
HIV-1 Tat and Heparan Sulfate Proteoglycans Orchestrate the Setup of in Cis and in Trans Cell-Surface Interactions Functional to Lymphocyte Trans-Endothelial Migration

 , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Experimental Studies
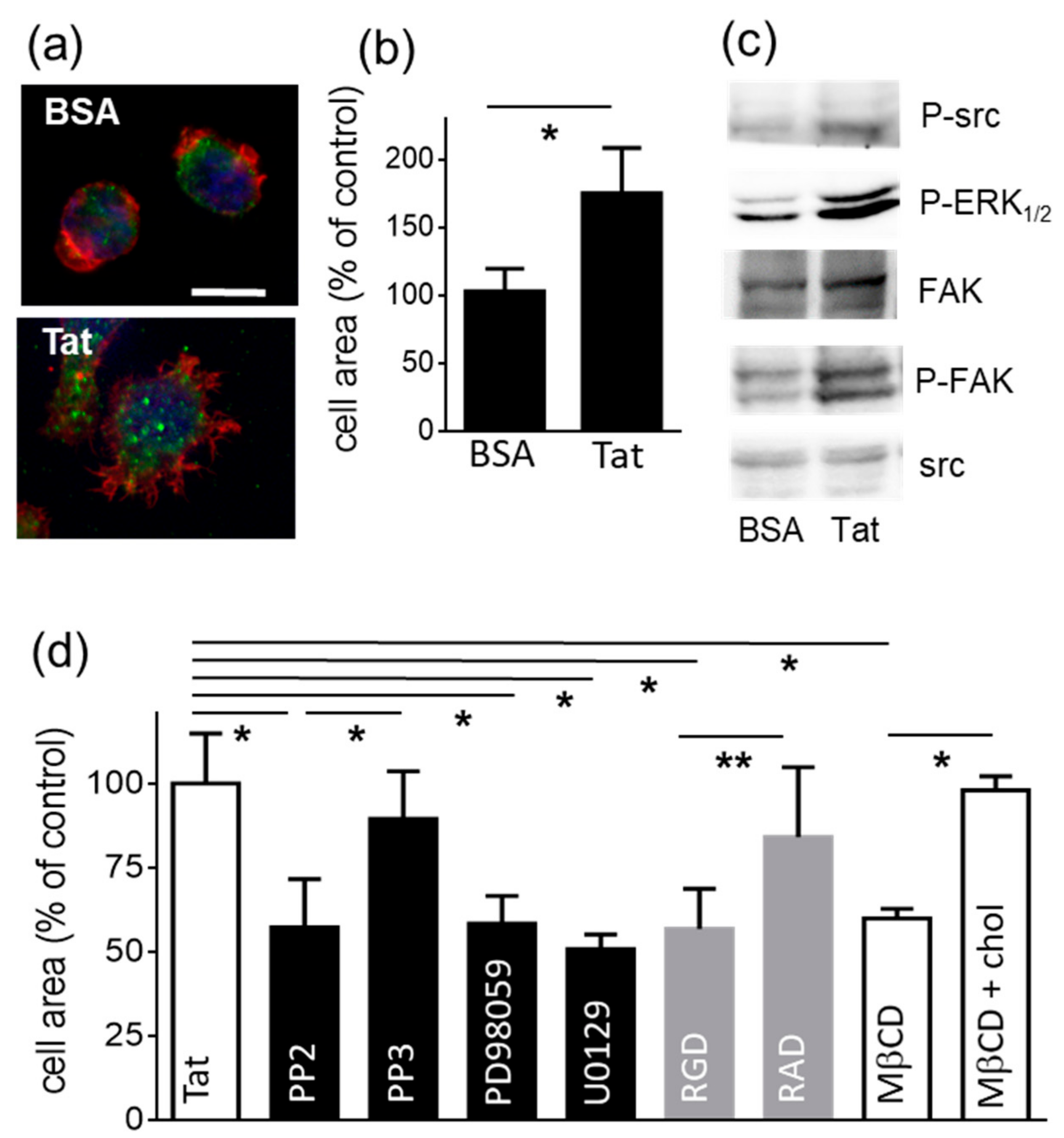
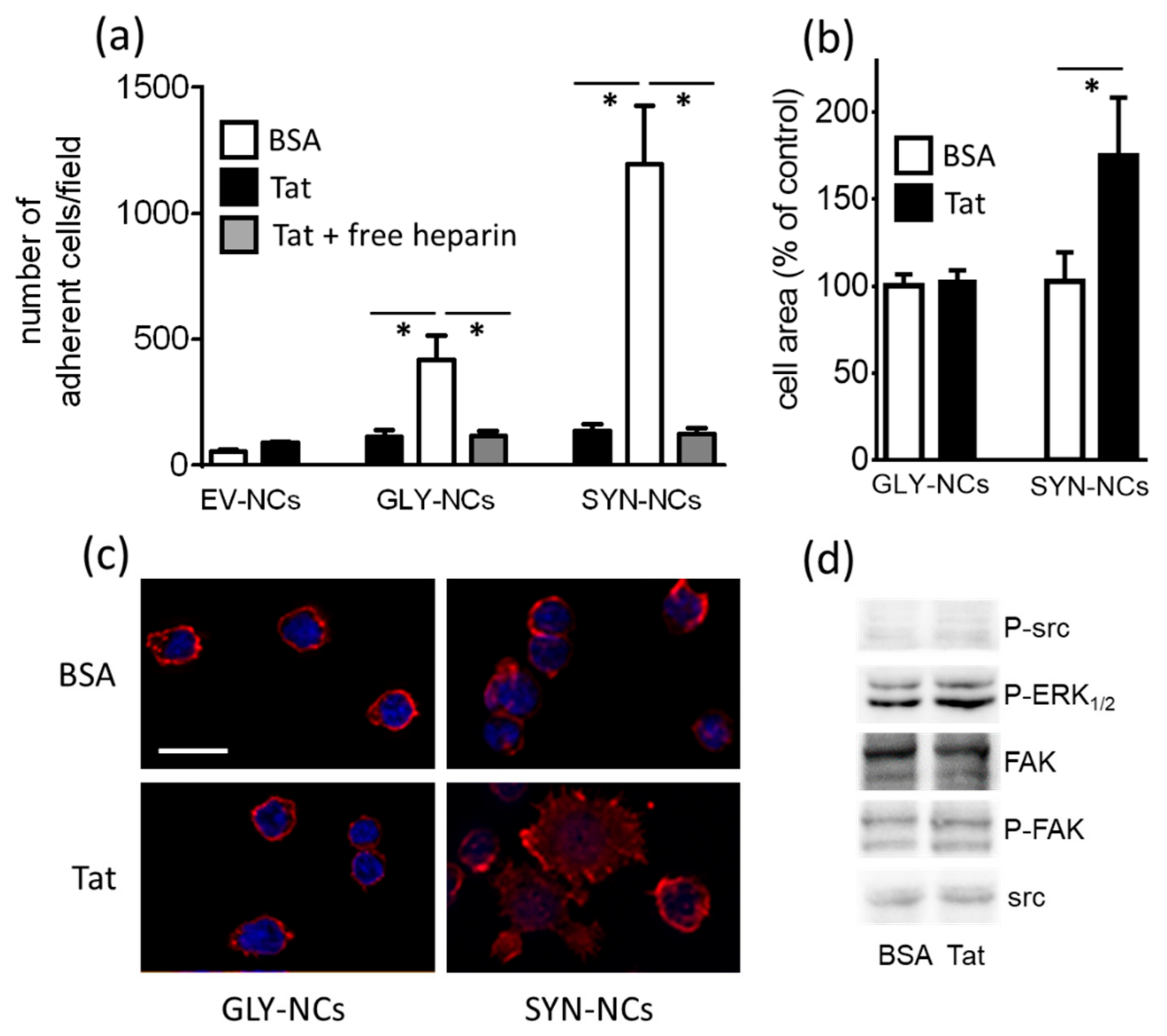
2.1.1. Lymphocyte Adhesion and Spreading onto Substrate-Immobilized Tat
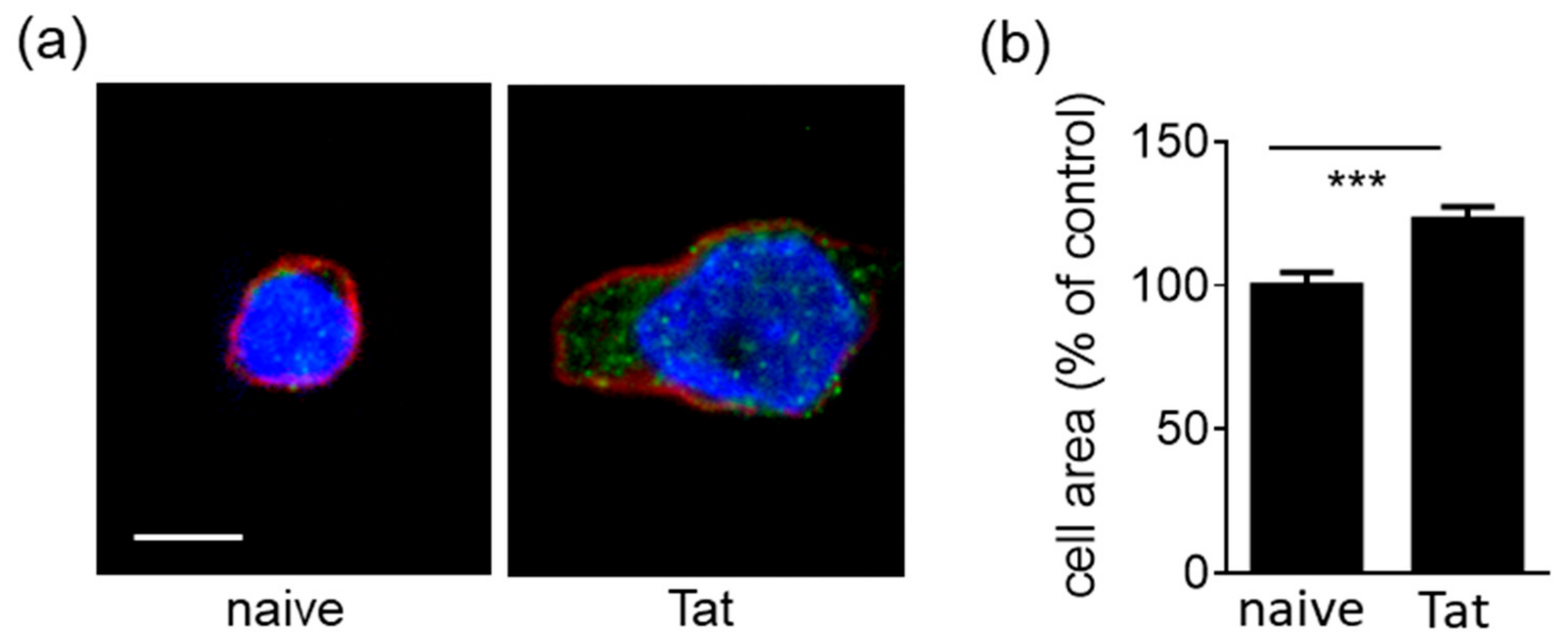
2.1.2. Tat-Presenting Lymphocytes Adhere and Spread onto the Endothelium
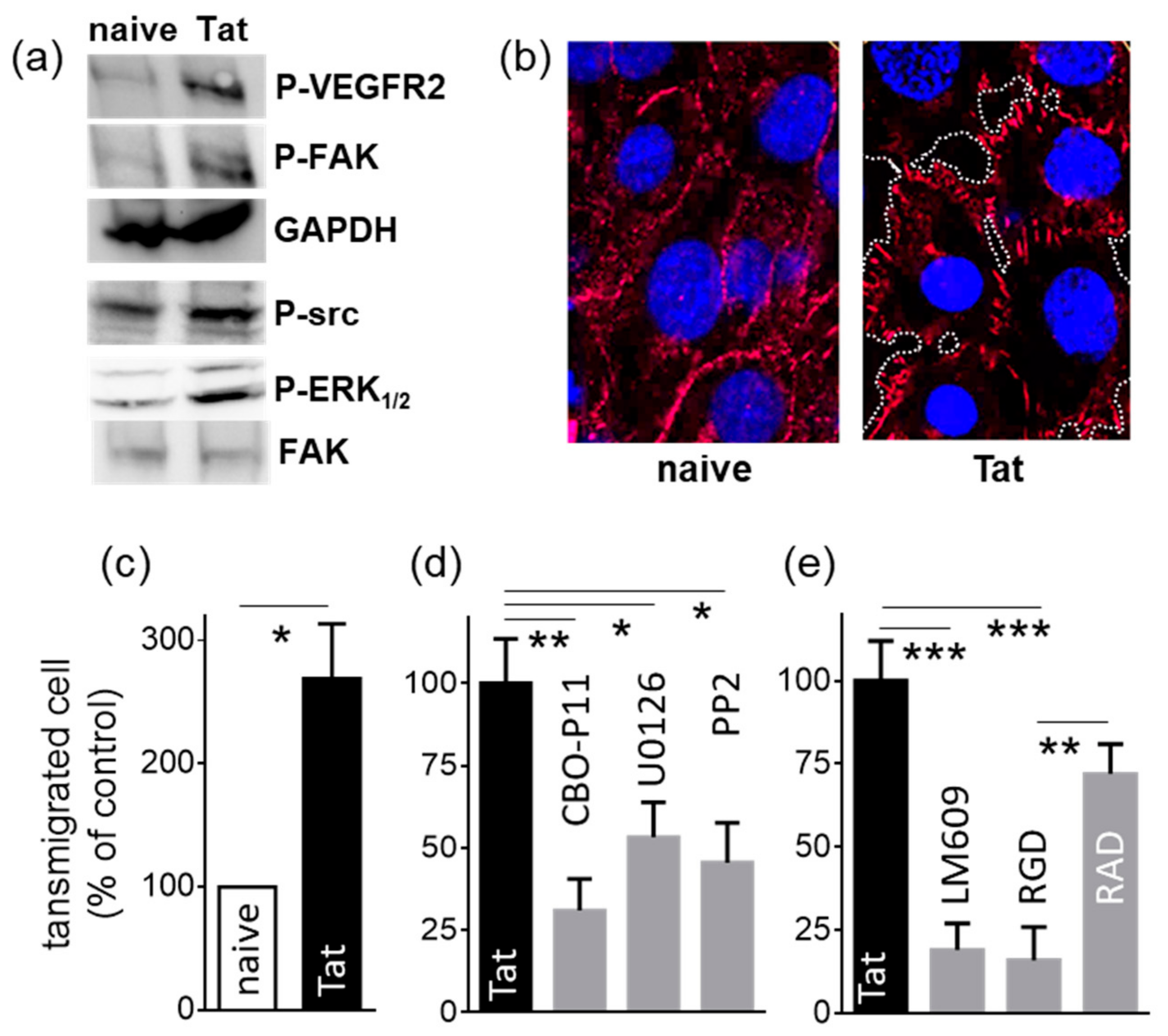
2.1.3. Tat-Presenting Lymphocytes Activate ECs
2.1.4. Tat-Presenting Lymphocytes Migrate across the Endothelium
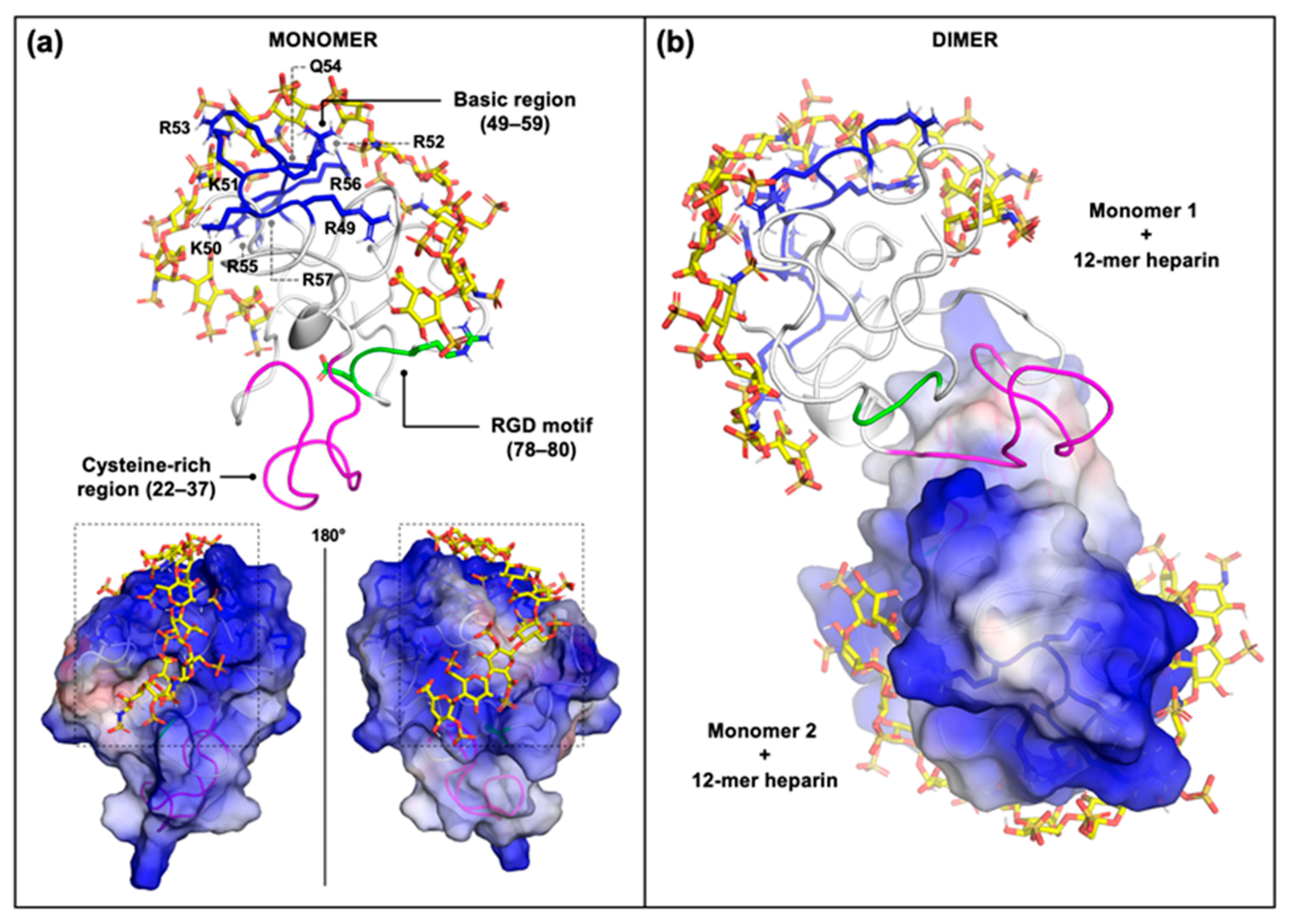
2.2. Computational Studies
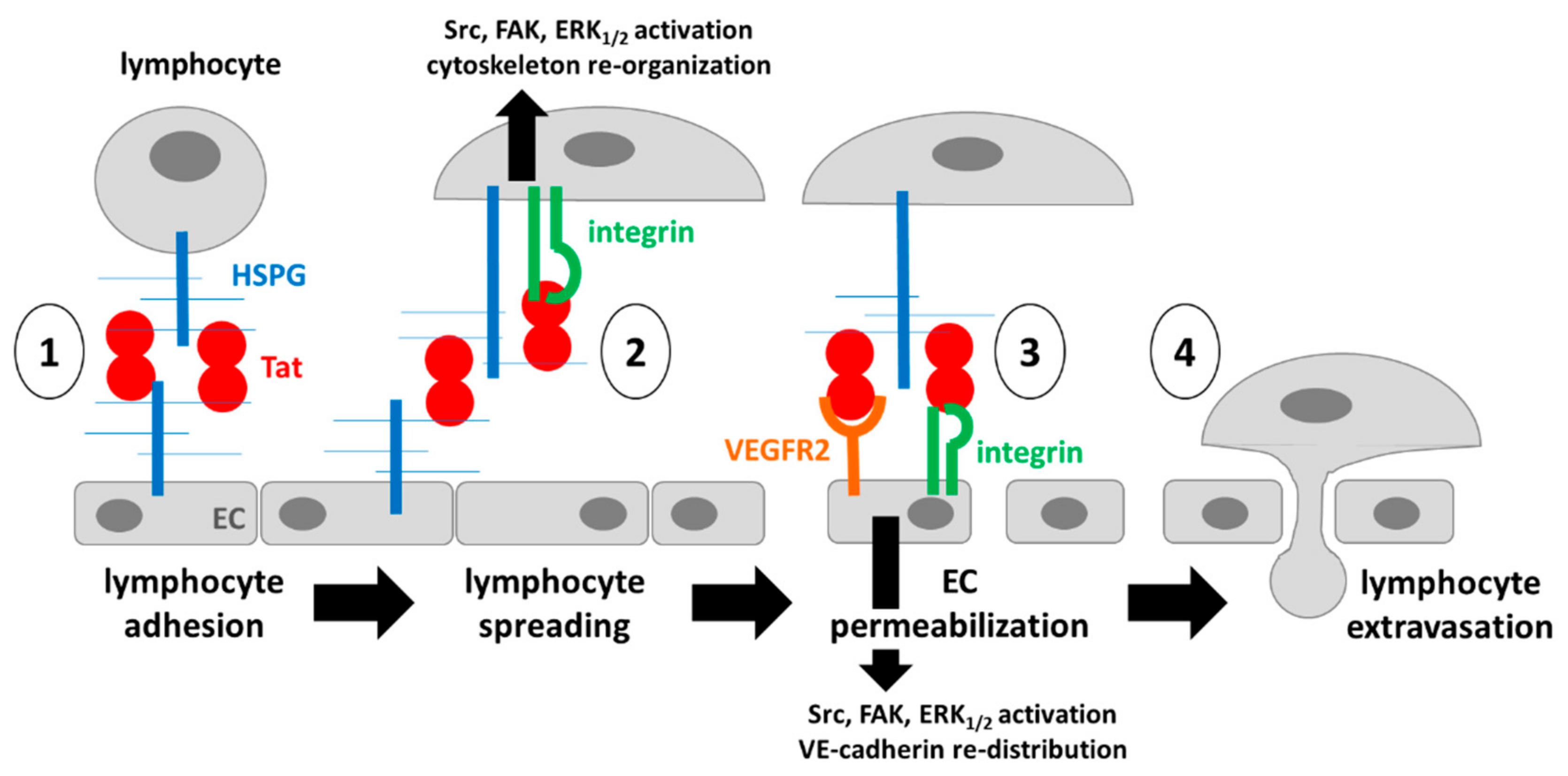
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rusnati, M.; Oreste, P.; Zoppetti, G.; Presta, M. Biotechnological Engineering of Heparin/Heparan Sulphate: A Novel Area of Multi-Target Drug Discovery. Curr. Pharm. Des. 2005, 11, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Gondelaud, F.; Ricard-Blum, S. Structures and interactions of syndecans. FEBS J. 2019, 286, 2994–3007. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Tiemann, M.; Schrader, C.; Janssen, D.; Wolf, E.; Vierbuchen, M.; Parwaresch, R.; Ernestus, K.; Plettenberg, A.; Stoehr, A.; et al. AIDS-related B-cell lymphoma (ARL): Correlation of prognosis with differentiation profiles assessed by immunophenotyping. Blood 2005, 106, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Reijmers, R.; Spaargaren, M.; Pals, S.T. Heparan sulfate proteoglycans in the control of B cell development and the pathogenesis of multiple myeloma. FEBS J. 2013, 280, 2180–2193. [Google Scholar] [CrossRef]
- Ajasin, D.; Eugenin, E.A. HIV-1 Tat: Role in Bystander Toxicity. Front. Cell. Infect. Microbiol. 2020, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Urbinati, C.; Nicoli, S.; Giacca, M.; David, G.; Fiorentini, S.; Caruso, A.; Alfano, M.; Cassetta, L.; Presta, M.; Rusnati, M. HIV-1 Tat and heparan sulfate proteoglycan interaction: A novel mechanism of lymphocyte adhesion and migration across the endothelium. Blood 2009, 114, 3335–3342. [Google Scholar] [CrossRef]
