
Coronavirus Infection-Associated Cell Death Signaling and Potential Therapeutic Targets




Abstract
1. Introduction
2. Life Cycle of SARS-CoV-2 and the Viral Proteins Involved in the Infection
3. Cell Death in Coronavirus Infections
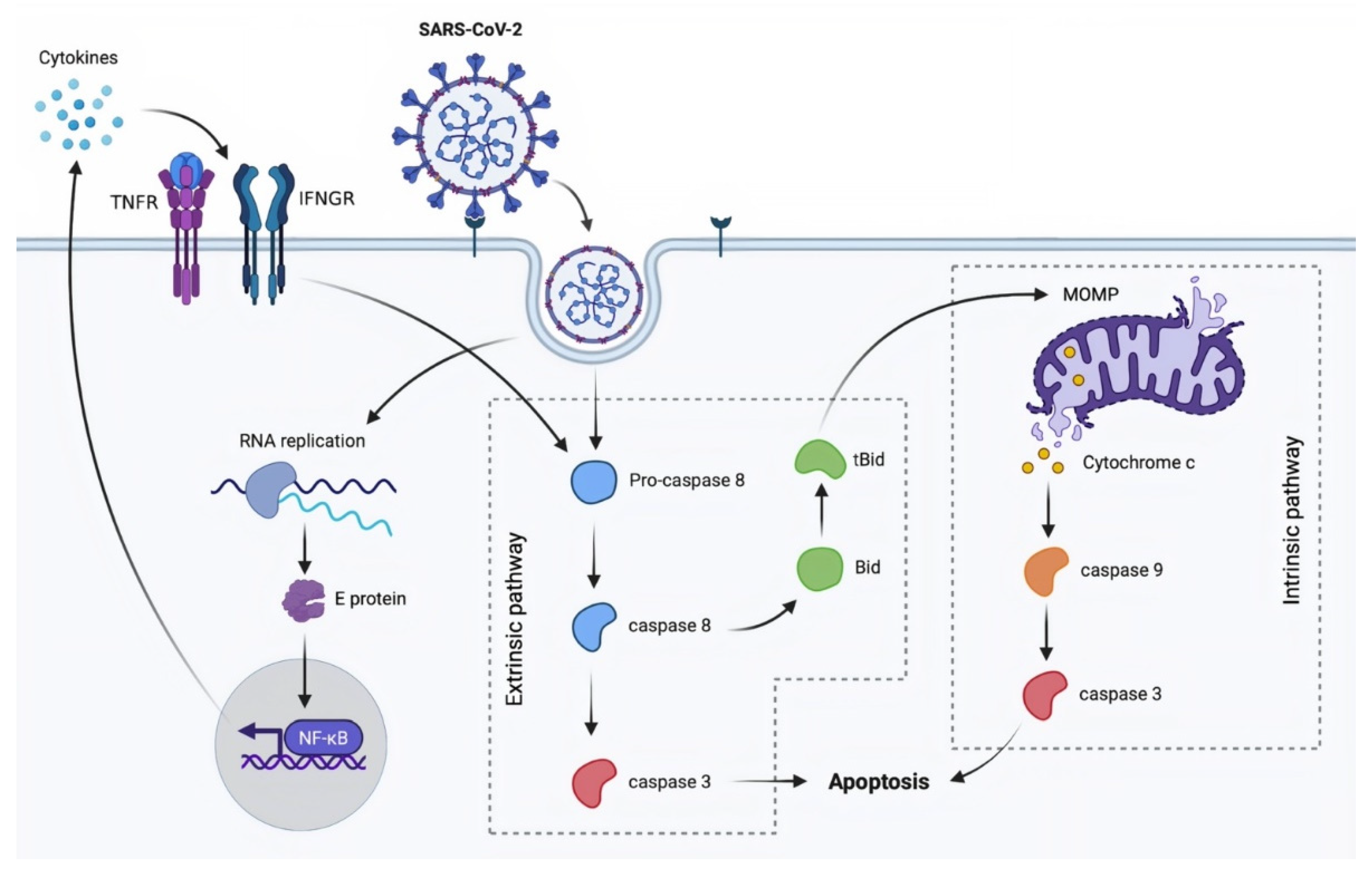
3.1. Apoptosis
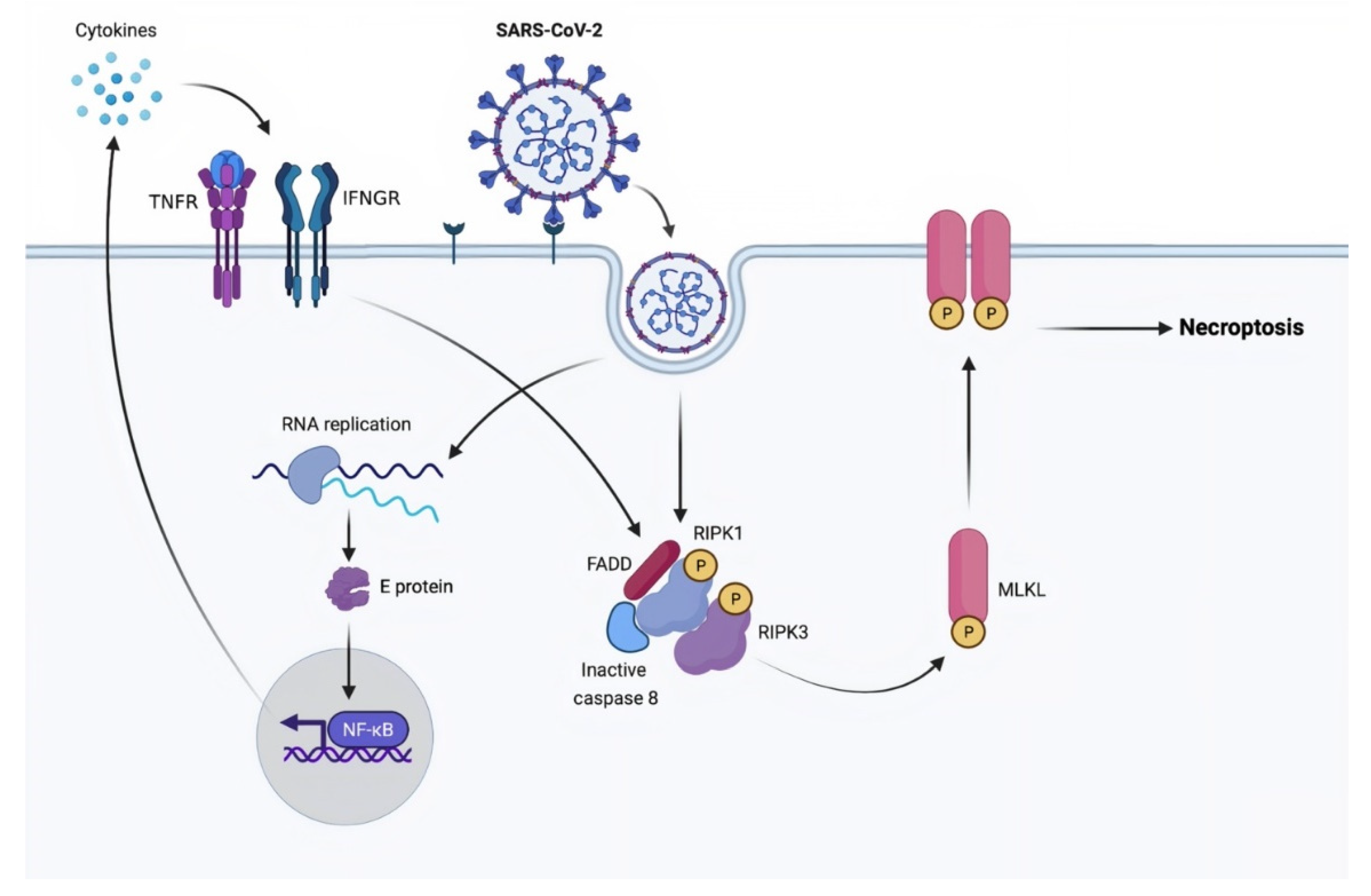
3.2. Necroptosis
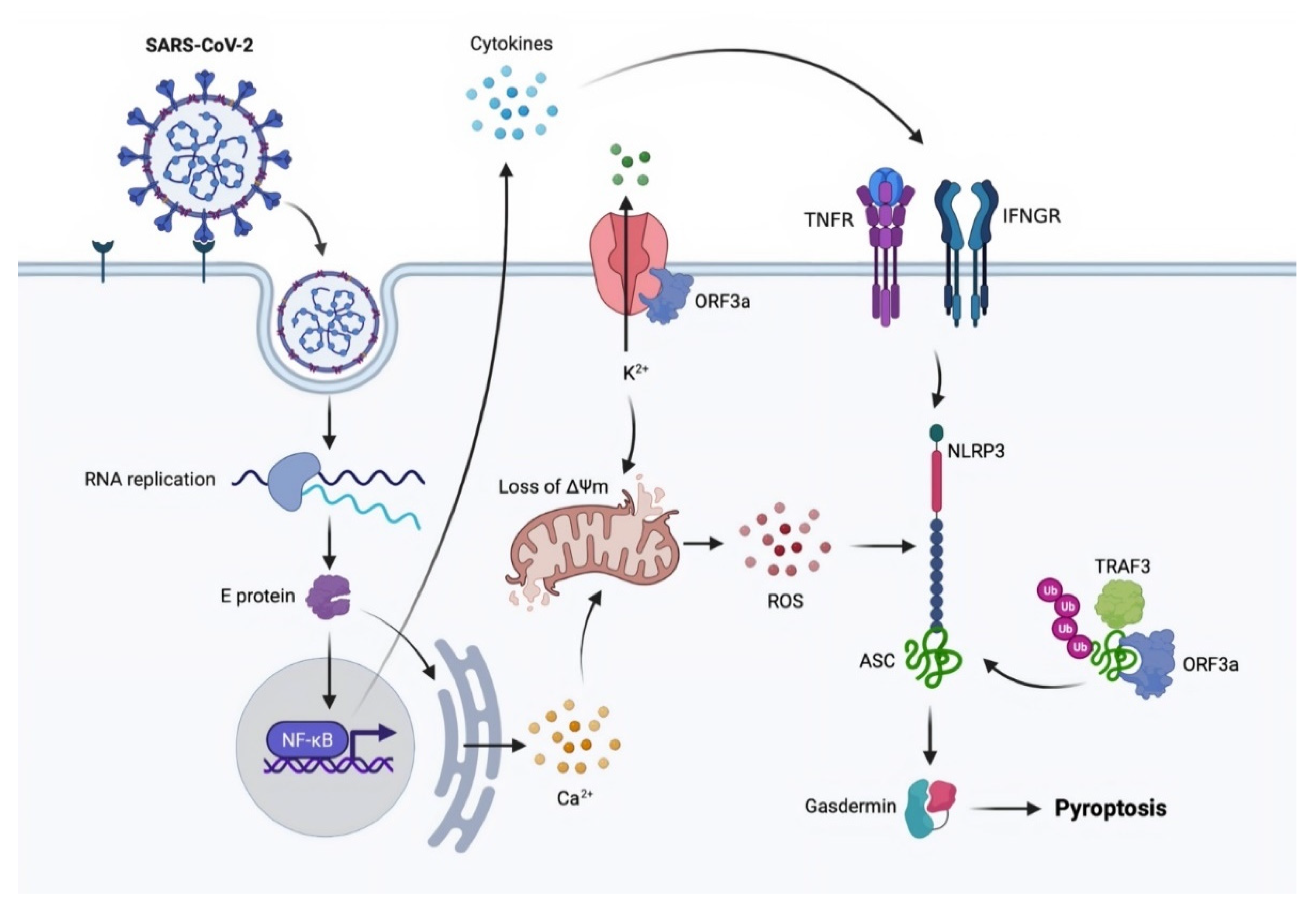
3.3. Pyroptosis
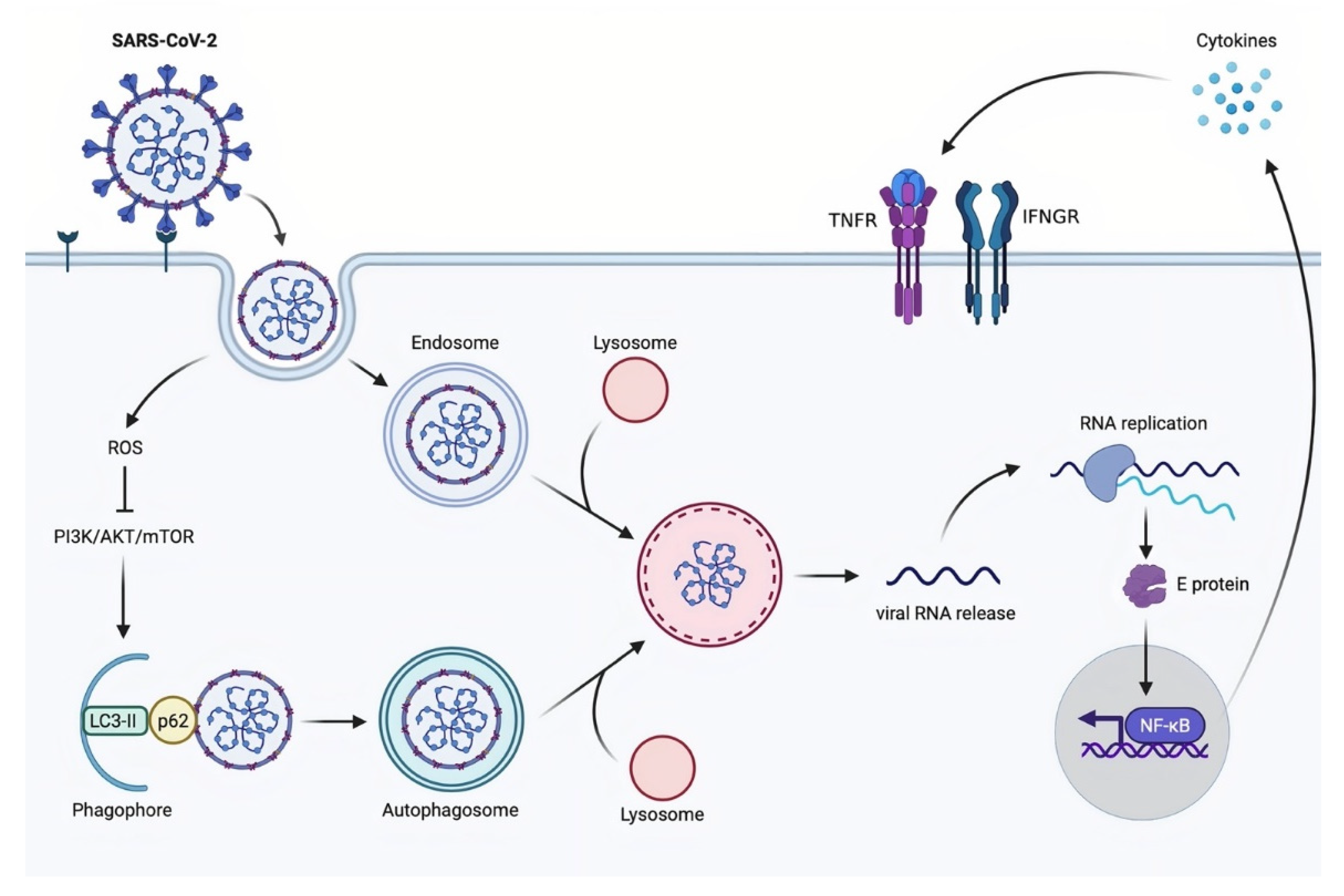
3.4. Autophagy
3.5. PANoptosis
4. The Possible Mechanisms of Cell Death Regulation in SARS-CoV-2 Infection
5. The Therapeutic Targets against Coronavirus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Compounds | Isolated From | Coronavirus Type | Target | IC50 | References |
|---|---|---|---|---|---|---|
| Alkaloids | Lycorine | Lycoris radiata | SARS-CoV | RdRp | 1.021 μM | [198] |
| SARS-CoV-2 | 0.878 μM | |||||
| Indigo | Isatis indigotica | SARS-CoV | 3CLpro | 752 μM | [181,188,199] | |
| Sinigrin | 217 μM | |||||
| Reserpine | Rauvolfia serpentina | SARS-CoV | 3CLpro, PLpro | 3.4 μM | [182] | |
| SARS-CoV-2 | 5.7 μM | [200] | ||||
| Cepharanthine | Stephania japonica | SARS-CoV-2 | S protein | 0.98 μM | [201,202] | |
| Flavonoids | Hesperetin | Isatis indigotica | SARS-CoV | 3CLpro | 60 μM | [188,203] |
