
In Silico Investigation of the Biological Implications of Complex DNA Damage with Emphasis in Cancer Radiotherapy through a Systems Biology Approach

 , , , , ,
, , , , , 
Abstract
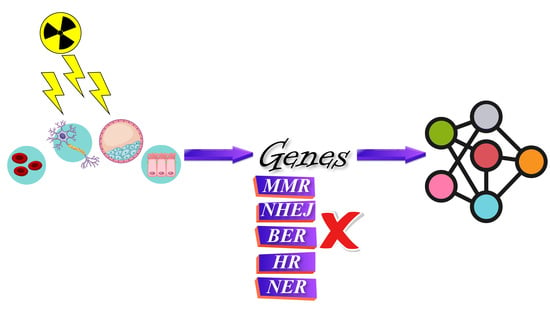
1. Introduction
2. Results and Discussion
2.1. Retrieval of Genes
2.2. Associations among Genes, Cancers and Non-Neoplastic Diseases
2.3. Prognostic Value of DNA Damage-Associated Genes in Diverse Cancers
3. Materials and Methods
3.1. Collection and Selection of Candidate Genes
3.1.1. DNA Repair Mechanisms
3.1.2. Cellular Response to DNA Damage
3.1.3. Complex DNA Damage
3.2. Gene Set Enrichment Analysis
3.3. Protein-Protein Interactions Networks
3.4. Gene-Disease Associations
3.5. Functional Networks
3.6. Survival Analysis
3.7. Transcriptome Response of Cells to Medium-to-High LET Radiation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ward, J.F. The complexity of DNA damage: Relevance to biological consequences. Int. J. Radiat. Biol. 1994, 66, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological consequences of radiation-induced DNA damage: Relevance to radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; McDaniel, L.D.; Schultz, R.A. The role of endogenous and exogenous DNA damage and mutagenesis. Curr. Opin. Genet. Dev. 2004, 14, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Aziz, K.; Nowsheen, S.; Pantelias, G.; Iliakis, G.; Gorgoulis, V.G.; Georgakilas, A.G. Targeting DNA damage and repair: Embracing the pharmacological era for successful cancer therapy. Pharmacol. Ther. 2012, 133, 334–350. [Google Scholar] [CrossRef]
- Ward, J.F. Some biochemical consequences of the spatial distribution of ionizing radiation-produced free radicals. Radiat. Res. 1981, 86, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Kamiya, K. Molecular nature of radiation injury and DNA repair disorders associated with radiosensitivity. Int. J. Hematol. 2012, 95, 239–245. [Google Scholar] [CrossRef]
- Mavragani, I.V.; Nikitaki, Z.; Souli, M.P.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A.G. Complex DNA damage: A route to radiation-induced genomic instability and carcinogenesis. Cancers 2017, 9, 91. [Google Scholar] [CrossRef]
- Hada, M.; Georgakilas, A.G. Formation of clustered DNA damage after high-let irradiation: A review. J. Radiat. Res. 2008, 49, 203–210. [Google Scholar] [CrossRef]
- Paul, D.; Ray, R.C.F. Chapter 2—The cell: The fundamental unit in systems biology. In Systems Biology in Toxicology and Environmental Health; Fry, R.C., Ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 11–42. [Google Scholar]
- Nikitaki, Z.; Michalopoulos, I.; Georgakilas, A.G. Molecular inhibitors of DNA repair: Searching for the ultimate tumor killing weapon. Future Med. Chem. 2015, 7, 1543–1558. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; O’Neill, P.; Stewart, R.D. Induction and repair of clustered DNA lesions: What do we know so far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef]
- Mavragani, I.V.; Nikitaki, Z.; Kalospyros, S.A.; Georgakilas, A.G. Ionizing radiation and complex DNA damage: From prediction to detection challenges and biological significance. Cancers 2019, 11, 1789. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G. Processing of DNA damage clusters in human cells: Current status of knowledge. Mol. Biosyst. 2008, 4, 30–35. [Google Scholar] [CrossRef]
- Gligorijevic, V.; Przulj, N. Methods for biological data integration: Perspectives and challenges. J. R. Soc. Interface 2015, 12, 20150571. [Google Scholar] [CrossRef]
- Song, R.; Sarnoski, E.A.; Acar, M. The systems biology of single-cell aging. Iscience 2018, 7, 154–169. [Google Scholar] [CrossRef]
- Zhao, X.M.; Tian, W.; Jiang, R.; Wan, J. Computational systems biology. Sci. World J. 2013, 2013, 350358. [Google Scholar] [CrossRef] [PubMed]
