
Computational Studies of Au(I) and Au(III) Anticancer MetalLodrugs: A Survey




{kind=link}
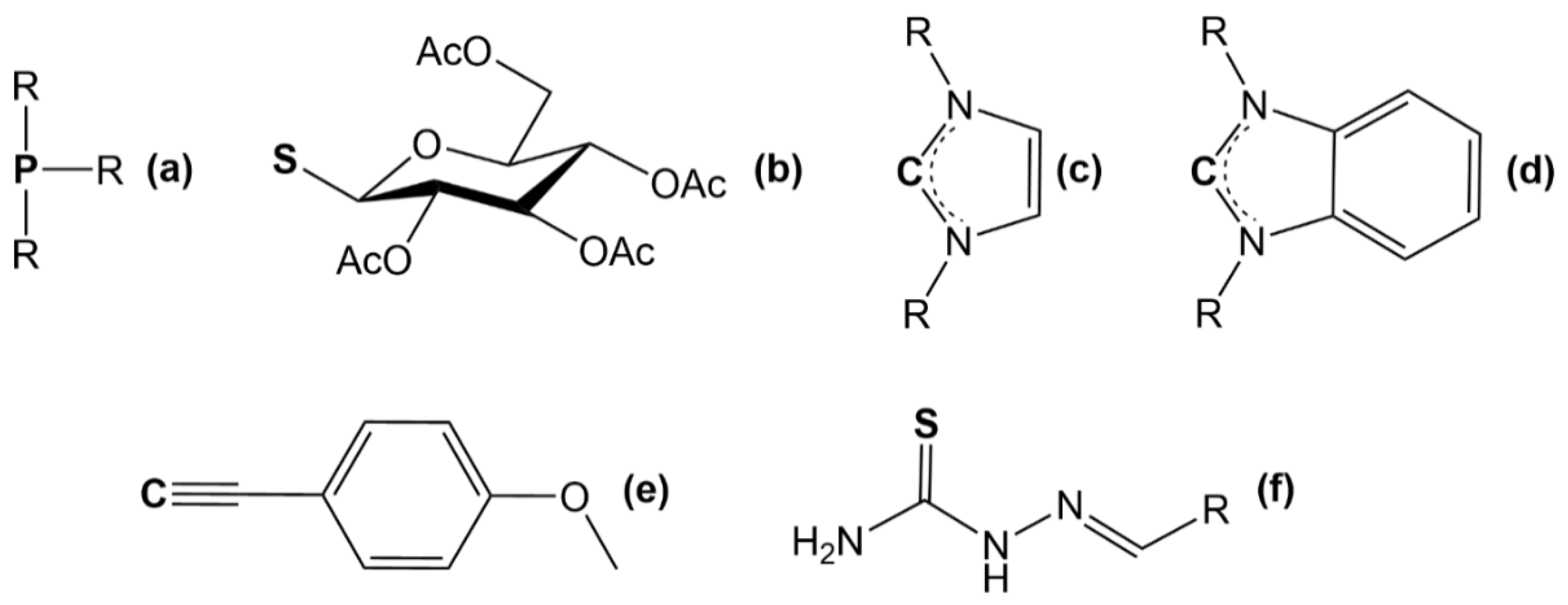{kind=link}
{kind=link}
{kind=link}
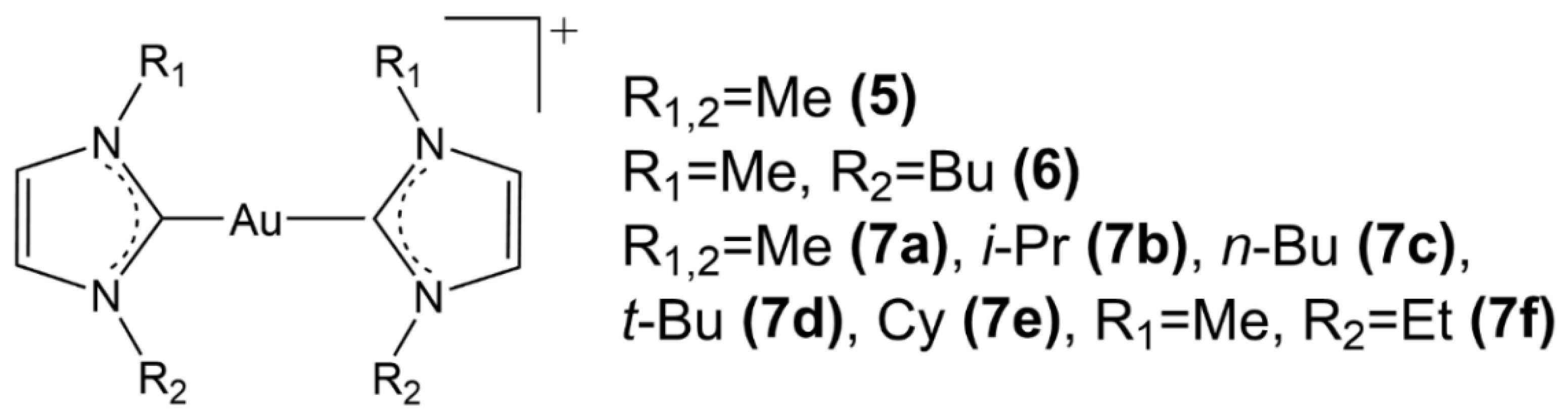{kind=link}
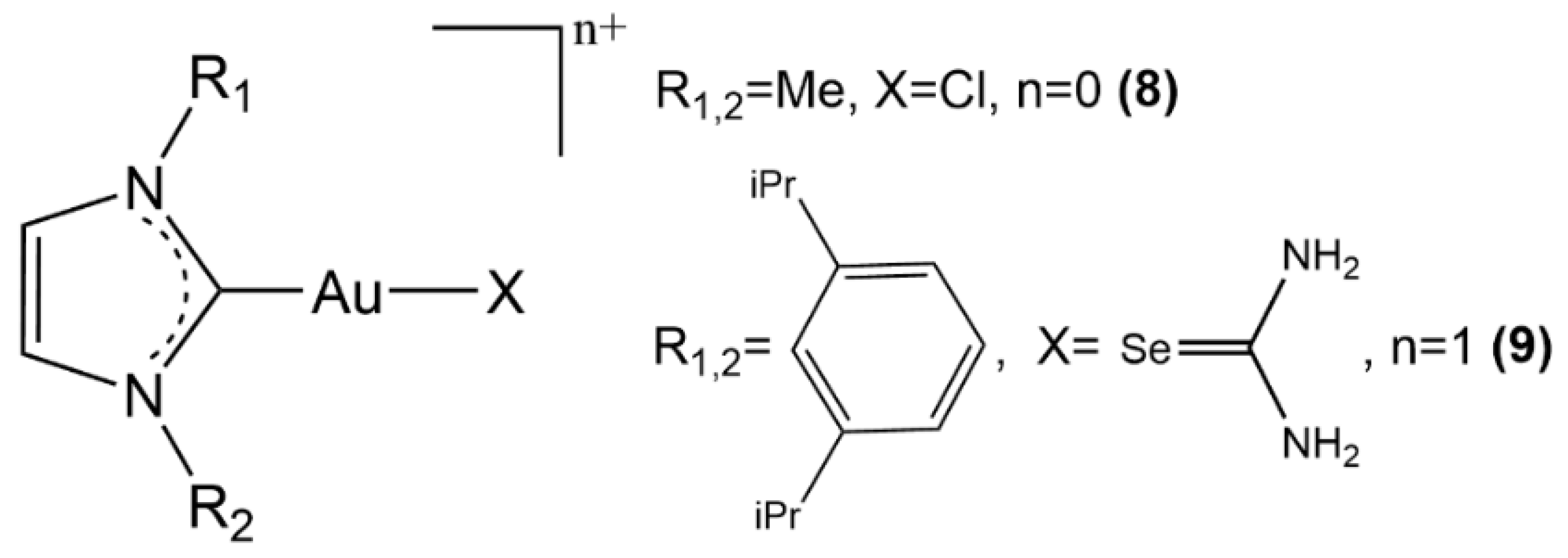{kind=link}
{kind=link}
{kind=link}
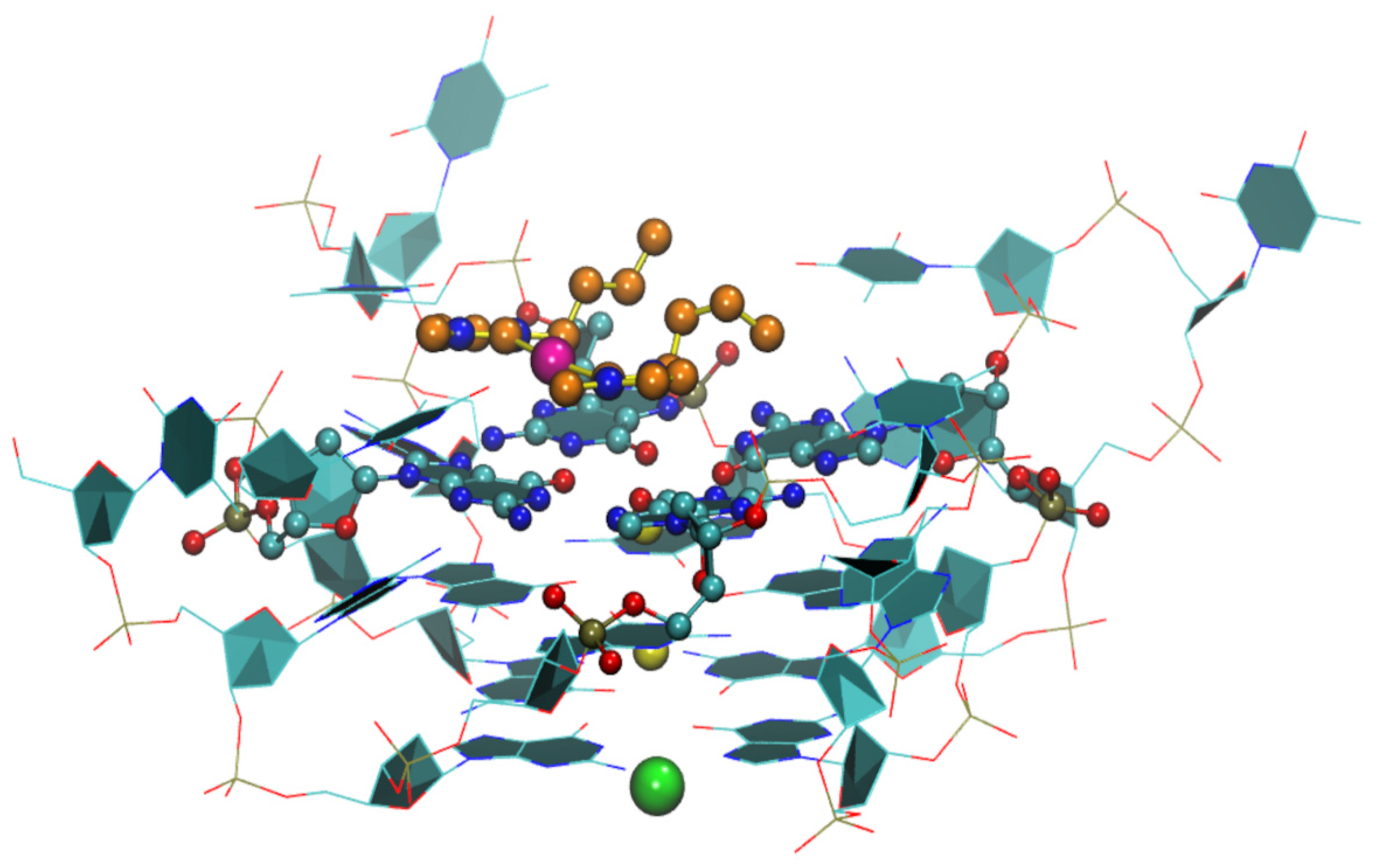{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Studies of Au(I) Metallodrugs
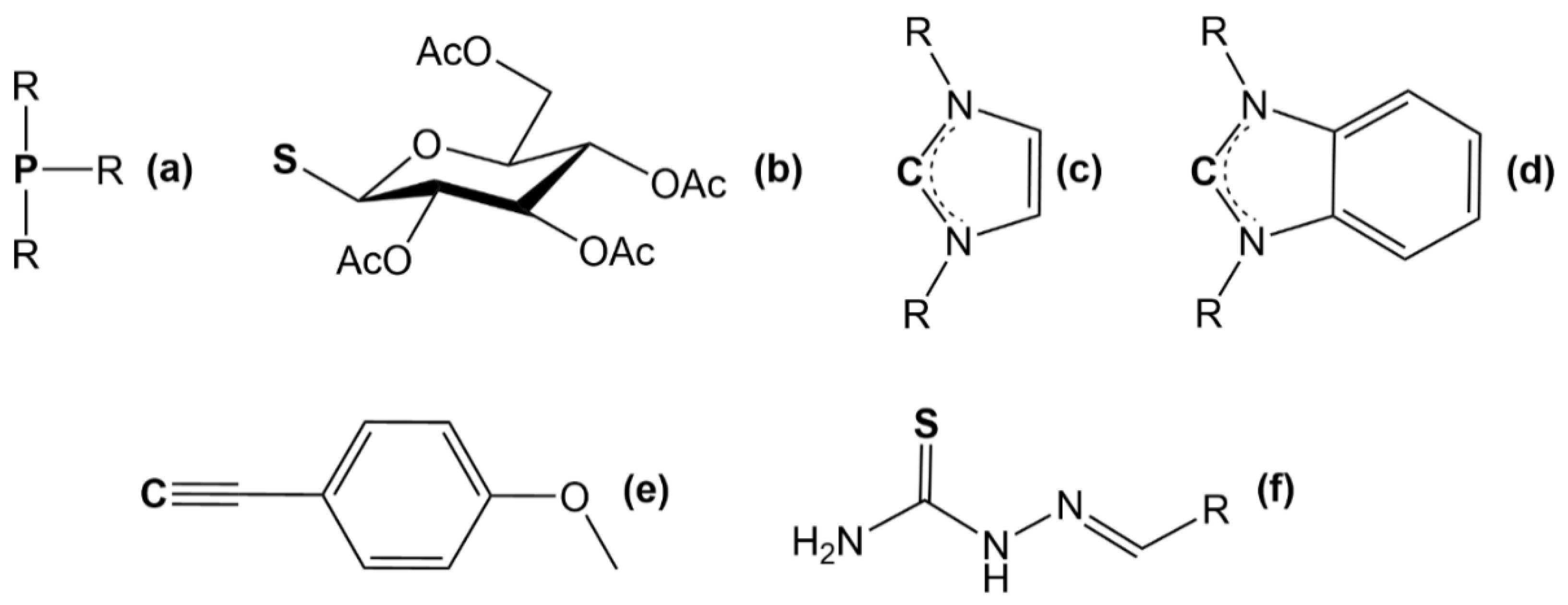
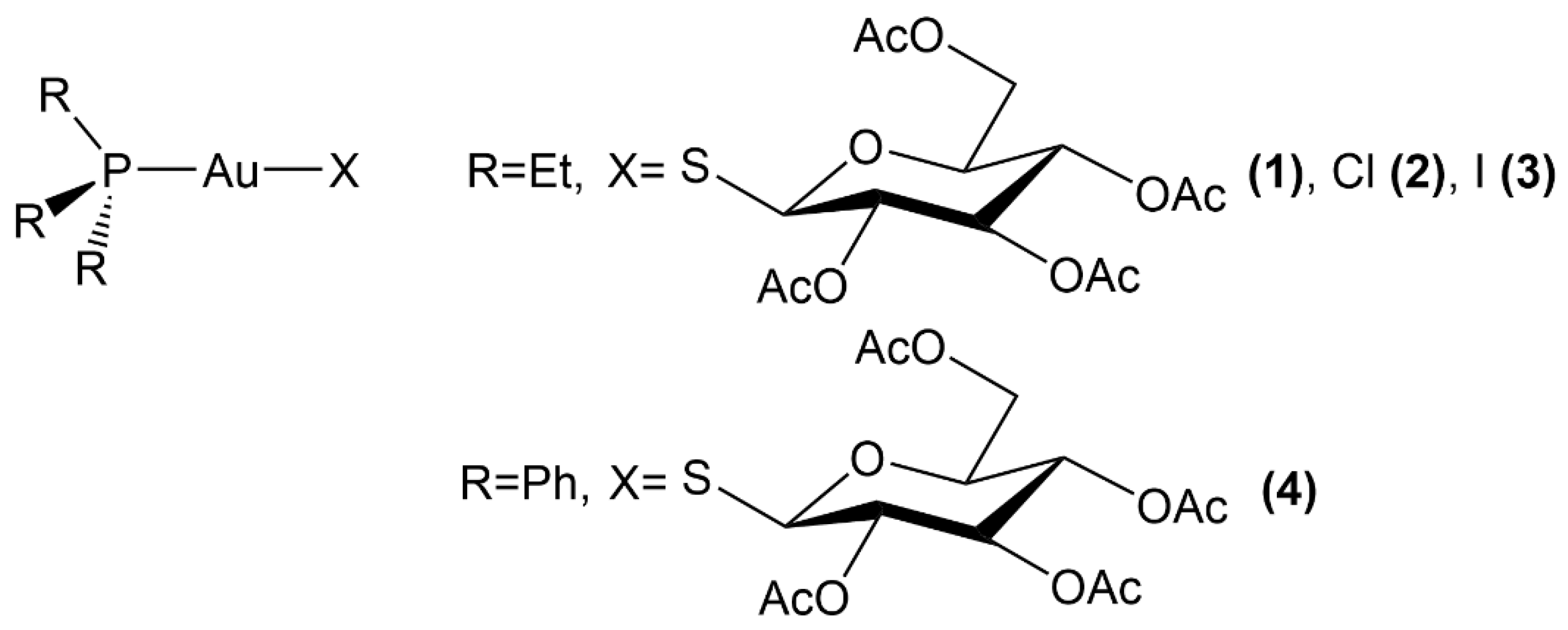
2.1. Auranofin and Auranofin Analogs
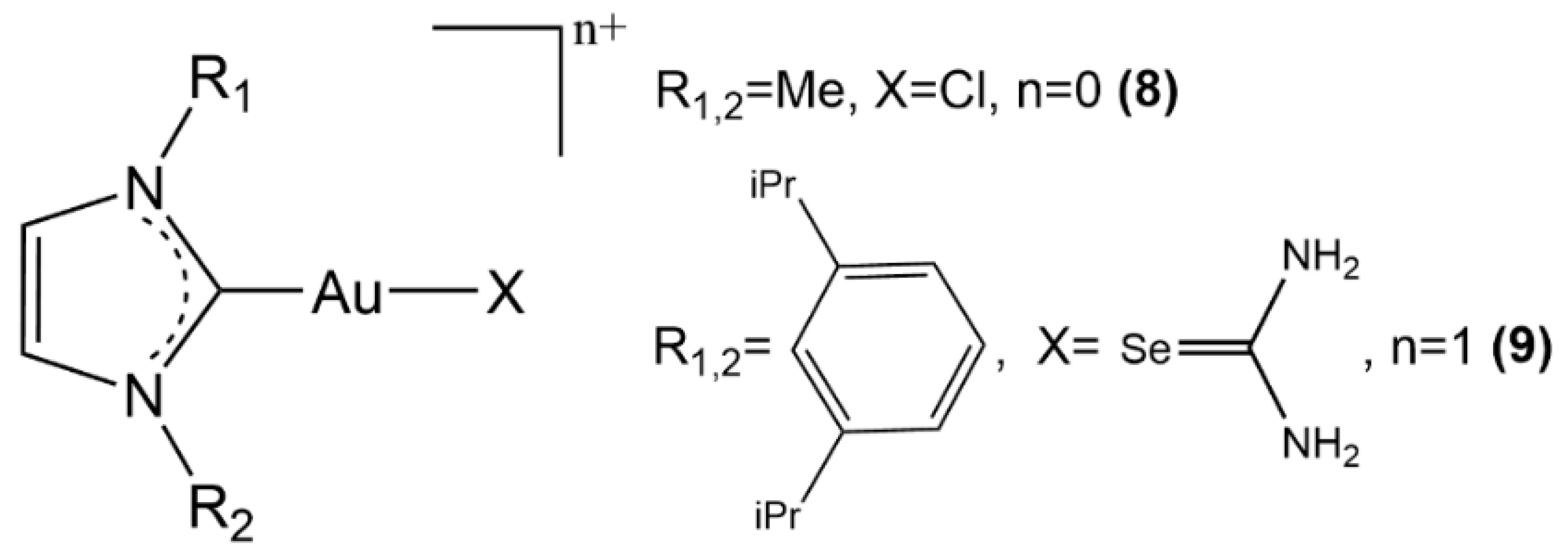
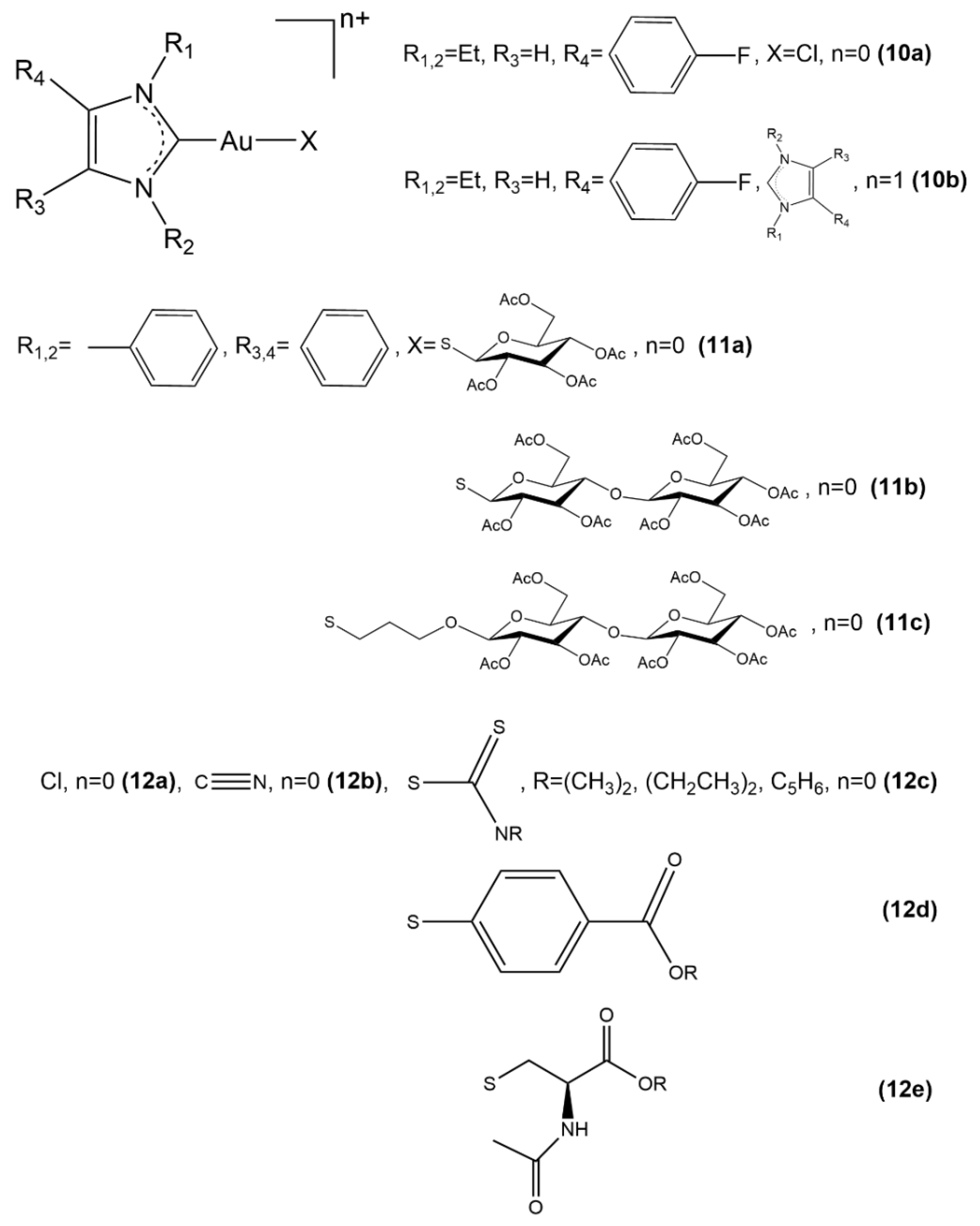
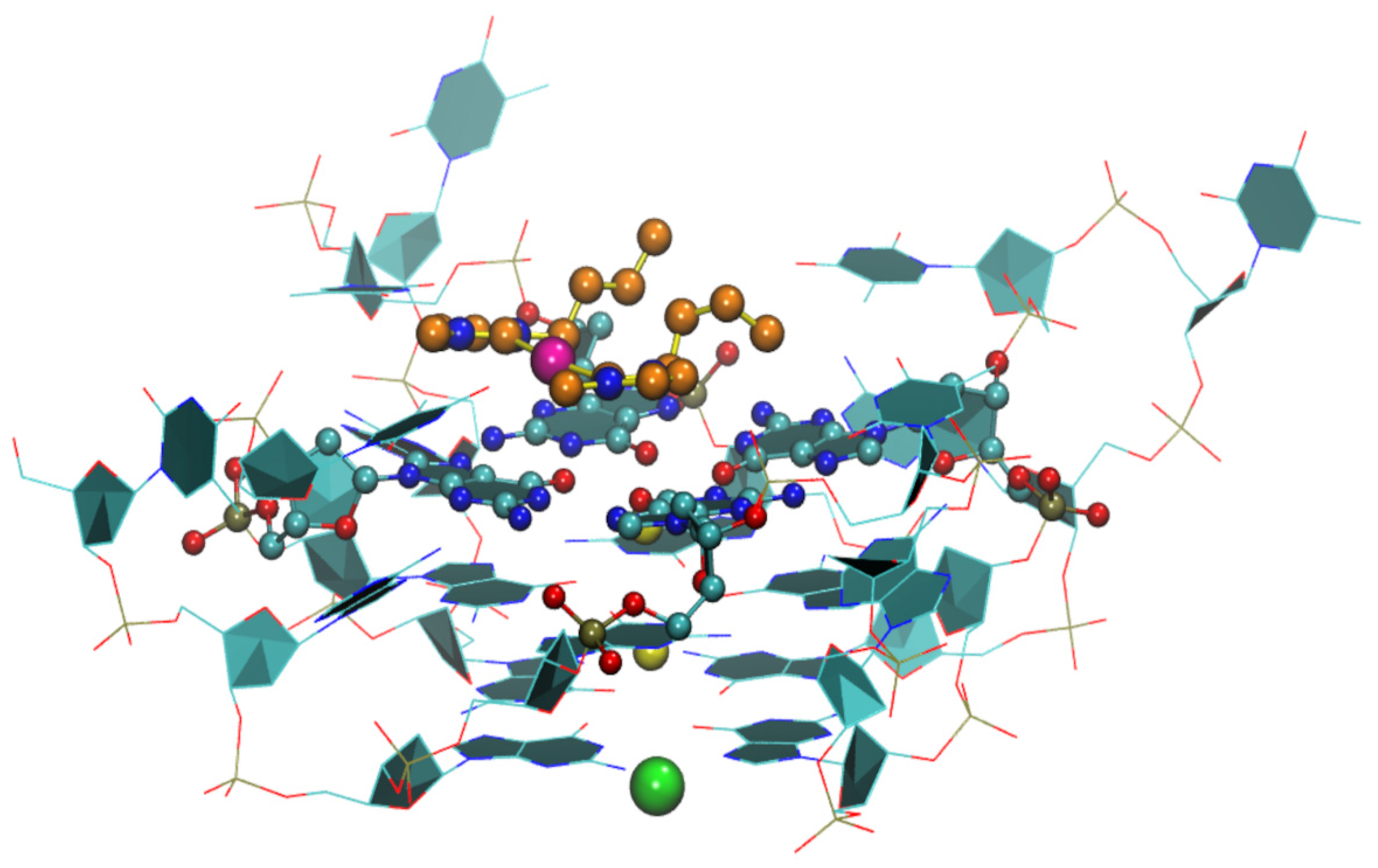
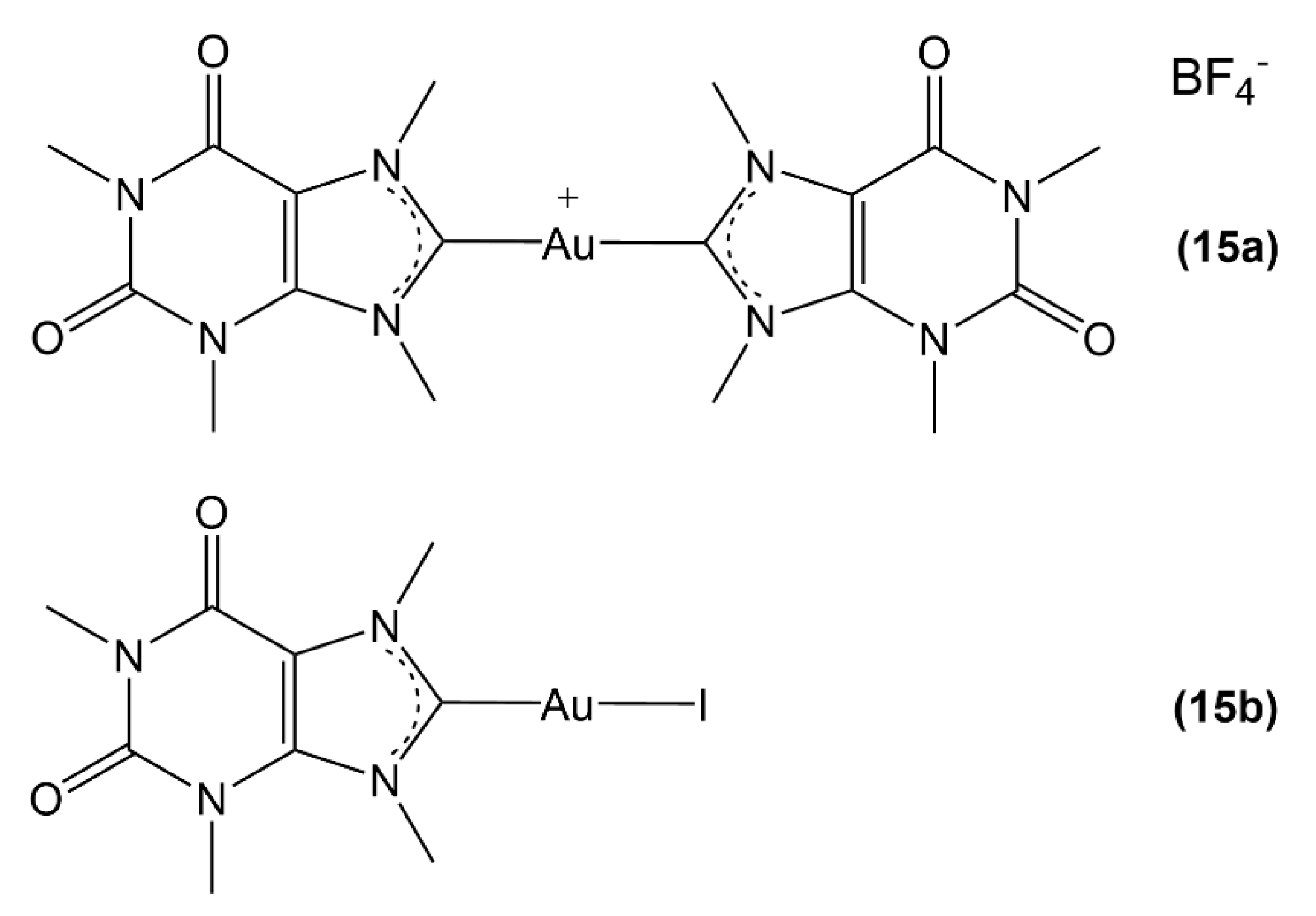
2.2. Au(I) Complexes with N-heterocyclic Carbene (NHC) Ligands
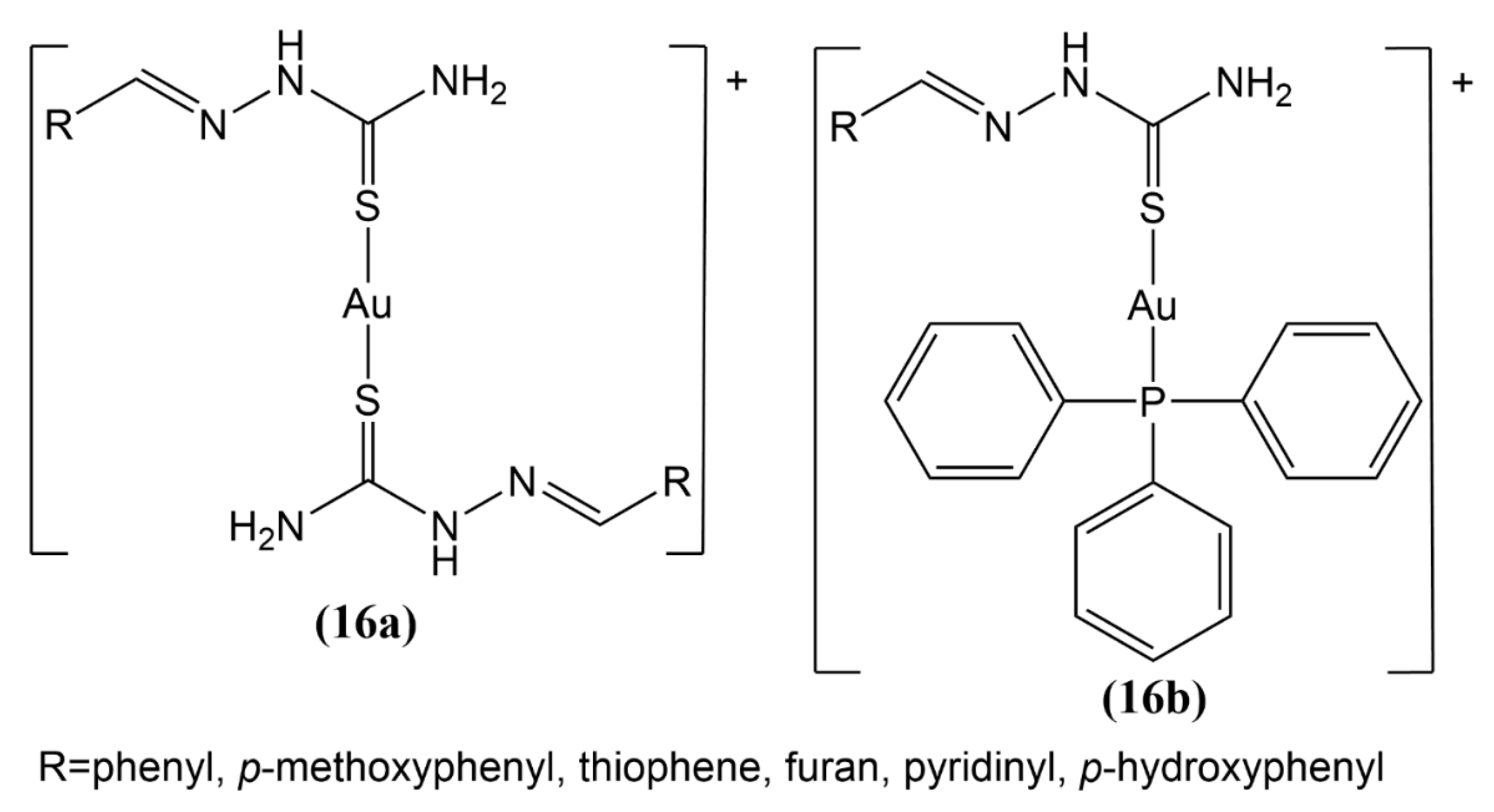
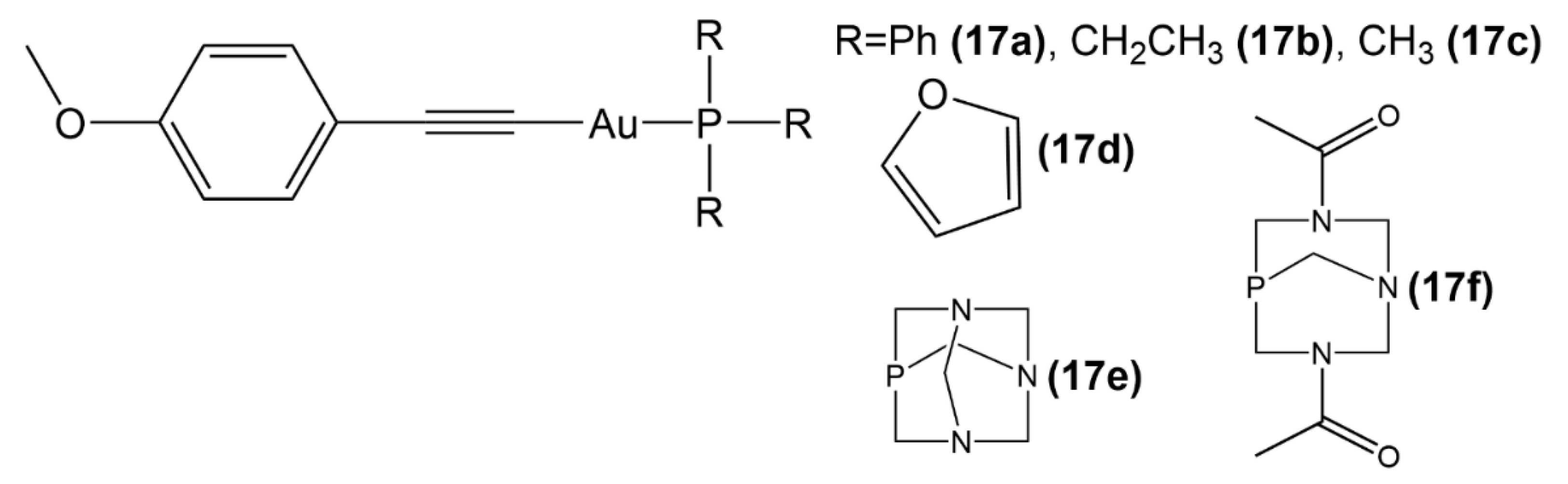
2.3. Au(I) Complexes with Other Structures
3. Computational Studies of Au(III) Metallodrugs
3.1. Interaction with Biologically Relevant Targets
3.1.1. Au(III) Complexes with Chelating N Donor Ligands
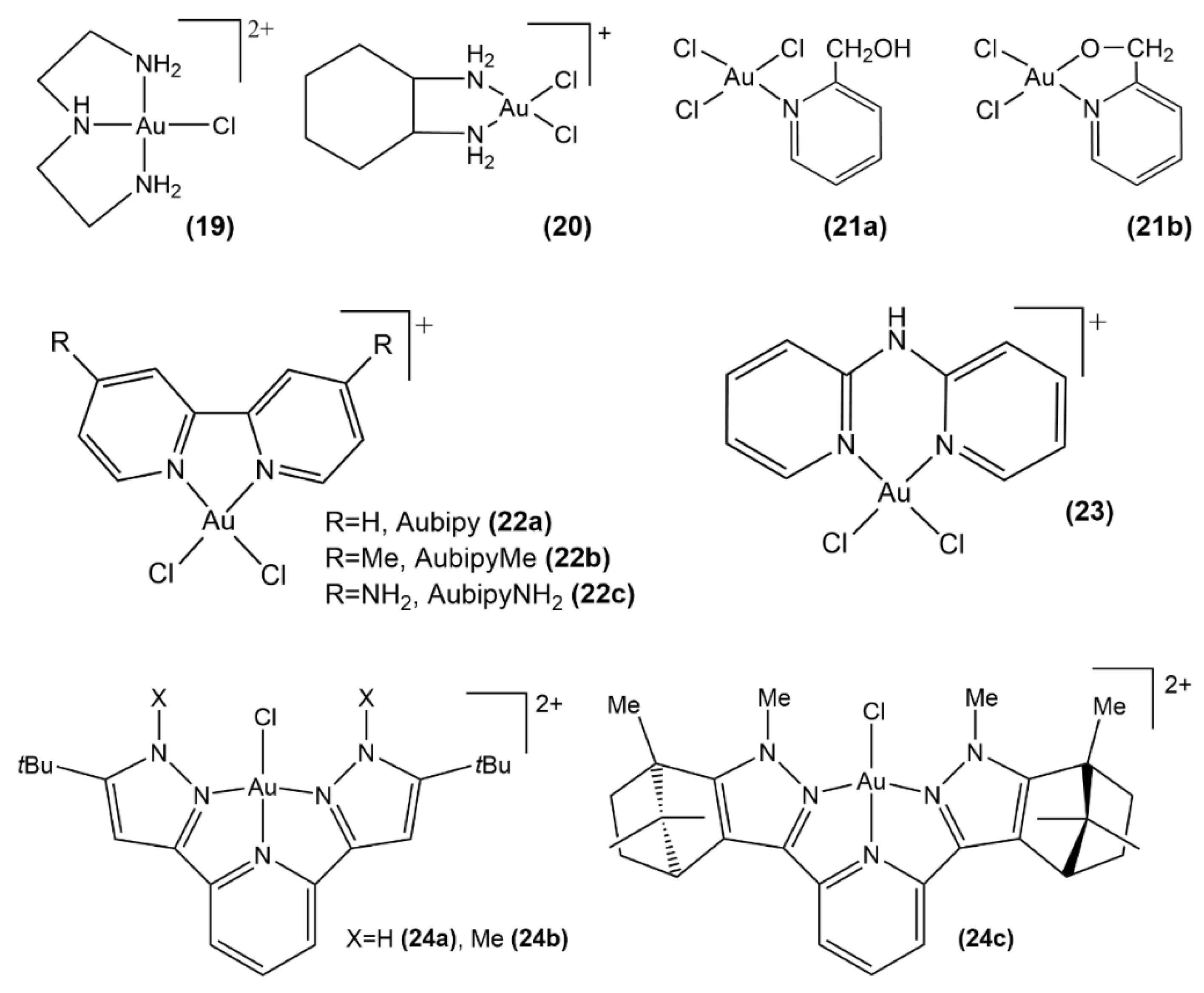
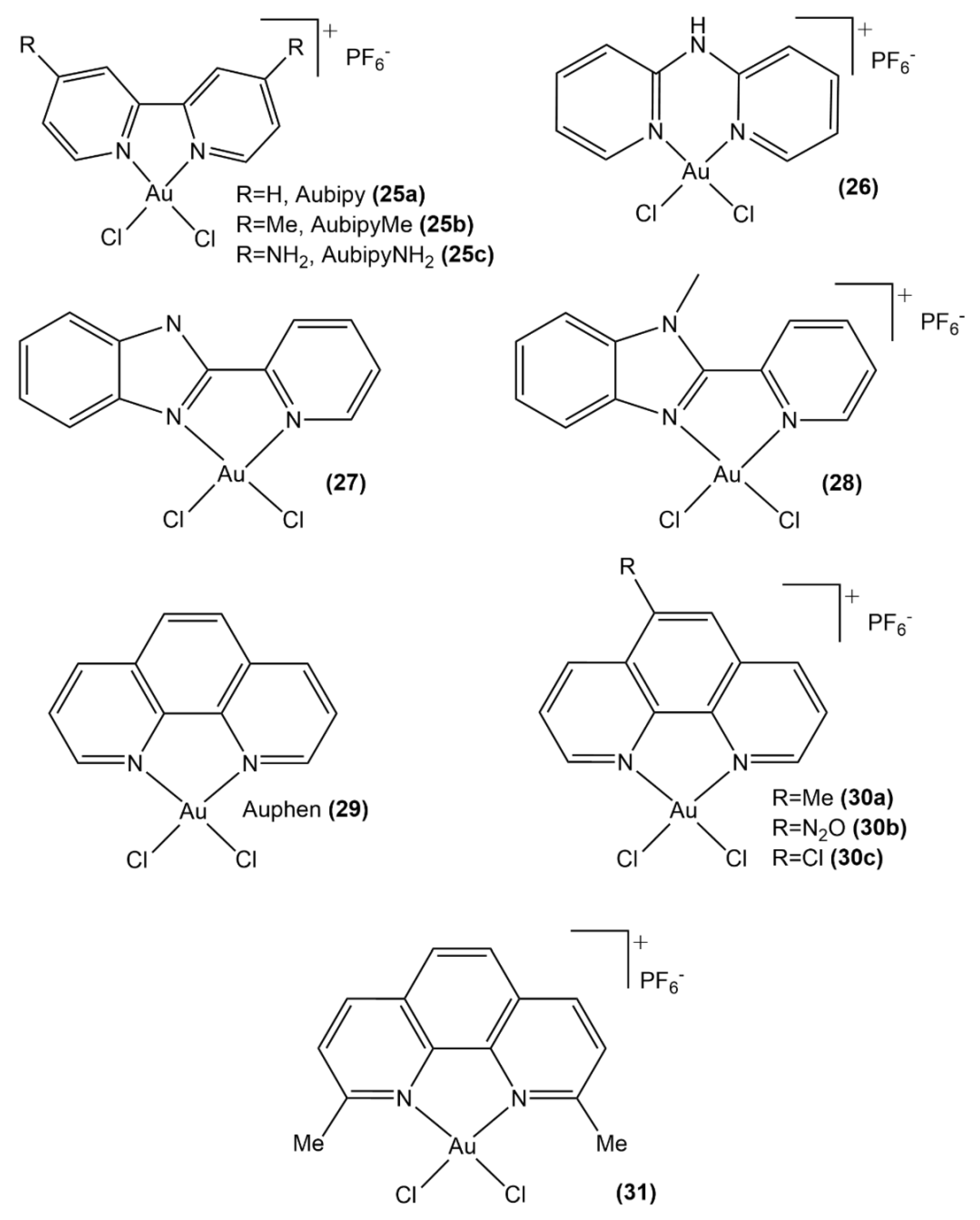
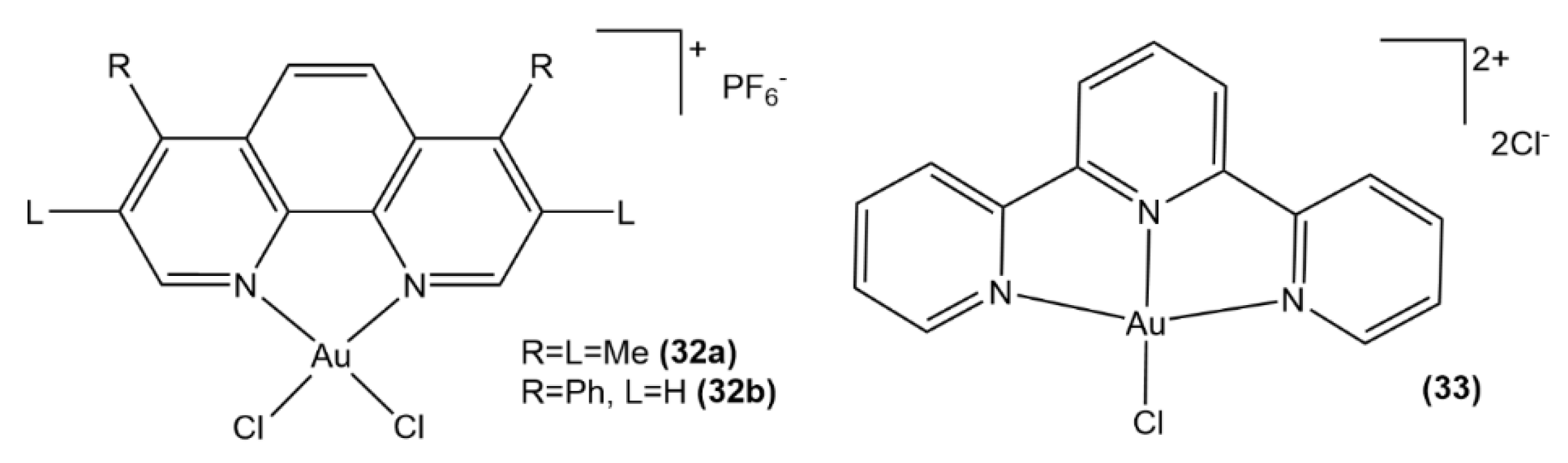
3.1.2. Au(III) Complexes with Cyclometalated Ligands
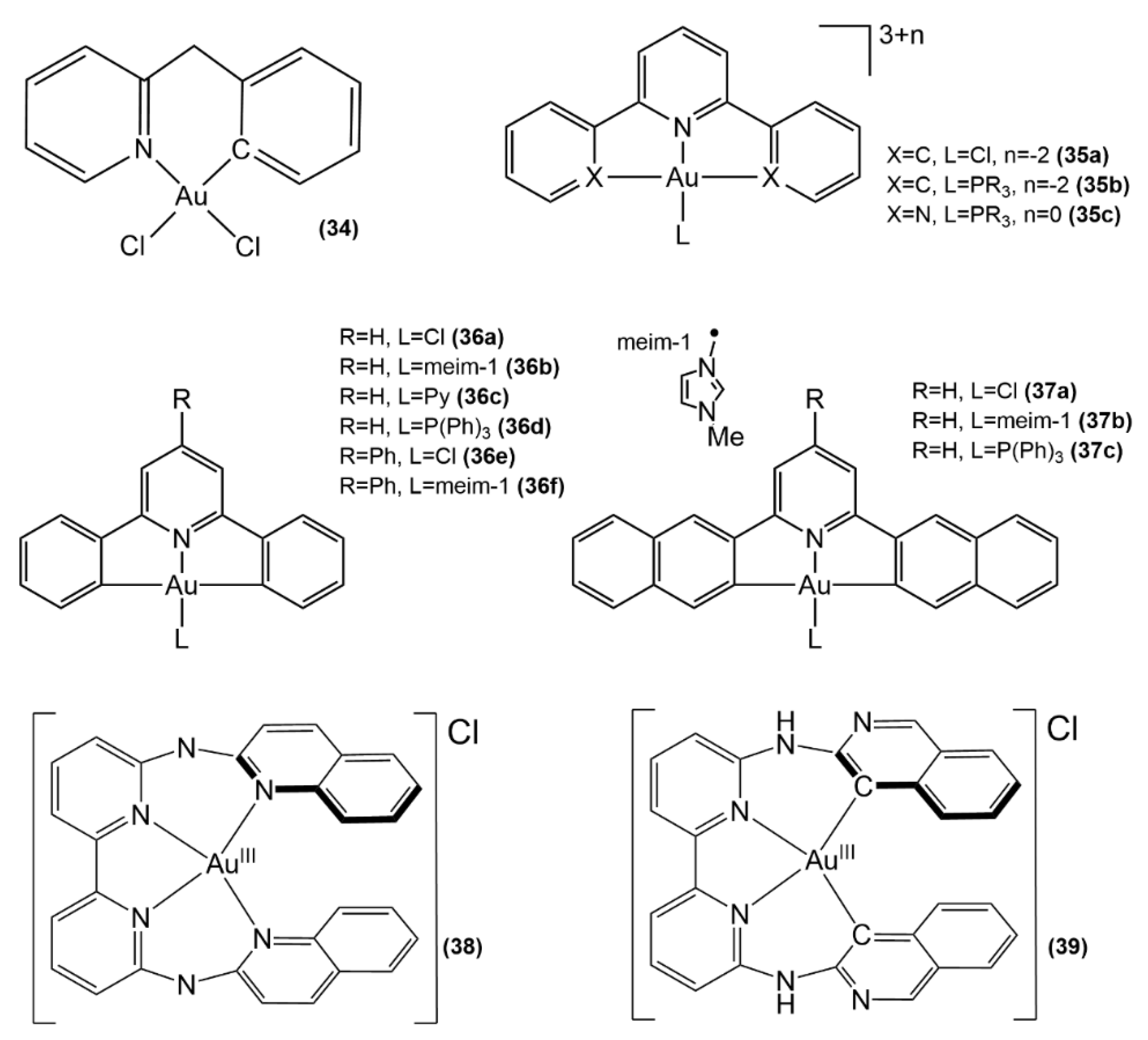
3.1.3. Au(III) Complexes with Cyclometalated Ligands
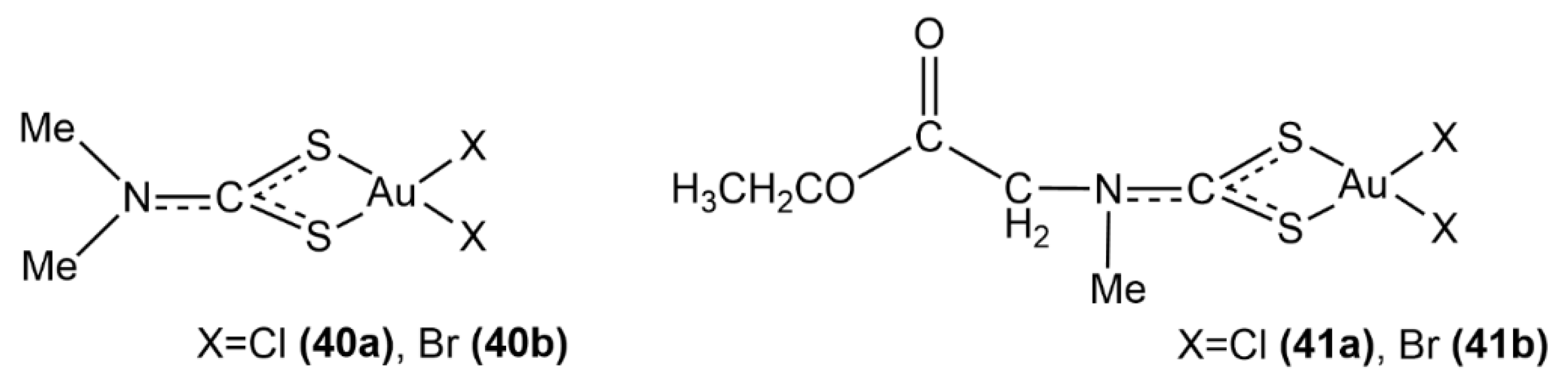

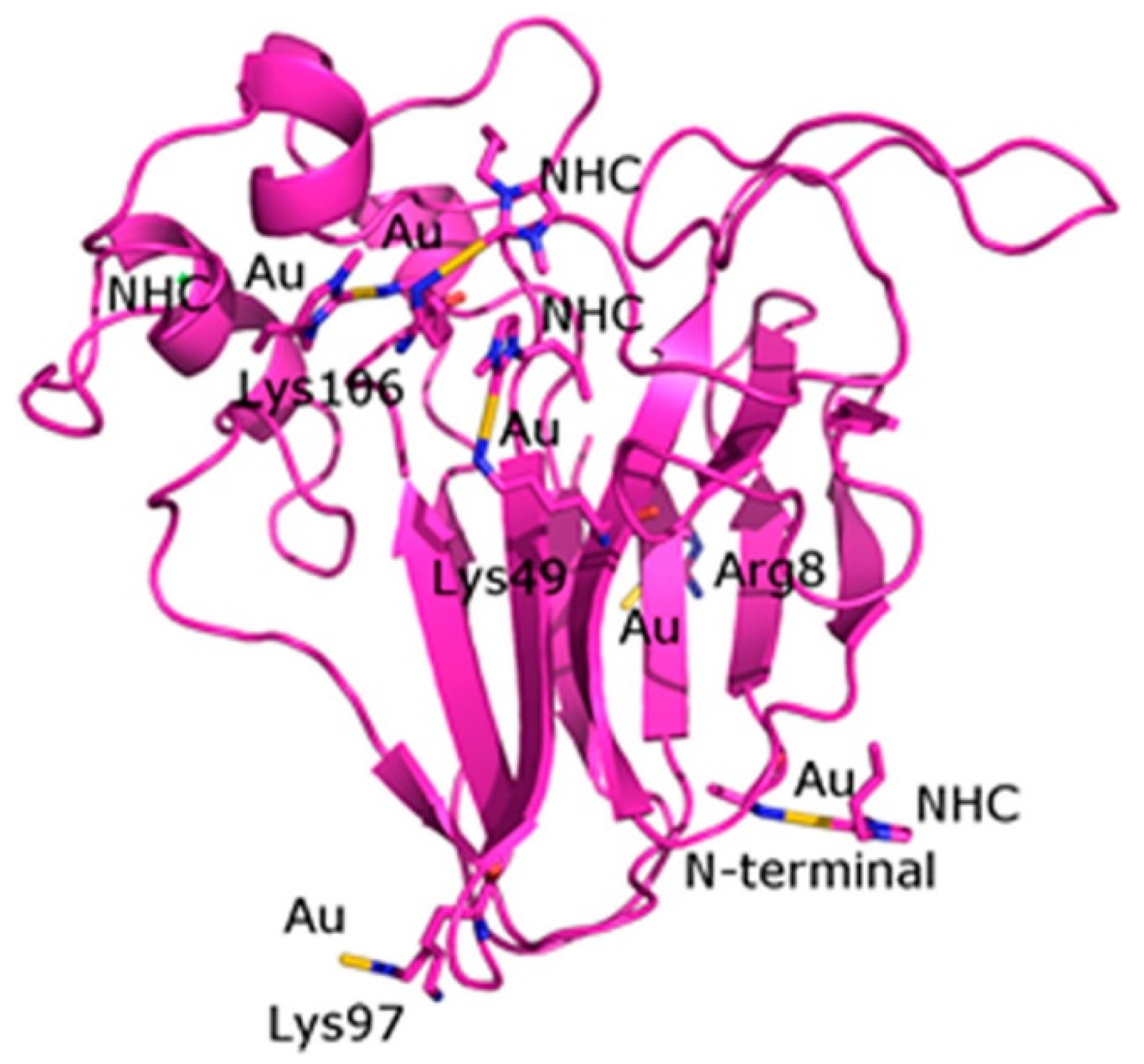
3.2. Redox Stability
4. Challenges, Strengths, and Limitations of Current Computational Approaches
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Pricker, S.P. Medical uses of gold compounds: Past, present and future. Gold Bull. 1996, 29, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Messori, L.; Marcon, G. Gold complexes in the treatment of rheumatoid arthritis. Met. Ions Biol. Syst. 2004, 41, 279–304. [Google Scholar]
- Ott, I. On the medicinal chemistry of gold complexes as anticancer drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Zou, T.; Lum, C.T.; Lok, C.N.; Zhang, J.J.; Che, C.M. Chemical biology of anticancer gold (III) and gold (I) complexes. Chem. Soc. Rev. 2015, 44, 8786–8801. [Google Scholar] [CrossRef] [PubMed]
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in drug design-a review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Spinello, A.; Saha, A.; Magistrato, A. Frontiers of metal-coordinating drug design. Expert Opin. Drug Dis. 2021, 16, 497–511. [Google Scholar] [CrossRef]
- Lee, R.F.; Menin, L.; Patiny, L.; Ortiz, D.; Dyson, P.J. Versatile tool for the analysis of metal–protein interactions reveals the promiscuity of metallodrug–protein interactions. Anal. Chem. 2017, 89, 11985–11989. [Google Scholar] [CrossRef]
- Sullivan, M.P.; Cziferszky, M.; Tolbatov, I.; Truong, D.; Mercadante, D.; Re, N.; Gust, R.; Goldstone, D.C.; Hartinger, C.G. Probing the paradigm of promiscuity for N-heterocyclic carbene complexes and their protein adduct formation. Angew. Chem. Int. Ed. 2021, 60, 19928. [Google Scholar] [CrossRef] [PubMed]
- Tolbatov, I.; Marrone, A.; Paciotti, R.; Re, N.; Coletti, C. Multilayered Modelling of the Metallation of Biological Targets. In International Conference on Computational Science and Its Applications; Springer: Cham, Swizerland, 2021; pp. 398–412. [Google Scholar]
- Paciotti, R.; Tolbatov, I.; Graziani, V.; Marrone, A.; Re, N.; Coletti, C. Insights on the activity of platinum-based anticancer complexes through computational methods. In AIP Conference Proceedings; AIP Publishing LLC: Melville, NY, USA, 2018; Volume 2040, No. 1; p. 020019. [Google Scholar]
