
Artemisinin and Derivatives-Based Hybrid Compounds: Promising Therapeutics for the Treatment of Cancer and Malaria




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
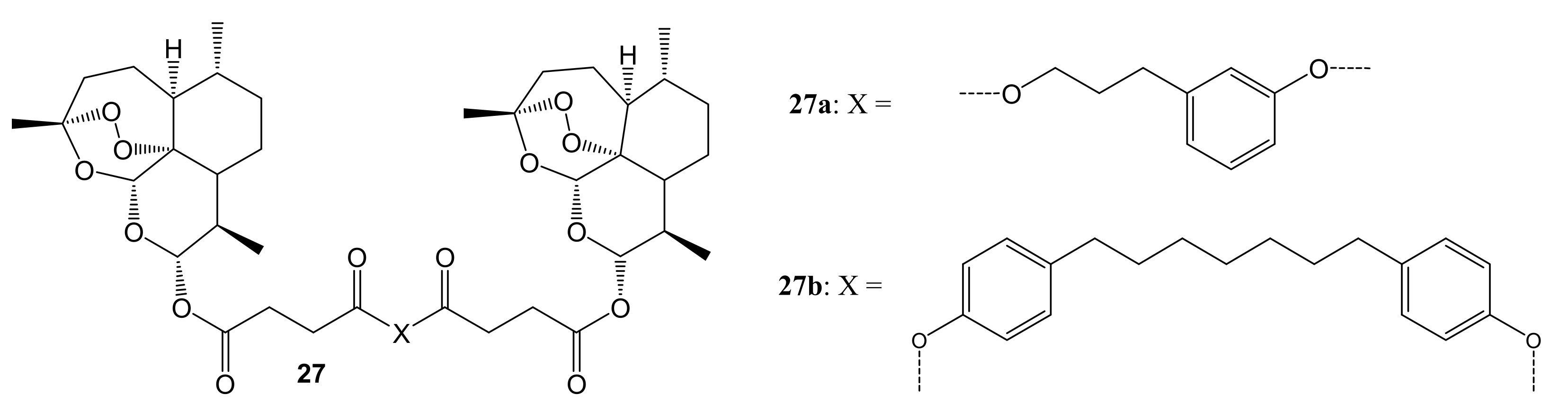{kind=link}
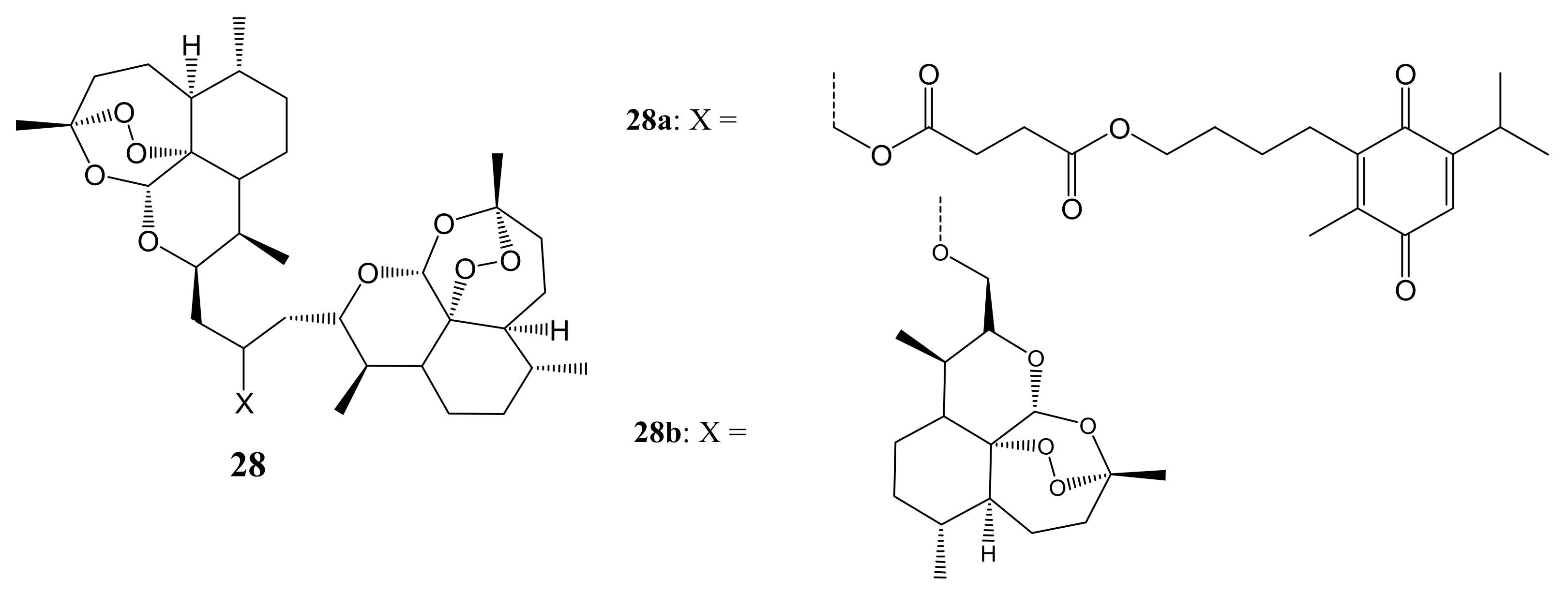{kind=link}
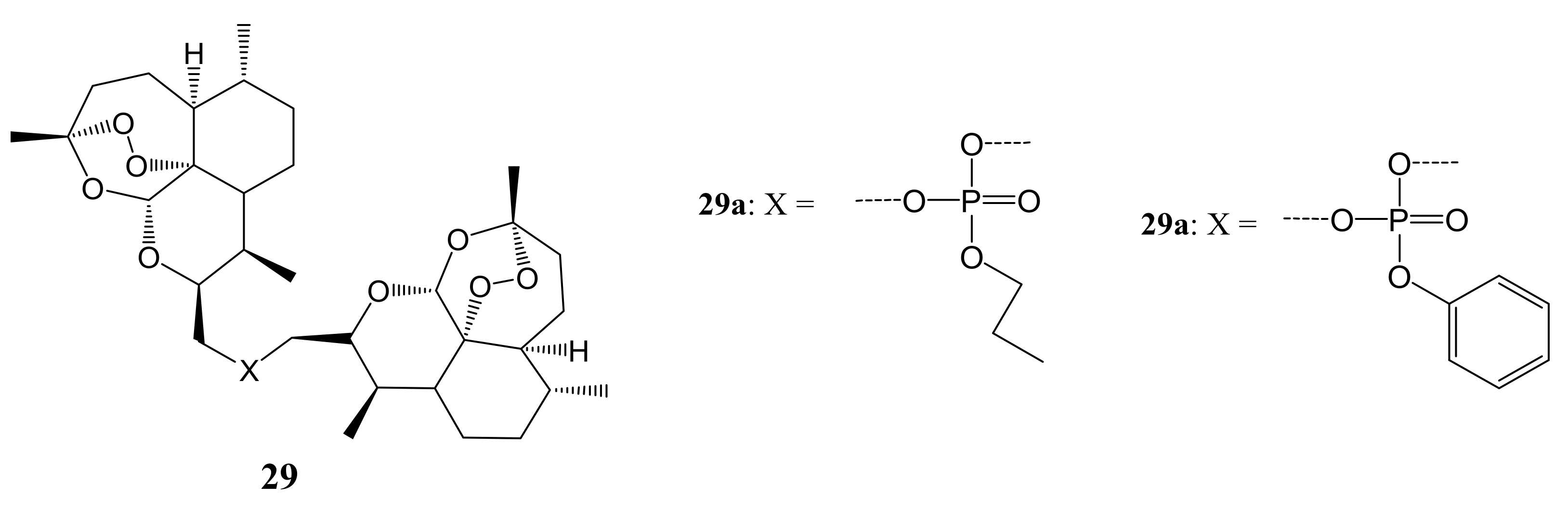{kind=link}
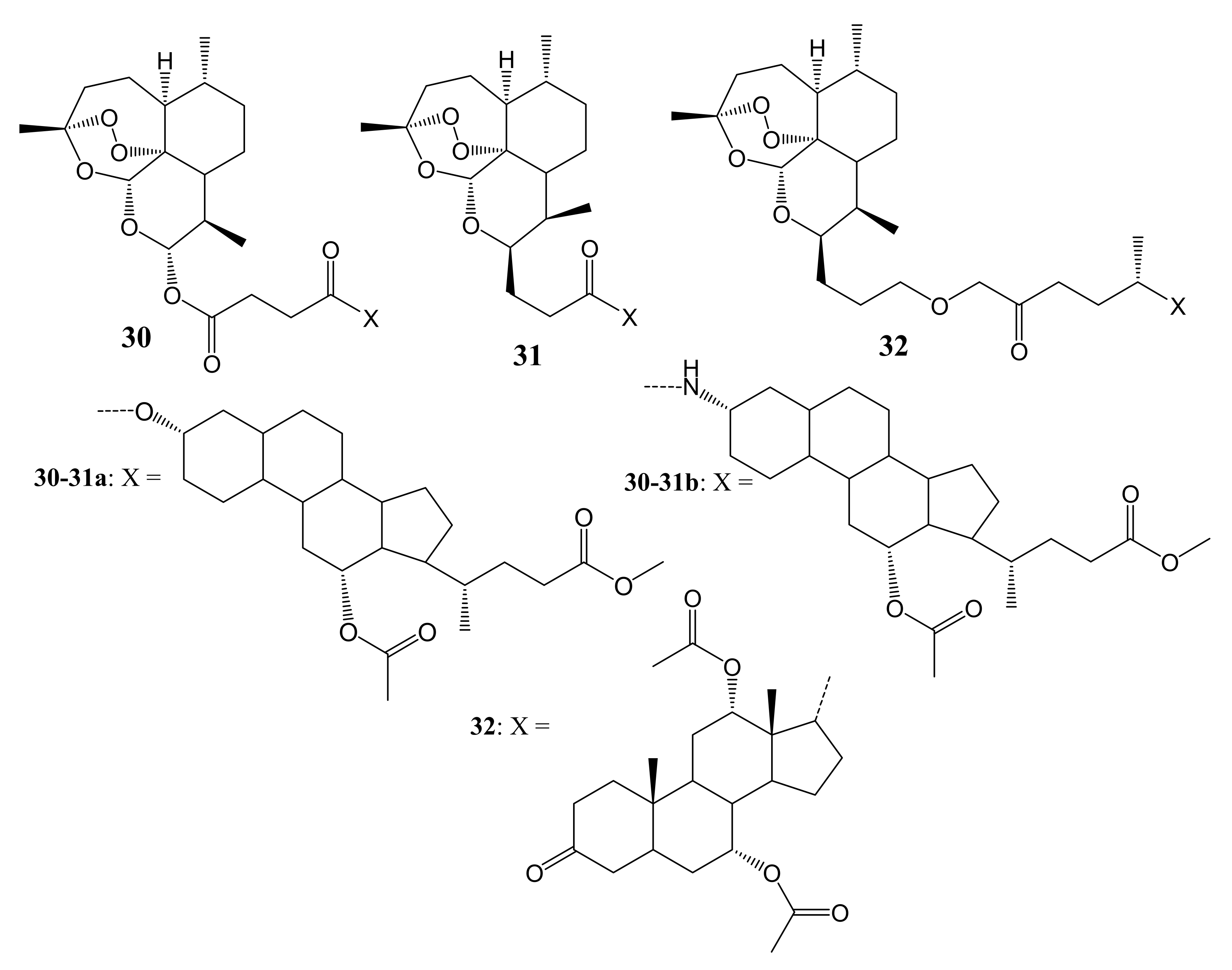{kind=link}
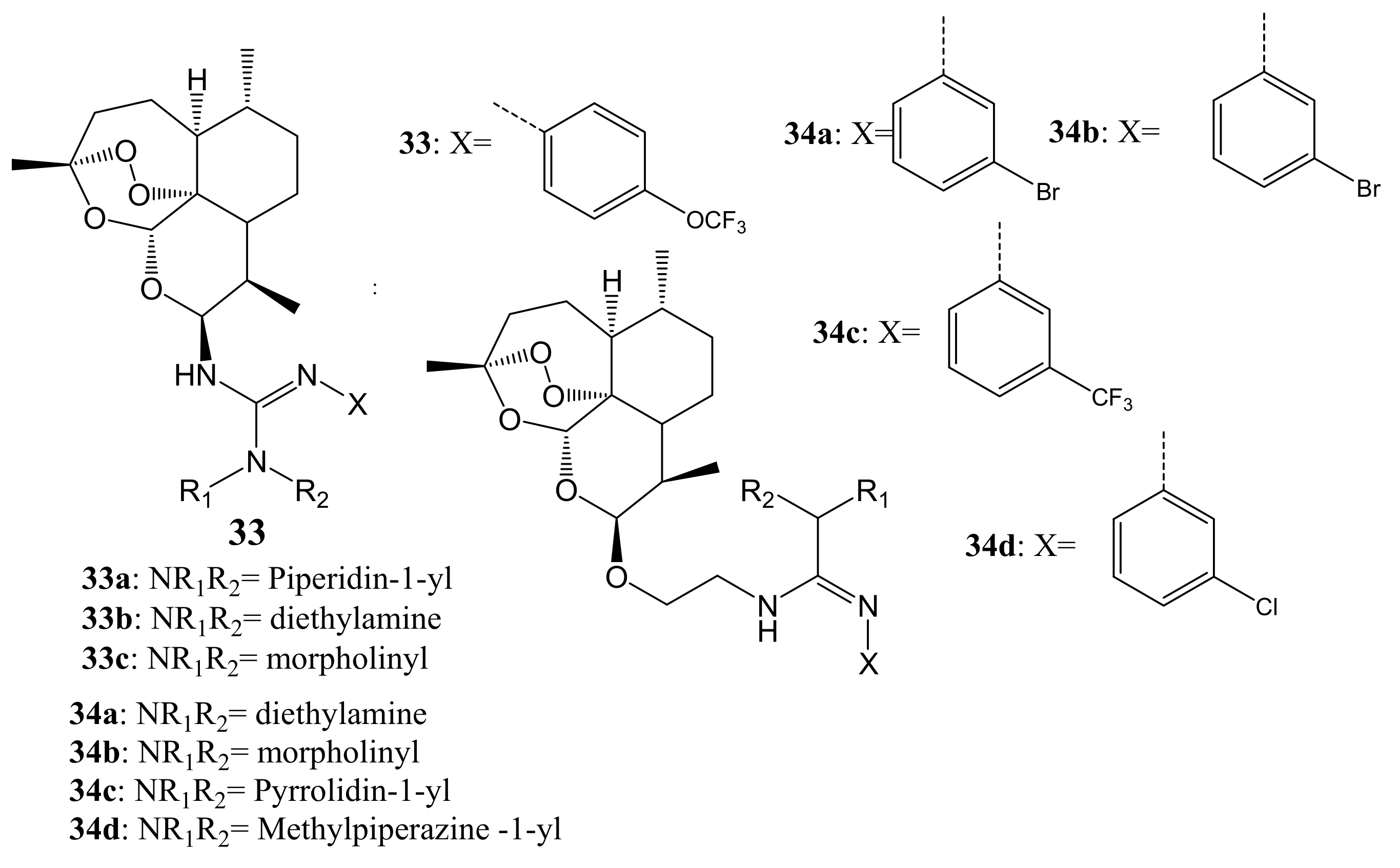{kind=link}
{kind=link}
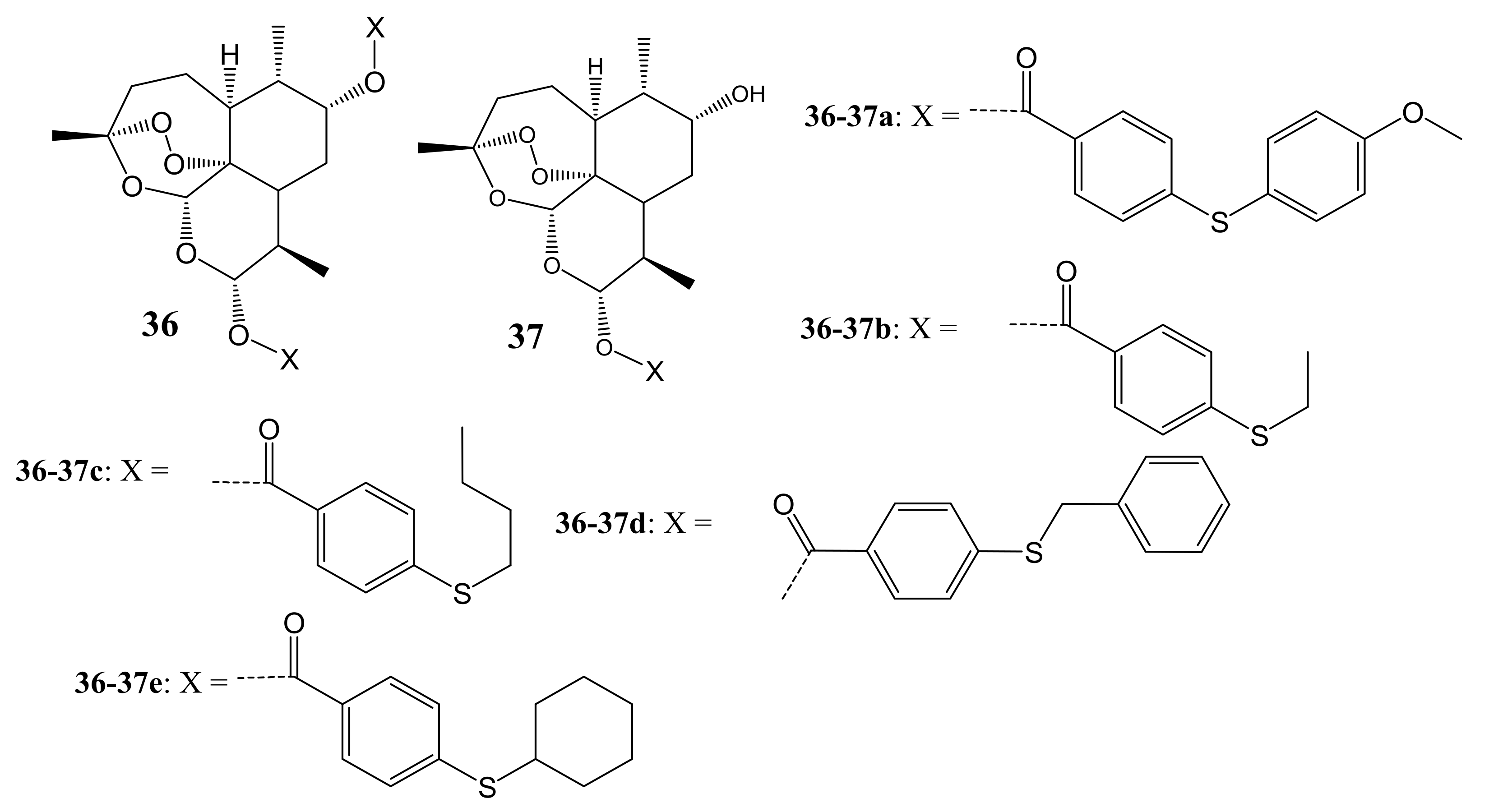{kind=link}
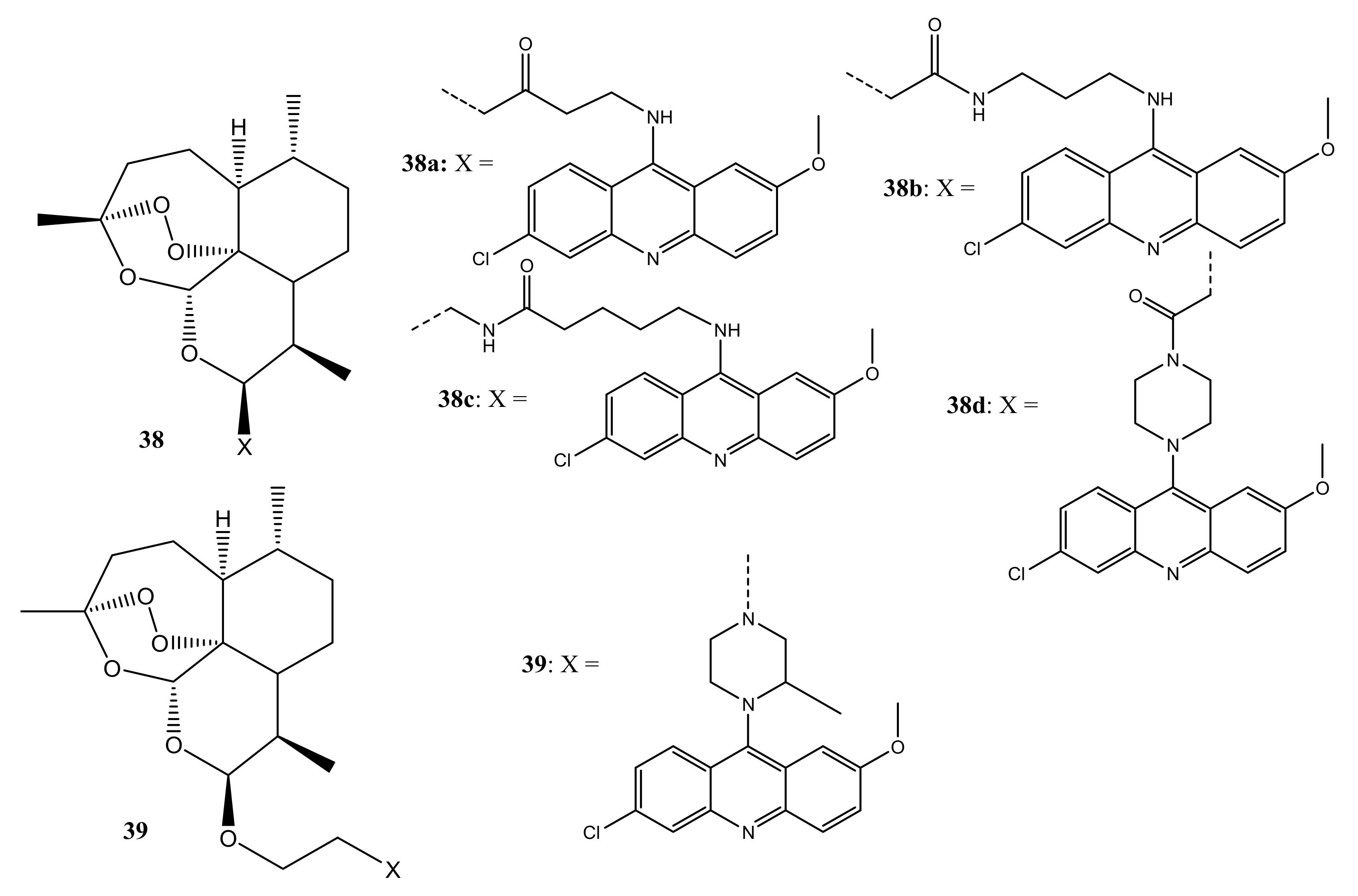{kind=link}
{kind=link}
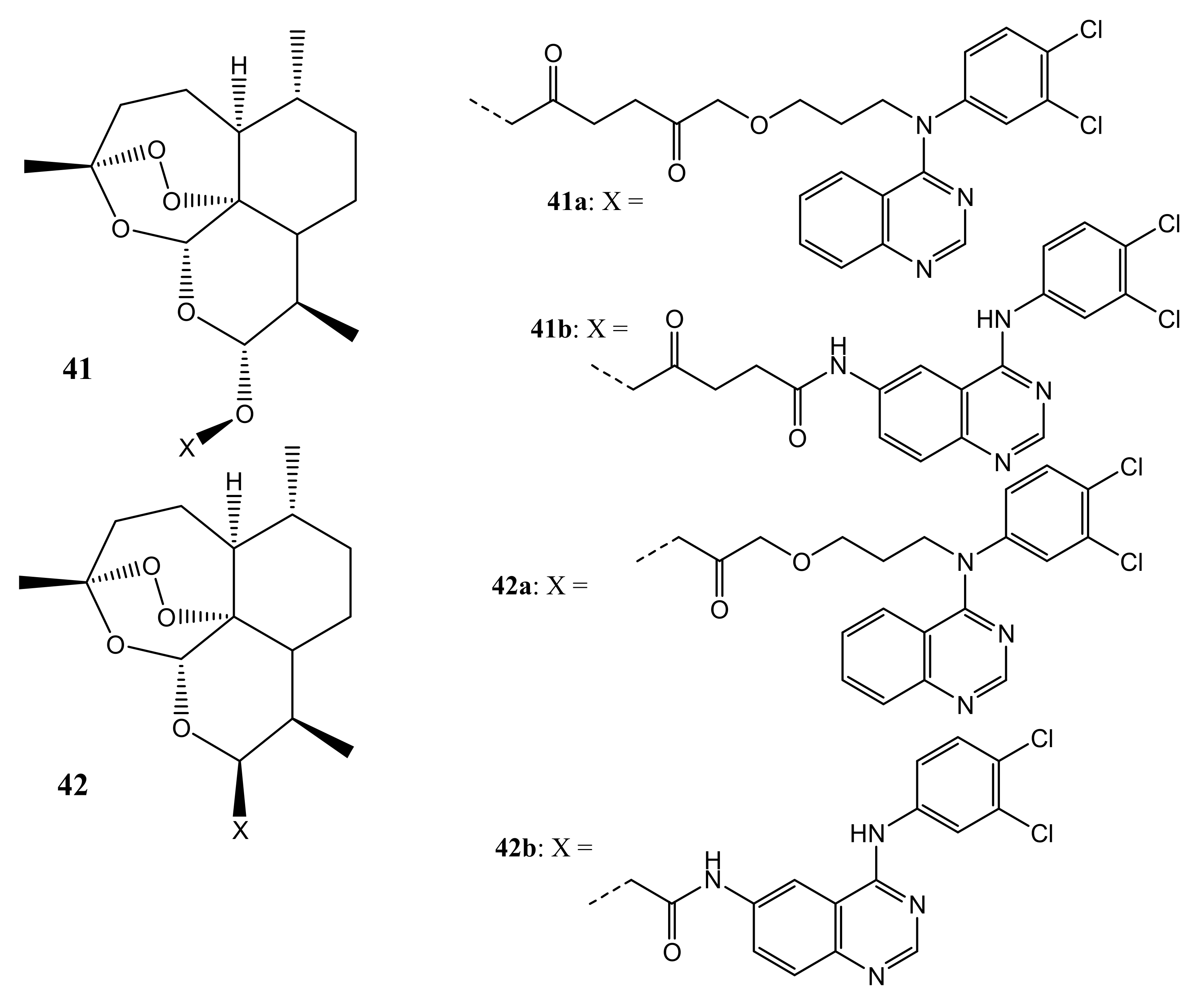{kind=link}
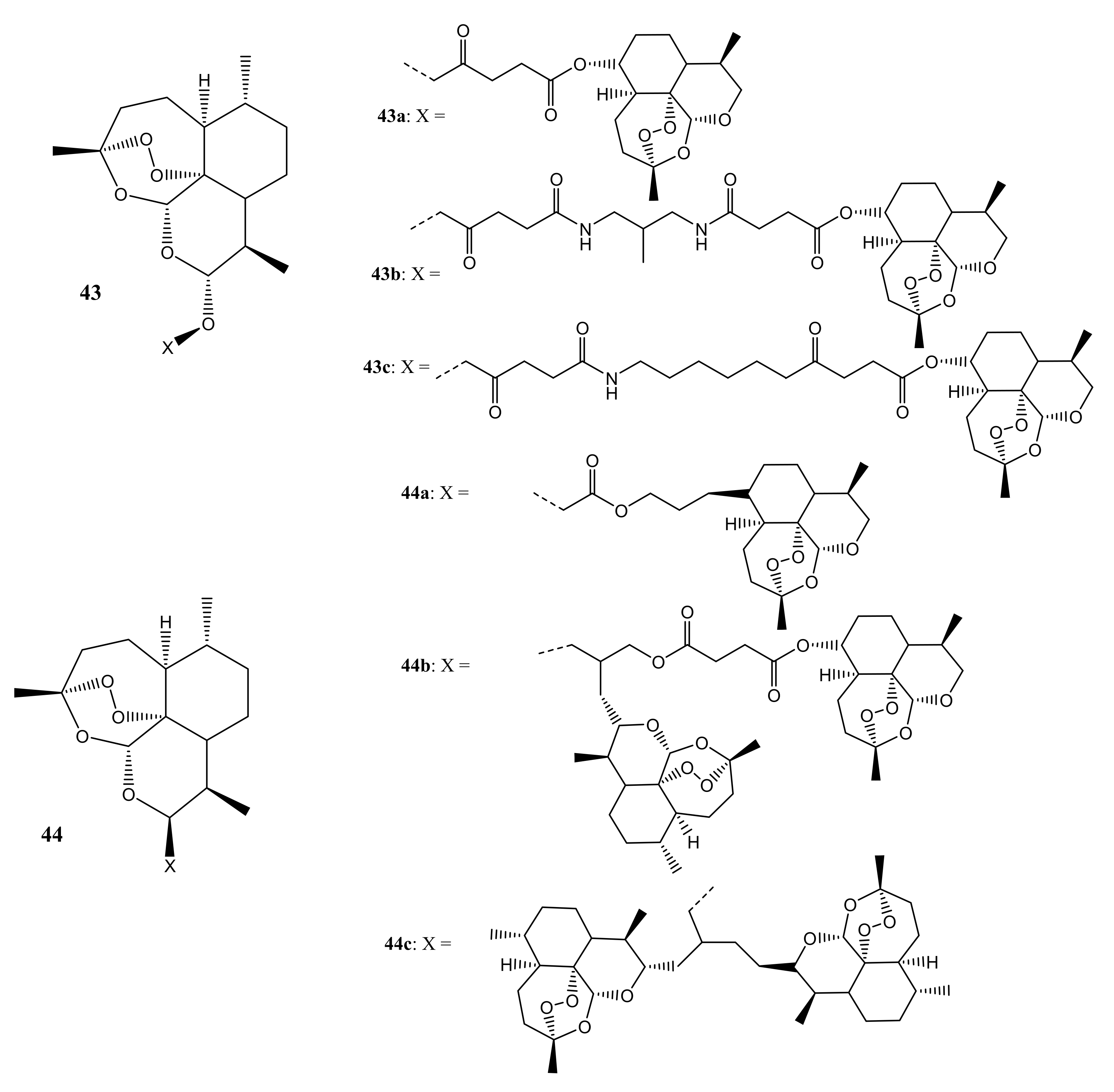{kind=link}
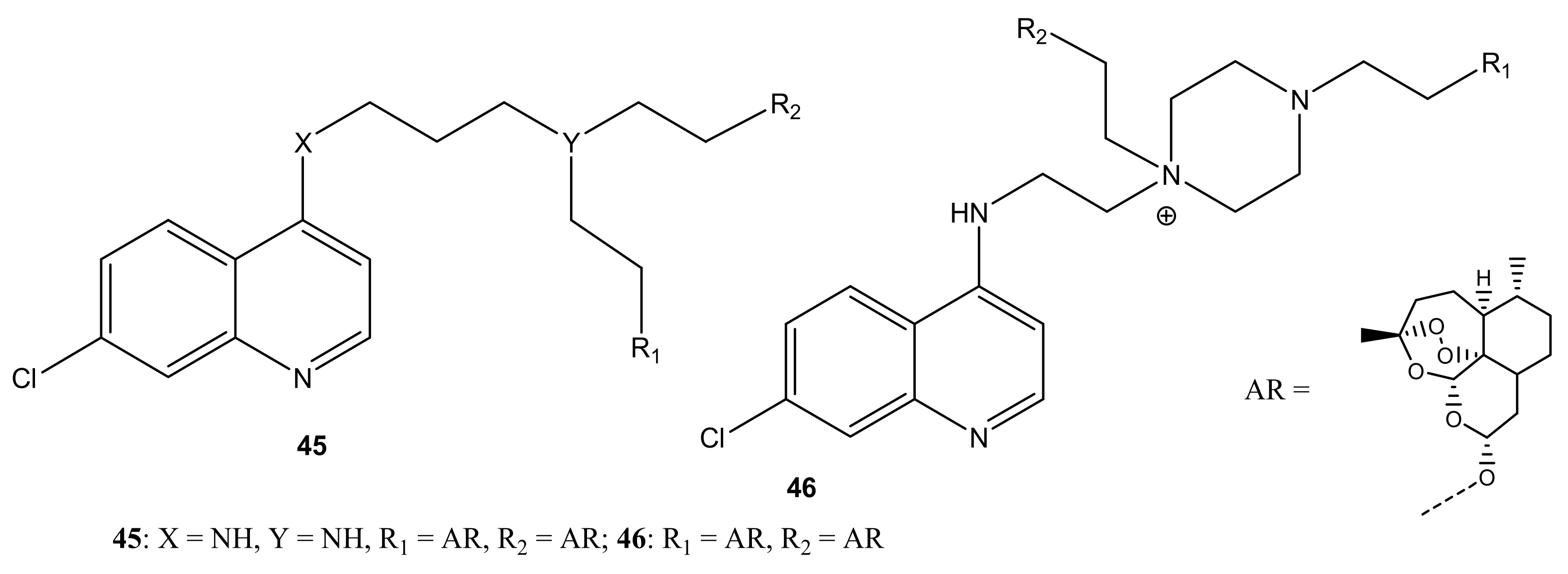{kind=link}
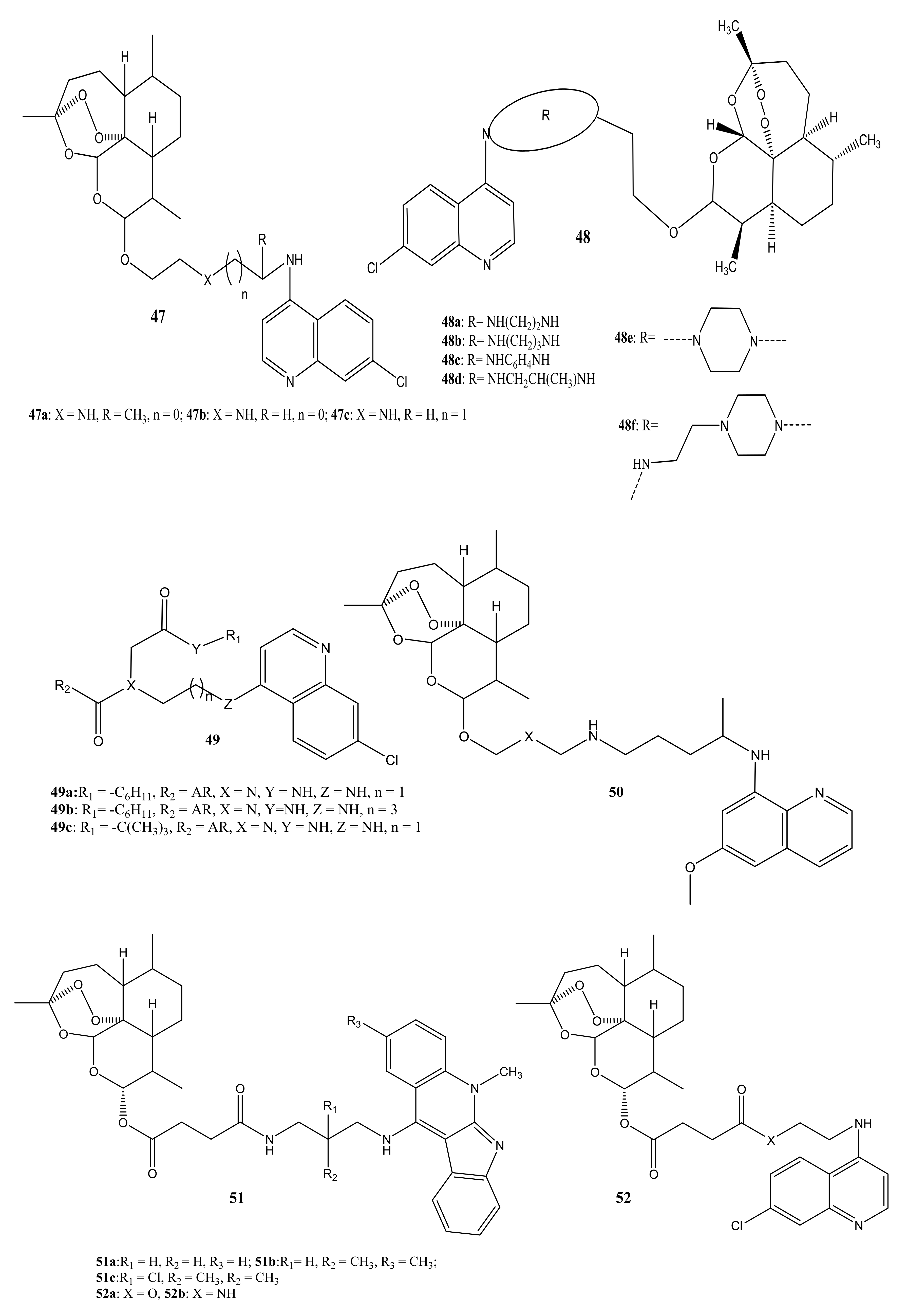{kind=link}
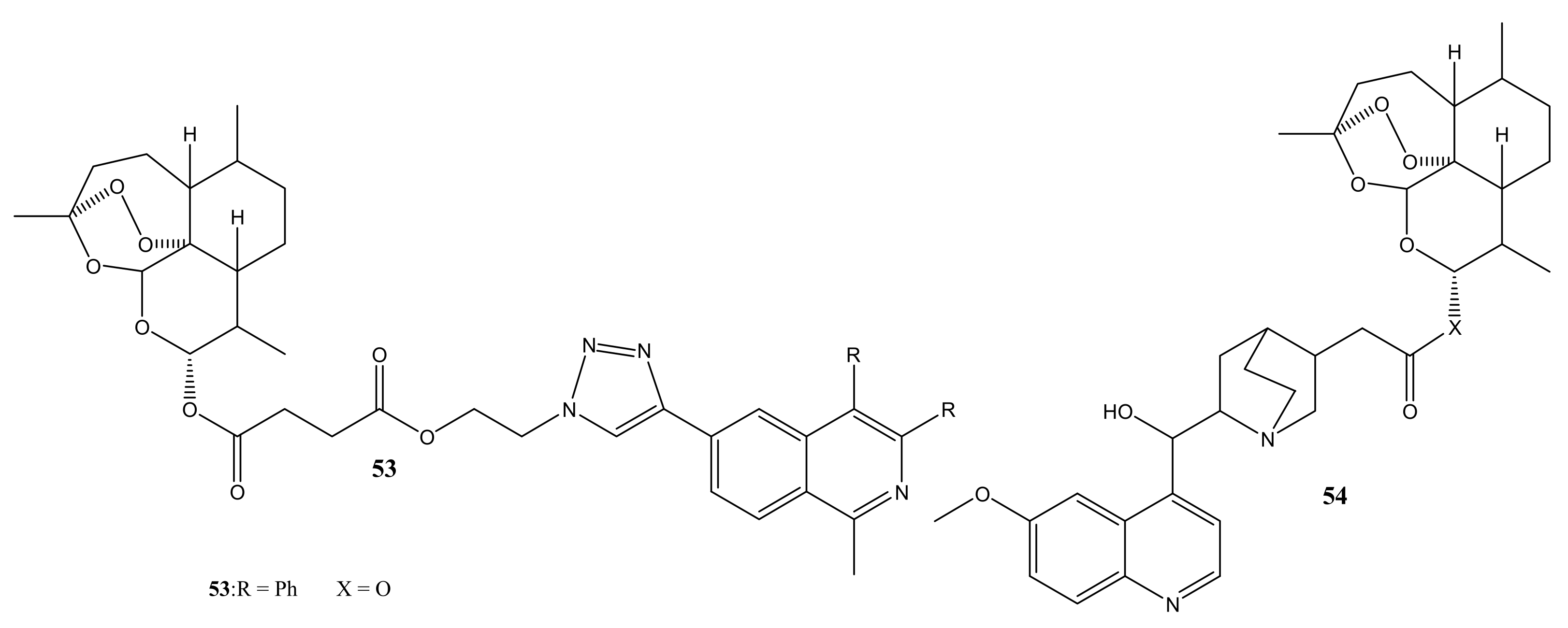{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mechanisms of Artemisinin
2.1. Mechanism of Artemisinin in Cancer
2.2. Mechanism of Artemisinin in Malaria
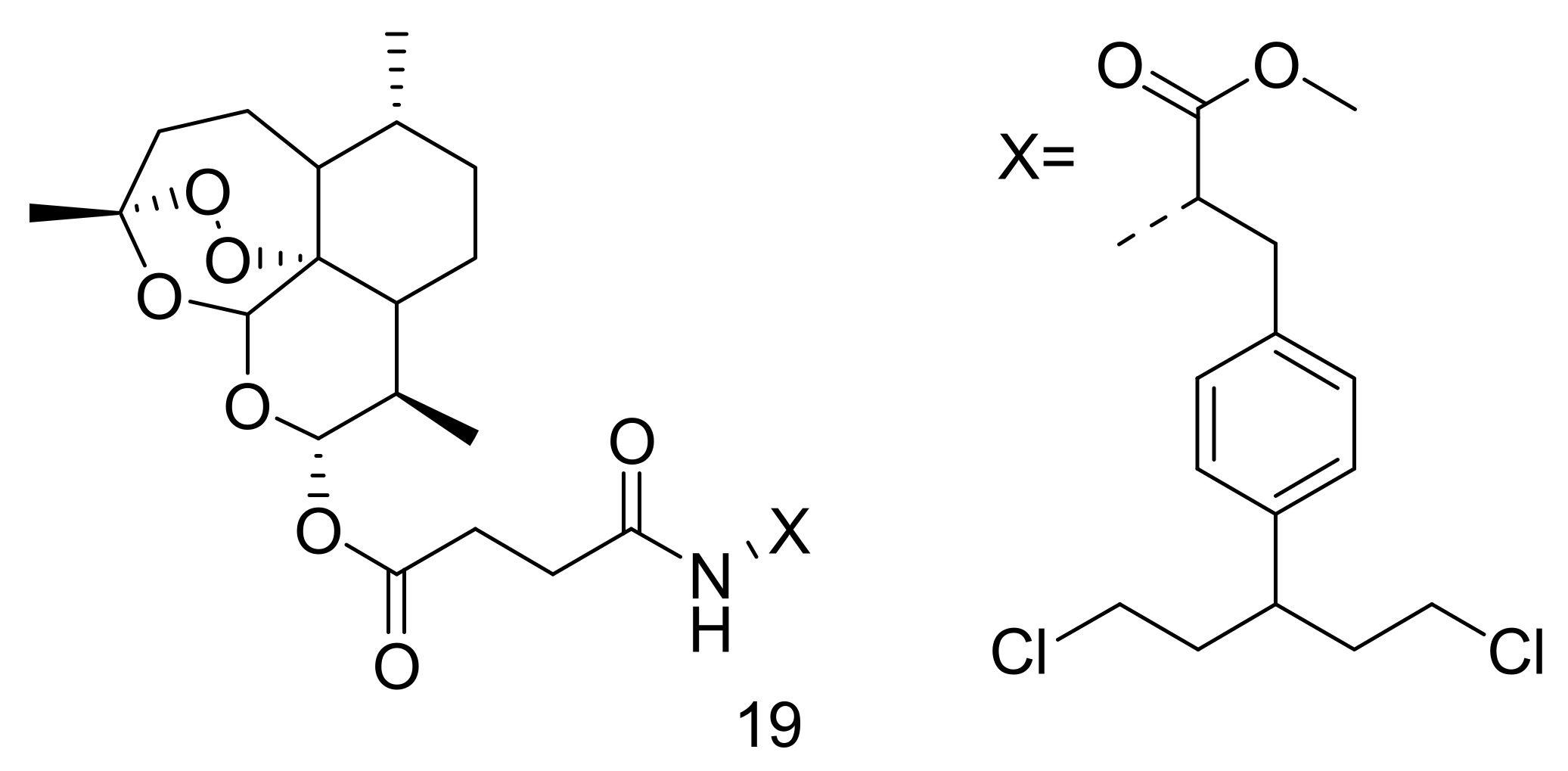
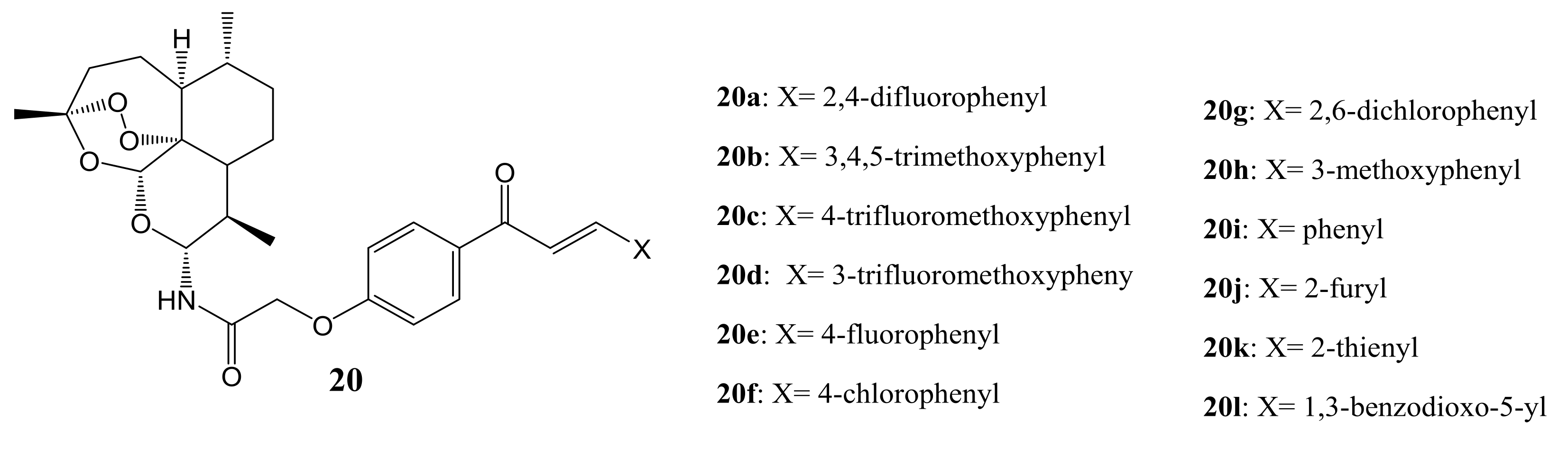
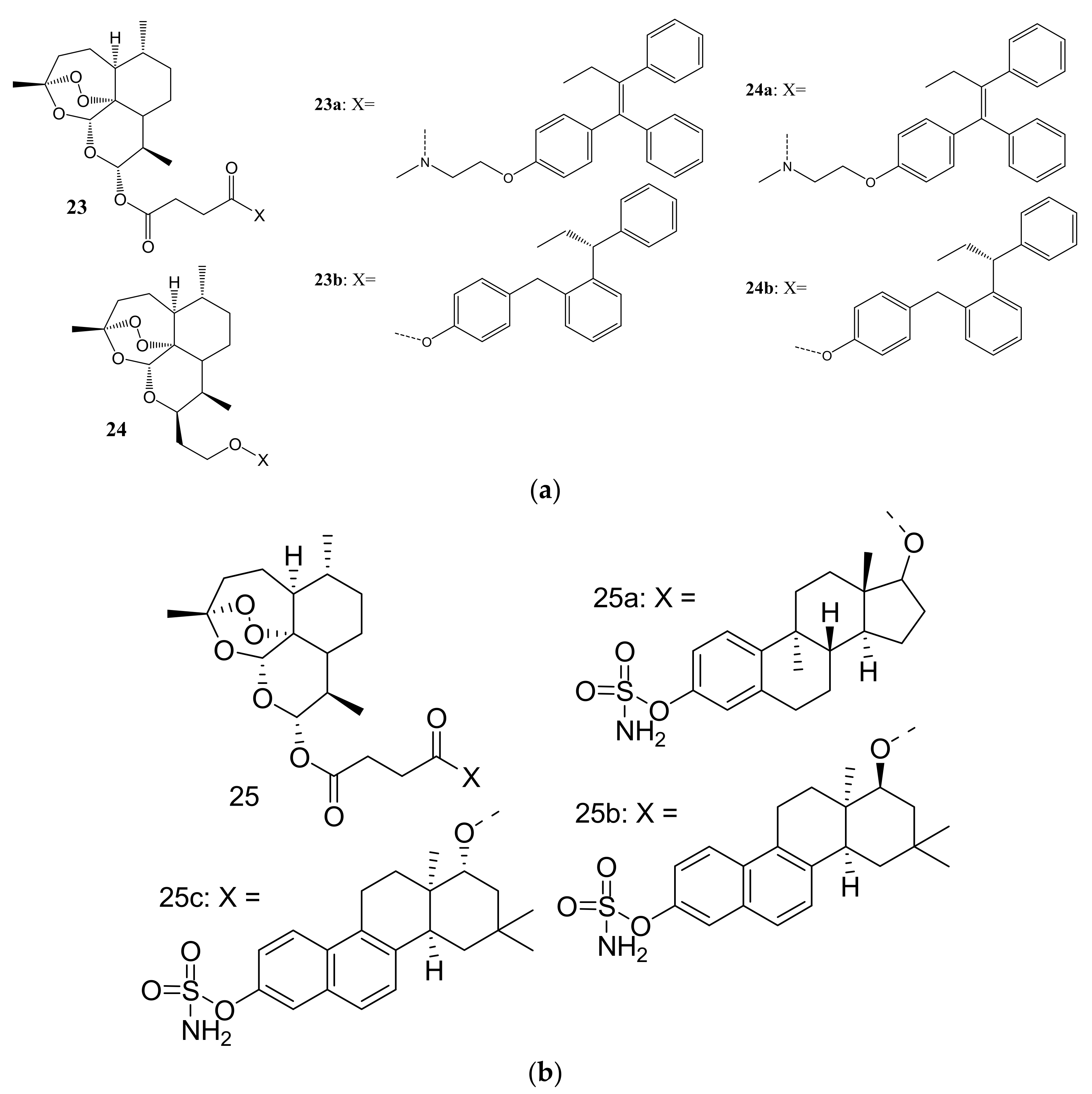
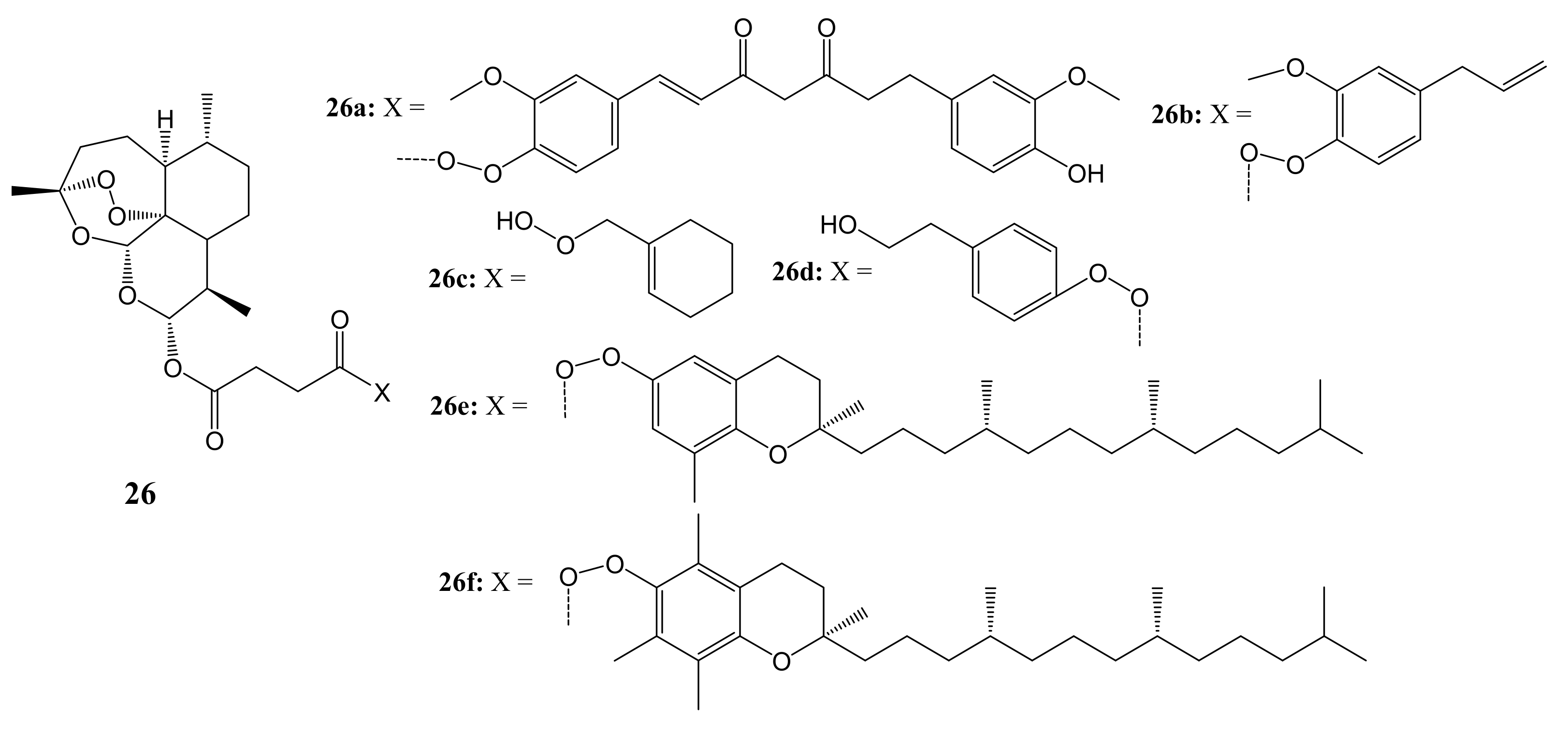
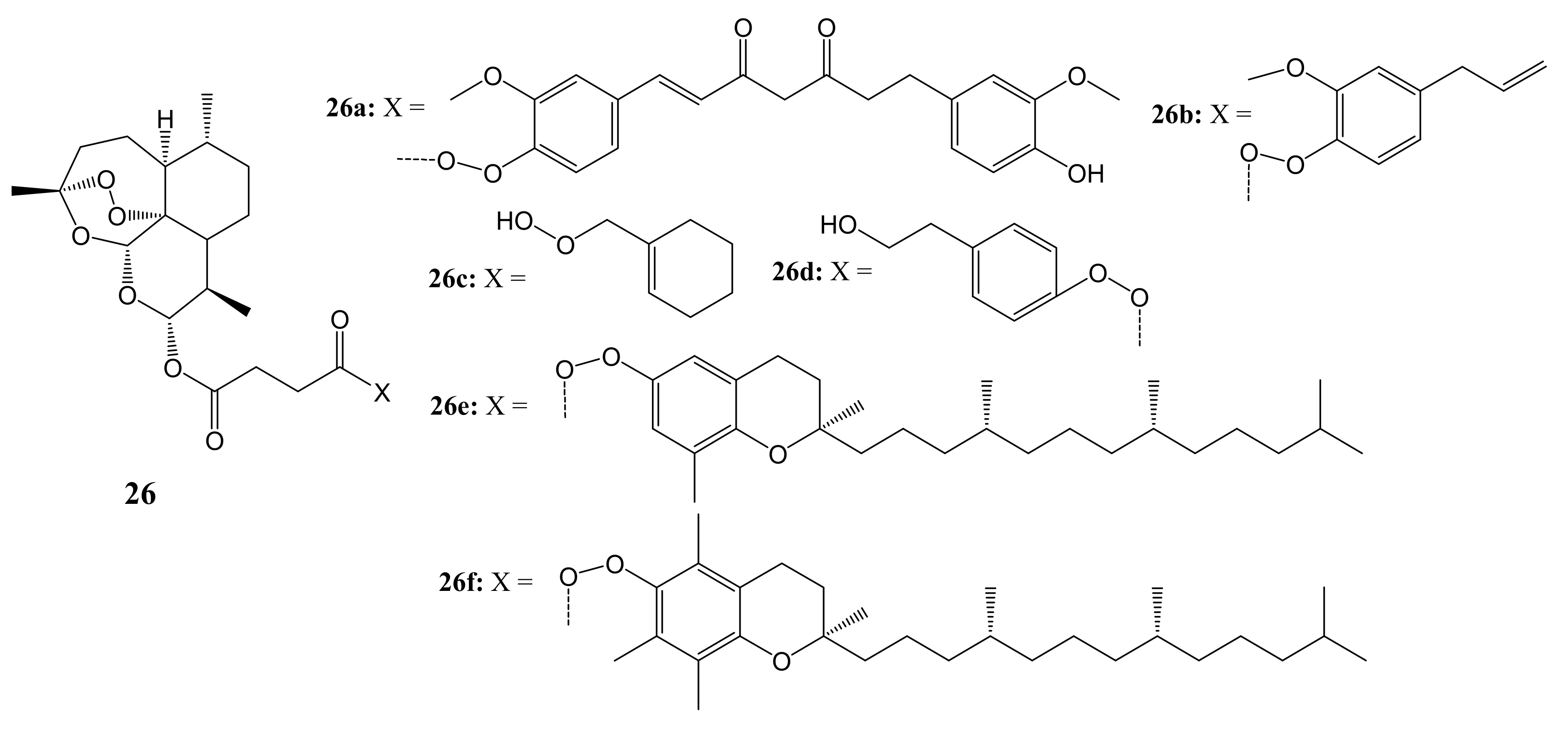
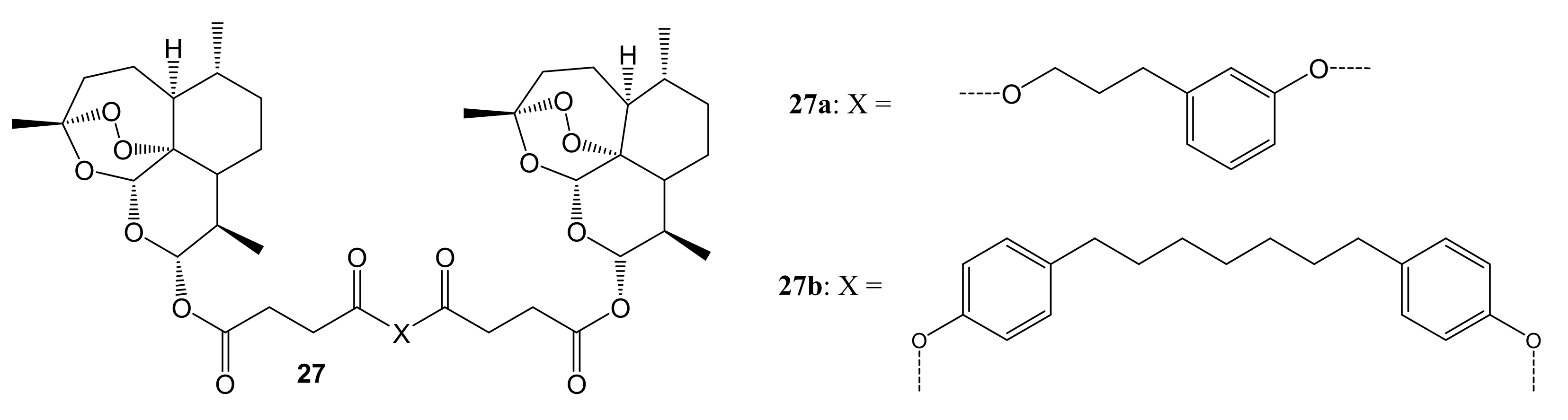
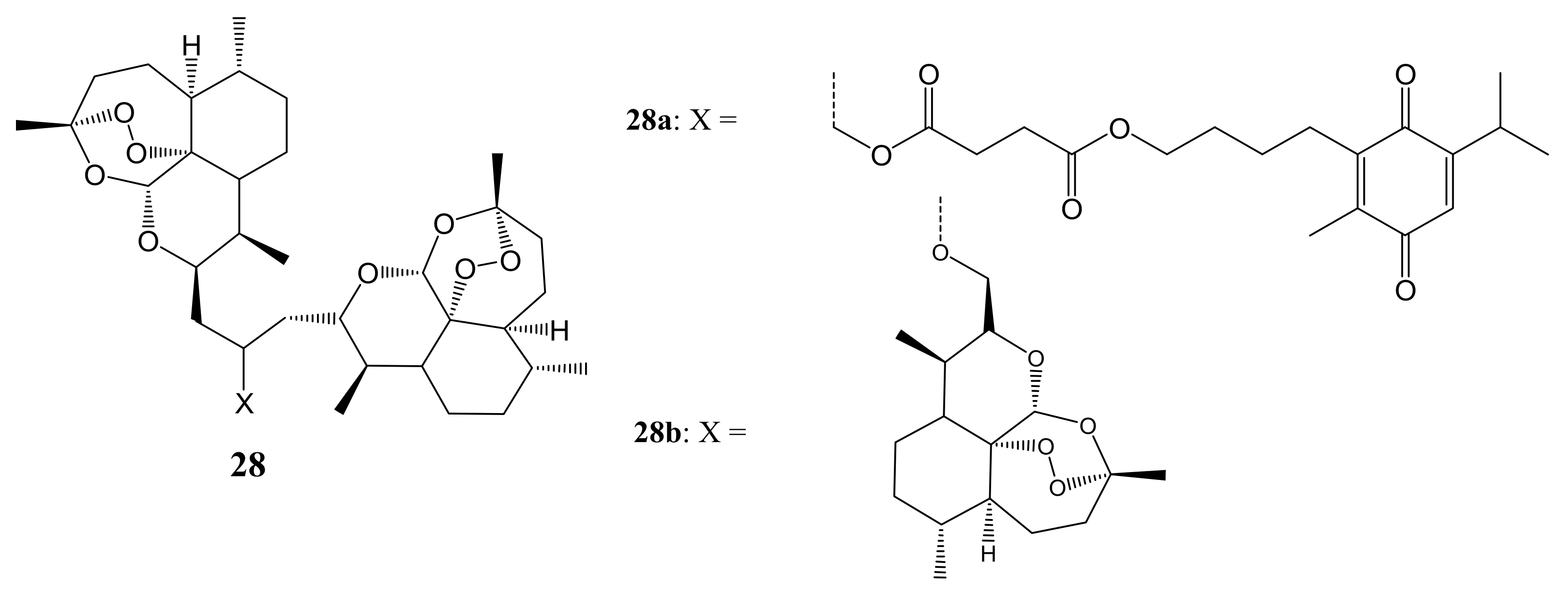
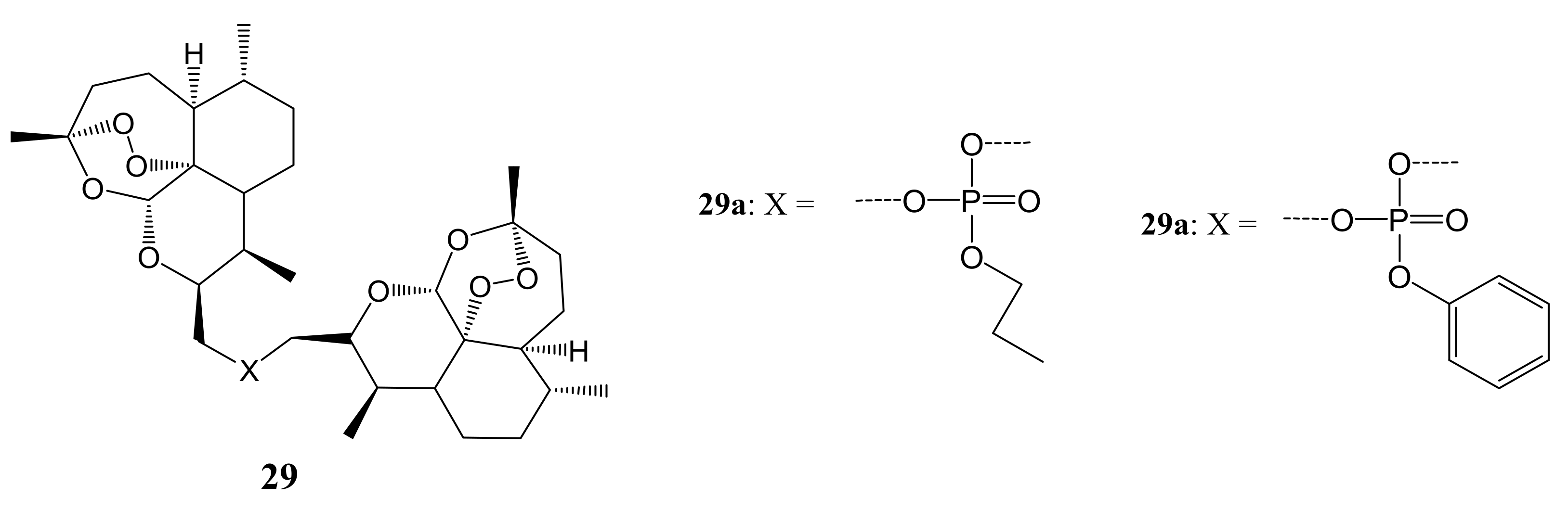
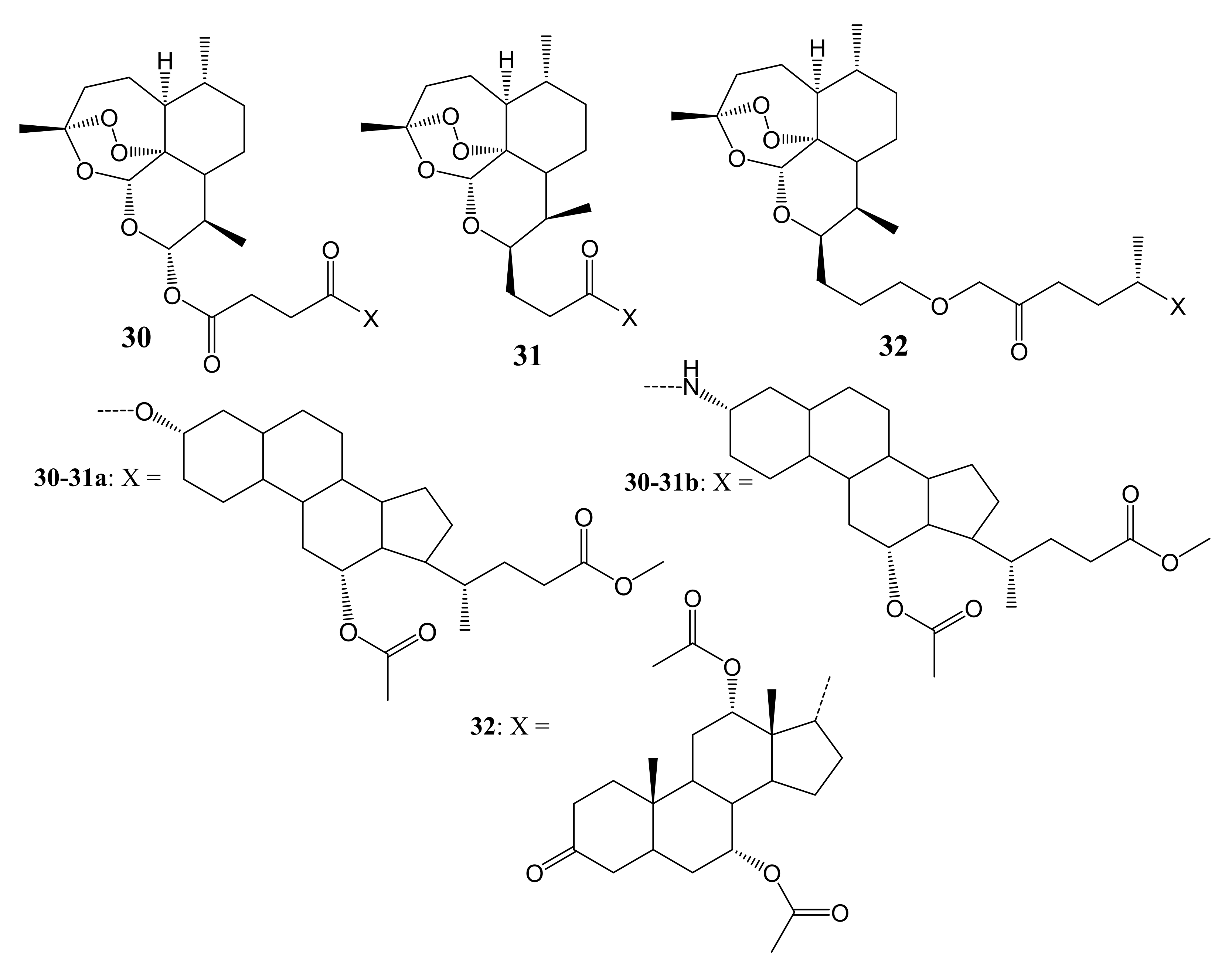
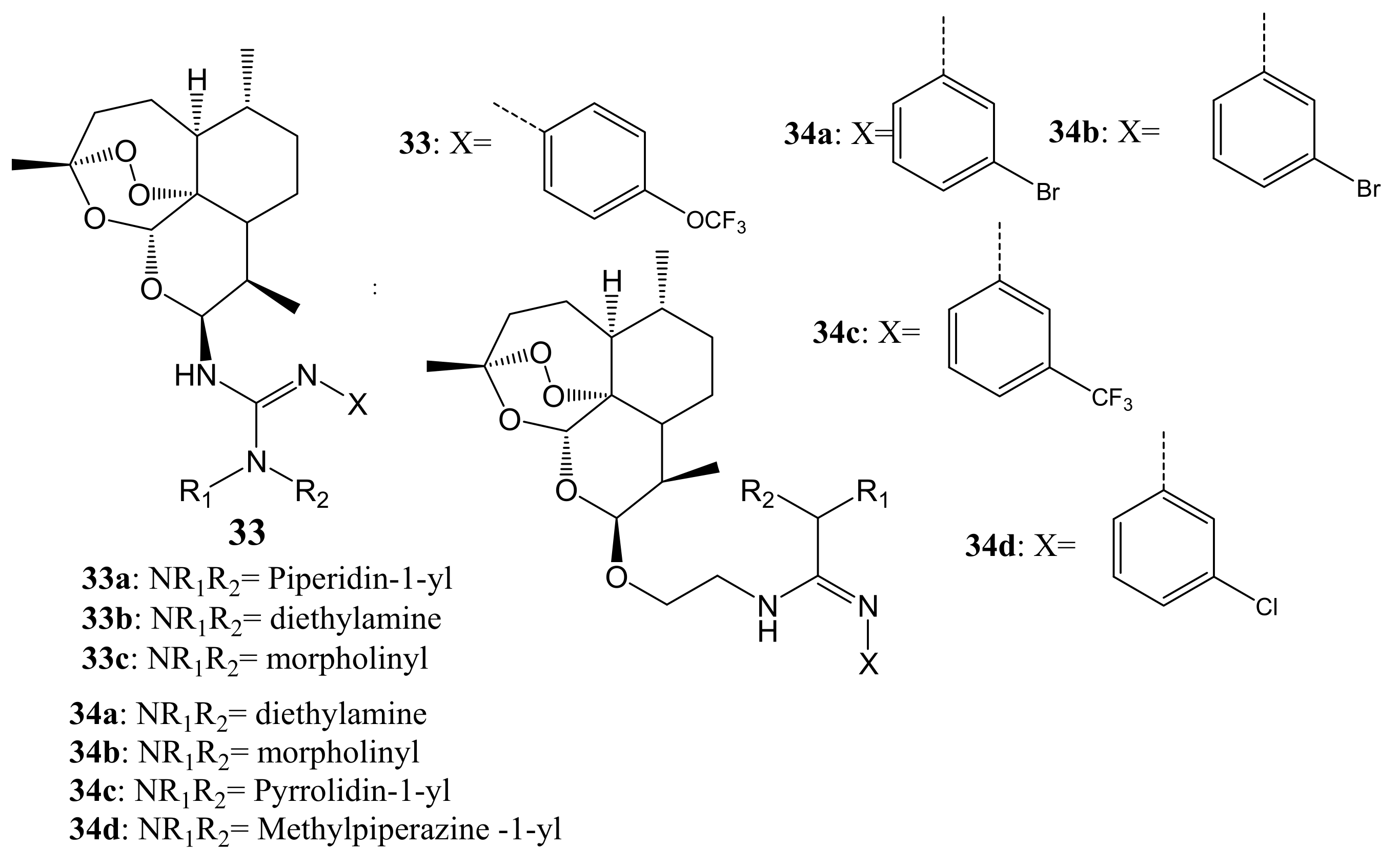
3. Artemisinin-Based Hybrid Compounds for Cancer Therapy
4. Artemisinin-Based Hybrid Compounds for Malaria Therapy
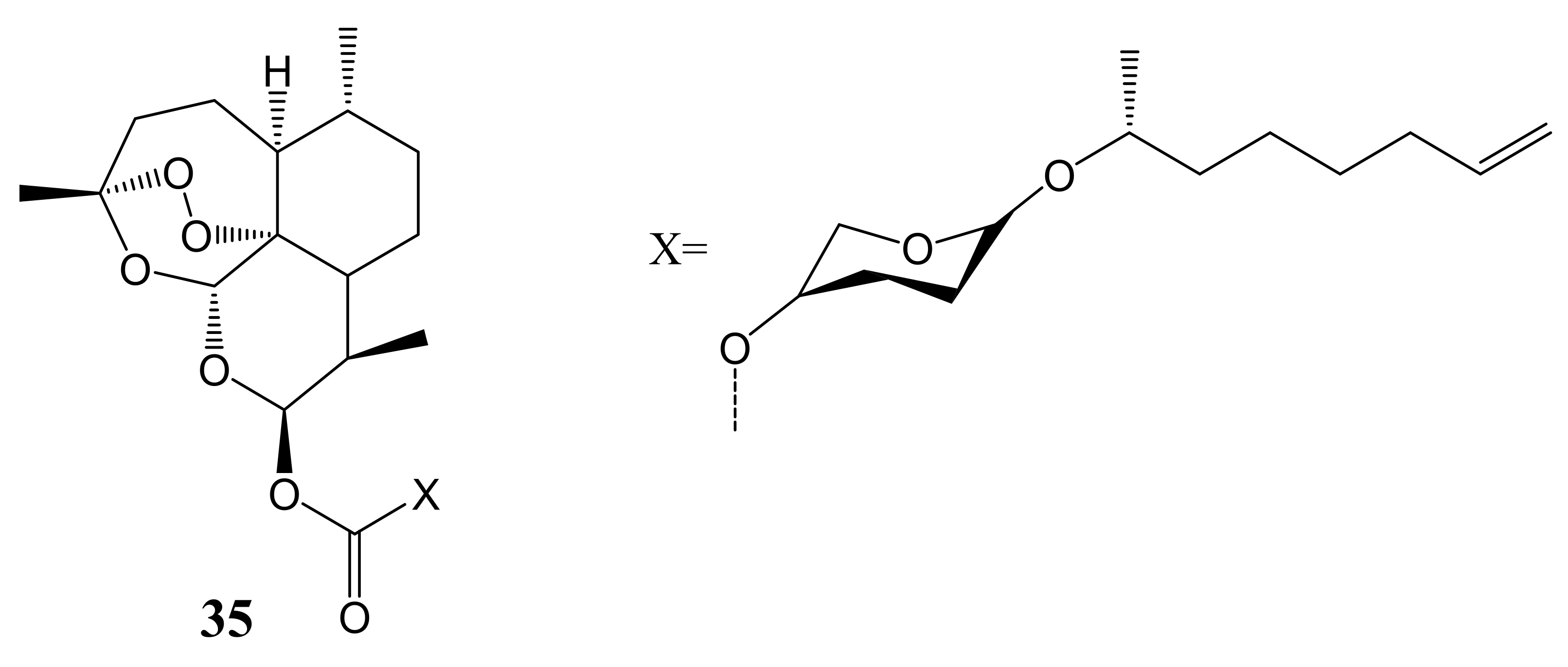
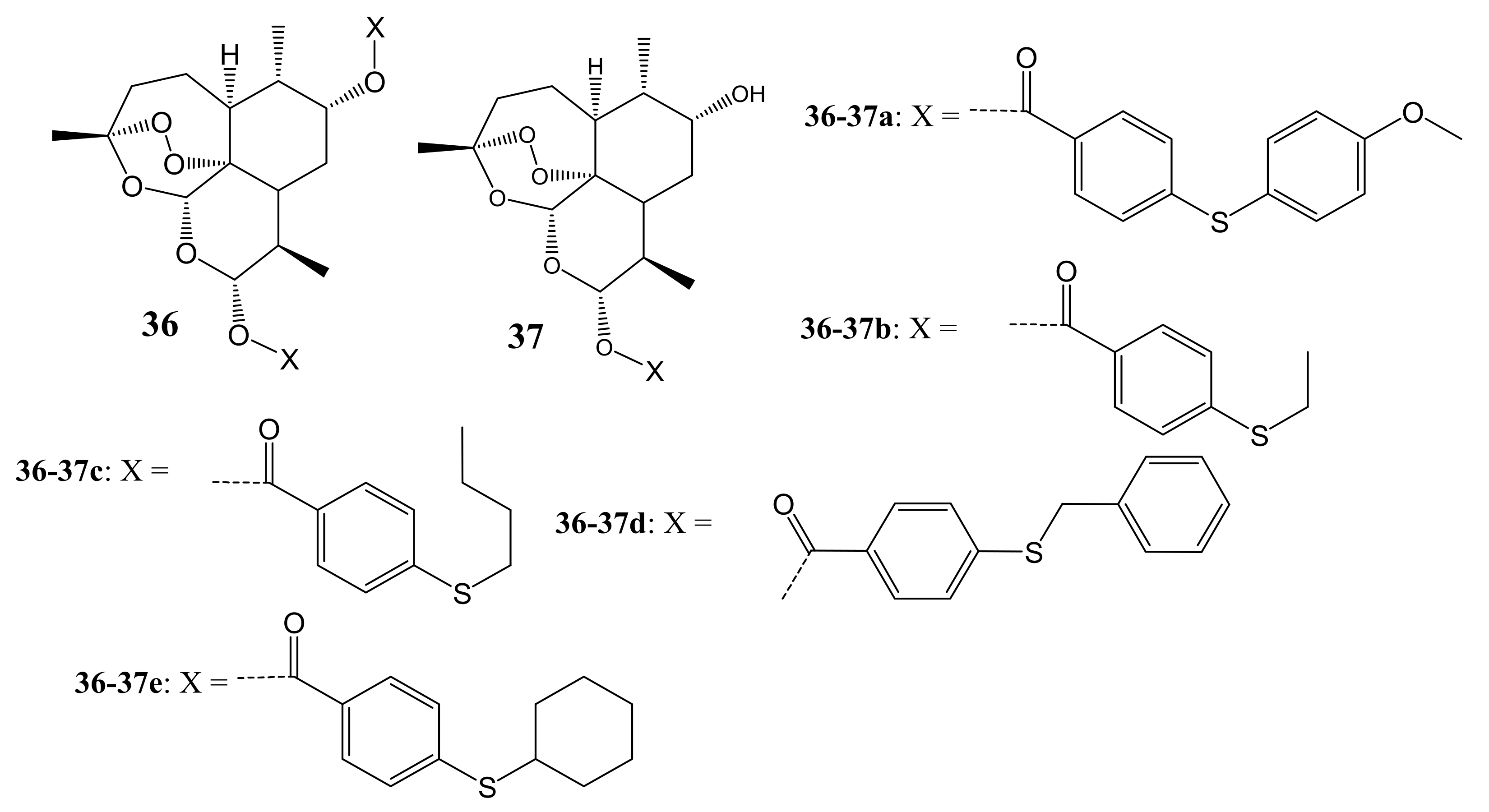
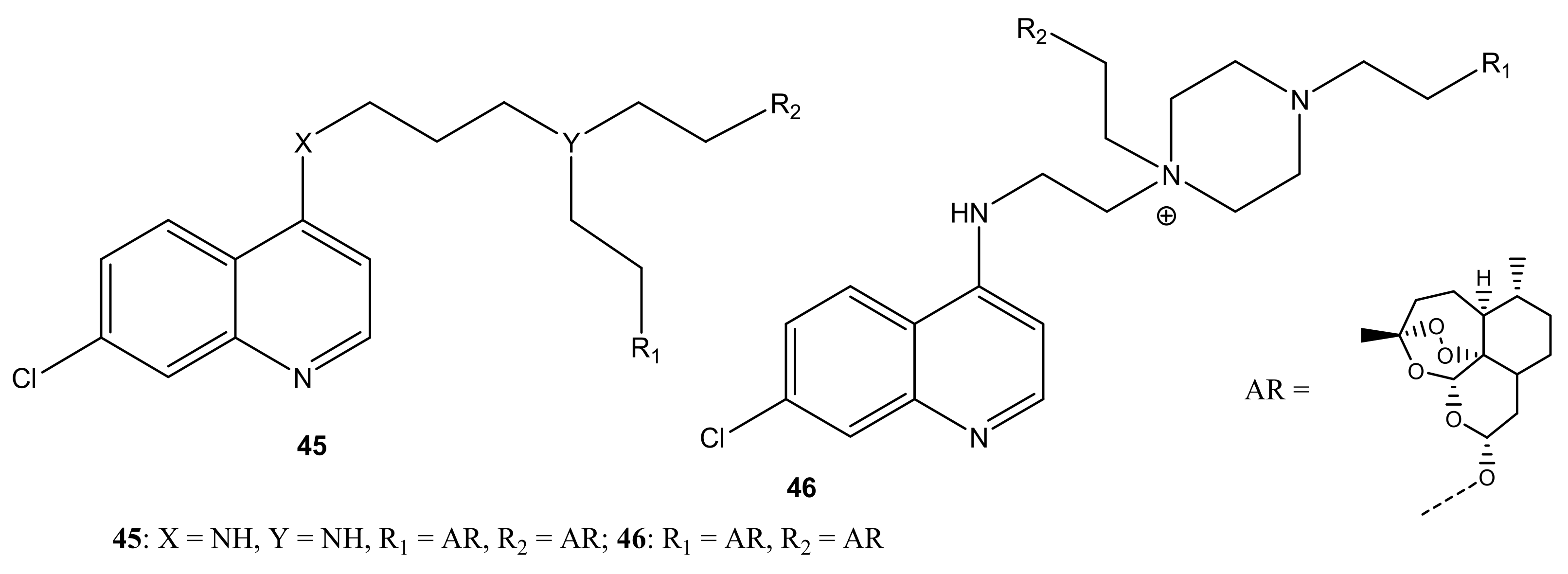
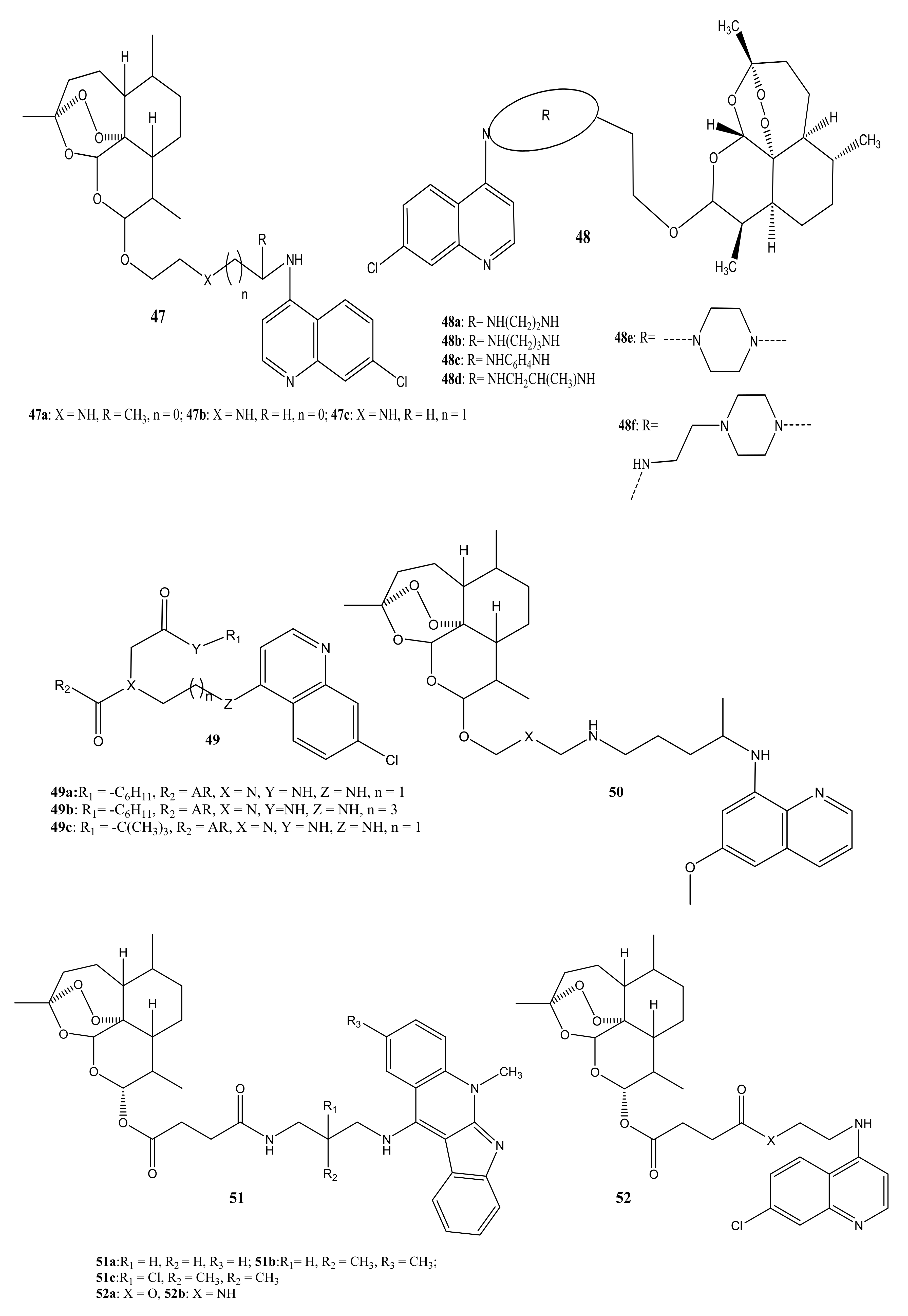
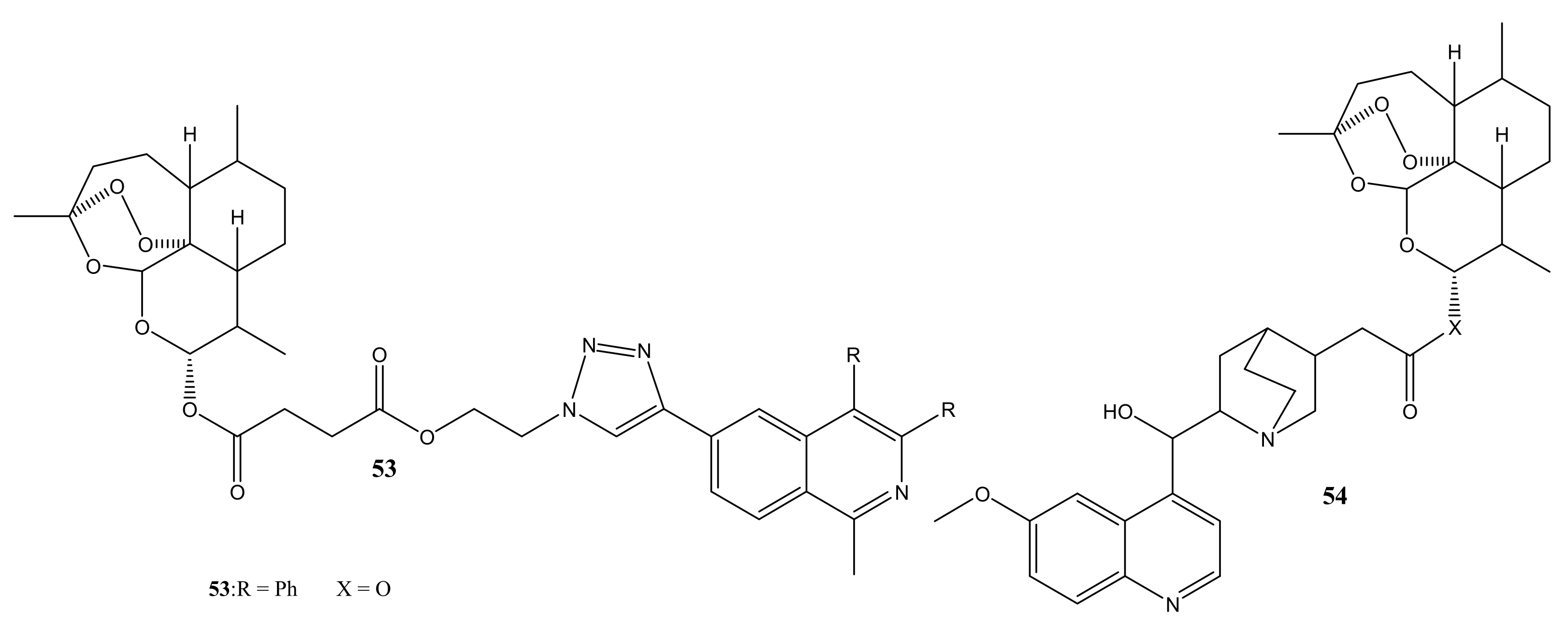
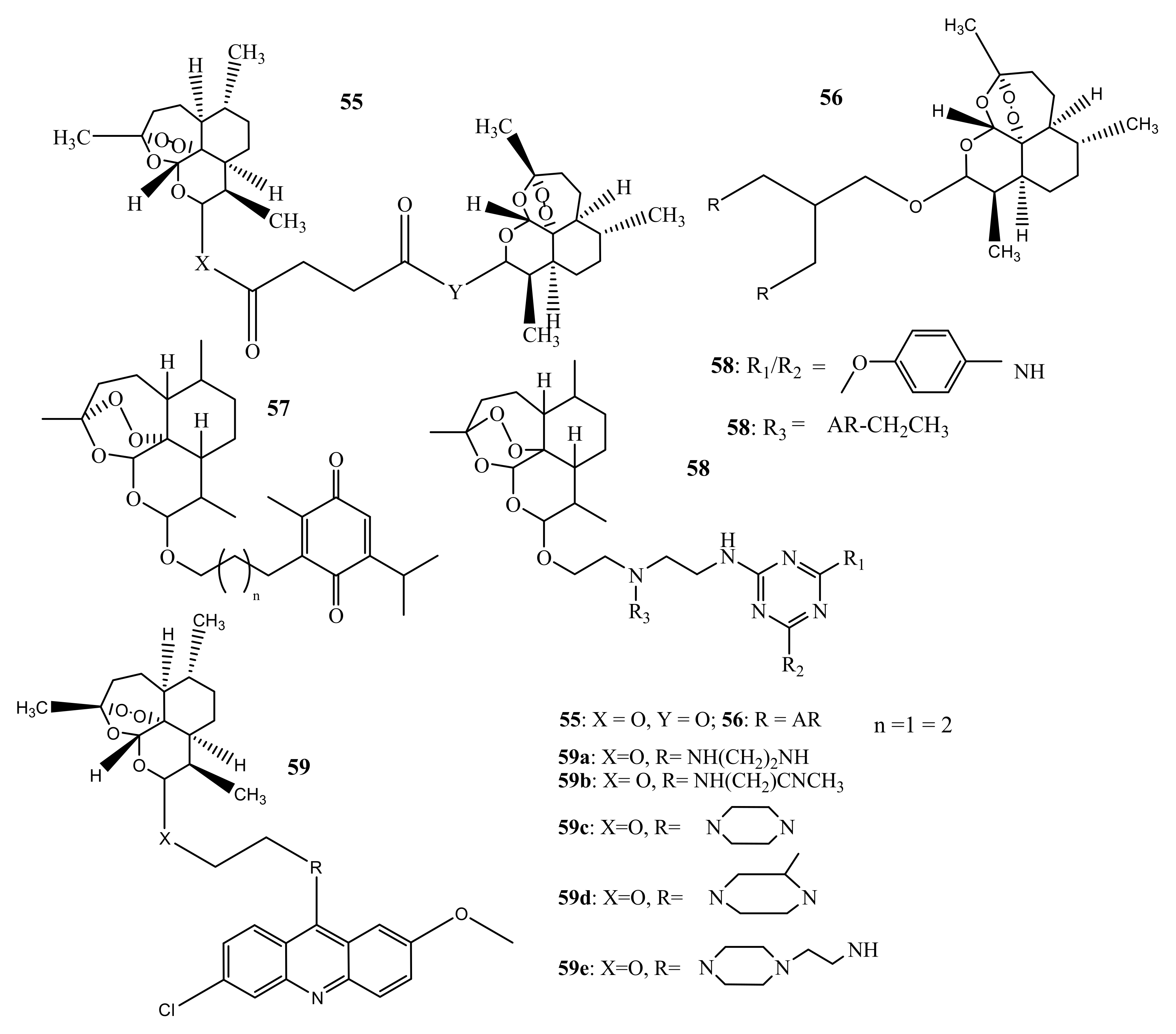
4.1. Artemisinin-Quinoline Hybrids
4.2. Ferrocene-Artemisinin Hybrids
4.3. Artemisinin Hybrids Containing Other Antimalarials
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Van Eijk, A.M.; Choubey, S.; Barla, P.; Haque, M.A.; Nandini, P.; Acharya, S.; Sullivan, S.A.; Mohanty, S.; Satpathi, S.; Carlton, J.M. Malaria in Sundargarh district, Odisha, India: Epidemiological and behavioral aspects from surveys. Acta Trop. 2020, 211, 105647. [Google Scholar] [CrossRef] [PubMed]
- Nqoro, X.; Naki, T.; Aderibigbe, B.A. Quinoline-based hybrid compounds with antimalarial activity. Molecules 2017, 22, 2268. [Google Scholar] [CrossRef] [Green Version]