| SARS-CoV-2 | S protein | 16.88 mM | [204] | |||
| Amentoflavone | Selaginella sinensis, Torreya nucifera | SARS-CoV | 3CLpro | 8.3 μM | [205,206] | |
| Myricetin | Myricaceae, Anacardiaceae, Polygonaceae, Pinaceae, Primulaceae | SARS-CoV | Nsp13, 3CLpro | 2.71 μM | [197,207] | |
| Scutellarein | 0.86 μM | |||||
| Scutellarein | Scutellaria | SARS-CoV-2 | 3CLpro | 5.8 μM | [208] | |
| Baicalin | Scutellaria baicalensis | SARS-CoV-2 | 3CLpro | 83.4 µM | [208] | |
| Baicalein | 0.39 μM | |||||
| Dihydromyricetin | Eriocaulon buergerianum | SARS-CoV-2 | 3CLpro | 1.24 μM | [208] | |
| Quercetagetin | Polygoni avicularis | SARS-CoV-2 | 3CLpro | 2.86 μM | ||
| Myricetin | Ampelopsis japonica | SARS-CoV-2 | 3CLpro | 1.20 μM | ||
| Scutellarein | Scutellaria, Erigeronti | SARS-CoV-2 | 3CLpro | 5.8 μM | ||
| Tannic acid | Camellia sinensis | SARS-CoV | - | 3 μM | [209] | |
| SARS-CoV-2 | Mpro, TMPRSS2 | 2.31-13.4 μM | [210] | |||
| Theaflavin | Camellia sinensis | SARS-CoV-2 | 3CLpro | 8.44 μg/ml | [211] | |
| Epigallocatechin gallate (EGCG) | 7.58 μg/ml | |||||
| Papyriflavonol | Broussonetia papyrifera | SARS-CoV | PLpro | 3.7 μM | [205,212,213] | |
| C-5-alkyl group (prenyl)-substituted flavan | 3CLpro | 52.7 μM | ||||