- Barresi, E.; Tolbatov, I.; Marzo, T.; Zappelli, E.; Marrone, A.; Re, N.; Pratesi, A.; Martini, C.; Taliani, S.; Da Settimo, F.; et al. Two mixed valence diruthenium (II, III) isomeric complexes show different anticancer properties. Dalton Trans. 2021, 50, 9643–9647. [Google Scholar] [CrossRef] [PubMed]
- Tolbatov, I.; Coletti, C.; Marrone, A.; Re, N. Reactivity of arsenoplatin complex versus water and thiocyanate: A DFT benchmark study. Theor. Chem. Acc. 2020, 139, 1–11. [Google Scholar] [CrossRef]
- Barresi, E.; Tolbatov, I.; Pratesi, A.; Notarstefano, V.; Baglini, E.; Daniele, S.; Taliani, S.; Re, N.; Giorgini, E.; Martini, C.; et al. A mixed-valence diruthenium (II, III) complex endowed with high stability: From experimental evidence to theoretical interpretation. Dalton Trans. 2020, 49, 14520–14527. [Google Scholar] [CrossRef] [PubMed]
- Tolbatov, I.; Marzo, T.; Cirri, D.; Gabbiani, C.; Coletti, C.; Marrone, A.; Paciotti, R.; Messori, L.; Re, N. Reactions of cisplatin and cis-[PtI2(NH3)2] with molecular models of relevant protein side-chains: A comparative analysis. J. Inorg. Biochem. 2020, 209, 111096. [Google Scholar] [CrossRef] [PubMed]
- Urig, S.; Fritz-Wolf, K.; Réau, R.; Herold-Mende, C.; Tóth, K.; Davioud-Charvet, E.; Becker, K. Undressing of phosphine gold(I) complexes as irreversible inhibitors of human disulfide reductases. Angew. Chem. Int. Ed. 2006, 45, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Bhabak, K.P.; Bhuyan, B.J.; Mugesh, G. Bioinorganic and medicinal chemistry: Aspects of gold (I)-protein complexes. Dalton Trans. 2011, 40, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Messori, L.; Scaletti, F.; Massai, L.; Cinellu, M.A.; Gabbiani, C.; Vergara, A.; Merlino, A. The mode of action of anticancer gold-based drugs: A structural perspective. Chem. Comm. 2013, 49, 10100–10102. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, G.; Massai, L.; Messori, L.; Cinellu, M.A.; Merlino, A. Structural evidences for a secondary gold binding site in the hydrophobic box of lysozyme. Biometals 2015, 28, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraro, G.; Gabbiani, C.; Merlino, A. First crystal structure for a gold carbene–protein adduct. Bioconjug. Chem. 2016, 27, 1584–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, B.; Casini, A. A golden future in medicinal inorganic chemistry: The promise of anticancer gold organometallic compounds. Dalton Trans. 2014, 43, 4209–4219. [Google Scholar] [CrossRef] [PubMed]
- Oehninger, L.; Rubbiani, R.; Ott, I. N-Heterocyclic carbene metal complexes in medicinal chemistry. Dalton Trans. 2013, 42, 3269–3284. [Google Scholar] [CrossRef]
- Carlos Lima, J.; Rodriguez, L. Phosphine-gold (I) compounds as anticancer agents: General description and mechanisms of action. Anticancer Agents Med. Chem. 2011, 11, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Onodera, T.; Momose, I.; Kawada, M. Potential anticancer activity of auranofin. Chem. Pharmaceut. Bull. 2019, 67, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Maddelein, M.L.; Sun, R.W.Y.; Gornitzka, H.; Cuvillier, O.; Hemmert, C. Pharmacomodulation on gold-NHC complexes for anticancer applications–is lipophilicity the key point? Eur. J. Med. Chem. 2018, 157, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Jiang, G.; Xu, Z.; Zhao, S.; Liu, W. Advances in alkynyl gold complexes for use as potential anticancer agents. Coord. Chem. Rev. 2020, 423, 213492. [Google Scholar] [CrossRef]
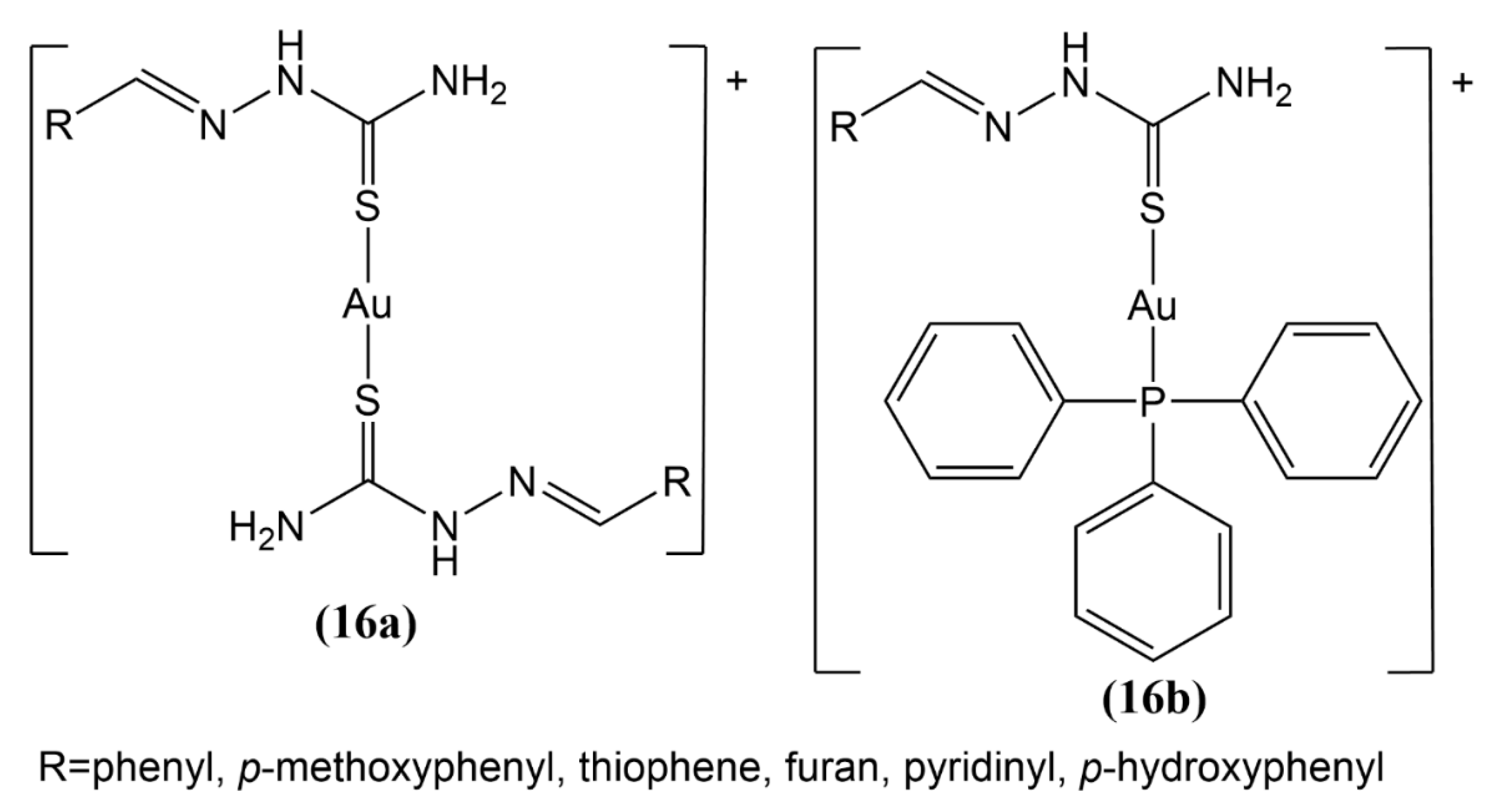
- Tavares, T.T.; Azevedo, G.C.; Garcia, A.; Carpanez, A.G.; Lewer, P.M.; Paschoal, D.; Müller, B.L.; Dos Santos, H.F.; Matos, R.C.; Silva, H.; et al. Gold (I) complexes with aryl-thiosemicarbazones: Molecular modeling, synthesis, cytotoxicity and TrxR inhibition. Polyhedron 2017, 132, 95–104. [Google Scholar] [CrossRef]