- Peter, S.; Aderibigbe, B.A. Ferrocene-based compounds with antimalaria/anticancer activity. Molecules 2019, 24, 3604. [Google Scholar] [CrossRef] [Green Version]
- Zaw, M.T.; Lin, Z. Human Plasmodium knowlesi infections in South-East Asian countries. J. Microbiol. Immunol. Infect. 2019, 52, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Wangdi, K.; Gatton, M.L.; Kelly, G.C.; Banwell, C.; Dev, V.; Clements, A.C.A. Malaria elimination in India and regional implications. Lancet Infect. Dis. 2016, 16, e214–e224. [Google Scholar] [CrossRef]
- Chaves, L.F.; Rojas, M.R.; Jiménez, S.D.; Prado, M.; Rodríguez, R.M. Housing quality improvement is associated with malaria transmission reduction in Costa Rica. Socio-Econ. Plan. Sci. 2021, 74, 100951. [Google Scholar] [CrossRef]
- Monroe, A.; Moore, S.; Koenker, H.; Lynch, M.; Ricotta, E. Measuring and characterizing nighttime human behaviour as it relates to residual malaria transmission in sub-Saharan Africa: A review of the published literature. Malar. J. 2019, 18, 6. [Google Scholar] [CrossRef] [Green Version]
- Coban, C. The host targeting effect of chloroquine in malaria. Curr. Opin. Immunol. 2020, 66, 98–107. [Google Scholar] [CrossRef]
- Coban, C.; Lee, M.S.J.; Ishii, K.J. Tissue-specific immunopathology during malaria infection. Nat. Rev. Immunol. 2018, 18, 266–278. [Google Scholar] [CrossRef]
- Khoury, D.S.; Zaloumis, S.G.; Grigg, M.J.; Haque, A.; Davenport, M.P. Malaria Parasite Clearance: What Are We Really Measuring? Trends Parasit. 2020, 36, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Sun, Z.; Kong, F.; Xiao, J. Artemisinin-derived hybrids and their anticancer activity. Eur. J. Med. Chem. 2020, 188, 112044. [Google Scholar] [CrossRef]
- Sung, H.; Ferlat, J.; Rebecca, M.E.; Siegel, M.P.H.; Laversanne, M.; Soaerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Cancer J. Clin. 2021, 74, 209–249. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakara, A.B.; Sundaram, C.; Harikumar, K.B.; Tharakan, S.T.; Lai, O.S.; Sung, B.; Aggarwal, B.B. Cancer is a preventable disease that requires major lifestyle changes. Pharm. Res. 2008, 25, 2097–2116. [Google Scholar] [CrossRef]
- Efferth, T. Beyond malaria: The inhibition of viruses by artemisinin-type compounds. Biotechnol. Adv. 2018, 36, 1730–1737. [Google Scholar] [CrossRef]
- Ma, N.; Zhang, Z.; Liao, F.; Jiang, T.; Tu, Y. The birth of artemisinin. Pharmacol. Therap. 2020, 216, 107658. [Google Scholar] [CrossRef]