- Mooney, K.M.; Morgan, A.E.; Mc Auley, M.T. Aging and computational systems biology. Wiley Interdiscip. Reviews. Syst. Biol. Med. 2016, 8, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Ostaszewski, M.; Kalliolias, G.D.; Chiocchia, G.; Olaso, R.; Petit-Teixeira, E.; Helikar, T.; Niarakis, A. Computational systems biology approach for the study of rheumatoid arthritis: From a molecular map to a dynamical model. Genom. Comput. Biol. 2018, 4, e100050. [Google Scholar] [CrossRef][Green Version]
- Finley, S.D.; Chu, L.H.; Popel, A.S. Computational systems biology approaches to anti-angiogenic cancer therapeutics. Drug Discov. Today 2015, 20, 17–197. [Google Scholar] [CrossRef]
- Rai, S.; Raj, U.; Varadwaj, P.K. Systems biology: A powerful tool for drug development. Curr. Top. Med. Chem. 2018, 18, 1745–1754. [Google Scholar] [CrossRef]
- Durmus, S.; Cakir, T.; Ozgur, A.; Guthke, R. A review on computational systems biology of pathogen-host interactions. Front. Microbiol. 2015, 6, 235. [Google Scholar] [PubMed]
- Cancer Stat Facts: Leukemia—Acute Myeloid Leukemia (AML). Available online: https://seer.cancer.gov/statfacts/html/amyl.html (accessed on 7 June 2021).
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2021. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of bcl-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- A current view on inflammation. Nat. Immunol. 2017, 18, 825. [CrossRef] [PubMed]
- Bennett, J.M.; Reeves, G.; Billman, G.E.; Sturmberg, J.P. Inflammation-nature’s way to efficiently respond to all types of challenges: Implications for understanding and managing “the epidemic” of chronic diseases. Front. Med. 2018, 5, 316. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Wan, D.; Jiang, W.; Hao, J. Research advances in how the cgas-sting pathway controls the cellular inflammatory response. Front. Immunol. 2020, 11, 615. [Google Scholar] [CrossRef]
- Pavlopoulou, A.; Karaca, E.; Balestrazzi, A.; Georgakilas, A.G. In silico phylogenetic and structural analyses of plant endogenous danger signaling molecules upon stress. Oxidative Med. Cell. Longev. 2019, 2019, 8683054. [Google Scholar] [CrossRef]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, sting-dependent proinflammatory gene induction necessitates canonical nf-kappab activation through tbk1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. Intogen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef]
- Ogle, K.S.; Swanson, G.M.; Woods, N.; Azzouz, F. Cancer and comorbidity: Redefining chronic diseases. Cancer 2000, 88, 653–663. [Google Scholar] [CrossRef]
- Delp, M.D.; Charvat, J.M.; Limoli, C.L.; Globus, R.K.; Ghosh, P. Apollo lunar astronauts show higher cardiovascular disease mortality: Possible deep space radiation effects on the vascular endothelium. Sci. Rep. 2016, 6, 29901. [Google Scholar] [CrossRef]
- Pavlopoulou, A.; Bagos, P.G.; Koutsandrea, V.; Georgakilas, A.G. Molecular determinants of radiosensitivity in normal and tumor tissue: A bioinformatic approach. Cancer Lett. 2017, 403, 37–47. [Google Scholar] [CrossRef]
- Di Nanni, N.; Bersanelli, M.; Milanesi, L.; Mosca, E. Network diffusion promotes the integrative analysis of multiple omics. Front. Genet. 2020, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- Akl, H.; Vervloessem, T.; Kiviluoto, S.; Bittremieux, M.; Parys, J.B.; De Smedt, H.; Bultynck, G. A dual role for the anti-apoptotic bcl-2 protein in cancer: Mitochondria versus endoplasmic reticulum. Biochim. Biophys. Acta 2014, 1843, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xing, X.; Liu, Q.; Wang, Z.; Xin, Y.; Zhang, P.; Hu, C.; Liu, Y. Hypoxia-induced autophagy reduces radiosensitivity by the hif-1alpha/mir-210/bcl-2 pathway in colon cancer cells. Int. J. Oncol. 2015, 46, 750–756. [Google Scholar] [CrossRef]
