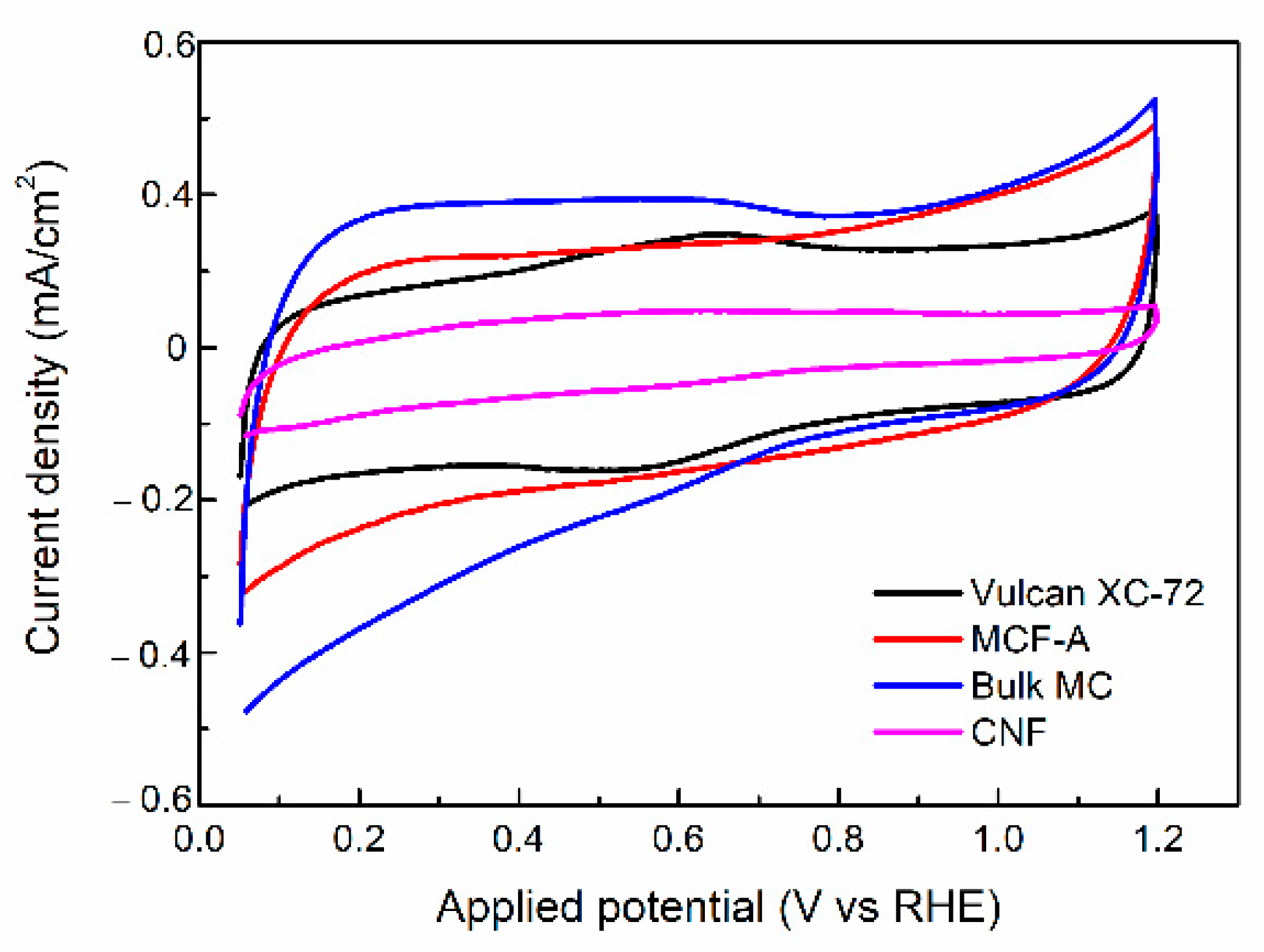
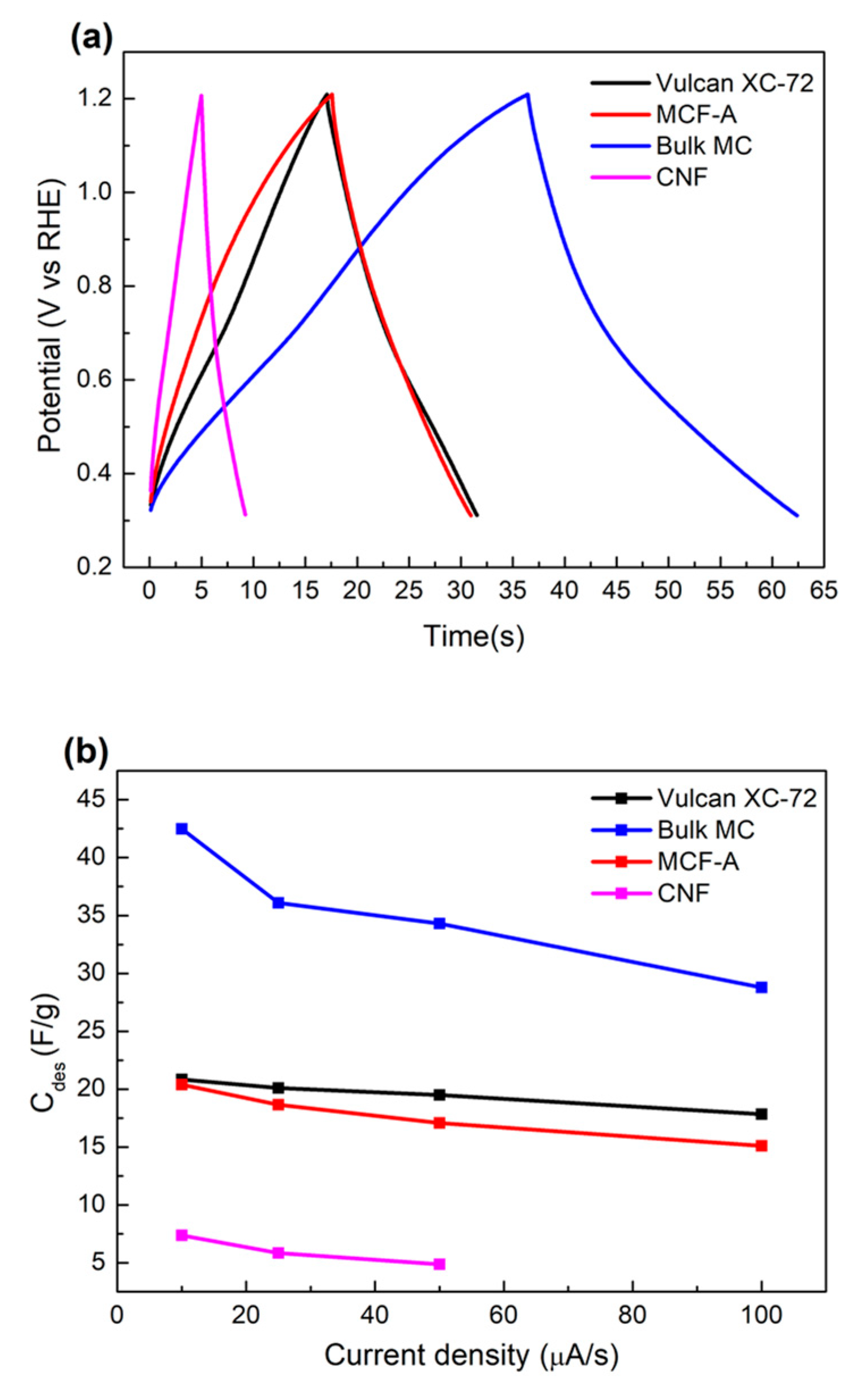
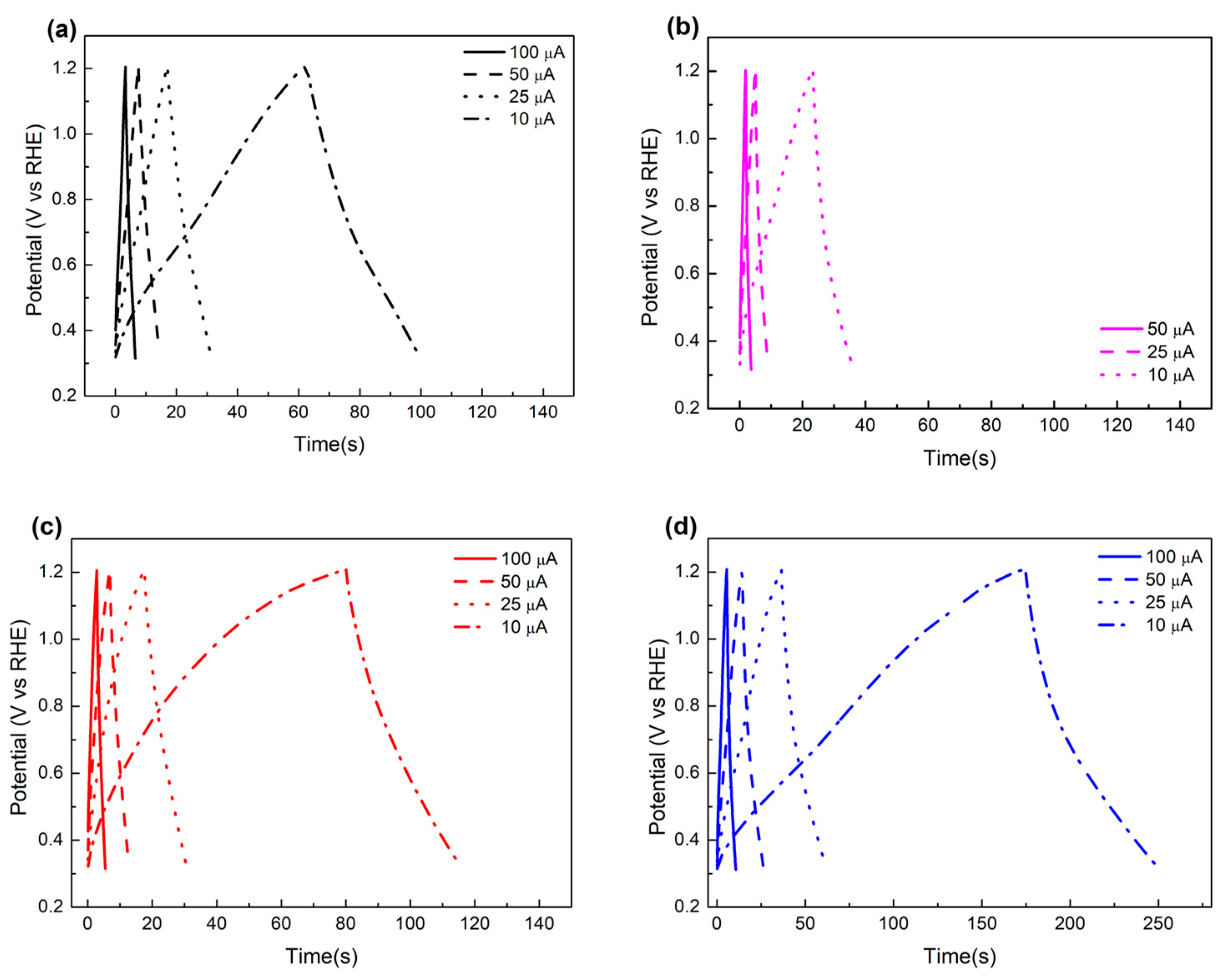
2.4. Electrochemical Characterization of MCF-A Powder
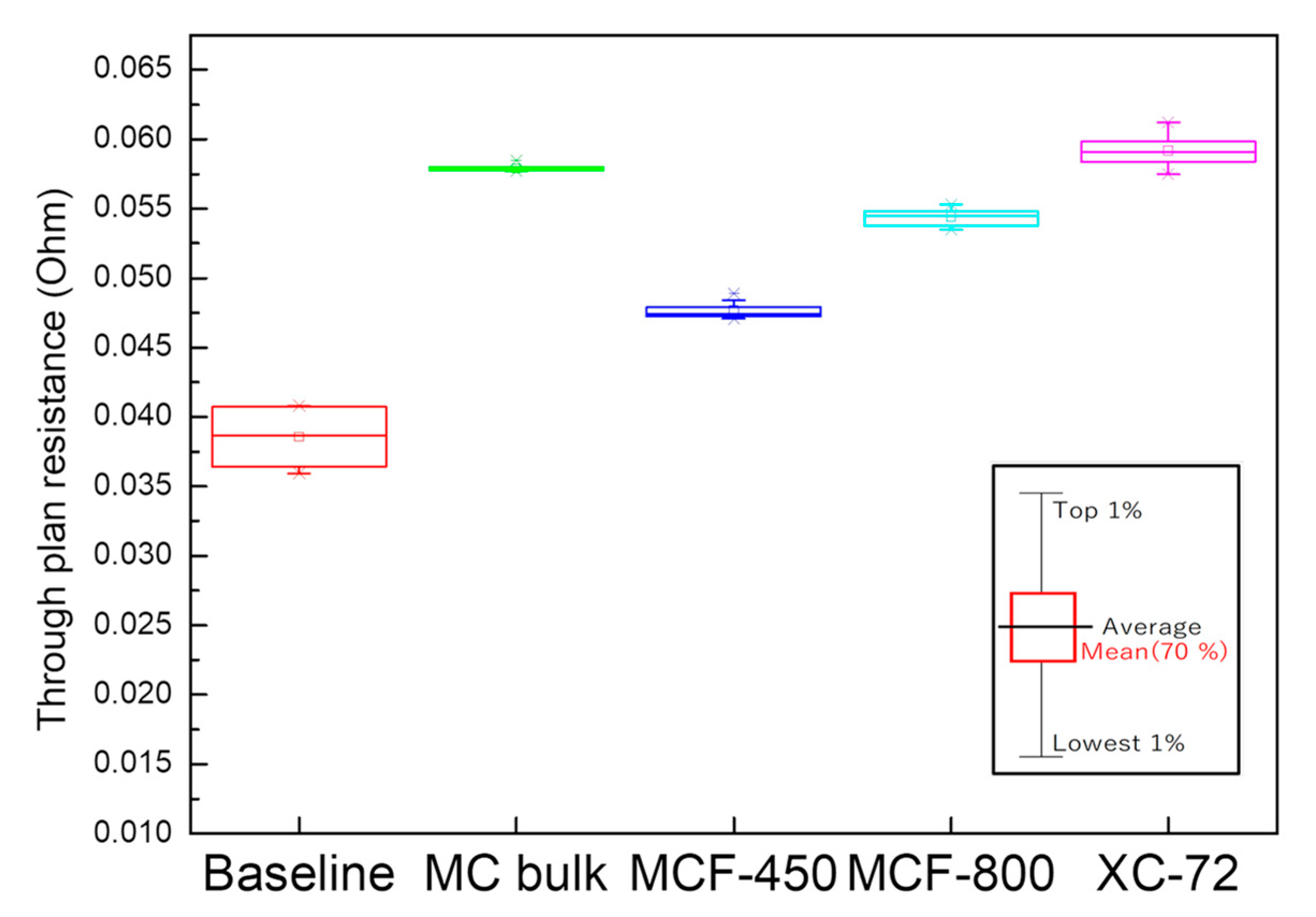
2.5. Characterization of MCF-A Layer
3. Materials and Methods
3.1. Materials
3.2. Preparation of MCF Precursor Solutions for ESD
3.3. ESD Process of MCF Precursor Solutions
3.4. Calcination Process of MCF Precursors
3.5. Material Characterization
3.6. Electrochemical Characterization
3.7. Evaluation of the Resistance toward Compression
3.8. Evaluation of the Through-Plane Electrical Resistance
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Auer, E.; Freund, A.; Pietsch, J.; Tacke, T. Carbons as supports for industrial precious metal catalysts. Appl. Catal. A Gen. 1998, 173, 129–271. [Google Scholar] [CrossRef]
- Rodríguez-Reinoso, F. The role of carbon materials in heterogeneous catalysis. Carbon 1998, 36, 159–175. [Google Scholar] [CrossRef]
- Serp, P.; Corrias, M.; Kalck, P. Carbon nanotubes and nanofibers in catalysis. Appl. Catal. A Gen. 2003, 253, 337–358. [Google Scholar] [CrossRef]
- Schlögl, R. Carbons. In Preparation of Solid Catalysts; Ertl, G., Knözinger, H., Weitkamp, J., Eds.; Wiley & Sons: Weinheim, Germany, 1999; pp. 150–241. [Google Scholar]
- Jüntgen, H. Activated carbon as catalyst support: A review of new research results. Fuel 1986, 65, 1436–1446. [Google Scholar] [CrossRef]
