
Enriching the Arsenal of Pharmacological Tools against MICAL2

 ,
,  ,
,  , , ,
, , , 
Abstract
:
1. Introduction
2. Results and Discussion
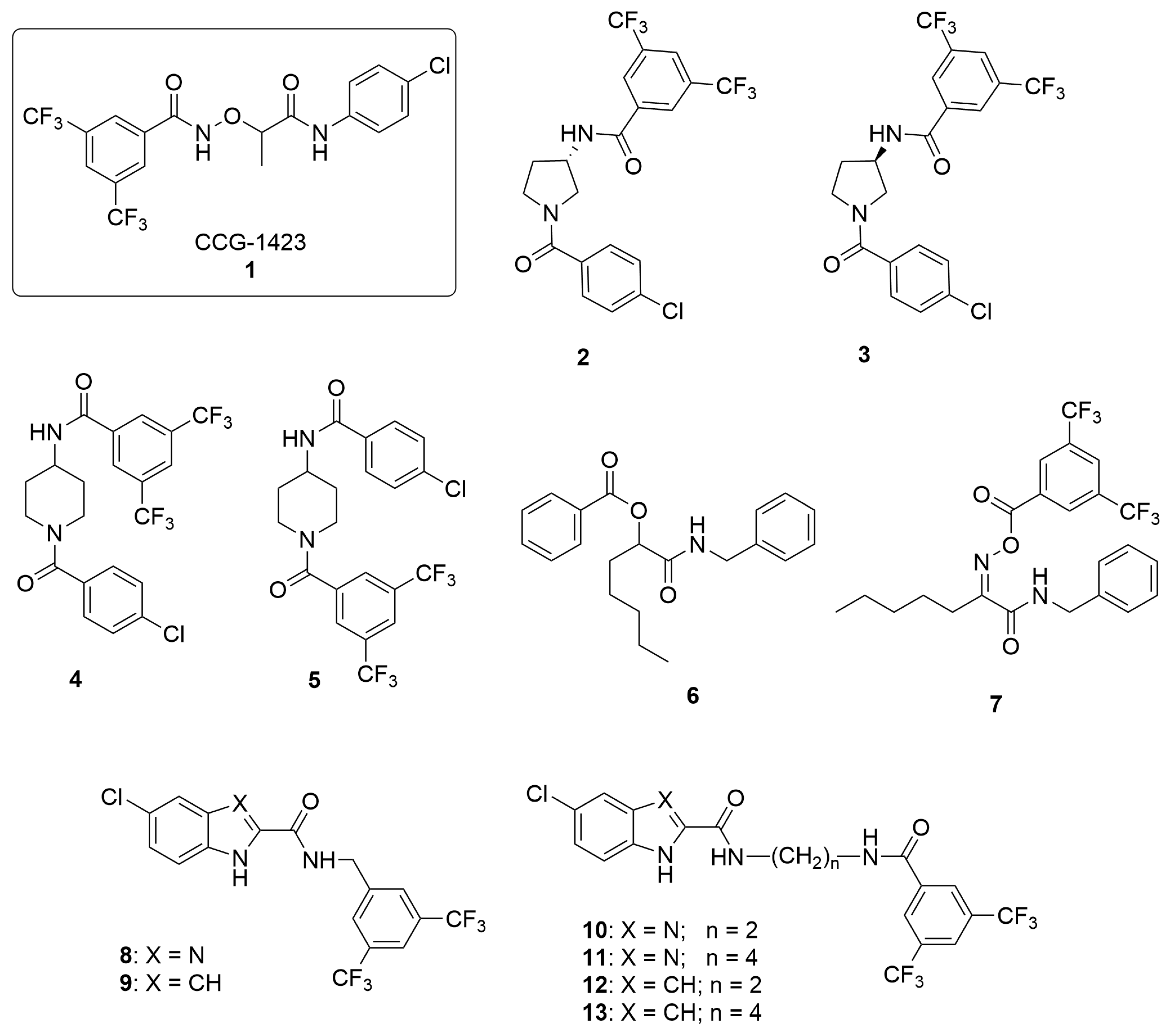
2.1. Design and Synthesis
2.2. Protein Expression
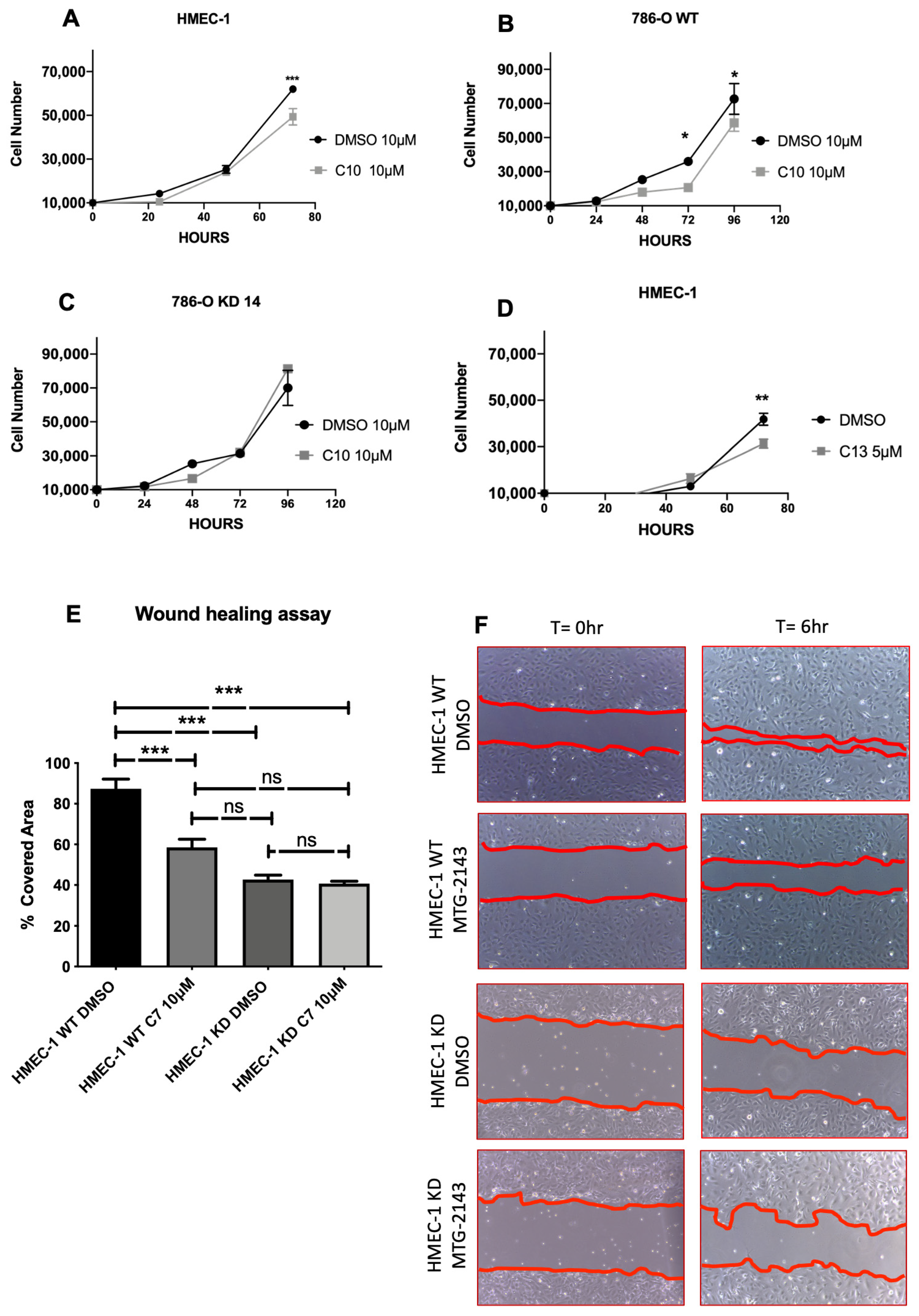
2.3. Biological Assays
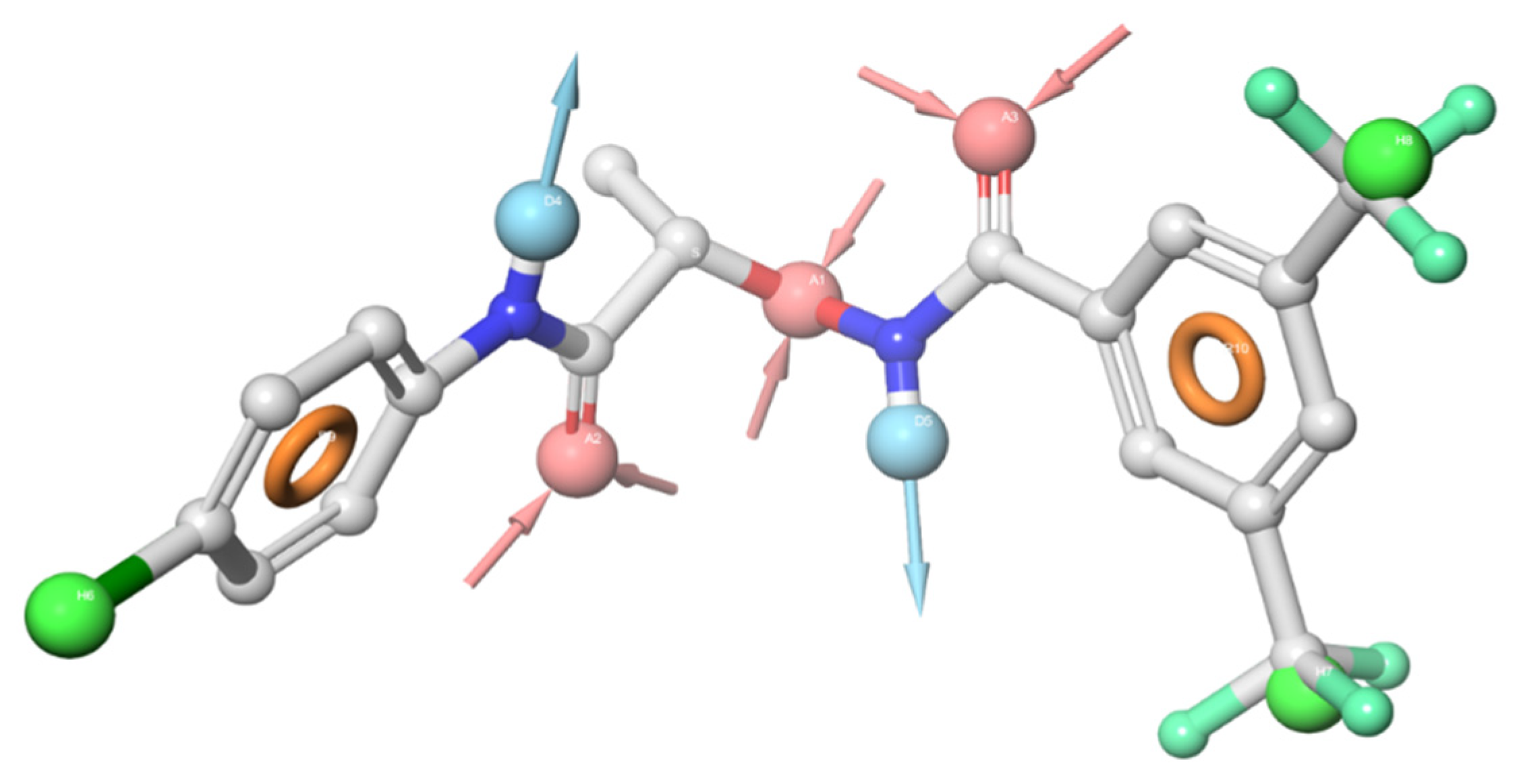
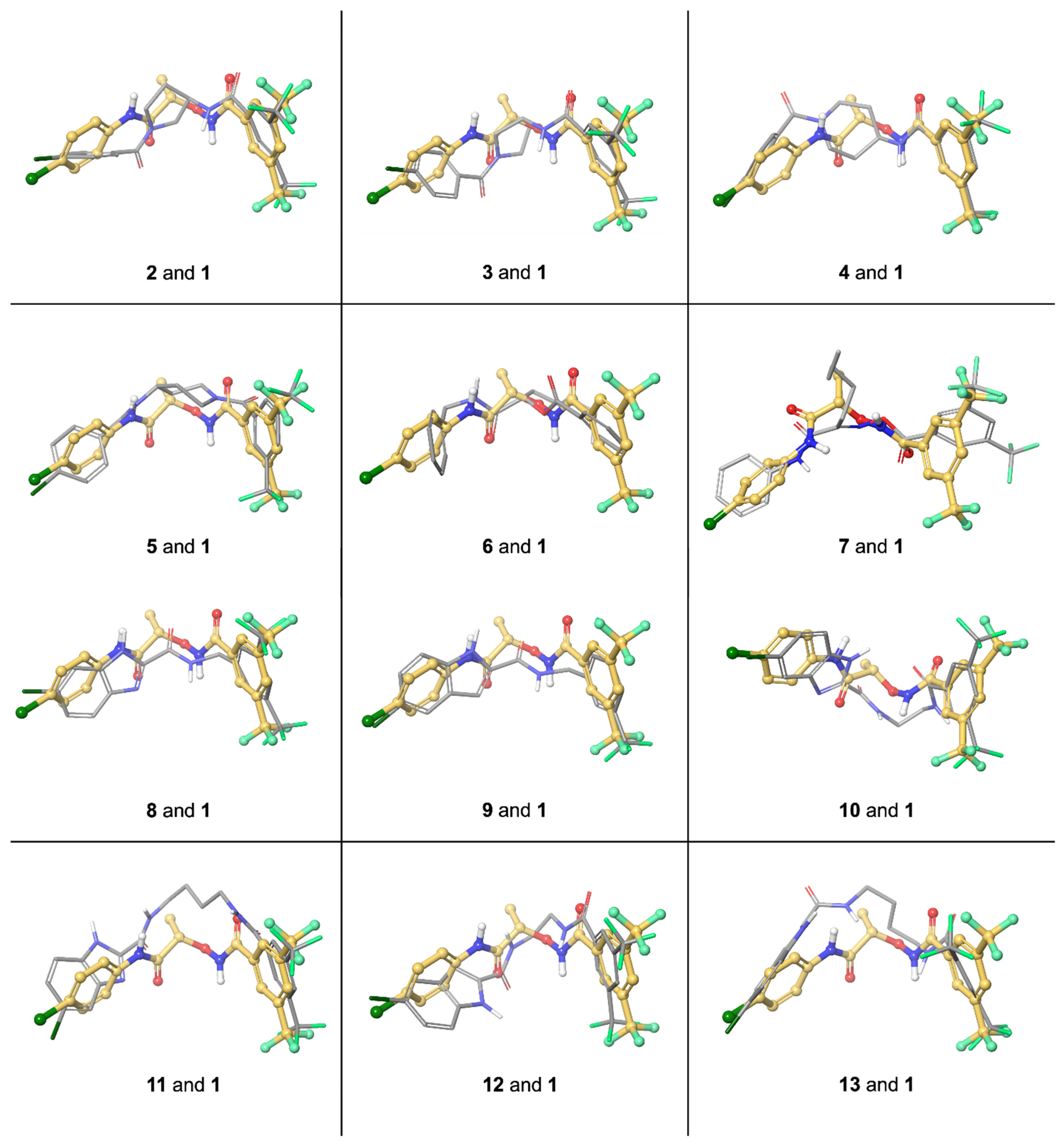
3. Molecular Modeling
4. Discussion and Conclusions
5. Materials and Methods
5.1. Chemistry and General Methods
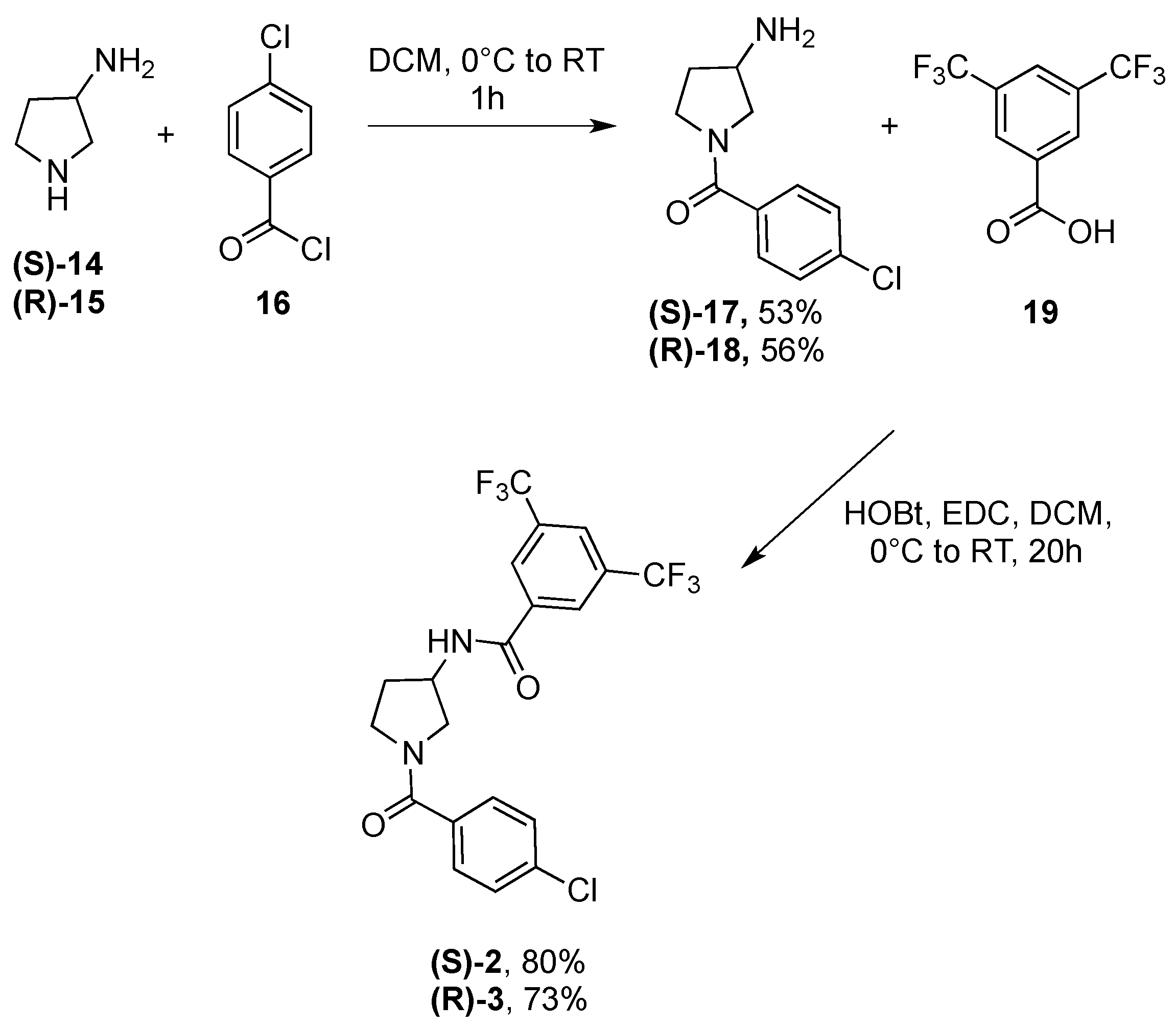
5.1.1. General Procedure for the Synthesis of Compounds 2, 3