- AbdelKhalek, A.; Abutaleb, N.S.; Mohammad, H.; Seleem, M.N. Antibacterial and antivirulence activities of auranofin against Clostridium difficile. Int. J. Antimicrob. 2019, 53, 54–62. [Google Scholar] [CrossRef]
- Mbouaka, A.L.; Leitsch, D.; Koehsler, M.; Walochnik, J. Antimicrobial effect of auranofin against Acanthamoeba spp. Int. J. Antimicrob. 2021, 58, 106425. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, N.P.; Patterson, T.F.; Srinivasan, A.; Chaturvedi, A.K.; Fothergill, A.W.; Wormley, F.L.; Ramasubramanian, A.K.; Lopez-Ribot, J.L. Repurposing auranofin as an antifungal: In vitro activity against a variety of medically important fungi. Virulence 2017, 8, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Hu, J.; Wu, S.; Wang, L.; Cao, X.; Zhang, X.; Dai, B.; Cao, M.; Shao, R.; Zhang, R.; et al. Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells. Oncotarget 2016, 7, 3548. [Google Scholar] [CrossRef] [Green Version]
- Yue, S.; Luo, M.; Liu, H.; Wei, S. Recent advances of gold compounds in anticancer immunity. Front. Chem. 2020, 8, 543. [Google Scholar] [CrossRef]
- Pratesi, A.; Cirri, D.; Ciofi, L.; Messori, L. Reactions of auranofin and its pseudohalide derivatives with serum albumin investigated through ESI-Q-TOF MS. Inorg. Chem. 2018, 57, 10507–10510. [Google Scholar] [CrossRef]
- Liu, N.; Guo, Z.; Xia, X.; Liao, Y.; Zhang, F.; Huang, C.; Liu, Y.; Deng, X.; Jiang, L.; Wang, X.; et al. Auranofin lethality to prostate cancer includes inhibition of proteasomal deubiquitinases and disrupted androgen receptor signaling. Eur. J. Pharmacol. 2019, 846, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hwangbo, H.; Ji, S.Y.; Kim, M.Y.; Kim, S.Y.; Lee, H.; Kim, G.Y.; Kim, S.; Cheong, J.; Choi, Y.H. Anti-inflammatory effect of auranofin on palmitic acid and LPS-induced inflammatory response by modulating TLR4 and NOX4-mediated NF-κB signaling pathway in RAW264.7 Macrophages. Int. J. Mol. Sci. 2021, 22, 5920. [Google Scholar] [CrossRef]
- Zhang, X.; Selvaraju, K.; Saei, A.A.; D’Arcy, P.; Zubarev, R.A.; Arnér, E.S.; Linder, S. Repurposing of auranofin: Thioredoxin reductase remains a primary target of the drug. Biochimie 2019, 162, 46–54. [Google Scholar] [CrossRef]
- Landini, I.; Massai, L.; Cirri, D.; Gamberi, T.; Paoli, P.; Messori, L.; Mini, E.; Nobili, S. Structure-activity relationships in a series of auranofin analogues showing remarkable antiproliferative properties. J. Inorg. Biochem. 2020, 208, 111079. [Google Scholar] [CrossRef]
- Marzo, T.; Cirri, D.; Pollini, S.; Prato, M.; Fallani, S.; Cassetta, M.I.; Novelli, A.; Rossolini, G.M.; Messori, L. Auranofin and its analogues show potent antimicrobial activity against multidrug-resistant pathogens: Structure-activity relationships. ChemMedChem 2018, 13, 2448–2454. [Google Scholar] [CrossRef] [PubMed]
- Marzo, T.; Cirri, D.; Gabbiani, C.; Gamberi, T.; Magherini, F.; Pratesi, A.; Guerri, A.; Biver, T.; Binacchi, F.; Stefanini, M.; et al. Auranofin, Et3PauCl, and Et3PAuI are highly cytotoxic on colorectal cancer cells: A chemical and biological study. ACS Med. Chem. Lett. 2017, 8, 997–1001. [Google Scholar] [CrossRef]
- Howell, J.A. DFT investigation of the interaction between gold (I) complexes and the active site of thioredoxin reductase. J. Organomet. Chem. 2009, 694, 868–873. [Google Scholar] [CrossRef]
- Dos Santos, H.F. Reactivity of auranofin with S-, Se-and N-containing amino acids. Comput. Theor. Chem. 2014, 1048, 95–101. [Google Scholar] [CrossRef]