- Yang, J.; He, Y.; Li, Y.; Zhang, X.; Wong, Y.; Shen, S.; Zhong, T.; Zhang, J.; Liu, Q.; Wang, J. Advances in the research on the targets of anti-malaria actions of artemisinin. Pharmacol. Ther. 2020, 216, 107697. [Google Scholar] [CrossRef]
- Wang, J.; Xu, C.; Wong, Y.K.; Li, Y.; Liao, F.; Jiang, T.; Tu, Y. Artemisinin, the magic drug discovered from traditional Chinese medicine. Engineering 2019, 5, 32–39. [Google Scholar] [CrossRef]
- Nosten, F.; White, N.J. Artemisinin-based combination treatment of falciparum malaria. Am. J. Trop. Med. Hyg. 2007, 77, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xu, G.; Zhang, S.; Wang, D.; Prabha, P.S.; Zuo, Z. Antitumor Research on Artemisinin and Its Bioactive Derivatives. Nat. Prod. Bioprospect. 2018, 8, 303–319. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, R.I.; Foglio, M.A.; Olalla Saad, S.T. Artemisinin-type drugs for the treatment of hematological malignancies. Cancer Chemother. Pharmacol. 2020, 87, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Neill, P.M.O.; Barton, V.E.; Ward, S.A. The Molecular Mechanism of Action of Artemisinin—The Debate Continues. Molecules 2010, 15, 1705–1721. [Google Scholar]
- Sarder, A.; Pokharel, Y.R. Synthetic Derivatives of Artemisinin and Cancer. Int. J. Med. Biomed. Sci. 2016, 1, 12–16. [Google Scholar]
- Konstat-korzenny, E.; Ascencio-Aragin, J.; Niezen-lugo, S.; Vasquez-Lopez, R. Artemisinin and Its Synthetic Derivatives as a Possible Therapy for Cancer. Med. Sci. 2018, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Du, J.H.; Zhang, H.D.; Ma, Z.J.; Ji, K.M. Artesunate induces oncosis-like cell death in vitro and has antitumor activity against pancreatic cancer xenografts in vivo. Cancer Chemother. Pharmacol. 2010, 65, 895–902. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.D.; Tan, S.H.; Ng, S.; Shi, Y.; Zhou, J.; Tan, K.S.W.; Wong, W.S.F.; Shen, H.M. Artesunate induces cell death in human cancer cells via enhancing lysosomal function and lysosomal degradation of ferritin. J. Biol. Chem. 2014, 289, 33425–33441. [Google Scholar] [CrossRef] [Green Version]
- Ju, R.-J.; Cheng, L.; Peng, X.-M.; Wang, T.; Li, C.-Q.; Song, X.-L.; Liu, S.; Chao, J.-P.; Li, X.-T. Octreotide-modified liposomes containing daunorubicin and dihydroartemisinin for treatment of invasive breast cancer. Artif. Cells Nanomed. Biotechnol. 2018, 46, 616–628. [Google Scholar] [CrossRef] [Green Version]