| Xanthoangelol E | Angelica keiskei | SARS-CoV | PLpro, 3CLpro | 11.4 μM | [214] | |
| Terpenoids | Glycyrrhizin | Glycyrrhiza glabra | SARS-CoV-2 | Mpro | 0.44 mg/mL | [215] |
| Betulinic acid | Betula pendula, Pterocarpus santalinus | SARS-CoV | 3CLpro | 10 μM | [216] | |
| Savinin | 25 μM | |||||
| Betulinic acid | Olea europaea | SARS-CoV-2 | Mpro | 14.55 μM | [217] | |
| Andrographolide | Andrographis paniculata | SARS-CoV-2 | Mpro | 0.034 μM | [191,218] | |
| Diarylheptanoids | Hirsutenone | Alnus japonica | SARS-CoV | PLpro | 4.1 μM | [193] |
| Hirsutanonol | 7.8 μM | |||||
| Oregonin | 20.1 μM | |||||
| Rubranol | 12.3 μM | |||||
| Rubranoside B | Alnus japonica | SARS-CoV | PLpro | 8.0 μM | [193] | |
| Rubranoside A | 9.1 μM | |||||
| Panduratin A | Boesenbergia rotunda | SARS-CoV-2 | - | 0.81 μΜ | [219] | |
| Anthraquinones | Emodin | Rheum, Polygonaceae, Isatis indigotica | SARS-CoV | S protein | 200 μM | [179,220,221] |
| SARS-CoV-2 | Mpro, S protein, RdRp | - | [222] | |||
| Aloe-emodin | SARS-CoV | 3CLpro | 132 μM | [188] | ||
| SARS-CoV-2 | Mpro, S protein, RdRp | - | [222] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Roser, M.; Ritchie, H.; Ortiz-Ospina, E.; Hasell, J. Coronavirus Pandemic (COVID-19). Available online: Ourworldindata.org (accessed on 11 September 2021).