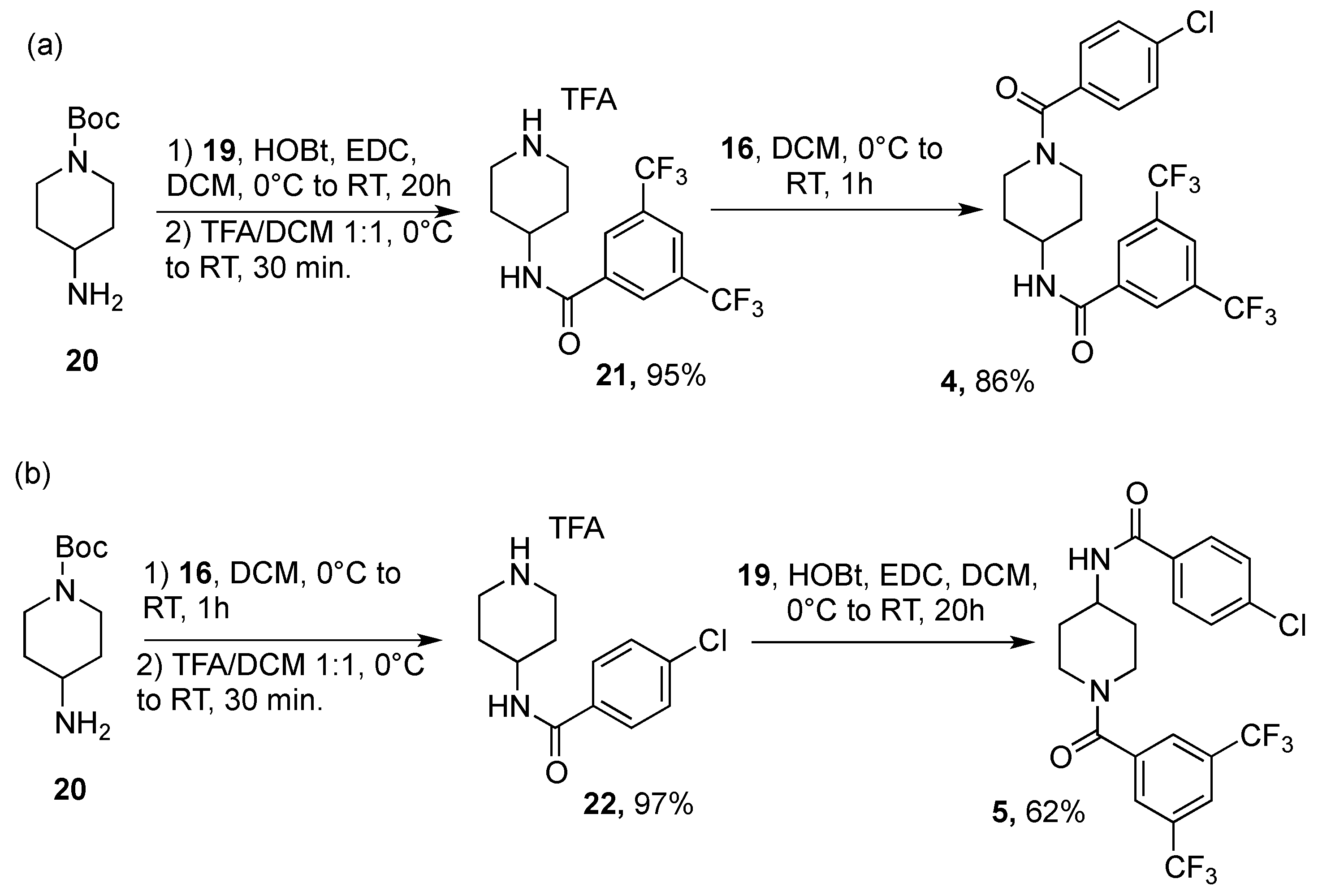
5.1.2. Synthesis of Compound 4
5.1.3. Synthesis of Compound 5
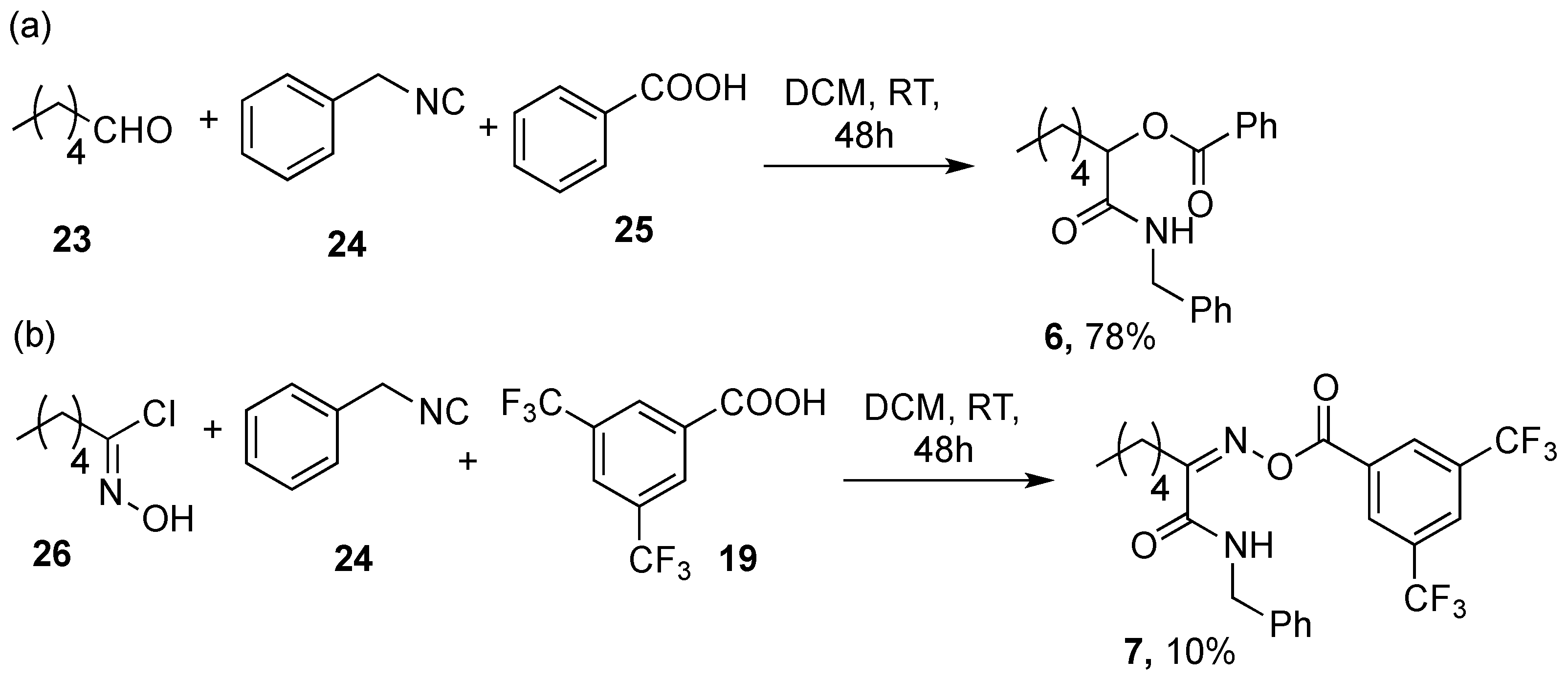
5.1.4. Synthesis of Compound 6
5.1.5. Synthesis of Compound 7
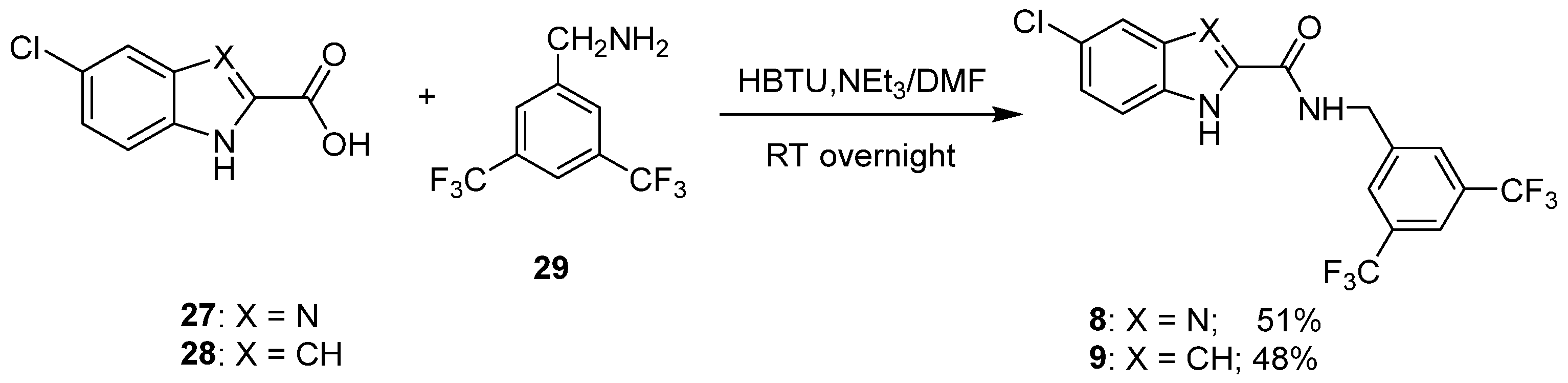
5.1.6. General Procedure for the Synthesis of Compounds 8 and 9
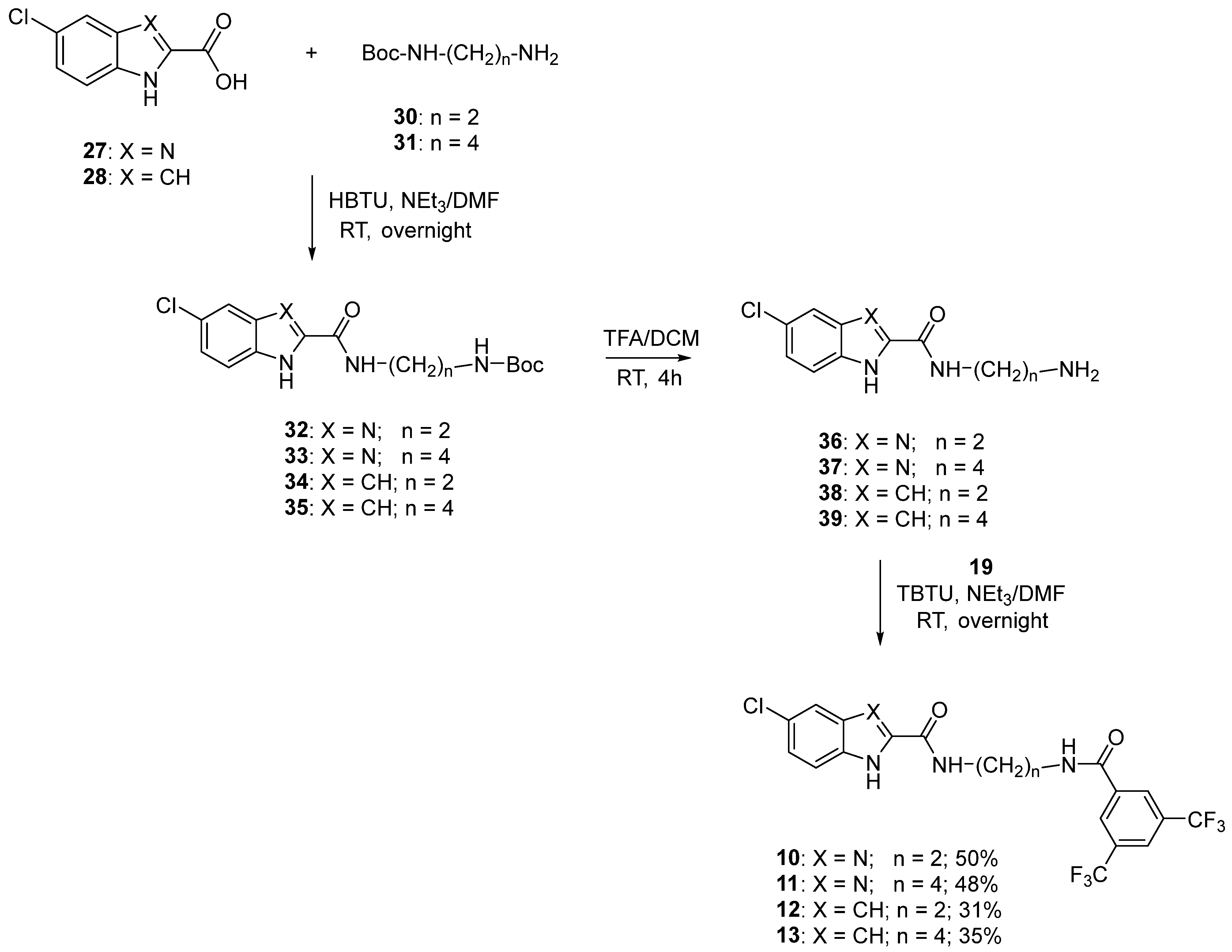
5.1.7. General Procedure for the Synthesis of Compounds 10–13
5.1.8. General Procedure for the Synthesis of Compounds 32–35
5.1.9. General Procedure for the Synthesis of Compounds 36–39
5.2. Cell Lines, Culture Procedure