- Van Huijsduijnen, R.H.; Guy, R.; Chibale, K.; Haynes, R.; Peitz, I.; Kelter, G.; Phillips, M.A.; Vennerstrom, J.L.; Yuthavong, Y.; Wells, T.N.C. Anticancer Properties of Distinct Antimalarial Drug Classes. PLoS ONE 2013, 8, e82962. [Google Scholar]
- Lam, N.S.; Long, X.; Wong, J.W.; Griffin, R.C.; Doery, J.C.G. Artemisinin and its derivatives: A potential treatment for leukemia. Anticancer Drugs 2018, 30, 1–18. [Google Scholar] [CrossRef]
- Weifeng, T.; Feng, S.; Xiangji, L.; Changqing, S.; Zhiquan, Q.; Huazhong, Z.; Peining, Y.; Yong, Y.; Mengchao, W.; Xiaoqing, J.; et al. Artemisinin inhibits in vitro and in vivo invasion and metastasis of human hepatocellular carcinoma cells. Phytomedicine 2011, 18, 158–162. [Google Scholar] [CrossRef]
- Slezakova, S.; Ruda-kucerova, J. Anticancer Activity of Artemisinin and its Derivatives. Anticancer Res. 2017, 37, 5995–6003. [Google Scholar]
- Wong, Y.K.; Xu, C.; Kalesh, K.A.; Chengchao, X.; Lin, Q.; Wong, W.S.F.; Shen, H.-M.; Wang, J. Artemisinin as an anticancer drug: Recent advances in target profiling and mechanisms of action. Med. Res. Rev. 2017, 37, 1492–1517. [Google Scholar] [CrossRef]
- Aguiar, A.C.; Murce, E.; Cortopassi, W.A.; Pimentel, A.S.; Almeida, M.M.; Barros, D.C.; Guedes, J.S.; Meneghetti, M.R.; Krettli, A.U. Drugs and Drug Resistance Chloroquine analogues as antimalarial candidates with potent in vitro and in vivo activity. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 459–464. [Google Scholar] [CrossRef]
- Kumari, A.; Karnatak, M.; Singh, D.; Shankar, R.; Jat, J.L.; Sharma, S.; Yadav, D.; Shrivastava, R.; Verma, V.P. Current scenario of artemisinin and its analogues for antimalarial activity. Eur. J. Med. Chem. 2019, 163, 804–829. [Google Scholar] [CrossRef]
- Khoury, D.S.; Cao, P.; Zaloumis, S.G.; Davenport, M.P. Interdisciplinary Approaches to Malaria Consortium. Artemisinin resistance and the unique selection pressure of a short-acting antimalarial. Trends Parasitol. 2020, 36, 884–887. [Google Scholar] [CrossRef]
- Meshnick, S.R. Artemisinin: Mechanisms of action, resistance and toxicity. Int. J. Parasitol. 2002, 32, 1655–1660. [Google Scholar] [CrossRef]
- Hu, Y.; Li, N.; Zhang, J.; Wang, Y.; Chen, L.; Sun, J. Artemisinin-indole and artemisinin-imidazole hybrids: Synthesis, cytotoxic evaluation and reversal effects on multidrug resistance in MCF-7/ADR cells. Bioorg. Med. Chem. Lett. 2019, 29, 1138–1142. [Google Scholar] [CrossRef]