- Pierleoni, R.; Menotta, M.; Antonelli, A.; Sfara, C.; Serafini, G.; Dominici, S.; Laguardia, M.E.; Salis, A.; Damonte, G.; Banci, L.; et al. Effect of the redox state on HIV-1 tat protein multimerization and cell internalization and trafficking. Mol. Cell. Biochem. 2010, 345, 105–118. [Google Scholar] [CrossRef]
- Urbinati, C.E.; Ravelli, C.; Tanghetti, E.; Belleri, M.; Giacopuzzi, E.; Monti, E.; Presta, M.; Rusnati, M. Substrate-Immobilized HIV-1 Tat Drives VEGFR2/αvβ3–Integrin Complex Formation and Polarization in Endothelial Cells. Arter. Thromb. Vasc. Biol. 2012, 32, e25–e34. [Google Scholar] [CrossRef]
- Chi, D.; Henry, J.; Kelley, J.; Thorpe, R.; Smith, J.K.; Krishnaswamy, G. The Effects of HIV Infection on Endothelial Function. Endothelium 2000, 7, 223–242. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.R.; Rachel, G.; Parthasarathy, D. HIV Proteins and Endothelial Dysfunction: Implications in Cardiovascular Disease. Front. Cardiovasc. Med. 2018, 5, 185. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Presta, M. HIV-1 Tat protein and endothelium: From protein/cell interaction to AIDS-associated pathologies. Angiogenesis 2002, 5, 141–151. [Google Scholar] [CrossRef]
- Urbinati, C.E.; Grillo, E.; Chiodelli, P.; Tobia, C.; Caccuri, F.; Fiorentini, S.; David, G.; Rusnati, M. Syndecan-1 increases B-lymphoid cell extravasation in response to HIV-1 Tat via αvβ3/pp60src/pp125FAK pathway. Oncogene 2016, 36, 2609–2618. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.; Nava, B.; Caputi, M. Tat is a multifunctional viral protein that modulates cellular gene expression and functions. Oncotarget 2017, 8, 27569–27581. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.E.; Hallett, M.B. Neutrophil Cell Shape Change: Mechanism and Signalling during Cell Spreading and Phagocytosis. Int. J. Mol. Sci. 2019, 20, 1383. [Google Scholar] [CrossRef] [PubMed]
- Dewitt, S.; Hallett, M. Leukocyte membrane “expansion”: A central mechanism for leukocyte extravasation. J. Leukoc. Biol. 2007, 81, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Bredt, D.S.; Pabo, C.O. Tat Protein from Human Immunodeficiency Virus Forms a Metal-Linked Dimer. Science 1988, 240, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Chen, L.; Cotter, R.J.; Pabo, C.O. Dimerization of the tat protein from human immunodeficiency virus: A cysteine-rich peptide mimics the normal metal-linked dimer interface. Proc. Natl. Acad. Sci. USA 1988, 85, 6297–6300. [Google Scholar] [CrossRef]