- Di Sarra, F.; Fresch, B.; Bini, R.; Saielli, G.; Bagno, A. Reactivity of auranofin with selenols and thiols–implications for the anticancer activity of gold (I) compounds. Eur. J. Inorg. Chem. 2013, 2013, 2718–2727. [Google Scholar] [CrossRef]
- Shoeib, T.; Atkinson, D.W.; Sharp, B.L. Structural analysis of the anti-arthritic drug Auranofin: Its complexes with cysteine, selenocysteine and their fragmentation products. Inorg. Chim. Acta 2010, 363, 184–192. [Google Scholar] [CrossRef]
- Tolbatov, I.; Cirri, D.; Marchetti, L.; Marrone, A.; Coletti, C.; Re, N.; La Mendola, D.; Messori, L.; Marzo, T.; Gabbiani, C.; et al. Mechanistic insights into the anticancer properties of the auranofin analog Au(PEt3)I: A theoretical and experimental study. Front. Chem. 2020, 8, 812. [Google Scholar] [CrossRef]
- Madeira, J.M.; Gibson, D.L.; Kean, W.F.; Klegeris, A. The biological activity of auranofin: Implications for novel treatment of diseases. Inflammopharmacology 2012, 20, 297–306. [Google Scholar] [CrossRef]
- Gunatilleke, S.S.; De Oliveira, C.A.F.; McCammon, J.A.; Barrios, A.M. Inhibition of cathepsin B by Au (I) complexes: A kinetic and computational study. J. Biol. Inorg. Chem. 2008, 13, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Sayed, A.A.; Williams, D.L.; Boumis, G.; Brunori, M.; Dimastrogiovanni, D.; Miele, A.E.; Pauly, F.; Bellelli, A. Inhibition of Schistosoma mansoni thioredoxin-glutathione reductase by auranofin: Structural and kinetic aspects. J. Biol. Chem. 2009, 284, 28977–28985. [Google Scholar] [CrossRef] [Green Version]
- Caroli, A.; Simeoni, S.; Lepore, R.; Tramontano, A.; Via, A. Investigation of a potential mechanism for the inhibition of SmTGR by Auranofin and its implications for Plasmodium falciparum inhibition. Biochem. Biophys. Res. Comm. 2012, 417, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, H.D.; Verpoort, F. N-heterocyclic carbene transition metal complexes for catalysis in aqueous media. Chem. Soc. Rev. 2012, 41, 7032–7060. [Google Scholar] [CrossRef] [PubMed]
- Gautier, A.; Cisnetti, F. Advances in metal–carbene complexes as potent anti-cancer agents. Metallomics 2012, 4, 23–32. [Google Scholar] [CrossRef]
- Porchia, M.; Pellei, M.; Marinelli, M.; Tisato, F.; Del Bello, F.; Santini, C. New insights in Au-NHCs complexes as anticancer agents. Eur. J. Med. Chem. 2018, 146, 709–746. [Google Scholar] [CrossRef] [PubMed]
- Mora, M.; Gimeno, M.C.; Visbal, R. Recent advances in gold–NHC complexes with biological properties. Chem. Soc. Rev. 2019, 48, 447–462. [Google Scholar] [CrossRef]
- Rubbiani, R.; Schuh, E.; Meyer, A.; Lemke, J.; Wimberg, J.; Metzler-Nolte, N.; Meyer, F.; Mohr, F.; Ott, I. TrxR inhibition and antiproliferative activities of structurally diverse gold N-heterocyclic carbene complexes. MedChemComm 2013, 4, 942–948. [Google Scholar] [CrossRef]
- Magherini, F.; Fiaschi, T.; Valocchia, E.; Becatti, M.; Pratesi, A.; Marzo, T.; Massai, L.; Gabbiani, C.; Landini, I.; Nobili, S.; et al. Antiproliferative effects of two gold(I)-N-heterocyclic carbene complexes in A2780 human ovarian cancer cells: A comparative proteomic study. Oncotarget 2018, 9, 28042–28068. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, H.F.; Vieira, M.A.; Sánchez Delgado, G.Y.; Paschoal, D. Ligand exchange reaction of Au (I) RN-heterocyclic carbene complexes with cysteine. J. Phys. Chem. A 2016, 120, 2250–2259. [Google Scholar] [CrossRef]
- Tolbatov, I.; Coletti, C.; Marrone, A.; Re, N. Insight into the substitution mechanism of antitumor Au (I) N-heterocyclic carbene complexes by cysteine and selenocysteine. Inorg. Chem. 2020, 59, 3312–3320. [Google Scholar] [CrossRef] [PubMed]