- Tian, Y.; Liang, Z.; Xu, H.; Mou, Y.; Guo, C. Design, Synthesis and Cytotoxicity of Novel Dihydroartemisinin-Coumarin Hybrids via Click Chemistry. Molecules 2016, 21, 758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Hou, Z.; Tian, Y.; Mou, Y.; Guo, C. Design, synthesis, cytotoxicity and mechanism of novel dihydroartemisinin-coumarin hybrids as potential anti-cancer agents. Eur. J. Med. Chem. 2018, 151, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kong, L.; Liu, H.; Zhang, Y.; Zhang, L.; Liu, X.; Yuan, F.; Li, Y.; Zuo, Z. Design and synthesis of novel artemisinin derivatives with potent activities against colorectal cancer in vitro and in vivo. Eur. J. Med. Chem. 2019, 182, 111665. [Google Scholar] [CrossRef] [PubMed]
- An, R.; Lin, B.; Zhao, S.; Cao, C.; Wang, Y.; Cheng, X.; Liu, Y.; Guo, M.; Xu, H.; Wang, Y.; et al. Discovery of novel artemisinin-sulfonamide hybrids as potential carbonic anhydrase IX inhibitors with improved antiproliferative activities. Bioorg. Chem. 2020, 104, 104347. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, Y.; Liu, Y.; Zhang, X.; Chen, T.; Chen, K.; Ba, Q.; Li, J.; Liu, H.; Wang, H. Preclinical Efficacy and Safety Assessment of Artemisinin-Chemotherapeutic Agent Conjugates for Ovarian Cancer. EBioMedicine 2016, 14, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Zhai, X.; Ren, L.; Meng, H.; Liu, H.; Zhu, W.; Zhao, Y. Design, synthesis and antitumor activity of novel artemisinin derivatives using hybrid approach. Chem. Pharm. Bull. 2011, 59, 984–990. [Google Scholar] [CrossRef] [Green Version]
- Tien, D.D.; Giang, L.N.T.; Anh, D.T.T.; Dung, N.T.; Ha, T.N.; Ha, N.T.T.H.; Phuong, H.T.; Chinh, P.T.; Kiem, P.; Tuyen, N.V. Synthesis and Cytotoxic Evaluation of Artenisinin-triazole Hybrids. Nat. Prod. Commun. 2016, 11, 1789–1792. [Google Scholar] [PubMed]
- Ma, G.T.; Lee, S.K.; Park, K.-K.; Park, J.; Son, S.H.; Jung, M.; Chung, W.-Y. Artemisinin-Daumone Hybrid Inhibits Cancer Cell-Mediated Osteolysis by Targeting Cancer Cells and Osteoclasts. Cell. Physiol. Biochem. 2018, 49, 1460–1475. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, T.; Mai, C.; Bogautdinov, R.P.; Morozkina, S.N.; Shavva, A.G.; Friedrich, O.; Gilbert, D.F.; Tsogoeva, S.B. Synthesis of Tamoxifen-Artemisinin and Estrogen- Artemisinin Hybrids Highly Potent Against Breast and Prostate Cancer. ChemMedChem 2020, 15, 1473–1479. [Google Scholar] [CrossRef]