- Albini, A.; Ferrini, S.; Benelli, R.; Sforzini, S.; Giunciuglio, D.; Aluigi, M.G.; Proudfoot, A.E.I.; Alouani, S.; Wells, T.; Mariani, G.; et al. HIV-1 Tat protein mimicry of chemokines. Proc. Natl. Acad. Sci. USA 1998, 95, 13153–13158. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Tulipano, G.; Urbinati, C.E.; Tanghetti, E.; Giuliani, R.; Giacca, M.; Ciomei, M.; Corallini, A.; Presta, M. The Basic Domain in HIV-1 Tat Protein as a Target for Polysulfonated Heparin-mimicking Extracellular Tat Antagonists. J. Biol. Chem. 1998, 273, 16027–16037. [Google Scholar] [CrossRef] [PubMed]
- Mitola, S.; Soldi, R.; Zanon, I.; Barra, L.; Gutierrez, M.I.; Berkhout, B.; Giacca, M.; Bussolino, F. Identification of Specific Molecular Structures of Human Immunodeficiency Virus Type 1 Tat Relevant for Its Biological Effects on Vascular Endothelial Cells. J. Virol. 2000, 74, 344–353. [Google Scholar] [CrossRef]
- Cafaro, A.; Barillari, G.; Moretti, S.; Palladino, C.; Tripiciano, A.; Falchi, M.; Picconi, O.; Cossut, M.R.P.; Campagna, M.; Arancio, A.; et al. HIV-1 Tat Protein Enters Dysfunctional Endothelial Cells via Integrins and Renders Them Permissive to Virus Replication. Int. J. Mol. Sci. 2020, 22, 317. [Google Scholar] [CrossRef] [PubMed]
- Chiodelli, P.; Urbinati, C.E.; Mitola, S.; Tanghetti, E.; Rusnati, M. Sialic Acid Associated with αvβ3 Integrin Mediates HIV-1 Tat Protein Interaction and Endothelial Cell Proangiogenic Activation. J. Biol. Chem. 2012, 287, 20456–20466. [Google Scholar] [CrossRef]
- Urbinati, C.E.; Bugatti, A.; Giacca, M.; Schlaepfer, D.; Presta, M.; Rusnati, M. αvβ3-integrin-dependent activation of focal adhesion kinase mediates NF-κB activation and motogenic activity by HIV-1 Tat in endothelial cells. J. Cell Sci. 2005, 118, 3949–3958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Coomans, C.; David, G. Membrane Heparan Sulfate Proteoglycan-supported FGF2-FGFR1 Signaling. J. Biol. Chem. 2001, 276, 41921–41929. [Google Scholar] [CrossRef] [PubMed]
- Panetti, T.S. Tyrosine phosphorylation of paxillin, FAK, and p130CAS: Effects on cell spreading and migration. Front. Biosci. 2002, 7, d143–d150. [Google Scholar]
- Zhao, Y.; Lin, Y.; Zhang, H.; Mañas, A.; Tang, W.; Zhang, Y.; Wu, D.; Lin, A.; Xiang, J. Ubl4A is required for insulin-induced Akt plasma membrane translocation through promotion of Arp2/3-dependent actin branching. Proc. Natl. Acad. Sci. USA 2015, 112, 9644–9649. [Google Scholar] [CrossRef] [PubMed]