- Liu, H.; Song, C.; Zhang, L.; Zhang, J.; Wang, H.; Wilkinson, D.P. A review of anode catalysis in the direct methanol fuel cell. J. Power Sources 2006, 55, 95–110. [Google Scholar] [CrossRef]
- Candelaria, S.L.; Shao, Y.; Zhou, W.; Li, X.; Xiao, J.; Zhang, J.-G.; Wang, Y.; Liu, J.; Li, J.; Cao, G. Nanostructured carbon for energy storage and conversion. Nano Energy 2012, 1, 195–220. [Google Scholar] [CrossRef]
- Joo, S.H.; Choi, S.J.; Oh, I.; Kwak, J.; Liu, Z.; Terasaki, O.; Ryoo, R. Ordered nanoporous arrays of carbon supporting high dispersions of platinum nanoparticles. Nature 2001, 412, 167–172. [Google Scholar] [CrossRef]
- Tanaka, S.; Nishiyama, N.; Egashira, Y.; Ueyama, K. Synthesis of ordered mesoporous carbons with channel structure from an organic–organic nanocomposite. Chem. Commun. 2005, 16, 2125–2127. [Google Scholar] [CrossRef]
- Meng, Y.; Gu, D.; Zhang, F.; Shi, Y.; Yang, H.; Li, Z.; Yu, C.; Tu, B.; Zhao, D. Ordered mesoporous polymers and homologous carbon frameworks: Amphiphilic surfactant templating and direct transformation. Angew. Chem. Int. Ed. 2005, 44, 7053–7059. [Google Scholar] [CrossRef]
- Liang, C.; Hong, K.; Guiochon, G.A.; Mays, J.W.; Dai, S. Synthesis of a large-scale highly ordered porous carbon film by self-assembly of block copolymers. Angew. Chem. Int. Ed. 2004, 43, 5785–5789. [Google Scholar] [CrossRef]