- Hirano, T.; Murakami, M. COVID-19: A new virus, but a familiar receptor and cytokine release syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Prompetchara, E.; Ketloy, C.; Palaga, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac. J. Allergy Immunol. 2020, 38, 1–9. [Google Scholar]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.-L.; Zhou, P. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5, 235. [Google Scholar] [CrossRef]
- Chauhan, S. Comprehensive review of coronavirus disease 2019 (COVID-19). Biomed. J. 2020, 43, 334–340. [Google Scholar] [CrossRef]
- Tzotzos, S.J.; Fischer, B.; Fischer, H.; Zeitlinger, M. Incidence of ARDS and outcomes in hospitalized patients with COVID-19: A global literature survey. Crit. Care 2020, 24, 516. [Google Scholar] [CrossRef] [PubMed]
- Tarighi, P.; Eftekhari, S.; Chizari, M.; Sabernavaei, M.; Jafari, D.; Mirzabeigi, P. A review of potential suggested drugs for coronavirus disease (COVID-19) treatment. Eur. J. Pharmacol. 2021, 895, 173890. [Google Scholar] [CrossRef]
- Bartoszko, J.J.; Siemieniuk, R.A.; Kum, E.; Qasim, A.; Zeraatkar, D.; Ge, L.; Han, M.A.; Sadeghirad, B.; Agarwal, A.; Agoritsas, T. Prophylaxis against covid-19: Living systematic review and network meta-analysis. BMJ 2021, 373, n949. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.C. Traditional Chinese medicine and clinical pharmacology. Drug Discov. Eval. Methods Clin. Pharmacol. 2020, 455–482. [Google Scholar] [CrossRef]
- Chakravarti, R.; Singh, R.; Ghosh, A.; Dey, D.; Sharma, P.; Velayutham, R.; Roy, S.; Ghosh, D. A review on potential of natural products in the management of COVID-19. RSC Adv. 2021, 11, 16711–16735. [Google Scholar] [CrossRef]
- Ospanov, M.; Leó, F.; Janar, J.; Khan, I.A.; Ibrahim, M.A. Challenges and future directions of potential natural products leads against 2019-nCoV outbreak. Curr. Plant Biol. 2020, 24, 100180. [Google Scholar] [CrossRef]
- Musarra-Pizzo, M.; Pennisi, R.; Ben-Amor, I.; Mandalari, G.; Sciortino, M.T. Antiviral Activity Exerted by Natural Products against Human Viruses. Viruses 2021, 13, 828. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.J. Pathogenesis of COVID-19 from a cell biology perspective. Eur. Respir. Soc. 2020, 55, 2000607. [Google Scholar] [CrossRef]
- Yang, J.; Petitjean, S.J.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef]
- Dey, A.; Sen, S.; Maulik, U. Unveiling COVID-19-associated organ-specific cell types and cell-specific pathway cascade. Brief. Bioinform. 2021, 22, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of Structural and Non-Structural Proteins and Therapeutic Targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Trougakos, I.P.; Stamatelopoulos, K.; Terpos, E.; Tsitsilonis, O.E.; Aivalioti, E.; Paraskevis, D.; Kastritis, E.; Pavlakis, G.N.; Dimopoulos, M.A. Insights to SARS-CoV-2 life cycle, pathophysiology, and rationalized treatments that target COVID-19 clinical complications. J. Biomed. Sci. 2021, 28, 9. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Coronaviruses 2015, 1–23. [Google Scholar] [CrossRef]
- Salata, C.; Calistri, A.; Parolin, C.; Palu, G. Coronaviruses: A paradigm of new emerging zoonotic diseases. Pathog. Dis. 2019, 77, ftaa006. [Google Scholar] [CrossRef]
- Kannan, S.; Ali, P.S.S.; Sheeza, A.; Hemalatha, K. COVID-19 (Novel Coronavirus 2019)-recent trends. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2006–2011. [Google Scholar] [PubMed]
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef]
- Chen, Y.W.; Yiu, C.-P.B.; Wong, K.-Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 2020, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Lung, J.; Lin, Y.S.; Yang, Y.H.; Chou, Y.L.; Shu, L.H.; Cheng, Y.C.; Liu, H.T.; Wu, C.Y. The potential chemical structure of anti-SARS-CoV-2 RNA-dependent RNA polymerase. J. Med. Virol. 2020, 92, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, T.M.; Buchmeier, M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology 2001, 279, 371–374. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef]
- Kamitani, W.; Narayanan, K.; Huang, C.; Lokugamage, K.; Ikegami, T.; Ito, N.; Kubo, H.; Makino, S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 12885–12890. [Google Scholar] [CrossRef]
- Taylor, J.K.; Coleman, C.M.; Postel, S.; Sisk, J.M.; Bernbaum, J.G.; Venkataraman, T.; Sundberg, E.J.; Frieman, M.B. Severe acute respiratory syndrome coronavirus ORF7a inhibits bone marrow stromal antigen 2 virion tethering through a novel mechanism of glycosylation interference. J. Virol. 2015, 89, 11820–11833. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Harcourt, B.H.; Jukneliene, D.; Kanjanahaluethai, A.; Bechill, J.; Severson, K.M.; Smith, C.M.; Rota, P.A.; Baker, S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004, 78, 13600–13612. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- Shum, K.T.; Tanner, J.A. Differential inhibitory activities and stabilisation of DNA aptamers against the SARS coronavirus helicase. Chembiochem 2008, 9, 3037. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 255. [Google Scholar] [CrossRef] [PubMed]
- Imre, G. Cell death signalling in virus infection. Cell. Signal. 2020, 76, 109772. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Garron, T.M.; Chang, Q.; Su, Z.; Zhou, C.; Qiu, Y.; Gong, E.C.; Zheng, J.; Yin, Y.W.; Ksiazek, T. Cell-type apoptosis in lung during SARS-CoV-2 infection. Pathogens 2021, 10, 509. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.-Y.; Zhou, W.; Qiu, Y. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef]
- Bianchi, M.; Borsetti, A.; Ciccozzi, M.; Pascarella, S. SARS-Cov-2 ORF3a: Mutability and function. Int. J. Biol. Macromol. 2021, 170, 820–826. [Google Scholar] [CrossRef]