- Banon-Rodriguez, I.; De Guinoa, J.S.; Bernardini, A.; Ragazzini, C.; Fernandez, E.; Carrasco, Y.R.; E Jones, G.; Wandosell, F.; Anton, I.M. WIP Regulates Persistence of Cell Migration and Ruffle Formation in Both Mesenchymal and Amoeboid Modes of Motility. PLoS ONE 2013, 8, e70364. [Google Scholar] [CrossRef]
- Podyma-Inoue, K.A.; Hara-Yokoyama, M.; Shinomura, T.; Kimura, T.; Yanagishita, M. Syndecans Reside in Sphingomyelin-Enriched Low-Density Fractions of the Plasma Membrane Isolated from a Parathyroid Cell Line. PLoS ONE 2012, 7, e32351. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.D.; Fernandez-Borja, M. Leukocyte adhesion and polarization: Role of glycosylphosphatidylinositol-anchored proteins. BioArchitecture 2015, 5, 61–69. [Google Scholar] [CrossRef][Green Version]
- Previtera, M.L.; Peterman, K.; Shah, S.; Luzuriaga, J. Lipid Rafts Direct Macrophage Motility in the Tissue Microenvironment. Ann. Biomed. Eng. 2014, 43, 896–905. [Google Scholar] [CrossRef]
- Lebakken, C.S.; McQuade, K.J.; Rapraeger, A.C. Syndecan-1 Signals Independently of β1 Integrins during Raji Cell Spreading. Exp. Cell Res. 2000, 259, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Ria, R.; Vacca, A.; Ribatti, D.; Di Raimondo, F.; Merchionne, F.; Dammacco, F. Alpha(v)beta(3) integrin engagement enhances cell invasiveness in human multiple myeloma. Haematologica 2002, 87, 836–845. [Google Scholar]
- Jakobsson, L.; Kreuger, J.; Holmborn, K.; Lundin, L.; Eriksson, I.; Kjellén, L.; Claesson-Welsh, L. Heparan Sulfate in trans Potentiates VEGFR-Mediated Angiogenesis. Dev. Cell 2006, 10, 625–634. [Google Scholar] [CrossRef]
- Rusnati, M.; Urbinati, C.; Musulin, B.; Ribatti, D.; Albini, A.; Noonan, D.; Marchisone, C.; Waltenberger, J.; Presta, M. Activation of Endothelial Cell Mitogen Activated Protein Kinase ERK1/2by Extracellular HIV-1 Tat Protein. Endothelium 2001, 8, 65–74. [Google Scholar] [CrossRef]
- Gavard, J. Endothelial permeability and VE-cadherin. Cell Adhes. Migr. 2014, 8, 158–164. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Singh, S.K. HIV-1 Tat C phosphorylates VE-cadherin complex and increases human brain microvascular endothelial cell permeability. BMC Neurosci. 2014, 15, 80. [Google Scholar] [CrossRef]