- Taguchi, A.; Schüth, F. Ordered mesoporous materials in catalysis. Microporous Mesoporous Mater. 2005, 77, 1–45. [Google Scholar] [CrossRef]
- Staykov, A.; Ooishi, Y.; Ishihara, T. Immobilizing metal nanoparticles on single wall nanotubes. Effect of surface curvature. J. Phys. Chem. C. 2014, 118, 8907–8916. [Google Scholar] [CrossRef]
- Hayashi, A.; Notsu, H.; Kimijima, K.; Miyamoto, J.; Yagi, I. Preparation of Pt/mesoporous carbon (MC) electrode catalyst and its reactivity toward oxygen reduction. Electrochim. Acta. 2008, 53, 6117–6125. [Google Scholar] [CrossRef]
- Kimijima, K.; Hayashi, A.; Yagi, I. Preparation of a self-standing mesoporous carbon membrane with perpendicularly ordered pore structures. Chem. Commun. 2008, 44, 5809–5811. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Kimijima, K.; Miyamoto, J.; Yagi, I. Direct observation of well-dispersed Pt nanoparticles inside the pores of mesoporous carbon through the cross section of Pt/mesoporous carbon particles. Chem. Lett. 2009, 38, 346–347. [Google Scholar] [CrossRef]
- Hayashi, A.; Kimijima, K.; Miyamoto, J.; Yagi, I. Oxygen transfer and storage processes inside the mesopores of Pt/MC catalysts thin layer electrode. J. Phys. Chem. 2009, 113, 12149–122153. [Google Scholar]
- Minamida, Y.; Noda, Z.; Hayashi, A.; Sasaki, K. Development of MEAs with Pt/mesoporous carbon as a cathode catalyst. ECS Trans. 2014, 64, 137–144. [Google Scholar] [CrossRef]