- Issa, E.; Merhi, G.; Panossian, B.; Salloum, T.; Tokajian, S. SARS-CoV-2 and ORF3a: Nonsynonymous mutations, functional domains, and viral pathogenesis. Msystems 2020, 5, e00266-20. [Google Scholar] [CrossRef]
- Lu, W.; Zheng, B.-J.; Xu, K.; Schwarz, W.; Du, L.; Wong, C.K.; Chen, J.; Duan, S.; Deubel, V.; Sun, B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. USA 2006, 103, 12540–12545. [Google Scholar] [CrossRef]
- Scott, C.; Griffin, S. Viroporins: Structure, function and potential as antiviral targets. J. Gen. Virol. 2015, 96, 2000–2027. [Google Scholar] [CrossRef]
- Thompson, E.A.; Cascino, K.; Ordonez, A.A.; Zhou, W.; Vaghasia, A.; Hamacher-Brady, A.; Brady, N.R.; Sun, I.-H.; Wang, R.; Rosenberg, A.Z. Metabolic programs define dysfunctional immune responses in severe COVID-19 patients. Cell Rep. 2021, 34, 108863. [Google Scholar] [CrossRef]
- Castaño-Rodriguez, C.; Honrubia, J.M.; Gutiérrez-Álvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Báguena, C.; Queralt-Martín, M. Role of severe acute respiratory syndrome coronavirus viroporins E, 3a, and 8a in replication and pathogenesis. MBio 2018, 9, e02325-17. [Google Scholar] [CrossRef]
- Wang, F.; Nie, J.; Wang, H.; Zhao, Q.; Xiong, Y.; Deng, L.; Song, S.; Ma, Z.; Mo, P.; Zhang, Y. Characteristics of peripheral lymphocyte subset alteration in COVID-19 pneumonia. J. Infect. Dis. 2020, 221, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, I. Systems biology of death receptor networks: Live and let die. Cell Death Dis. 2014, 5, e1259. [Google Scholar] [CrossRef] [PubMed]
- Bellesi, S.; Metafuni, E.; Hohaus, S.; Maiolo, E.; Marchionni, F.; D’Innocenzo, S.; La Sorda, M.; Ferraironi, M.; Ramundo, F.; Fantoni, M. Increased CD95 (Fas) and PD-1 expression in peripheral blood T lymphocytes in COVID-19 patients. Br. J. Haematol. 2020, 191, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Chen, I.-Y.; Moriyama, M.; Chang, M.-F.; Ichinohe, T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef]
- Kanzawa, N.; Nishigaki, K.; Hayashi, T.; Ishii, Y.; Furukawa, S.; Niiro, A.; Yasui, F.; Kohara, M.; Morita, K.; Matsushima, K. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-κB activation. FEBS Lett. 2006, 580, 6807–6812. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef]
- Yap, J.K.; Moriyama, M.; Iwasaki, A. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. J. Immunol. 2020, 205, 307–312. [Google Scholar] [CrossRef]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Singh, K.D.; Karnik, S.S. Angiotensin receptors: Structure, function, signaling and clinical applications. J. Cell Signal. 2016, 1, 111. [Google Scholar]
- Mori, J.; Oudit, G.Y.; Lopaschuk, G.D. SARS-CoV-2 perturbs the renin-angiotensin system and energy metabolism. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E43–E47. [Google Scholar] [CrossRef]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 inflammasome in severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.; Kim, J. Autophagy: An essential degradation program for cellular homeostasis and life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Gutierrez, D.; Bauer, M.A.; Zimmermann, A.; Kainz, K.; Hofer, S.J.; Kroemer, G.; Madeo, F. Digesting the crisis: Autophagy and coronaviruses. Microb. Cell 2020, 7, 119. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-S.; Nabar, N.R.; Huang, N.-N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 2019, 10, 5770. [Google Scholar] [CrossRef]
- Li, F.; Li, J.; Wang, P.-H.; Yang, N.; Huang, J.; Ou, J.; Xu, T.; Zhao, X.; Liu, T.; Huang, X. SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ROS-suppressed PI3K/AKT/mTOR signaling. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2021, 1867, 166260. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.S. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 2021, 184, 149–168. [Google Scholar] [CrossRef]
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, aaz7548. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Lakhani, S.A.; Masud, A.; Kuida, K.; Porter, G.A.; Booth, C.J.; Mehal, W.Z.; Inayat, I.; Flavell, R.A. Caspases 3 and 7: Key mediators of mitochondrial events of apoptosis. Science 2006, 311, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef]
- Giampietri, C.; Starace, D.; Petrungaro, S.; Filippini, A.; Ziparo, E. Necroptosis: Molecular signalling and translational implications. Int. J. Cell Biol. 2014, 2014, 490275. [Google Scholar] [CrossRef]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.-G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef]
- Malireddi, R.S.; Tweedell, R.E.; Kanneganti, T.-D. PANoptosis components, regulation, and implications. Aging 2020, 12, 11163. [Google Scholar] [CrossRef]
- Malireddi, R.S.; Karki, R.; Sundaram, B.; Kancharana, B.; Lee, S.; Samir, P.; Kanneganti, T.-D. Inflammatory cell death, PANoptosis, mediated by cytokines in diverse cancer lineages inhibits tumor growth. Immunohorizons 2021, 5, 568–580. [Google Scholar] [CrossRef]
- Christgen, S.; Zheng, M.; Kesavardhana, S.; Karki, R.; Malireddi, R.; Banoth, B.; Place, D.E.; Briard, B.; Sharma, B.R.; Tuladhar, S. Identification of the PANoptosome: A molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 2020, 10, 237. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Malireddi, R.S.; Kanneganti, T.-D. Caspases in cell death, inflammation, and pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef]
- Malireddi, R.; Gurung, P.; Kesavardhana, S.; Samir, P.; Burton, A.; Mummareddy, H.; Vogel, P.; Pelletier, S.; Burgula, S.; Kanneganti, T.-D. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity–independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020, 217, e20191644. [Google Scholar] [CrossRef]