- Avraham, H.; Jiang, S.; Lee, T.-H.; Prakash, O.; Avraham, S. HIV-1 Tat-Mediated Effects on Focal Adhesion Assembly and Permeability in Brain Microvascular Endothelial Cells. J. Immunol. 2004, 173, 6228–6233. [Google Scholar] [CrossRef] [PubMed]
- Esser, S.; Lampugnani, M.; Corada, M.; Dejana, E.; Risau, W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 1998, 111, 1853–1865. [Google Scholar] [CrossRef] [PubMed]
- Khalfaoui-Bendriss, G.; Dussault, N.; Fernandez-Sauze, S.; Berenguer-Daizé, C.; Sigaud, R.; Delfino, C.; Cayol, M.; Metellus, P.; Chinot, O.; Mabrouk, K.; et al. Adrenomedullin blockade induces regression of tumor neovessels through interference with vascular endothelial-cadherin signalling. Oncotarget 2015, 6, 7536–7553. [Google Scholar] [CrossRef] [PubMed]
- Schulte, D.; Küppers, V.; Dartsch, N.; Broermann, A.; Li, H.; Zarbock, A.; Kamenyeva, O.; Kiefer, F.; Khandoga, A.; Massberg, S.; et al. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 2011, 30, 4157–4170. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.; Ninchoji, T.; Gordon, E.; André, H.; Dejana, E.; Vestweber, D.; Kvanta, A.; Claesson-Welsh, L. Vascular permeability in retinopathy is regulated by VEGFR2 Y949 signaling to VE-cadherin. eLife 2020, 9, e54056. [Google Scholar] [CrossRef] [PubMed]
- Zilberberg, L.; Shinkaruk, S.; Lequin, O.; Rousseau, B.; Hagedorn, M.; Costa, F.; Caronzolo, D.; Balke, M.; Canron, X.; Convert, O.; et al. Structure and Inhibitory Effects on Angiogenesis and Tumor Development of a New Vascular Endothelial Growth Inhibitor. J. Biol. Chem. 2003, 278, 35564–35573. [Google Scholar] [CrossRef]
- Dorland, Y.L.; Huveneers, S. Cell–cell junctional mechanotransduction in endothelial remodeling. Experientia 2016, 74, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Paiardi, G.; Milanesi, M.; Wade, R.; D’Ursi, P.; Rusnati, M. A Bittersweet Computational Journey among Glycosaminoglycans. Biomolecules 2021, 11, 739. [Google Scholar] [CrossRef] [PubMed]
- Péloponèse, J.-M.; Grégoire, C.; Opi, S.; Esquieu, D.; Sturgis, J.; Lebrun, É.; Meurs, É.; Collette, Y.; Olive, D.; Aubertin, A.-M.; et al. 1H-13C nuclear magnetic resonance assignment and structural characterization of HIV-1 Tat protein. Life Sci. 2000, 323, 883–894. [Google Scholar] [CrossRef]