- Botta, L.; Filippi, S.; Bizzarri, B.M.; Zippilli, C.; Meschini, R.; Pogni, R.; Baratto, M.C.; Villanova, L.; Saladino, R. Synthesis and Evaluation of Artemisinin-Based Hybrid and Dimer Derivatives as Antimelanoma Agents. ACS Omega 2019, 5, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Gruber, L.; Abdelfatah, S.; Fröhlich, T.; Reiter, C.; Klein, V.; Tsogoeva, S.B.; Efferth, T. Thymoquinone-Derived Hybrid Compounds. Molecules 2018, 23, 841. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Yu, Z.; Yang, X.; Hu, P.; He, Y. Synthesis of novel ring-contracted artemisinin dimers with potent anticancer activities. Eur. J. Med. Chem. 2018, 150, 829–840. [Google Scholar] [CrossRef]
- Letis, A.S.; Seo, E.; Nikolaropoulos, S.S.; Efferth, T.; Giannis, A.; Fousteris, M.A. Synthesis and cytotoxic activity of new artemisinin hybrid molecules against human leukemia cells. Bioorg. Med. Chem. 2017, 25, 3357–3367. [Google Scholar] [CrossRef]
- Xie, L.; Zhao, Y.; Zhai, X.; Li, P.; Liu, C.; Li, Y.; Gong, P. The Application of Tandem Aza-Wittig Reaction to Synthesize Artemisinin—Guanidine Hybrids and Their Anti-Tumor Activity. Arch. Pharm. 2011, 344, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.; Kim, M.; Chung, W.; Park, K.; Jung, M. Discovery of Artemisinin-Glycolipid Hybrids as Anti-oral Cancer Agents. Chem. Pharm. Bull. 2011, 59, 1471–1475. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wu, J.; Xu, T.; Yao, C.; Yu, B.; Liu, J. Synthesis and cytotoxicity of novel artemisinin derivatives containing sulfur atoms. Eur. J. Med. Chem. 2016, 123, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Mercer, A.E.; Stocks, P.A.; La Pensee, L.J.I.; Cosstick, R.; Park, B.K.; Kennedy, M.E.; Piantanida, I.; Ward, S.A.; Davies, J.; et al. Antitumour and antimalarial activity of artemisinin—Acridine hybrids. Bioorg. Med. Chem. Lett. 2009, 19, 2033–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joubert, J.P.; Smit, F.J.; du Plessis, L.; Smith, P.J.; N’Da, D.D. Synthesis and in vitro biological evaluation of aminoacridines and artemisinin–acridine hybrids. Eur. J. Pharm. Sci. 2014, 56, 16–27. [Google Scholar] [CrossRef]