- Malireddi, R.; Kesavardhana, S.; Kanneganti, T.-D. ZBP1 and TAK1: Master regulators of NLRP3 inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 2019, 9, 406. [Google Scholar] [CrossRef] [PubMed]
- Samir, P.; Malireddi, R.; Kanneganti, T.-D. The PANoptosome: A deadly protein complex driving pyroptosis, apoptosis, and necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 2020, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Kanneganti, T.D. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol. Rev. 2020, 297, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Kanneganti, T.-D. Newly Identified Function of Caspase-6 in ZBP1-mediated Innate Immune Responses, NLRP3 Inflammasome Activation, PANoptosis, and Host Defense. J. Cell. Immunol. 2020, 2, 341. [Google Scholar]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP 3 inflammasome assembly. EMBO J. 2019, 38, e101638. [Google Scholar] [CrossRef]
- Taabazuing, C.Y.; Okondo, M.C.; Bachovchin, D.A. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem. Biol. 2017, 24, 507–514. [Google Scholar] [CrossRef]
- Laing, A.G.; Lorenc, A.; Del Barrio, I.D.M.; Das, A.; Fish, M.; Monin, L.; Muñoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635. [Google Scholar] [CrossRef]
- Moulian, N.; Truffault, F.; Gaudry-Talarmain, Y.M.; Serraf, A.; Berrih-Aknin, S. In vivo and in vitro apoptosis of human thymocytes are associated with nitrotyrosine formation. Blood J. Am. Soc. Hematol. 2001, 97, 3521–3530. [Google Scholar] [CrossRef] [PubMed]
- Morse, J.S.; Lalonde, T.; Xu, S.; Liu, W.R. Learning from the past: Possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. Chembiochem 2020, 21, 730. [Google Scholar] [CrossRef]
- Chen, B.; Tian, E.-K.; He, B.; Tian, L.; Han, R.; Wang, S.; Xiang, Q.; Zhang, S.; El Arnaout, T.; Cheng, W. Overview of lethal human coronaviruses. Signal Transduct. Target. Ther. 2020, 5, 89. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Korteweg, C.; McNutt, M.A.; Gu, J. Pathogenetic mechanisms of severe acute respiratory syndrome. Virus Res. 2008, 133, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Nassar, A.; Ibrahim, I.M.; Amin, F.G.; Magdy, M.; Elgharib, A.M.; Azzam, E.B.; Nasser, F.; Yousry, K.; Shamkh, I.M.; Mahdy, S.M. A Review of Human Coronaviruses’ Receptors: The Host-Cell Targets for the Crown Bearing Viruses. Molecules 2021, 26, 6455. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schäfer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020, 71, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Fung, S.-Y.; Yuen, K.-S.; Ye, Z.-W.; Chan, C.-P.; Jin, D.-Y. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: Lessons from other pathogenic viruses. Emerg. Microbes Infect. 2020, 9, 558–570. [Google Scholar] [CrossRef]
- Chen, R.-F.; Chang, J.-C.; Yeh, W.-T.; Lee, C.-H.; Liu, J.-W.; Eng, H.-L.; Yang, K.D. Role of vascular cell adhesion molecules and leukocyte apoptosis in the lymphopenia and thrombocytopenia of patients with severe acute respiratory syndrome (SARS). Microbes Infect. 2006, 8, 122–127. [Google Scholar] [CrossRef]
- Garassino, M.C.; Ribas, A. At the crossroads: COVID-19 and immune-checkpoint blockade for cancer. Cancer Immunol. Res. 2021, 9, 261–264. [Google Scholar] [CrossRef]
- Tan, Y.-X.; Tan, T.H.; Lee, M.J.-R.; Tham, P.-Y.; Gunalan, V.; Druce, J.; Birch, C.; Catton, M.; Fu, N.Y.; Yu, V.C. Induction of apoptosis by the severe acute respiratory syndrome coronavirus 7a protein is dependent on its interaction with the Bcl-XL protein. J. Virol. 2007, 81, 6346–6355. [Google Scholar] [CrossRef]
- Chu, H.; Zhou, J.; Wong, B.H.-Y.; Li, C.; Chan, J.F.-W.; Cheng, Z.-S.; Yang, D.; Wang, D.; Lee, A.C.-Y.; Li, C. Middle East respiratory syndrome coronavirus efficiently infects human primary T lymphocytes and activates the extrinsic and intrinsic apoptosis pathways. J. Infect. Dis. 2016, 213, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.A.; Jha, B.K.; Silverman, R.H.; Hesselberth, J.R.; Barton, D.J. Ribonuclease L and metal-ion–independent endoribonuclease cleavage sites in host and viral RNAs. Nucleic Acids Res. 2014, 42, 5202–5216. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.H. Viral encounters with 2′, 5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zeqiraj, E.; Dong, B.; Jha, B.K.; Duffy, N.M.; Orlicky, S.; Thevakumaran, N.; Talukdar, M.; Pillon, M.C.; Ceccarelli, D.F. Dimeric structure of pseudokinase RNase L bound to 2-5A reveals a basis for interferon-induced antiviral activity. Mol. Cell 2014, 53, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Mbita, Z.; Hull, R.; Dlamini, Z. Human immunodeficiency virus-1 (HIV-1)-mediated apoptosis: New therapeutic targets. Viruses 2014, 6, 3181–3227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, L.; Jiang, D.; Wang, J.; Cong, X.; Fei, R. SARS-CoV nucleocapsid protein induced apoptosis of COS-1 mediated by the mitochondrial pathway. Artif. Cells Blood Substit. Biotechnol. 2007, 35, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wang, A.; Fang, Y.; Shu, T.; Wu, D.; Wang, C.; Huang, M.; Min, J.; Jin, L.; Zhou, W. SARS-CoV-2 Membrane Glycoprotein M Triggers Apoptosis with the Assistance of Nucleocapsid Protein N in Cells. Front. Cell. Infect. Microbiol. 2021, 11, 627. [Google Scholar] [CrossRef]
- Barhoumi, T.; Alghanem, B.; Shaibah, H.; Mansour, F.A.; Alamri, H.S.; Akiel, M.A.; Alroqi, F.; Boudjelal, M. SARS-CoV-2 Coronavirus Spike Protein-Induced Apoptosis, Inflammatory, and Oxidative Stress Responses in THP-1-Like-Macrophages: Potential Role of Angiotensin-Converting Enzyme Inhibitor (Perindopril). Front. Immunol. 2021, 12, 728896. [Google Scholar] [CrossRef]