- Bugatti, A.; Paiardi, G.; Urbinati, C.E.; Chiodelli, P.; Orro, A.; Uggeri, M.; Milanesi, L.; Caruso, A.; Caccuri, F.; D’Ursi, P.; et al. Heparin and heparan sulfate proteoglycans promote HIV-1 p17 matrix protein oligomerization: Computational, biochemical and biological implications. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.; Xin, X.; Zheng, M.; Wang, S.; Peng, S.; Li, J.; Wang, L.; Jiang, H.; Geng, M. A Triad of Lys12, Lys41, Arg78 Spatial Domain, a Novel Identified Heparin Binding Site on Tat Protein, Facilitates Tat-Driven Cell Adhesion. PLoS ONE 2008, 3, e2662. [Google Scholar] [CrossRef] [PubMed]
- Sury, K.; Perazella, M.A. The Changing Face of Human Immunodeficiency Virus-Mediated Kidney Disease. Adv. Chronic Kidney Dis. 2019, 26, 185–197. [Google Scholar] [CrossRef]
- McArthur, J.C. HIV dementia: An evolving disease. J. Neuroimmunol. 2004, 157, 3–10. [Google Scholar] [CrossRef]
- Kariyanna, P.T.; Yager, J.; Salciccioli, L.; Lazar, J.M.; Polman, D.J.; Chandrakumar, H.P.; McFarlane, I.M. HIV-associated Extra-cranial Arterial Aneurysms: A Systematic Review. Am. J. Med. Case Rep. 2020, 8, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Carr, E.R. HIV- and AIDS-Associated Cancers. Clin. J. Oncol. Nurs. 2013, 17, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of Cell Surface Heparan Sulfate Proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Tumova, S.; Woods, A.; Couchman, J.R. Heparan Sulfate Chains from Glypican and Syndecans Bind the Hep II Domain of Fibronectin Similarly Despite Minor Structural Differences. J. Biol. Chem. 2000, 275, 9410–9417. [Google Scholar] [CrossRef]
- Wu, S.; Frank, I.; Derby, N.; Martinelli, E.; Cheng, C.Y. HIV-1 establishes a sanctuary site in the testis by permeating the BTB through changes in cytoskeletal organization. Endocrinology 2021, 162, bqab156. [Google Scholar] [CrossRef]
- Lee, C.-S.; Kim, Y.G.; Cho, H.-J.; Park, J.; Jeong, H.; Lee, S.-E.; Lee, S.-P.; Kang, H.-J.; Kim, H.-S. Dipeptidyl Peptidase-4 Inhibitor Increases Vascular Leakage in Retina through VE-cadherin Phosphorylation. Sci. Rep. 2016, 6, 29393. [Google Scholar] [CrossRef] [PubMed]
- Hakanpaa, L.; Kiss, E.A.; Jacquemet, G.; Miinalainen, I.; Lerche, M.; Guzmán, C.; Mervaala, E.; Eklund, L.; Ivaska, J.; Saharinen, P. Targeting β1-integrin inhibits vascular leakage in endotoxemia. Proc. Natl. Acad. Sci. USA 2018, 115, E6467–E6476. [Google Scholar] [CrossRef] [PubMed]