- Wang, Q.; Yan, J.; Fan, Z.J. Carbon materials for high volumetric performance supercapacitors: Design, progress, challenges and opportunities. Energy Environ. Sci. 2016, 9, 729–762. [Google Scholar] [CrossRef]
- Zhang, L.L.; Zhao, X.S. Carbon-based materials as supercapacitor electrodes. Chem. Soc. Rev. 2009, 38, 2520–2531. [Google Scholar] [CrossRef]
- Liang, J.; Sun, Z.H.; Li, F.; Cheng, H.M. Carbon materials for Li–S batteries: Functional evolution and performance improvement. Energy Storage Mater. 2016, 2, 76–106. [Google Scholar] [CrossRef]
- Yang, C.; Yao, Y.; He, S.; Xie, H.; Hitz, E.; Hu, L. Ultrafine silver nanoparticles for seeded lithium deposition toward stable lithium metal anode. Adv. Mater. 2017, 29, 1702714. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, S.; Fujihara, K.; Teo, W.E.; Lim, T.C.; Ma, Z. Electrospinning Process. In An Introduction to Electrospinning and Nanofibers; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2005; pp. 90–154. [Google Scholar]
- Wang, H.Q.; Zhang, C.F.; Chen, Z.X.; Liu, H.K.; Guo, Z.P. Large-scale synthesis of ordered mesoporous carbon fiber and its application as cathode material for lithium–sulfur batteries. Carbon 2015, 81, 782–787. [Google Scholar] [CrossRef]
- Ghosh, S.; Yong, W.D.; Jin, E.M.; Polaki, S.R.; Jeong, S.M.; Jun, H. Mesoporous carbon nanofiber engineered for improved supercapacitor performance. Korean J. Chem. Eng. 2019, 36, 312–320. [Google Scholar] [CrossRef]