- Toy, H.I.; Karakulah, G.; Kontou, P.I.; Alotaibi, H.; Georgakilas, A.G.; Pavlopoulou, A. Investigating molecular determinants of cancer cell resistance to ionizing radiation through an integrative bioinformatics approach. Front. Cell Dev. Biol. 2021, 9, 620248. [Google Scholar] [CrossRef]
- Bates, J.E.; Chen, M.H.; Constine, L.S. Radiation-associated coronary disease in young cancer survivors: The beat goes on; we must preserve it. JACC. CardioOncol. 2021, 3, 393–396. [Google Scholar] [CrossRef]
- Duma, M.N.; Oszfolk, N.I.; Boeckh-Behrens, T.; Oechsner, M.; Zimmer, C.; Meyer, B.; Pfluger, P.T.; Combs, S.E. Positive correlation between blood glucose and radiotherapy doses to the central gustatory system in glioblastoma multiforme patients. Radiat. Oncol. 2019, 14, 97. [Google Scholar] [CrossRef] [PubMed]
- Baradaran-Ghahfarokhi, M. Radiation-induced kidney injury. J. Ren. Inj. Prev. 2012, 1, 49–50. [Google Scholar]
- Klaus, R.; Niyazi, M.; Lange-Sperandio, B. Radiation-induced kidney toxicity: Molecular and cellular pathogenesis. Radiat. Oncol. 2021, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lipshultz, S.E.; Schwartz, C.; Fajardo, L.F.; Coen, V.; Constine, L.S. Radiation-associated cardiovascular disease: Manifestations and management. Semin. Radiat. Oncol. 2003, 13, 346–356. [Google Scholar] [CrossRef]
- Koutroumpakis, E.; Palaskas, N.L.; Lin, S.H.; Abe, J.I.; Liao, Z.; Banchs, J.; Deswal, A.; Yusuf, S.W. Modern radiotherapy and risk of cardiotoxicity. Chemotherapy 2020, 65, 65–76. [Google Scholar] [CrossRef]
- Kountouras, J.; Zavos, C. Recent advances in the management of radiation colitis. World J. Gastroenterol. 2008, 14, 7289–7301. [Google Scholar] [CrossRef]
- Kim, J.; Jung, Y. Radiation-induced liver disease: Current understanding and future perspectives. Exp. Mol. Med. 2017, 49, e359. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, J.; Kraft, A.S. Bax-induced apoptotic cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 529–531. [Google Scholar] [CrossRef]
- Wang, K.; Tepper, J.E. Radiation therapy-associated toxicity: Etiology, management, and prevention. CA Cancer J. Clin. 2021, 71, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G.; Pavlopoulou, A.; Louka, M.; Nikitaki, Z.; Vorgias, C.E.; Bagos, P.G.; Michalopoulos, I. Emerging molecular networks common in ionizing radiation, immune and inflammatory responses by employing bioinformatics approaches. Cancer Lett. 2015, 368, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.M.; Gatti, R.A. Clinical radiation sensitivity with DNA repair disorders: An overview. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 1323–1331. [Google Scholar] [CrossRef]
- Balkwill, F.; Joffroy, C. Tnf: A tumor-suppressing factor or a tumor-promoting factor? Future Oncol. 2010, 6, 1833–1836. [Google Scholar] [CrossRef]
- Cory, S.; Huang, D.C.; Adams, J.M. The bcl-2 family: Roles in cell survival and oncogenesis. Oncogene 2003, 22, 8590–8607. [Google Scholar] [CrossRef]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin d as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Qie, S.; Diehl, J.A. Cyclin d1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef]
- Luu Hoang, K.N.; Anstee, J.E.; Arnold, J.N. The diverse roles of heme oxygenase-1 in tumor progression. Front. Immunol. 2021, 12, 658315. [Google Scholar] [CrossRef] [PubMed]
- Kyriakou, I.; Tremi, I.; Georgakilas, A.G.; Emfietzoglou, D. Microdosimetric investigation of the radiation quality of low-medium energy electrons using geant4-DNA. Appl. Radiat. Isot. 2021, 172, 109654. [Google Scholar] [CrossRef]
- Cammarata, F.P.; Forte, G.I.; Broggi, G.; Bravata, V.; Minafra, L.; Pisciotta, P.; Calvaruso, M.; Tringali, R.; Tomasello, B.; Torrisi, F.; et al. Molecular investigation on a triple negative breast cancer xenograft model exposed to proton beams. Int. J. Mol. Sci. 2020, 21, 6337. [Google Scholar] [CrossRef] [PubMed]