- Tolbatov, I.; Marzo, T.; Coletti, C.; La Mendola, D.; Storchi, L.; Re, N.; Marrone, A. Reactivity of antitumor coinage metal-based N-heterocyclic carbene complexes with cysteine and selenocysteine protein sites. J. Inorg. Biochem. 2021, 223, 111533. [Google Scholar] [CrossRef]
- Liu, W.; Gust, R. Metal N-heterocyclic carbene complexes as potential antitumor metallodrugs. Chem. Soc. Rev. 2013, 42, 755–773. [Google Scholar] [CrossRef]
- Tolbatov, I.; Coletti, C.; Marrone, A.; Re, N. Reactivity of gold (I) monocarbene complexes with protein targets: A theoretical study. Int. J. Mol. Sci. 2019, 20, 820. [Google Scholar] [CrossRef] [Green Version]
- Paciotti, R.; Tolbatov, I.; Marrone, A.; Storchi, L.; Re, N.; Coletti, C. Computational investigations of bioinorganic complexes: The case of calcium, gold and platinum ions. In AIP Conference Proceedings; AIP Publishing LLC: Melville, NY, USA, 2019; Volume 2186, No. 1; p. 030011. [Google Scholar]
- Schmidt, C.; Albrecht, L.; Balasupramaniam, S.; Misgeld, R.; Karge, B.; Brönstrup, M.; Prokop, A.; Baumann, K.; Reichl, S.; Ott, I. A gold (I) biscarbene complex with improved activity as a TrxR inhibitor and cytotoxic drug: Comparative studies with different gold metallodrugs. Metallomics 2019, 11, 533–545. [Google Scholar] [CrossRef]
- Seliman, A.A.; Altaf, M.; Odewunmi, N.A.; Kawde, A.N.; Zierkiewicz, W.; Ahmad, S.; Altuwaijri, S.; Isab, A.A. Synthesis, X-ray structure, DFT calculations and anticancer activity of a selenourea coordinated gold (I)-carbene complex. Polyhedron 2017, 137, 197–206. [Google Scholar] [CrossRef]
- Dada, O.; Sánchez-Sanz, G.; Tacke, M.; Zhu, X. Synthesis and anticancer activity of novel NHC-gold (I)-sugar complexes. Tetrahedron Lett. 2018, 59, 2904–2908. [Google Scholar] [CrossRef]
- Curran, D.; Dada, O.; Müller-Bunz, H.; Rothemund, M.; Sánchez-Sanz, G.; Schobert, R.; Zhu, X.; Tacke, M. Synthesis and cytotoxicity studies of novel NHC*-gold (I) complexes derived from lepidiline A. Molecules 2018, 23, 2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubbiani, R.; Can, S.; Kitanovic, I.; Alborzinia, H.; Stefanopoulou, M.; Kokoschka, M.; Mönchgesang, S.; Sheldrick, W.S.; Wölfl, S.; Ott, I. Comparative in vitro evaluation of N-heterocyclic carbene gold (I) complexes of the benzimidazolylidene type. J. Med. Chem. 2011, 54, 8646–8657. [Google Scholar] [CrossRef] [PubMed]
- Rubbiani, R.; Salassa, L.; De Almeida, A.; Casini, A.; Ott, I. Cytotoxic gold (I) N-heterocyclic carbene complexes with phosphane ligands as potent enzyme inhibitors. ChemMedChem 2014, 9, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Guarra, F.; Marzo, T.; Ferraroni, M.; Papi, F.; Bazzicalupi, C.; Gratteri, P.; Pescitelli, G.; Messori, L.; Biver, T.; Gabbiani, C. Interaction of a gold (I) dicarbene anticancer drug with human telomeric DNA G-quadruplex: Solution and computationally aided X-ray diffraction analysis. Dalton Trans. 2018, 47, 16132–16138. [Google Scholar] [CrossRef] [PubMed]
- Wragg, D.; De Almeida, A.; Bonsignore, R.; Kühn, F.E.; Leoni, S.; Casini, A. On the mechanism of Gold/NHC compounds binding to DNA G-quadruplexes: Combined metadynamics and biophysical methods. Angew. Chem. 2018, 130, 14732–14736. [Google Scholar] [CrossRef]
- Maji, B.; Bhattacharya, S. Advances in the molecular design of potential anticancer agents via targeting of human telomeric DNA. ChemComm 2014, 50, 6422–6438. [Google Scholar]