- Chandrasekar, A.; Maynes, M.; Natesampillai, S.; Shweta, F.; Badley, A.; Cummins, N. SARS-CoV-2 spike protein induces monocyte apoptosis and interleukin-8 production. Top. Antivir. Med. 2021, 29, 61. [Google Scholar]
- Ivanisenko, N.V.; Seyrek, K.; Kolchanov, N.A.; Ivanisenko, V.A.; Lavrik, I.N. The role of death domain proteins in host response upon SARS-CoV-2 infection: Modulation of programmed cell death and translational applications. Cell Death Discov. 2020, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Hillert, L.K.; Ivanisenko, N.V.; Busse, D.; Espe, J.; König, C.; Peltek, S.E.; Kolchanov, N.A.; Ivanisenko, V.A.; Lavrik, I.N. Dissecting DISC regulation via pharmacological targeting of caspase-8/c-FLIP L heterodimer. Cell Death Differ. 2020, 27, 2117–2130. [Google Scholar] [CrossRef]
- Seyrek, K.; Ivanisenko, N.V.; Richter, M.; Hillert, L.K.; König, C.; Lavrik, I.N. Controlling cell death through post-translational modifications of DED proteins. Trends Cell Biol. 2020, 30, 354–369. [Google Scholar] [CrossRef]
- Ma, Y.; Walsh, M.J.; Bernhardt, K.; Ashbaugh, C.W.; Trudeau, S.J.; Ashbaugh, I.Y.; Jiang, S.; Jiang, C.; Zhao, B.; Root, D.E. CRISPR/Cas9 screens reveal Epstein-Barr virus-transformed B cell host dependency factors. Cell Host Microbe 2017, 21, 580–591. [Google Scholar] [CrossRef]
- Safa, A.R.; Pollok, K.E. Targeting the anti-apoptotic protein c-FLIP for cancer therapy. Cancers 2011, 3, 1639–1671. [Google Scholar] [CrossRef]
- Fiore, A.; Ugel, S.; De Sanctis, F.; Sandri, S.; Fracasso, G.; Trovato, R.; Sartoris, S.; Solito, S.; Mandruzzato, S.; Vascotto, F. Induction of immunosuppressive functions and NF-κB by FLIP in monocytes. Nat. Commun. 2018, 9, 5193. [Google Scholar] [CrossRef] [PubMed]
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.-L.; Schröter, M. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386, 517–521. [Google Scholar] [CrossRef]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.-L.; Schröter, M.; Burns, K.; Mattmann, C. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef]
- Sadek, J.; Wuo, M.G.; Rooklin, D.; Hauenstein, A.; Hong, S.H.; Gautam, A.; Wu, H.; Zhang, Y.; Cesarman, E.; Arora, P.S. Modulation of virus-induced NF-κB signaling by NEMO coiled coil mimics. Nat. Commun. 2020, 11, 1786. [Google Scholar] [CrossRef]
- Golks, A.; Brenner, D.; Krammer, P.H.; Lavrik, I.N. The c-FLIP–NH2 terminus (p22-FLIP) induces NF-κB activation. J. Exp. Med. 2006, 203, 1295–1305. [Google Scholar] [CrossRef]
- Shisler, J.L. Viral and cellular FLICE-inhibitory proteins: A comparison of their roles in regulating intrinsic immune responses. J. Virol. 2014, 88, 6539–6541. [Google Scholar] [CrossRef][Green Version]
- Katayama, R.; Ishioka, T.; Takada, S.; Takada, R.; Fujita, N.; Tsuruo, T.; Naito, M. Modulation of Wnt signaling by the nuclear localization of cellular FLIP-L. J. Cell Sci. 2010, 123, 23–28. [Google Scholar] [CrossRef]
- Chang, D.W.; Xing, Z.; Pan, Y.; Algeciras-Schimnich, A.; Barnhart, B.C.; Yaish-Ohad, S.; Peter, M.E.; Yang, X. c-FLIPL is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002, 21, 3704–3714. [Google Scholar] [CrossRef]
- Fricker, N.; Beaudouin, J.; Richter, P.; Eils, R.; Krammer, P.H.; Lavrik, I.N. Model-based dissection of CD95 signaling dynamics reveals both a pro-and antiapoptotic role of c-FLIPL. J. Cell Biol. 2010, 190, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Musiu, C.; Caligola, S.; Fiore, A.; Lamolinara, A.; Frusteri, C.; Del Pizzo, F.D.; De Sanctis, F.; Canè, S.; Adamo, A.; Hofer, F. Fatal cytokine release syndrome by an aberrant FLIP/STAT3 axis. Cell Death Differ. 2021, 27, 6083–6094. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhao, H.; Jin, M.; Zhu, H.; Shan, B.; Geng, J.; Dziedzic, S.A.; Amin, P.; Mifflin, L.; Naito, M.G. Modulating TRADD to restore cellular homeostasis and inhibit apoptosis. Nature 2020, 587, 133–138. [Google Scholar] [CrossRef]
- Ben-Hail, D.; Begas-Shvartz, R.; Shalev, M.; Shteinfer-Kuzmine, A.; Gruzman, A.; Reina, S.; De Pinto, V.; Shoshan-Barmatz, V. Novel compounds targeting the mitochondrial protein VDAC1 inhibit apoptosis and protect against mitochondrial dysfunction. J. Biol. Chem. 2016, 291, 24986–25003. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 2009, 122, 1906–1916. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Ide, T.; Yanagida, T.; Tsujimoto, Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 2000, 275, 12321–12325. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ye, F.; Wu, A.; Yang, R.; Pan, M.; Sheng, J.; Zhu, W.; Mao, L.; Wang, M.; Xia, Z. Comparative transcriptome analysis reveals the intensive early stage responses of host cells to SARS-CoV-2 infection. Front. Microbiol. 2020, 11, 2881. [Google Scholar] [CrossRef]
- Miller, S.C.; Huang, R.; Sakamuru, S.; Shukla, S.J.; Attene-Ramos, M.S.; Shinn, P.; Van Leer, D.; Leister, W.; Austin, C.P.; Xia, M. Identification of known drugs that act as inhibitors of NF-κB signaling and their mechanism of action. Biochem. Pharmacol. 2010, 79, 1272–1280. [Google Scholar] [CrossRef]
- Mofers, A.; Selvaraju, K.; Gubat, J.; D’Arcy, P.; Linder, S. Identification of proteasome inhibitors using analysis of gene expression profiles. Eur. J. Pharmacol. 2020, 889, 173709. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small molecule NF-κB pathway inhibitors in clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]
- Acerta Pharma, B. A Phase 2, Open Label, Randomized Study of the Efficacy and Safety of Acalabrutinib with Best Supportive Care versus Best Supportive Care in Subjects Hospitalized with COVID-19. 2020. Available online: https://pesquisa.bvsalud.org/global-literature-on-novel-coronavirus-2019-ncov/resource/pt/ictrp-PER-018-20 (accessed on 1 October 2021).