- Frohlich, T.; Reiter, C.; Saeed, M.E.M.; Hutterer, C.; Hahn, F.; Leidenberger, M.; Friedrich, O.; Kappes, B.; Marschall, M.; Thomas, E.; et al. Synthesis of Thymoquinone—Artemisinin Hybrids: New Potent. ACS Med. Chem. Lett. 2018, 9, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, T.; Reiter, C.; Ibrahim, M.M.; Beutel, J.; Hutterer, C.; Zeittra, I.; Bahsi, H.; Leidenberger, M.; Friedrich, O.; Kappes, B.; et al. Synthesis of Novel Hybrids of Quinazoline and Artemisinin with High Activities against Plasmodium falciparum, Human Cytomegalovirus, and Leukemia Cells. ACS Omega 2017, 2, 2422–2431. [Google Scholar] [CrossRef] [Green Version]
- Reiter, C.; Fröhlich, T.; Gruber, L.; Hutterer, C.; Marschall, M.; Voigtländer, C.; Friedrich, O.; Kappes, B.; Efferth, T.; Tsogoeva, S.B. Highly potent artemisinin-derived dimers and trimers: Synthesis and evaluation of their antimalarial, antileukemia and antiviral activities. Bioorg. Med. Chem. 2015, 23, 5452–5458. [Google Scholar] [CrossRef]
- Lombard, M.C.; N’Da, D.D.; Breytenbach, J.C.; Kolesnikova, N.I.; Van Ba, C.T.; Wein, S.; Norman, J.; Denti, P.; Vial, H.; Wiesner, L. Antimalarial and anticancer activities of artemisinin-quinoline hybrid-dimers and pharmacokinetic properties in mice. Eur. J. Pharm. Sci. 2012, 47, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Lombard, M.C.; N’Da, D.D.; Ba, C.T.; Van, W.S.; Norman, J. Potent in vivo anti-malarial activity and representative snapshot pharmacokinetic evaluation of artemisinin-quinoline hybrids. Malar. J. 2013, 12, 71. [Google Scholar] [CrossRef] [Green Version]
- Lombard, M.C.; N’Da, D.D.; Breytenbach, J.C.; Smith, P.J.; Lategan, C.A. Synthesis, in vitro antimalarial and cytotoxicity of artemisinin-aminoquinoline hybrids. Bioorg. Med. Chem. Lett. 2011, 21, 1683–1686. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.S.; Guantai, E.M.; Nell, M.; van Rensburg, C.E.; Ncokazi, K.; Egan, T.J.; Hoppe, H.C.; Chibale, K. Effects of highly active novel artemisinin-chloroquinoline hybrid compounds on β-hematin formation, parasite morphology and endocytosis in Plasmodium falciparum. Biochem. Pharmacol. 2011, 82, 236–247. [Google Scholar] [CrossRef]
- Capela, R.; Cabal, G.G.; Rosenthal, P.J.; Gut, J.; Mota, M.M.; Moreira, R.; Lopes, F.; Prudêncio, M. Design and Evaluation of Primaquine-Artemisinin Hybrids as a Multistage Antimalarial Strategy. Antimicrob. Agents Chemother. 2011, 55, 4698–4706. [Google Scholar] [CrossRef]
- Wang, N.; Wicht, K.; Shaban, E.; Ngoc, T.A.; Wang, M.; Hayashi, I.; Hossain, M.I.; Takemasa, Y.; Kaiser, M.; Sayed, I.; et al. Synthesis and evaluation of artesunate—Indoloquinoline hybrids as antimalarial drug candidates. MedChemComm 2014, 5, 927–931. [Google Scholar] [CrossRef]