- Ma, C.; Song, Y.; Shi, J.; Zhang, D.; Zhai, X.; Zhong, M.; Guo, Q.; Liu, L. Preparation and one-step activation of microporous carbon nanofibers for use as supercapacitor electrodes. Carbon 2016, 51, 290–300. [Google Scholar] [CrossRef]
- Wang, H.; Niu, H.; Wang, H.; Wang, W.; Jin, X.; Wang, H.; Zhou, H.; Lin, T. Micro-meso porous structured carbon nanofibers with ultra-high surface area and large supercapacitor electrode capacitance. J. Power Sources 2021, 482, 228986. [Google Scholar] [CrossRef]
- Ozaki, J.; Endo, N.; Ohizumi, W.; Igarashi, K.; Nakahara, M.; Oya, A. Novel preparation method for the production of mesoporous carbon fiber from a polymer blend. Carbon 1997, 35, 1031–1033. [Google Scholar] [CrossRef]
- Tanaka, S.; Doi, A.; Nakatani, N.; Katayama, Y.; Miyake, Y. Synthesis of ordered mesoporous carbon films, powders, and fibers by direct triblock-copolymer-templating method using an ethanol/water system. Carbon 2009, 47, 2688–2698. [Google Scholar] [CrossRef]
- Liang, C.; Dai, S. Synthesis of mesoporous carbon materials via enhanced hydrogen-bonding interaction. J. Am. Chem. Soc. 2006, 128, 5316–5317. [Google Scholar] [CrossRef]
- Son, W.K.; Youk, J.H.; Lee, T.S.; Park, W.H. Effect of pH on electrospinning of poly(vinyl alcohol). Mater. Lett. 2005, 59, 1571–1575. [Google Scholar]