- Eum, S.Y.; Jaraki, D.; Bertrand, L.; András, I.E.; Toborek, M. Disruption of epithelial barrier by quorum-sensing N-3-(oxododecanoyl)-homoserine lactone is mediated by matrix metalloproteinases. Am. J. Physiol. Liver Physiol. 2014, 306, G992–G1001. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.G.; Kotanidou, A.; Dimopoulou, I.; Orfanos, S.E. Endothelial Damage in Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8793. [Google Scholar] [CrossRef] [PubMed]
- Malavige, G.N.; Ogg, G.S. Pathogenesis of vascular leak in dengue virus infection. Immunology 2017, 151, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Vrancken, K.; Vervaeke, P.; Balzarini, J.; Liekens, S. Viruses as key regulators of angiogenesis. Rev. Med. Virol. 2011, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Loret, E.P. What does the structure-function relationship of the HIV-1 Tat protein teach us about developing an AIDS vaccine? Retrovirology 2009, 6, 50. [Google Scholar] [CrossRef] [PubMed]
- Pantano, S.; Carloni, P. Comparative analysis of HIV-1 Tat variants. Proteins Struct. Funct. Bioinform. 2004, 58, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Kunihara, T.; Hayashi, Y.; Arai, M. Conformational diversity in the intrinsically disordered HIV-1 Tat protein induced by zinc and pH. Biochem. Biophys. Res. Commun. 2018, 509, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Mediouni, S.; Chinthalapudi, K.; Ekka, M.K.; Usui, I.; Jablonski, J.A.; Clementz, M.A.; Mousseau, G.; Nowak, J.; Macherla, V.R.; Beverage, J.N.; et al. Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat. mBio 2019, 10, e02662-18. [Google Scholar] [CrossRef] [PubMed]
- Rusnati, M.; Tulipano, G.; Spillmann, D.; Tanghetti, E.; Oreste, P.; Zoppetti, G.; Giacca, M.; Presta, M. Multiple Interactions of HIV-I Tat Protein with Size-defined Heparin Oligosaccharides. J. Biol. Chem. 1999, 274, 28198–28205. [Google Scholar] [CrossRef] [PubMed]
- Monini, P.; Cafaro, A.; Srivastava, I.K.; Moretti, S.; Sharma, V.A.; Andreini, C.; Chiozzini, C.; Ferrantelli, F.; Cossut, M.R.P.; Tripiciano, A.; et al. HIV-1 Tat Promotes Integrin-Mediated HIV Transmission to Dendritic Cells by Binding Env Spikes and Competes Neutralization by Anti-HIV Antibodies. PLoS ONE 2012, 7, e48781. [Google Scholar] [CrossRef]