- Andermark, V.; Göke, K.; Kokoschka, M.; El Maaty, M.A.A.; Lum, C.T.; Zou, T.; Sun, R.W.-Y.; Aguiló, E.; Oehninger, L.; Rodríguez, L.; et al. Alkynyl gold (I) phosphane complexes: Evaluation of structure–activity-relationships for the phosphane ligands, effects on key signaling proteins and preliminary in-vivo studies with a nanoformulated complex. J. Inorg. Biochem. 2016, 160, 140–148. [Google Scholar] [CrossRef] [Green Version]
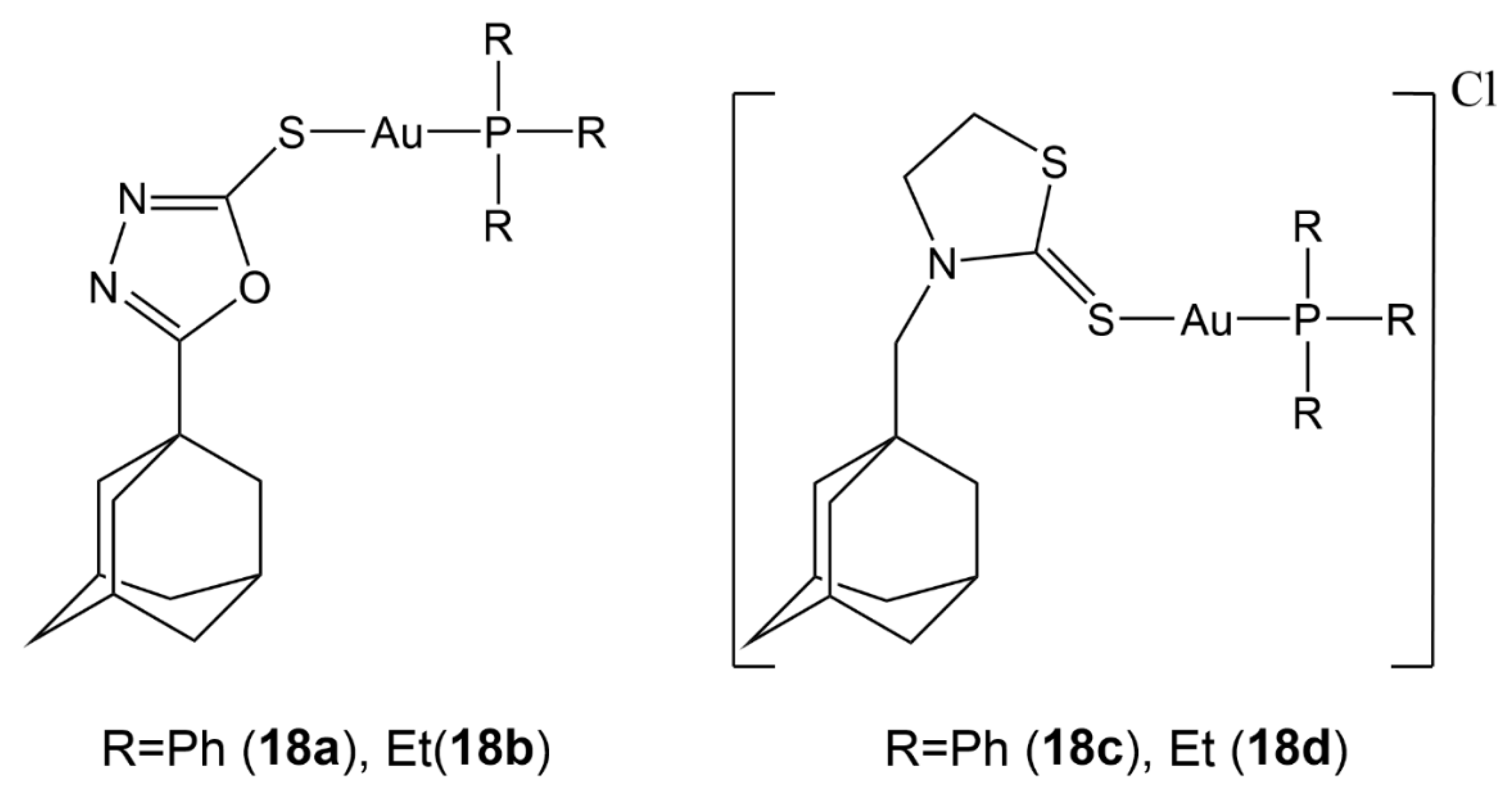
- Garcia, A.; Machado, R.C.; Grazul, R.M.; Lopes, M.T.P.; Corrêa, C.C.; Dos Santos, H.F.; de Almeida, M.V.; Silva, H. Novel antitumor adamantane–azole gold (I) complexes as potential inhibitors of thioredoxin reductase. J. Biol. Inorg. Chem. 2016, 21, 275–292. [Google Scholar] [CrossRef]
- Casini, A.; Messori, L. Molecular mechanisms and proposed targets for selected anticancer gold compounds. Curr. Top. Med. Chem. 2011, 11, 2647–2660. [Google Scholar] [CrossRef]
- Gabbiani, C.; Casini, A.; Messori, L. Gold(III) compounds as anticancer drugs. Gold Bull. 2007, 40, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, B.; Williams, M.R.; Bochmann, M. Gold (III) complexes for antitumor applications: An overview. Chem. Eur. J. 2018, 24, 11840–11851. [Google Scholar] [CrossRef]
- Casini, A.; Hartinger, C.; Gabbiani, C.; Mini, E.; Dyson, P.J.; Keppler, B.K.; Messori, L. Gold(III) compounds as anticancer agents: Relevance of gold–protein interactions for their mechanism of action. J. Inorg. Biochem. 2008, 102, 564–575. [Google Scholar] [CrossRef]
- Tiekink, E.R. Gold derivatives for the treatment of cancer. Crit. Rev. Oncol. 2002, 42, 225–248. [Google Scholar] [CrossRef]
- Messori, L.; Scaletti, F.; Massai, L.; Cinellu, M.A.; Russo Krauss, I.; Di Martino, G.; Vergara, A.; Paduano, L.; Merlino, A. Interactions of gold-based drugs with proteins: Crystal structure of the adduct formed between ribonuclease A and a cytotoxic gold(III) compound. Metallomics 2014, 6, 233–236. [Google Scholar] [CrossRef]
- Djeković, A.; Petrović, B.; Bugarčić, Ž.D.; Puchta, R.; van Eldik, R. Kinetics and mechanism of the reactions of Au(III) complexes with some biologically relevant molecules. Dalton Trans. 2012, 41, 3633–3641. [Google Scholar] [CrossRef]
- He, Y.; Zhou, L. A theoretical study on pyridine gold (III) complexes AuCl3 (Hpm) and AuCl2 (pm) targeting purine bases and cysteine. Comput. Theor. Chem. 2016, 1093, 20–28. [Google Scholar] [CrossRef]
- Radisavljević, S.; Petrović, B. Gold(III) complexes: An overview on their kinetics, interactions with DNA/BSA, cytotoxic activity, and computational calculations. Front. Chem. 2020, 8, 379. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277. [Google Scholar] [CrossRef] [Green Version]
- Verkman, A.S. Aquaporins in clinical medicine. Ann. Rev. Med. 2012, 63, 303–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribatti, D.; Ranieri, G.; Annese, T.; Nico, B. Aquaporins in cancer. Biochim. Biophys. Acta (BBA)-Gen. Subjects 2014, 1840, 1550–1553. [Google Scholar] [CrossRef] [PubMed]
- Aikman, B.; De Almeida, A.; Meier-Menches, S.M.; Casini, A. Aquaporins in cancer development: Opportunities for bioinorganic chemistry to contribute novel chemical probes and therapeutic agents. Metallomics 2018, 10, 696–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, A.P.; Ciancetta, A.; De Almeida, A.; Marrone, A.; Re, N.; Soveral, G.; Casini, A. Aquaporin inhibition by gold (III) compounds: New insights. ChemMedChem 2013, 8, 1086–1092. [Google Scholar] [CrossRef]
- De Almeida, A.; Mósca, A.F.; Wragg, D.; Wenzel, M.; Kavanagh, P.; Barone, G.; Leoni, S.; Soveral, G.; Casini, A. The mechanism of aquaporin inhibition by gold compounds elucidated by biophysical and computational methods. ChemComm 2017, 53, 3830–3833. [Google Scholar] [CrossRef]