- Bravata, V.; Cammarata, F.P.; Minafra, L.; Pisciotta, P.; Scazzone, C.; Manti, L.; Savoca, G.; Petringa, G.; Cirrone, G.A.P.; Cuttone, G.; et al. Proton-irradiated breast cells: Molecular points of view. J. Radiat. Res. 2019, 60, 451–465. [Google Scholar] [CrossRef]
- Konings, K.; Vandevoorde, C.; Belmans, N.; Vermeesen, R.; Baselet, B.; Walleghem, M.V.; Janssen, A.; Isebaert, S.; Baatout, S.; Haustermans, K.; et al. The combination of particle irradiation with the hedgehog inhibitor gant61 differently modulates the radiosensitivity and migration of cancer cells compared to x-ray irradiation. Front. Oncol. 2019, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, A.G. Role of DNA damage and repair in detrimental effects of ionizing radiation. Radiation 2021, 1, 1–4. [Google Scholar] [CrossRef]
- Lee, Y.K.; Hong, N.; Park, S.H.; Shin, D.Y.; Lee, C.R.; Kang, S.W.; Lee, J.; Jeong, J.J.; Nam, K.H.; Chung, W.Y.; et al. The relationship of comorbidities to mortality and cause of death in patients with differentiated thyroid carcinoma. Sci. Rep. 2019, 9, 11435. [Google Scholar] [CrossRef] [PubMed]
- Sarfati, D.; Koczwara, B.; Jackson, C. The impact of comorbidity on cancer and its treatment. CA Cancer J. Clin. 2016, 66, 337–350. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Smedley, D.; Haider, S.; Durinck, S.; Pandini, L.; Provero, P.; Allen, J.; Arnaiz, O.; Awedh, M.H.; Baldock, R.; Barbiera, G.; et al. The biomart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015, 43, W589–W598. [Google Scholar] [CrossRef]
- Zhang, J.; Haider, S.; Baran, J.; Cros, A.; Guberman, J.M.; Hsu, J.; Liang, Y.; Yao, L.; Kasprzyk, A. Biomart: A data federation framework for large collaborative projects. Database 2011, 2011, bar038. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, R.J.; Kahari, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl biomarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef]
- Sharma, S.; Ciufo, S.; Starchenko, E.; Darji, D.; Chlumsky, L.; Karsch-Mizrachi, I.; Schoch, C.L. The NCBI biocollections database. Database 2018, 2018, bay006. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. Genbank. Nucleic Acids Res. 2021, 49, D92–D96. [Google Scholar] [CrossRef]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S.; Ami, G.O.H.; Web Presence Working Group. Amigo: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. Webgestalt 2019: Gene set analysis toolkit with revamped uis and apis. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kirov, S.; Snoddy, J. Webgestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005, 33, W741–W748. [Google Scholar] [CrossRef]
- Kutmon, M.; Riutta, A.; Nunes, N.; Hanspers, K.; Willighagen, E.L.; Bohler, A.; Melius, J.; Waagmeester, A.; Sinha, S.R.; Miller, R.; et al. Wikipathways: Capturing the full diversity of pathway knowledge. Nucleic Acids Res. 2016, 44, D488–D494. [Google Scholar] [CrossRef] [PubMed]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Ryan, A.M.; Digles, D.; Lopes, E.N.; Ehrhart, F.; et al. Wikipathways: Connecting communities. Nucleic Acids Res. 2021, 49, D613–D621. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The string database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Pinero, J.; Bravo, A.; Queralt-Rosinach, N.; Gutierrez-Sacristan, A.; Deu-Pons, J.; Centeno, E.; Garcia-Garcia, J.; Sanz, F.; Furlong, L.I. Disgenet: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]
- Pinero, J.; Ramirez-Anguita, J.M.; Sauch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The disgenet knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar]