- Tyagi, M.; Rusnati, M.; Presta, M.; Giacca, M. Internalization of HIV-1 Tat Requires Cell Surface Heparan Sulfate Proteoglycans. J. Biol. Chem. 2001, 276, 3254–3261. [Google Scholar] [CrossRef] [PubMed]
- Chiodelli, P.; Mitola, S.; Ravelli, C.; Oreste, P.; Rusnati, M.; Presta, M. Heparan Sulfate Proteoglycans Mediate the Angiogenic Activity of the Vascular Endothelial Growth Factor Receptor-2 Agonist Gremlin. Arter. Thromb. Vasc. Biol. 2011, 31, e116–e127. [Google Scholar] [CrossRef]
- Tanghetti, E.; Ria, R.; Dell’Era, P.; Urbinati, C.E.; Rusnati, M.; Ennas, M.G.; Presta, M. Biological activity of substrate-bound basic fibroblast growth factor (FGF2): Recruitment of FGF receptor-1 in endothelial cell adhesion contacts. Oncogene 2002, 21, 3889–3897. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed]
- Desta, I.T.; Porter, K.A.; Xia, B.; Kozakov, D.; Vajda, S. Performance and Its Limits in Rigid Body Protein-Protein Docking. Structure 2020, 28, 1071–1081.e3. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbinati, C.; Milanesi, M.; Lauro, N.; Bertelli, C.; David, G.; D’Ursi, P.; Rusnati, M.; Chiodelli, P. HIV-1 Tat and Heparan Sulfate Proteoglycans Orchestrate the Setup of in Cis and in Trans Cell-Surface Interactions Functional to Lymphocyte Trans-Endothelial Migration. Molecules 2021, 26, 7488. https://doi.org/10.3390/molecules26247488
Urbinati C, Milanesi M, Lauro N, Bertelli C, David G, D’Ursi P, Rusnati M, Chiodelli P. HIV-1 Tat and Heparan Sulfate Proteoglycans Orchestrate the Setup of in Cis and in Trans Cell-Surface Interactions Functional to Lymphocyte Trans-Endothelial Migration. Molecules. 2021; 26(24):7488. https://doi.org/10.3390/molecules26247488
Chicago/Turabian StyleUrbinati, Chiara, Maria Milanesi, Nicola Lauro, Cinzia Bertelli, Guido David, Pasqualina D’Ursi, Marco Rusnati, and Paola Chiodelli. 2021. "HIV-1 Tat and Heparan Sulfate Proteoglycans Orchestrate the Setup of in Cis and in Trans Cell-Surface Interactions Functional to Lymphocyte Trans-Endothelial Migration" Molecules 26, no. 24: 7488. https://doi.org/10.3390/molecules26247488
APA StyleUrbinati, C., Milanesi, M., Lauro, N., Bertelli, C., David, G., D’Ursi, P., Rusnati, M., & Chiodelli, P. (2021). HIV-1 Tat and Heparan Sulfate Proteoglycans Orchestrate the Setup of in Cis and in Trans Cell-Surface Interactions Functional to Lymphocyte Trans-Endothelial Migration. Molecules, 26(24), 7488. https://doi.org/10.3390/molecules26247488