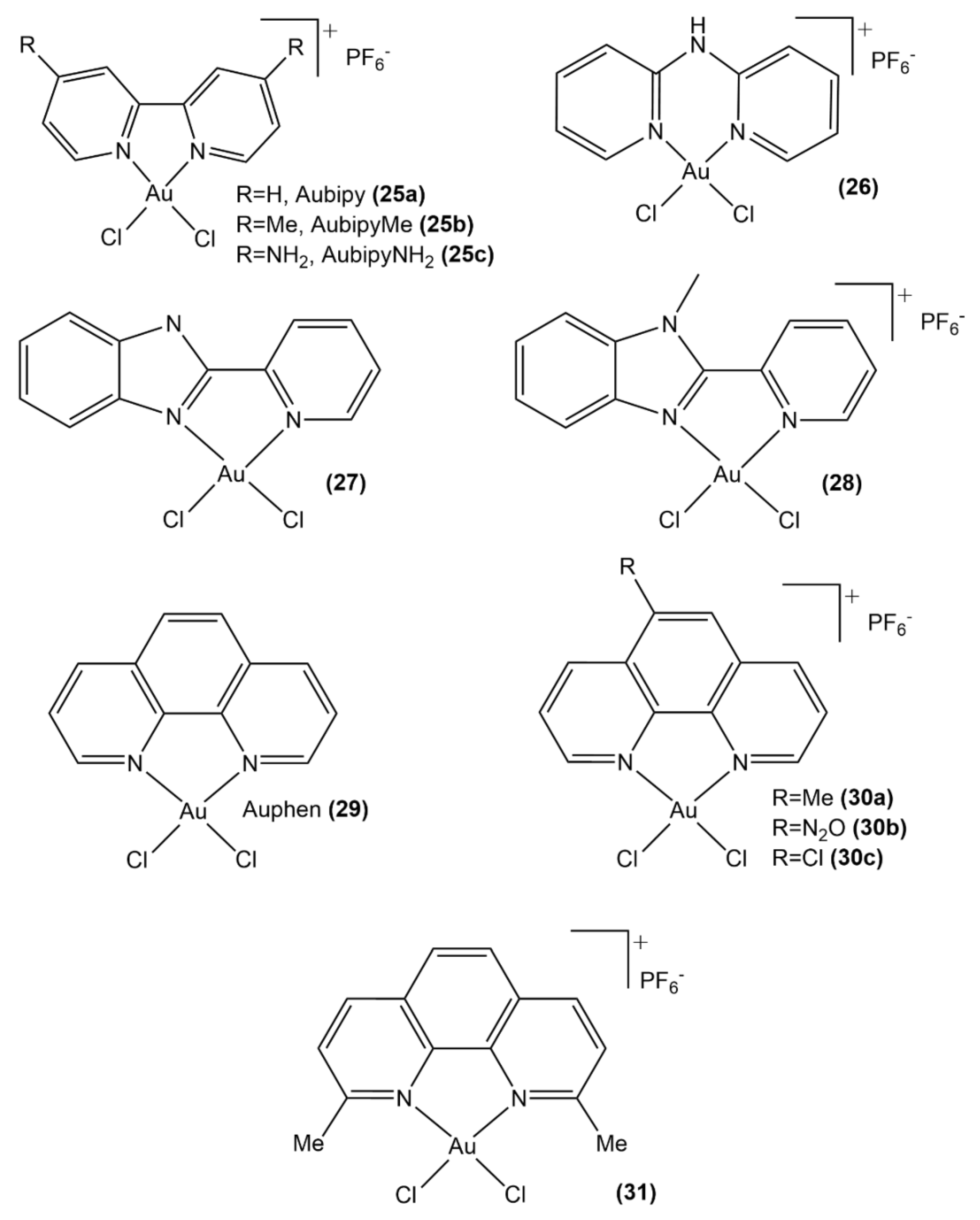
- Graziani, V.; Marrone, A.; Re, N.; Coletti, C.; Platts, J.A.; Casini, A. A multilevel theoretical study to disclose the binding mechanisms of gold (III) bipyridyl compounds as selective aquaglyceroporin inhibitors. Chem. Eur. J. 2017, 23, 13802–13813. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.P.; Marrone, A.; Ciancetta, A.; Galán Cobo, A.; Echevarría, M.; Moura, T.F.; Re, N.; Casini, A.; Soveral, G. Targeting aquaporin function: Potent inhibition of aquaglyceroporin-3 by a gold-based compound. PLoS ONE 2012, 7, e37435. [Google Scholar]
- Wenzel, M.N.; Mósca, A.F.; Graziani, V.; Aikman, B.; Thomas, S.R.; De Almeida, A.; Platts, J.A.; Re, N.; Coletti, C.; Marrone, A.; et al. Insights into the mechanisms of aquaporin-3 inhibition by gold (III) complexes: The importance of non-coordinative adduct formation. Inorg. Chem. 2019, 58, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Cutillas, N.; Yellol, G.S.; de Haro, C.; Vicente, C.; Rodríguez, V.; Ruiz, J. Anticancer cyclometalated complexes of platinum group metals and gold. Coord. Chem. Rev. 2013, 257, 2784–2797. [Google Scholar] [CrossRef]
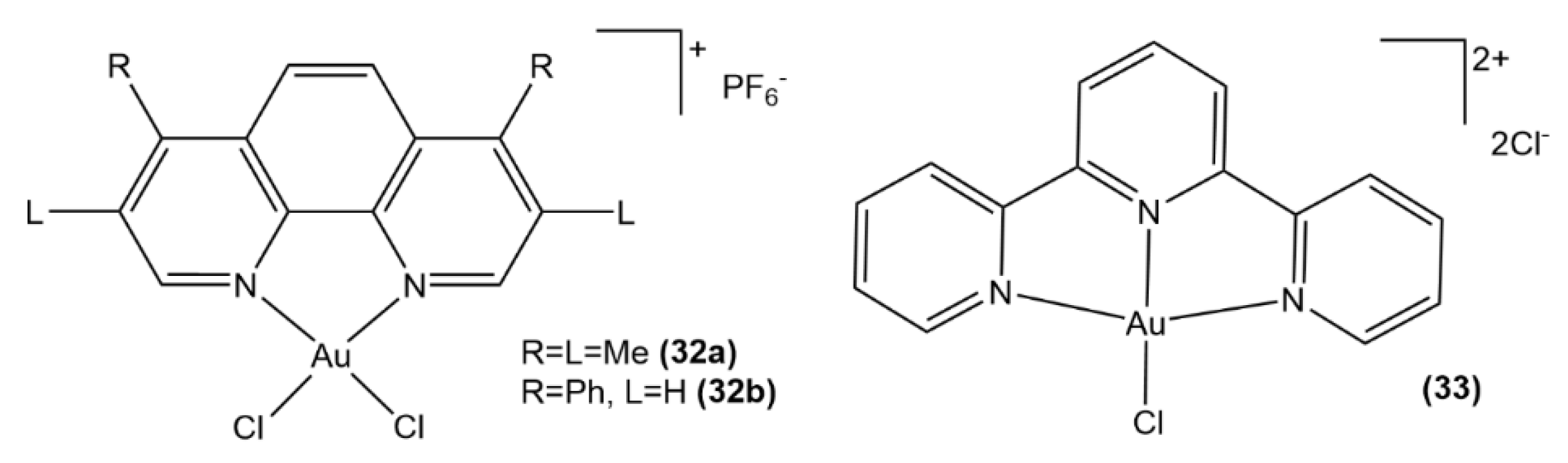
- Li, C.K.L.; Sun, R.W.Y.; Kui, S.C.F.; Zhu, N.; Che, C.M. Anticancer cyclometalated [AuIIIm (C^N^C)mL]n+ compounds: Synthesis and cytotoxic properties. Chem. Eur. J. 2006, 12, 5253–5266. [Google Scholar] [CrossRef]
- Kumar, R.; Nevado, C. Cyclometalated gold (III) complexes: Synthesis, reactivity, and physicochemical properties. Angew. Chem. Int. Ed. 2017, 56, 1994–2015. [Google Scholar] [CrossRef]
- Bindoli, A.; Rigobello, M.P.; Scutari, G.; Gabbiani, C.; Casini, A.; Messori, L. Thioredoxin reductase: A target for gold compounds acting as potential anticancer drugs. Coord. Chem. Rev. 2009, 253, 1692–1707. [Google Scholar] [CrossRef]
- Jia, J.J.; Geng, W.S.; Wang, Z.Q.; Chen, L.; Zeng, X.S. The role of thioredoxin system in cancer: Strategy for cancer therapy. Cancer Chemother. Pharmacol. 2019, 84, 453–470. [Google Scholar] [CrossRef]
- Tang, M.C.; Li, L.K.; Lai, S.L.; Cheung, W.L.; Ng, M.; Wong, C.Y.; Chan, M.-Y.; Yam, V.W.W. Design strategy towards horizontally oriented luminescent tetradentate-ligand-containing gold(III) systems. Angew. Chem. Int. Ed. 2020, 59, 21023–21031. [Google Scholar] [CrossRef]
- Delgado, G.S.; Paschoal, D.; Dos Santos, H.F. Reactivity of the [Au(C^N^C)Cl] complex in the presence of H2O and N-, S- and Se-containing nucleophiles: A DFT study. J. Biol. Inorg. Chem. 2018, 23, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Delgado, G.Y.S.; Arvellos, J.F.; Paschoal, D.F.; Dos Santos, H.F. Role of the enzymatic environment in the reactivity of the AuIII-C^N^C anticancer complexes. Inorg. Chem. 2021, 60, 3181–3195. [Google Scholar] [CrossRef]
- Delgado, G.Y.S.; Paschoal, D.; de Oliveira, M.A.; Dos Santos, H.F. Structure and redox stability of [Au(III)(X^N^X)PR3] complexes (X = C or N) in aqueous solution: The role of phosphine auxiliary ligand. J. Inorg. Biochem. 2019, 200, 110804. [Google Scholar] [CrossRef]
- Delgado, G.S.; Paschoal, D.; Dos Santos, H.F. Predicting standard reduction potential for anticancer Au (III)-complexes: A DFT study. Comput. Theor. Chem. 2017, 1108, 86–92. [Google Scholar] [CrossRef]
- Zhou, X.Q.; Carbo-Bague, I.; Siegler, M.A.; Hilgendorf, J.; Basu, U.; Ott, I.; Liu, R.; Zhang, L.; Ramu, V.; IJzerman, A.P.; et al. Rollover cyclometalation vs nitrogen coordination in tetrapyridyl anticancer gold (III) complexes: Effect on protein interaction and toxicity. JACS Au 2021, 1, 380–395. [Google Scholar] [CrossRef]