5.2.1. Manual Counting
5.2.2. 2D Migration Assay (Wound Healing Assay, WHA)
5.3. Statistical Analysis
5.4. Molecular Modeling
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Terman, J.R.; Mao, T.; Pasterkamp, R.J.; Yu, H.H.; Kolodkin, A.L. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell 2002, 109, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Nakamoto, T.; Ogawa, S.; Seo, S.; Matsumura, T.; Tachibana, K.; Morimoto, C.; Hirai, H. MICAL, a Novel CasL Interacting Molecule, Associates with Vimentin. J. Biol. Chem. 2002, 277, 14933–14941. [Google Scholar] [CrossRef] [Green Version]
- Ashida, S.; Furihata, M.; Katagiri, T.; Tamura, K.; Anazawa, Y.; Yoshioka, H.; Miki, T.; Fujioka, T.; Shuin, T.; Nakamura, Y.; et al. Expression of Novel Molecules, MICAL2-PV (MICAL2 Prostate Cancer Variants), Increases with High Gleason Score and Prostate Cancer Progression. Clin. Cancer Res. 2006, 12, 2767–2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadella, M.; Bianchet, M.; Gabelli, S.; Barrila, J.; Amzel, L.M. Structure and activity of the axon guidance protein MICAL. Proc. Natl. Acad. Sci. USA 2005, 102, 