- Rogers, F.B. Medical subject headings. Bull. Med. Libr. Assoc. 1963, 51, 114–116. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. Cytohubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef]
- Li, C.; Tang, Z.; Zhang, W.; Ye, Z.; Liu, F. Gepia2021: Integrating multiple deconvolution-based analysis into gepia. Nucleic Acids Res. 2021, 49, W242–W246. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. Gepia: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An integrated tcga pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type * | BAX | TNF | BCL2 | CCND1 | HMOX1 |
|---|---|---|---|---|---|
| Adrenocortical carcinoma (ACC) | x | x | x | x | |
| Bladder urothelial carcinoma (BLCA) | x | x | x | ||
| Breast invasive carcinoma (BRCA) | x | x | x | ||
| Cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) | x | x | |||
| Cholangio carcinoma (CHOL) | x | x | x | ||
| Colon adenocarcinoma (COAD) | x | x | x | ||
| Lymphoid neoplasm diffuse large B-cell lymphoma (DLBC) | x | x | x | x | |
| Esophageal carcinoma (ESCA) | x | x | x | ||
| Glioblastoma multiforme (GBM) | x | x | |||
| Head and neck squamous cell carcinoma (HNSC) | x | x | x | ||
| Kidney chromophobe (KICH) | x | x | |||
| Kidney renal clear cell carcinoma (KIRC) | x | ||||
| Kidney renal papillary cell carcinoma (KIRP) | x | x | |||
| Acute myeloid leukemia (LAML) | x | x | |||
| Brain lower grade glioma (LGG) | x | x | |||
| Liver hepatocellular carcinoma (LIHC) | x | x | x | x | |
| Lung adenocarcinoma (LUAD) | x | x | x | x | |
| Lung squamous cell carcinoma (LUSC) | x | x | |||
| Mesothelioma (MESO) | x | x | x | ||
| Ovarian serous cystadenocarcinoma (OV) | x | x | |||
| Pancreatic adenocarcinoma (PAAD) | x | x | x | ||
| Pheochromocytoma and paraganglioma (PCPG) | x | x | x | ||
| Prostate adenocarcinoma (PRAD) | x | x | x | ||
| Rectum adenocarcinoma (READ) | x | x | x | x | |
| Sarcoma (SARC) | x | x | x | ||
| Skin cutaneous melanoma (SKCM) | x | ||||
| Stomach adenocarcinoma (STAD) | x | x | x | ||
| Testicular germ cell tumors (TGCT) | x | x | x | x | |
| Thyroid carcinoma (THCA) | x | x | |||
| Thymoma (THYM) | x | x | x | x | |
| Uterine corpus endometrial carcinoma (UCEC) | x | x | x | x | |
| Uterine carcinosarcoma (UCS) | x | x | x | x | |
| Uveal melanoma (UVM) | x | x | x | x |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlopoulou, A.; Asfa, S.; Gioukakis, E.; Mavragani, I.V.; Nikitaki, Z.; Takan, I.; Pouget, J.-P.; Harrison, L.; Georgakilas, A.G. In Silico Investigation of the Biological Implications of Complex DNA Damage with Emphasis in Cancer Radiotherapy through a Systems Biology Approach. Molecules 2021, 26, 7602. https://doi.org/10.3390/molecules26247602
Pavlopoulou A, Asfa S, Gioukakis E, Mavragani IV, Nikitaki Z, Takan I, Pouget J-P, Harrison L, Georgakilas AG. In Silico Investigation of the Biological Implications of Complex DNA Damage with Emphasis in Cancer Radiotherapy through a Systems Biology Approach. Molecules. 2021; 26(24):7602. https://doi.org/10.3390/molecules26247602
Chicago/Turabian StylePavlopoulou, Athanasia, Seyedehsadaf Asfa, Evangelos Gioukakis, Ifigeneia V. Mavragani, Zacharenia Nikitaki, Işıl Takan, Jean-Pierre Pouget, Lynn Harrison, and Alexandros G. Georgakilas. 2021. "In Silico Investigation of the Biological Implications of Complex DNA Damage with Emphasis in Cancer Radiotherapy through a Systems Biology Approach" Molecules 26, no. 24: 7602. https://doi.org/10.3390/molecules26247602
APA StylePavlopoulou, A., Asfa, S., Gioukakis, E., Mavragani, I. V., Nikitaki, Z., Takan, I., Pouget, J.-P., Harrison, L., & Georgakilas, A. G. (2021). In Silico Investigation of the Biological Implications of Complex DNA Damage with Emphasis in Cancer Radiotherapy through a Systems Biology Approach. Molecules, 26(24), 7602. https://doi.org/10.3390/molecules26247602