- Tashima, D.; Yamamoto, E.; Kai, N.; Fujikawa, D.; Sakai, G.; Otsubo, M.; Kijima, T. Double layer capacitance of high surface area carbon nanospheres derived from resorcinol–formaldehyde polymers. Carbon 2011, 49, 4848–4857. [Google Scholar] [CrossRef]
- Ghosh, S.; Barg, S.; Jeong, S.M.; Ostrikov, K. Heteroatom-doped and oxygen-functionalized nanocarbons for high-performance supercapacitors. Adv. Energy Mater. 2020, 10, 2001239–2001283. [Google Scholar] [CrossRef]
- Stachewicz, U.; Stone, C.A.; Willis, C.R.; Barber, A.H. Charge assisted tailoring of chemical functionality at electrospun nanofiber surfaces. J. Mater. Chem. 2012, 22, 22935–22941. [Google Scholar] [CrossRef]
- Badhe, Y.; Balasubramanian, K. Nanoencapsulated core and shell electrospun fibers of resorcinol formaldehyde. Ind. Eng. Chem. Res. 2015, 54, 7614–7622. [Google Scholar] [CrossRef]
- Imaizumi, S.; Matsumoto, H.; Suzuki, K.; Minagawa, M.; Kimura, M.; Tanioka, A. Phenolic resin-based carbon thin fibers prepared by electrospinning: Additive effects of poly(vinyl butyral) and electrolytes. Polymer. J. 2009, 42, 1124–1128. [Google Scholar] [CrossRef]
- Teng, M.; Qiao, J.; Li, F.; Bera, P.K. Electrospun mesoporous carbon nanofibers produced from phenolic resin and their use in the adsorption of large dye molecules. Carbon 2012, 50, 2877–2886. [Google Scholar] [CrossRef]
- Durairaj, R.B. Resorcinol Based Resins and Applications. In Resorcinol Chemistry, Technology and Applications; Springer: Berlin, Germany, 2005; pp. 179–261. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pore Diameter (nm) | Surface Area (m2/g) | Notes | |