16830–16835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebold, C.; Berrow, N.; Walter, T.; Harlos, K.; Owens, R.; Stuart, D.; Terman, J.R.; Kolodkin, A.L.; Pasterkamp, J.; Jones, Y. High-resolution structure of the catalytic region of MICAL (molecule interacting with CasL), a multidomain flavoenzyme-signaling molecule. Proc. Natl. Acad. Sci. USA 2005, 102, 16836–16841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundquist, M.R.; Storaska, A.J.; Liu, T.-C.; Larsen, S.D.; Evans, T.; Neubig, R.R.; Jaffrey, S.R. Redox Modification of Nuclear Actin by MICAL-2 Regulates SRF Signaling. Cell 2014, 156, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Mariotti, S.; Barravecchia, I.; Vindigni, C.; Pucci, A.; Balsamo, M.; Libro, R.; Senchenko, V.; Dmitriev, A.; Jacchetti, E.; Cecchini, M.; et al. MICAL2 is a novel human cancer gene controlling mesenchymal to epithelial transition involved in cancer growth and invasion. Oncotarget 2015, 7, 1808–1825. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.R.; Chapeaublanc, E.; Kirkwood, L.; Nicolle, R.; Benhamou, S.; Lebret, T.; Allory, Y.; Southgate, J.; Radvanyi, F.; Goud, B. Deregulation of Rab and Rab Effector Genes in Bladder Cancer. PLoS ONE 2012, 7, e39469. [Google Scholar] [CrossRef] [PubMed]
- Hellweg, R.; Mooneyham, A.; Chang, Z.; Shetty, M.; Emmings, E.; Iizuka, Y.; Clark, C.; Starr, T.; Abrahante, J.; Schütz, F.; et al. RNA Sequencing of Carboplatin- and Paclitaxel-Resistant Endometrial Cancer Cells Reveals New Stratification Markers and Molecular Targets for Cancer Treatment. Horm. Cancer 2018, 9, 326–337. [Google Scholar] [CrossRef]
- Expression of MICAL2 in Renal Cancer—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000116117-MICAL2/pathology/renal+cancer (accessed on 10 December 2021).