- Lawal, M.M.; Lawal, I.A.; Klink, M.J.; Tolufashe, G.F.; Ndagi, U.; Kumalo, H.M. Density functional theory study of gold (III)-dithiocarbamate complexes with characteristic anticancer potentials. J. Inorg. Biochem. 2020, 206, 111044. [Google Scholar] [CrossRef] [PubMed]
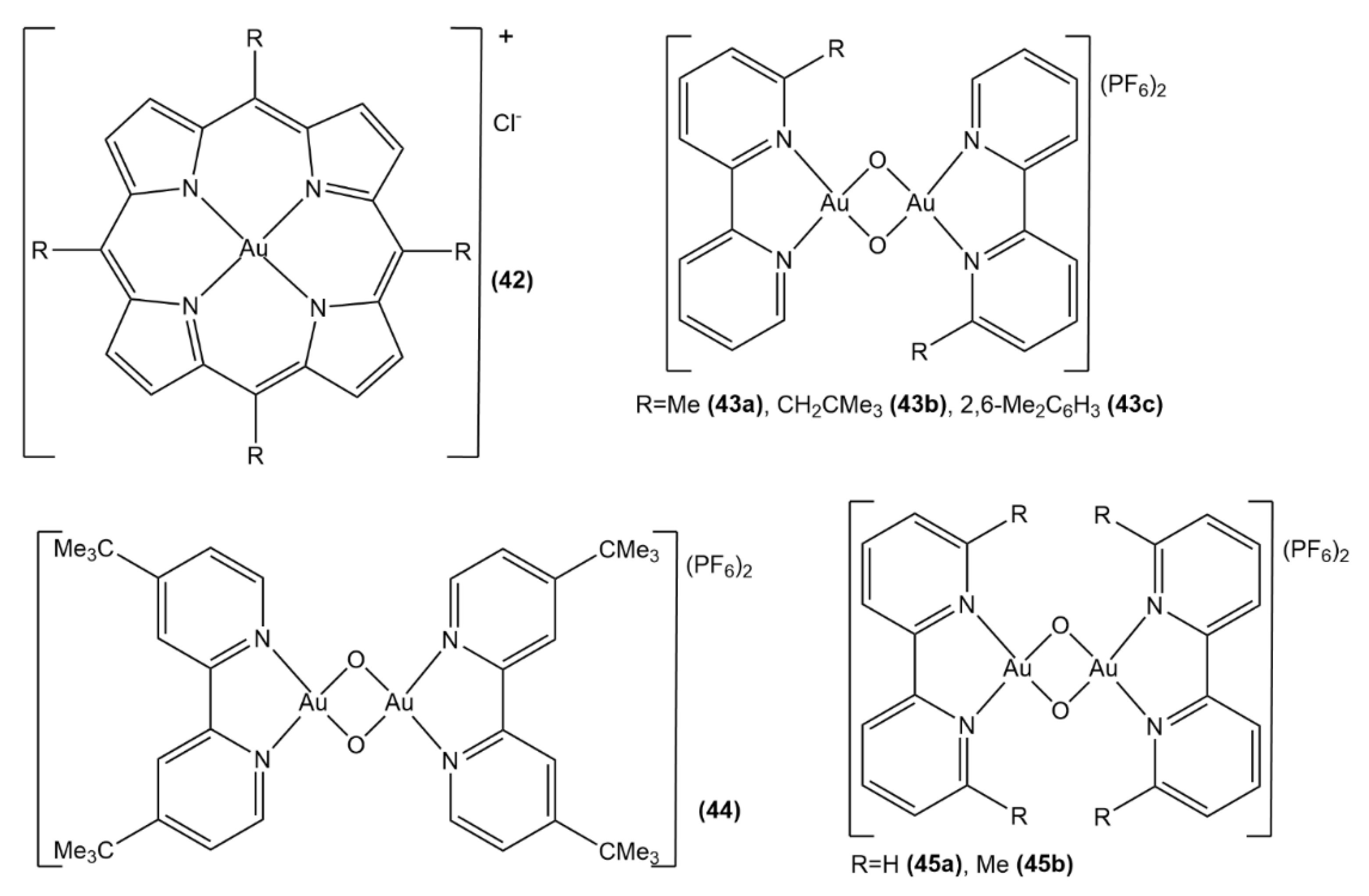
- Che, C.M.; Sun, R.W.Y.; Yu, W.Y.; Ko, C.B.; Zhu, N.; Sun, H. Gold (III) porphyrins as a new class of anticancer drugs: Cytotoxicity, DNA binding and induction of apoptosis in human cervix epitheloid cancer cells. Chem. Comm. 2003, 1718–1719. [Google Scholar] [CrossRef]
- Sun, R.W.Y.; Che, C.M. The anti-cancer properties of gold (III) compounds with dianionic porphyrin and tetradentate ligands. Coord. Chem. Rev. 2009, 253, 1682–1691. [Google Scholar] [CrossRef]
- Sun, R.W.Y.; Li, C.K.L.; Ma, D.L.; Yan, J.J.; Lok, C.N.; Leung, C.H.; Zhu, N.; Che, C.M. Stable anticancer gold (III)–porphyrin complexes: Effects of porphyrin structure. Chem. Eur. J. 2010, 16, 3097–3113. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Cinellu, M.A.; Minghetti, G.; Gabbiani, C.; Coronnello, M.; Mini, E.; Messori, L. Structural and solution chemistry, antiproliferative effects, and DNA and protein binding properties of a series of dinuclear gold (III) compounds with bipyridyl ligands. J. Med. Chem. 2006, 49, 5524–5531. [Google Scholar] [CrossRef]
- Gabbiani, C.; Casini, A.; Messori, L.; Guerri, A.; Cinellu, M.A.; Minghetti, G.; Corsini, M.; Rosani, C.; Zanello, P.; Arca, M. Structural characterization, solution studies, and DFT calculations on a series of binuclear gold (III) oxo complexes: Relationships to biological properties. Inorg. Chem. 2008, 47, 2368–2379. [Google Scholar] [CrossRef] [PubMed]
- Messori, L.; Abbate, F.; Marcon, G.; Orioli, P.; Fontani, M.; Mini, E.; Mazzei, T.; Carotti, S.; O’Connell, T.; Zanello, P. Gold (III) complexes as potential antitumor agents: Solution chemistry and cytotoxic properties of some selected gold (III) compounds. J. Med. Chem. 2000, 43, 3541–3548. [Google Scholar] [CrossRef]
- Clavier, H.; Nolan, S.P. Percent buried volume for phosphine and N-heterocyclic carbene ligands: Steric properties in organometallic chemistry. ChemComm 2010, 46, 841–861. [Google Scholar]
- Tolbatov, I.; Marrone, A. Reaction of dirhodium and diruthenium paddlewheel tetraacetate complexes with nucleophilic protein sites: A computational study. Inorg. Chim. Acta 2021, 530, 120684. [Google Scholar] [CrossRef]
- Tolbatov, I.; Marrone, A. Computational strategies to model the interaction and the reactivity of biologically-relevant transition metal complexes. Inorg. Chim. Acta 2021, 530, 120686. [Google Scholar] [CrossRef]

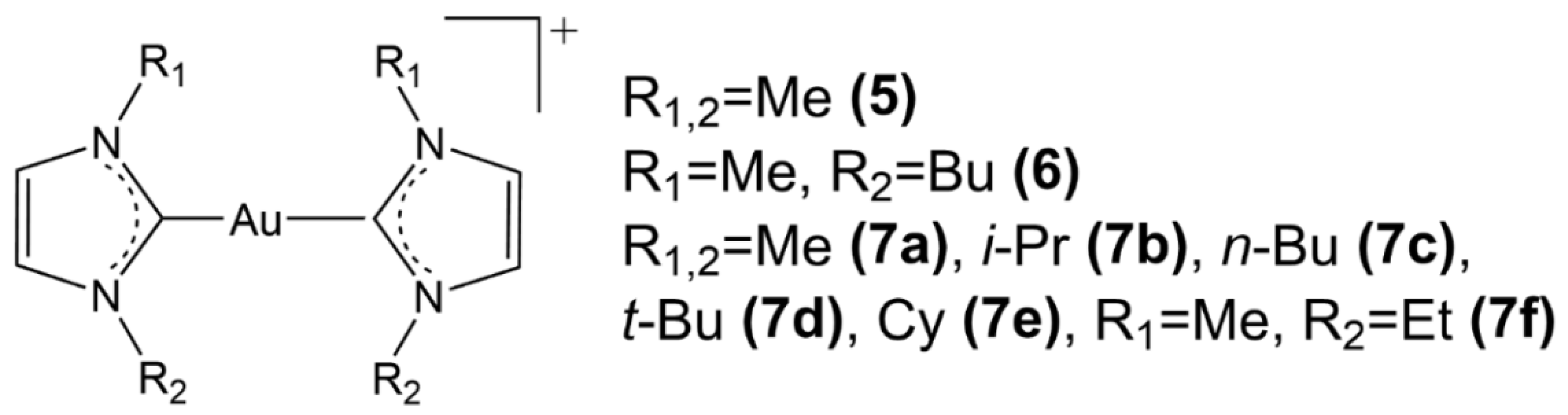
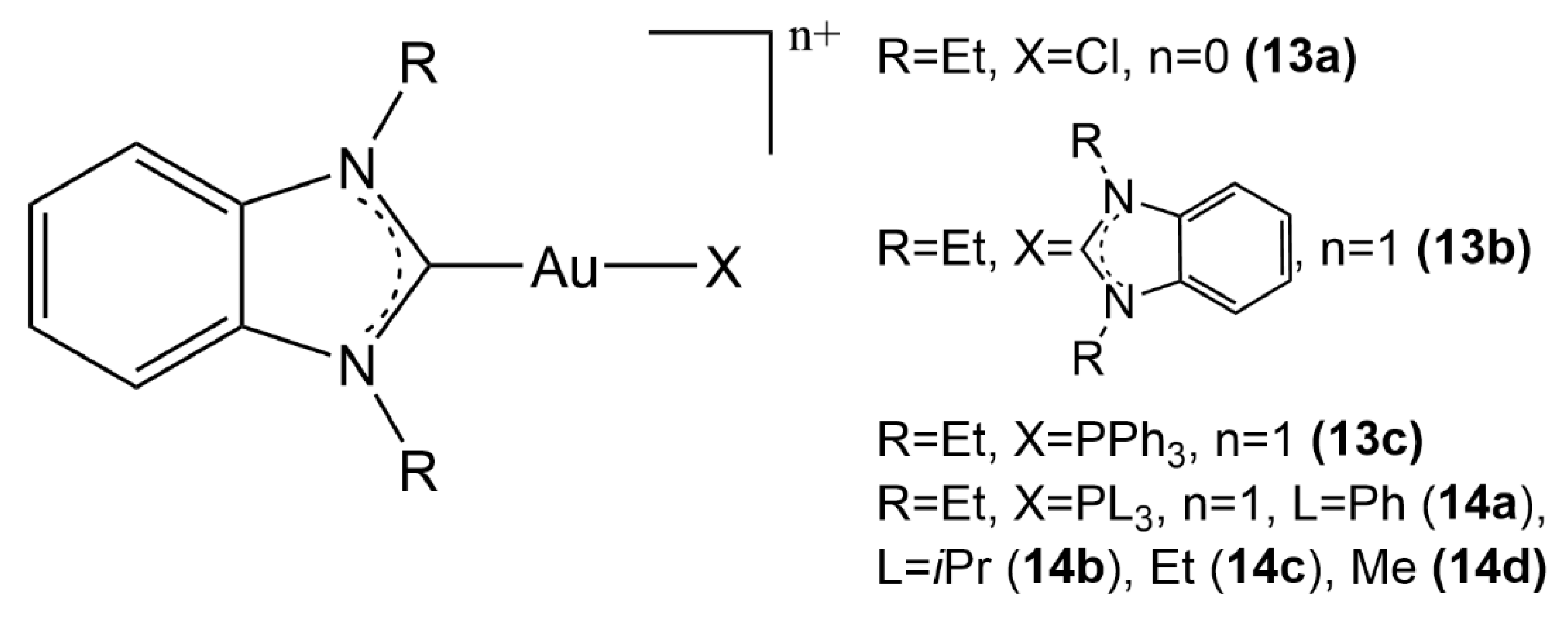

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolbatov, I.; Marrone, A.; Coletti, C.; Re, N. Computational Studies of Au(I) and Au(III) Anticancer MetalLodrugs: A Survey. Molecules 2021, 26, 7600. https://doi.org/10.3390/molecules26247600
Tolbatov I, Marrone A, Coletti C, Re N. Computational Studies of Au(I) and Au(III) Anticancer MetalLodrugs: A Survey. Molecules. 2021; 26(24):7600. https://doi.org/10.3390/molecules26247600
Chicago/Turabian StyleTolbatov, Iogann, Alessandro Marrone, Cecilia Coletti, and Nazzareno Re. 2021. "Computational Studies of Au(I) and Au(III) Anticancer MetalLodrugs: A Survey" Molecules 26, no. 24: 7600. https://doi.org/10.3390/molecules26247600
APA StyleTolbatov, I., Marrone, A., Coletti, C., & Re, N. (2021). Computational Studies of Au(I) and Au(III) Anticancer MetalLodrugs: A Survey. Molecules, 26(24), 7600. https://doi.org/10.3390/molecules26247600