- Çapcı, A.; Lorion, M.M.; Wang, H.; Simon, N.; Leidenberger, M.; Borges Silva, M.C.; Moreira, D.R.; Zhu, Y.; Meng, Y.; Chen, J.Y.; et al. Artemisinin—(Iso) quinoline Hybrids by C− H Activation and Click Chemistry: Combating Multidrug-Resistant Malaria. Angew. Chem. Int. Ed. 2019, 58, 13066–13079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, J.J.; Coughlan, D.; Heneghan, N.; Bell, A. A novel artemisinin—Quinine hybrid with potent antimalarial activity. Bioorg. Med. Chem. Lett. 2007, 17, 3599–3602. [Google Scholar] [CrossRef]
- De Lange, C.; Coertzen, D.; Smit, F.J.; Wentzel, J.F.; Wong, H.; Birkholtz, L.; Haynes, R.K.; N’Da, D.D. Synthesis, antimalarial activities and cytotoxicities of amino-artemisinin-1,2-disubstituted ferrocene hybrids. Bioorg. Med. Chem. 2018, 19, 3161–3163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloete, T.T.; Kock, C.D.; Smith, P.J.; N’Da, D.D. Synthesis, in vitro antiplasmodial activity and cytotoxicity of a series of artemisinin e triazine hybrids and hybrid-dimers. Eur. J. Med. Chem. 2014, 76, 470–481. [Google Scholar] [CrossRef] [PubMed]
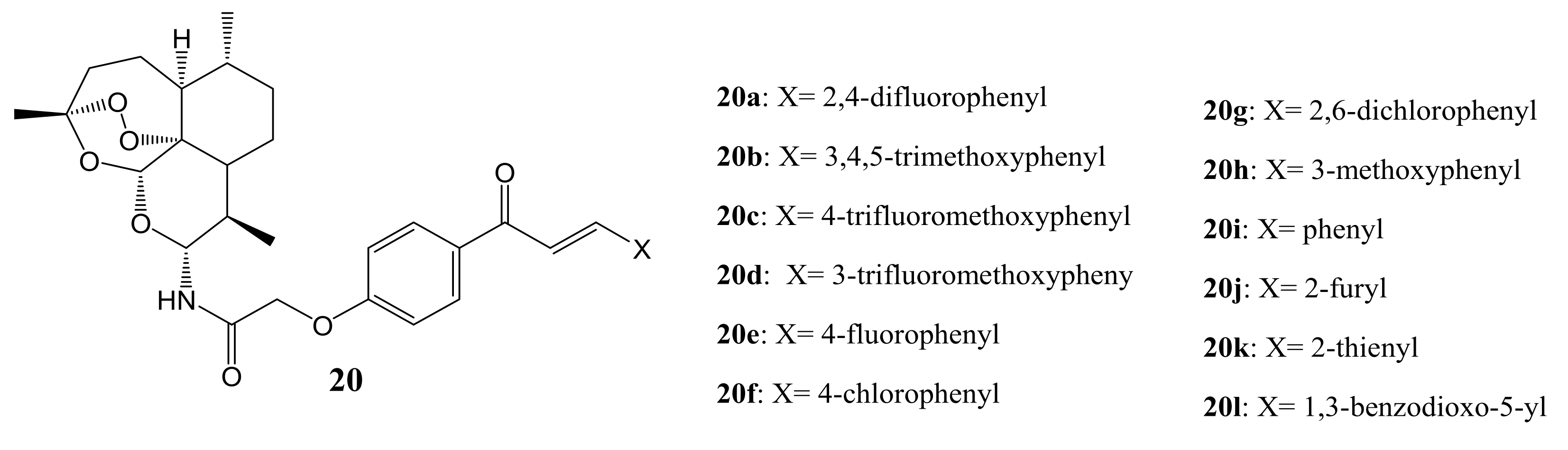
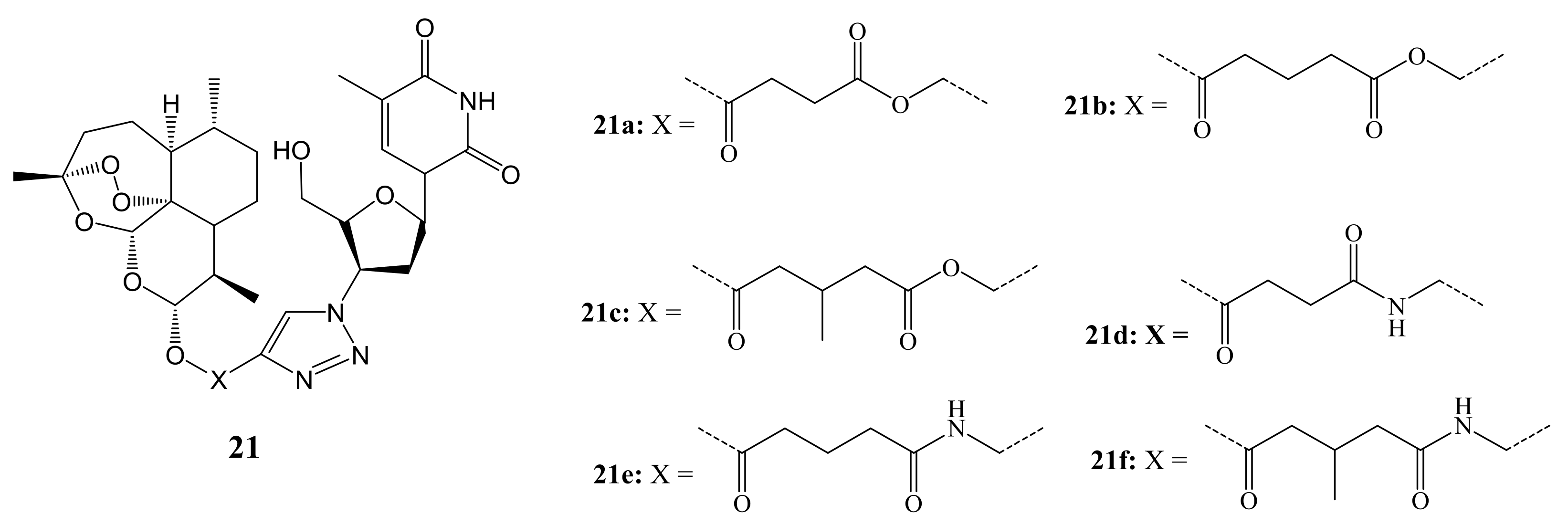
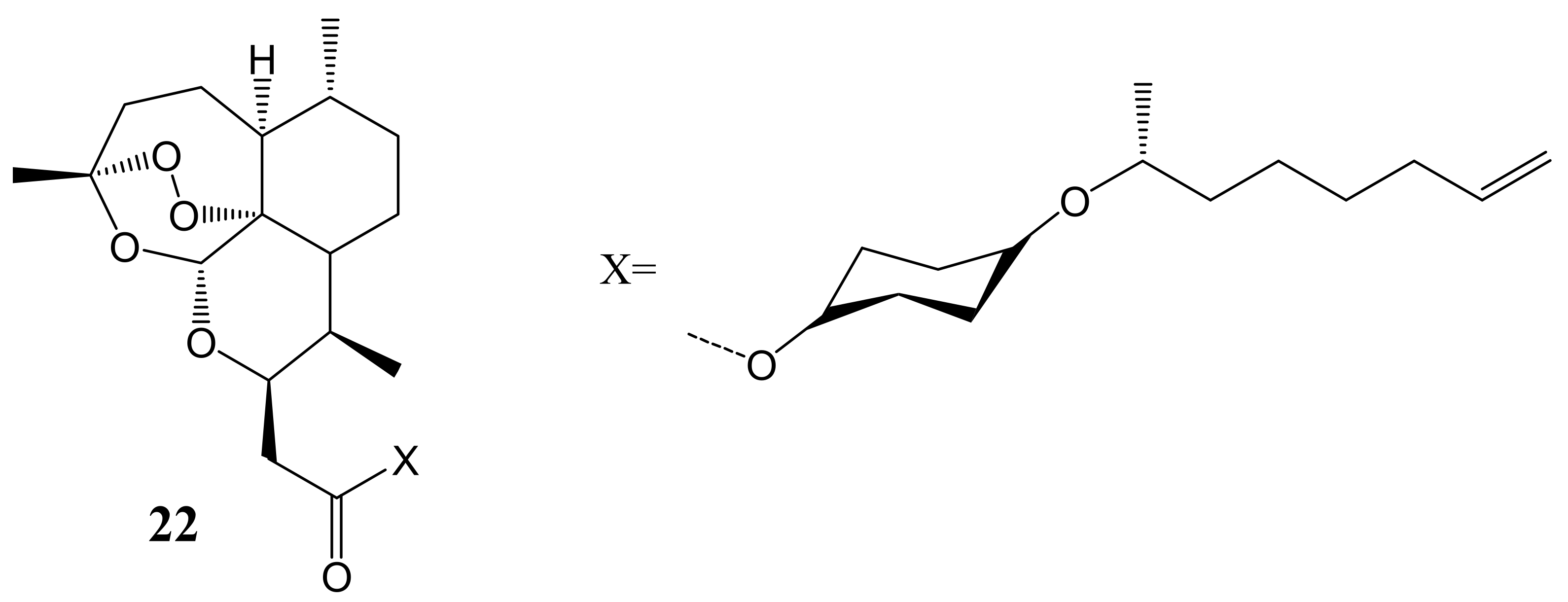
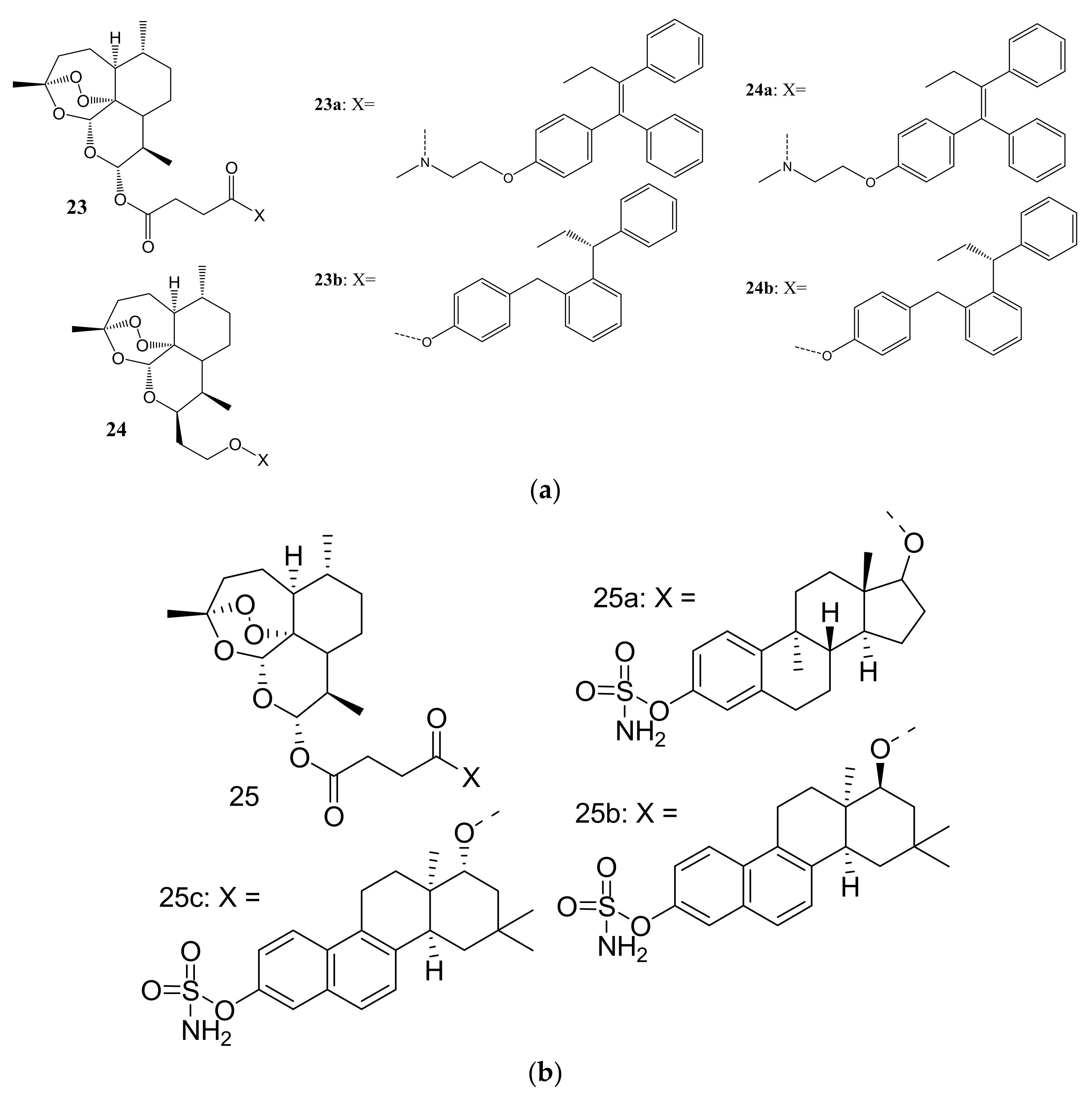
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peter, S.; Jama, S.; Alven, S.; Aderibigbe, B.A. Artemisinin and Derivatives-Based Hybrid Compounds: Promising Therapeutics for the Treatment of Cancer and Malaria. Molecules 2021, 26, 7521. https://doi.org/10.3390/molecules26247521
Peter S, Jama S, Alven S, Aderibigbe BA. Artemisinin and Derivatives-Based Hybrid Compounds: Promising Therapeutics for the Treatment of Cancer and Malaria. Molecules. 2021; 26(24):7521. https://doi.org/10.3390/molecules26247521
Chicago/Turabian StylePeter, Sijongesonke, Siphesihle Jama, Sibusiso Alven, and Blessing A. Aderibigbe. 2021. "Artemisinin and Derivatives-Based Hybrid Compounds: Promising Therapeutics for the Treatment of Cancer and Malaria" Molecules 26, no. 24: 7521. https://doi.org/10.3390/molecules26247521
APA StylePeter, S., Jama, S., Alven, S., & Aderibigbe, B. A. (2021). Artemisinin and Derivatives-Based Hybrid Compounds: Promising Therapeutics for the Treatment of Cancer and Malaria. Molecules, 26(24), 7521. https://doi.org/10.3390/molecules26247521