- Wang, Y.; Deng, W.; Zhang, Y.; Sun, S.; Zhao, S.; Chen, Y.; Zhao, X.; Liu, L.; Du, J. MICAL2 promotes breast cancer cell migration by maintaining epidermal growth factor receptor (EGFR) stability and EGFR/P38 signalling activation. Acta Physiol. 2018, 222, e12920. [Google Scholar] [CrossRef] [PubMed]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Vesely, P.; Heneberg, P.; Čermák, V.; Petruzelka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics—Anti-metastatic and Anti-invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef] [Green Version]
- Barravecchia, I.; Mariotti, S.; Pucci, A.; Scebba, F.; De Cesari, C.; Bicciato, S.; Tagliafico, E.; Tenedini, E.; Vindigni, C.; Cecchini, M.; et al. MICAL2 is expressed in cancer associated neo-angiogenic capillary endothelia and it is required for endothelial cell viability, motility and VEGF response. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 2111–2124. [Google Scholar] [CrossRef]
- Folkman, J.; Merler, E.; Abernathy, C.; Williams, G. ISOLATION OF A TUMOR FACTOR RESPONSIBLE FOR ANGIOGENESIS. J. Exp. Med. 1971, 133, 275–288. [Google Scholar] [CrossRef] [Green Version]
- Somchai, P.; Phongkitkarun, K.; Kueanjinda, P.; Jamnongsong, S.; Vaeteewoottacharn, K.; Luvira, V.; Okada, S.; Jirawatnotai, S.; Sampattavanich, S. Novel Analytical Platform For Robust Identification of Cell Migration Inhibitors. Sci. Rep. 2020, 10, 931. [Google Scholar] [CrossRef] [PubMed]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3. [Google Scholar] [CrossRef] [PubMed]
- Dömling, A.; Ugi, I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. Engl. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and Biology Of Multicomponent Reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passerini, M.; Simone, L. Gazz. Chim. Ital. 1921, 51, 126–129.
- Zhu, Y.; Wen, S.; Yin, G.; Hong, D.; Lu, P.; Wang, Y. Tandem Reaction of Propargyl Alcohol and N-Sulfonylhydrazone: Synthesis of Dihydropyrazole and Its Utility in the Preparation of 3,3-Diarylacrylonitrile. Org. Lett. 2011, 13, 3553–3555. [Google Scholar] [CrossRef]
- Wong, P.P.; Bodrug, N.; Hodivala-Dilke, K.M. Exploring Novel Methods for Modulating Tumor Blood Vessels in Cancer Treatment. Curr. Biol. 2016, 26, R1161–R1166. [Google Scholar] [CrossRef]
- Laduron, F.; Tamborowski, V.; Moens, L.; Horvath, A.; De Smaele, D.; Leurs, S. Efficient and Scalable Method for the Selective Alkylation and Acylation of Secondary Amines in the Presence of Primary Amines. Org. Process Res. Dev. 2005, 9, 102–104. [Google Scholar] [CrossRef]
- Ades, E.W.; Candal, F.J.; A Swerlick, R.; George, V.G.; Summers, S.; Bosse, D.C.; Lawley, T.J. HMEC-1: Establishment of an Immortalized Human Microvascular Endothelial Cell Line. J. Investig. Dermatol. 1992, 99, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.D.; Elliott, A.Y.; Stein, N.; Fraley, E.E. In vitro cultivation of human renal cell cancer. Vitr. Cell. Dev. Biol. Anim. 1978, 14, 779–786. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| N° conformers | 15 | 15 | 15 | 47 | 21 | 9 | 30 | 2 | 9 | 8 |
| Average Energy in kJ/mol | 22.7 | 22.53 | 20.7 | 17.22 | 16.191 | 15.95 | 20.99 | 24.91 | 20.9 | 20.71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barravecchia, I.; Barresi, E.; Russo, C.; Scebba, F.; De Cesari, C.; Mignucci, V.; De Luca, D.; Salerno, S.; La Pietra, V.; Giustiniano, M.; et al. Enriching the Arsenal of Pharmacological Tools against MICAL2. Molecules 2021, 26, 7519. https://doi.org/10.3390/molecules26247519
Barravecchia I, Barresi E, Russo C, Scebba F, De Cesari C, Mignucci V, De Luca D, Salerno S, La Pietra V, Giustiniano M, et al. Enriching the Arsenal of Pharmacological Tools against MICAL2. Molecules. 2021; 26(24):7519. https://doi.org/10.3390/molecules26247519
Chicago/Turabian StyleBarravecchia, Ivana, Elisabetta Barresi, Camilla Russo, Francesca Scebba, Chiara De Cesari, Valerio Mignucci, Davide De Luca, Silvia Salerno, Valeria La Pietra, Mariateresa Giustiniano, and et al. 2021. "Enriching the Arsenal of Pharmacological Tools against MICAL2" Molecules 26, no. 24: 7519. https://doi.org/10.3390/molecules26247519
APA StyleBarravecchia, I., Barresi, E., Russo, C., Scebba, F., De Cesari, C., Mignucci, V., De Luca, D., Salerno, S., La Pietra, V., Giustiniano, M., Pelliccia, S., Brancaccio, D., Donati, G., Da Settimo, F., Taliani, S., Angeloni, D., & Marinelli, L. (2021). Enriching the Arsenal of Pharmacological Tools against MICAL2. Molecules, 26(24), 7519. https://doi.org/10.3390/molecules26247519