|---|---|---|---|
| MCF-A | 4.5 | 370 | pH = 2.8 |
| MCF-WA | 3.8 | 340 | pH = 4.6 |
| MCF-B | 2 | 670 | pH = 7.6 |
| Bulk MC | 7.5 | 635 |
| Highest Temperature in the Heat Treatment | H Atom (%) | C Atom (%) |
|---|---|---|
| 400 °C | 4.03 | 76.63 |
| 700 °C | 1.21 | 81.24 |
| 800 °C | 0.91 | 86.80 |
| 900 °C | 0.76 | 86.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, T.-W.; Nagayama, M.; Matsuda, J.; Sasaki, K.; Hayashi, A. Mesoporous Carbon Fibers with Tunable Mesoporosity for Electrode Materials in Energy Devices. Molecules 2021, 26, 724. https://doi.org/10.3390/molecules26030724
Huang T-W, Nagayama M, Matsuda J, Sasaki K, Hayashi A. Mesoporous Carbon Fibers with Tunable Mesoporosity for Electrode Materials in Energy Devices. Molecules. 2021; 26(3):724. https://doi.org/10.3390/molecules26030724
Chicago/Turabian StyleHuang, Ting-Wei, Mayumi Nagayama, Junko Matsuda, Kazunari Sasaki, and Akari Hayashi. 2021. "Mesoporous Carbon Fibers with Tunable Mesoporosity for Electrode Materials in Energy Devices" Molecules 26, no. 3: 724. https://doi.org/10.3390/molecules26030724
APA StyleHuang, T.-W., Nagayama, M., Matsuda, J., Sasaki, K., & Hayashi, A. (2021). Mesoporous Carbon Fibers with Tunable Mesoporosity for Electrode Materials in Energy Devices. Molecules, 26(3), 724. https://doi.org/10.3390/molecules26030724