- Roschewski, M.; Lionakis, M.S.; Sharman, J.P.; Roswarski, J.; Goy, A.; Monticelli, M.A.; Roshon, M.; Wrzesinski, S.H.; Desai, J.V.; Zarakas, M.A. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci. Immunol. 2020, 5, eabd0110. [Google Scholar] [CrossRef]
- Agree, I. Karyopharm to Evaluate Low Dose Selinexor as a Potential Treatment for Hospitalized Patients with COVID-19. Available online: https://stockhouse.com/news/press-releases/2020/04/07/karyopharm-to-evaluate-low-dose-selinexor-as-a-potential-treatment-for (accessed on 2 October 2021).
- Cao, L.; Mu, W. Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacol. Res. 2020, 163, 105297. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, W.; Yan, Y.; Gong, T.; Han, J.; Tian, Z.; Zhou, R. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat. Immunol. 2014, 15, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lv, X.; Hu, B.; Shao, Z.; Wang, B.; Ma, K.; Lin, H.; Cui, M. RIPK1/RIPK3/MLKL-mediated necroptosis contributes to compression-induced rat nucleus pulposus cells death. Apoptosis 2017, 22, 626–638. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Soares, V.C.; de Azevedo-Quintanilha, I.G.; Dias, S.D.S.G.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; Mattos, M.; de Freitas, C.S.; Temerozo, J.R.; Teixeira, L. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Diao, M.-Y.; Zhu, Y.; Yang, J.; Xi, S.-S.; Wen, X.; Gu, Q.; Hu, W. Hypothermia protects neurons against ischemia/reperfusion-induced pyroptosis via m6A-mediated activation of PTEN and the PI3K/Akt/GSK-3β signaling pathway. Brain Res. Bull. 2020, 159, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Sengupta, S.; Kundu, R. Tiliacora racemosa leaves induce oxidative stress mediated DNA damage leading to G2/M phase arrest and apoptosis in cervical cancer cells SiHa. J. Ethnopharmacol. 2021, 269, 113686. [Google Scholar] [CrossRef]
- Kashyap, D.; Garg, V.K.; Tuli, H.S.; Yerer, M.B.; Sak, K.; Sharma, A.K.; Kumar, M.; Aggarwal, V.; Sandhu, S.S. Fisetin and quercetin: Promising flavonoids with chemopreventive potential. Biomolecules 2019, 9, 174. [Google Scholar] [CrossRef]
- Bonam, S.R.; Muller, S.; Bayry, J.; Klionsky, D.J. Autophagy as an emerging target for COVID-19: Lessons from an old friend, chloroquine. Autophagy 2020, 16, 2260–2266. [Google Scholar] [CrossRef]
- Gao, J.; Tian, Z.; Yang, X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 2020, 14, 72–73. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef]
- Jaffe, S. Regulators split on antimalarials for COVID-19. Lancet 2020, 395, 1179. [Google Scholar] [CrossRef]
- Ferner, R.E.; Aronson, J.K. Chloroquine and hydroxychloroquine in covid-19. Br. Med. J. Publ. Group 2020, 369, m1432. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Al-Bari, M.A.A. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol. Res. Perspect. 2017, 5, e00293. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, F.; Comunale, M.A.; Mehta, A.; Sehgal, M.; Jain, P.; Cuconati, A.; Lin, H.; Block, T.M.; Chang, J. Inhibition of endoplasmic reticulum-resident glucosidases impairs severe acute respiratory syndrome coronavirus and human coronavirus NL63 spike protein-mediated entry by altering the glycan processing of angiotensin I-converting enzyme 2. Antimicrob. Agents Chemother. 2015, 59, 206–216. [Google Scholar] [CrossRef]
- Katia, F.; Myriam, D.P.; Ucciferri, C.; Auricchio, A.; Di Nicola, M.; Marchioni, M.; Eleonora, C.; Emanuela, S.; Cipollone, F.; Vecchiet, J. Efficacy of canakinumab in mild or severe COVID-19 pneumonia. Immun. Inflamm. Dis. 2021, 9, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Mei, M.; Tan, X. Current Strategies of Antiviral Drug Discovery for COVID-19. Front. Mol. Biosci. 2021, 8, 310. [Google Scholar] [CrossRef]
- Jordan, S.C.; Zakowski, P.; Tran, H.P.; Smith, E.A.; Gaultier, C.; Marks, G.; Zabner, R.; Lowenstein, H.; Oft, J.; Bluen, B. Compassionate use of tocilizumab for treatment of SARS-CoV-2 pneumonia. Clin. Infect. Dis. 2020, 71, 3168–3173. [Google Scholar] [CrossRef]
- Falcone, M.; Tiseo, G.; Valoriani, B.; Barbieri, C.; Occhineri, S.; Mazzetti, P.; Vatteroni, M.L.; Suardi, L.R.; Riccardi, N.; Pistello, M. Efficacy of bamlanivimab/etesevimab and casirivimab/imdevimab in preventing progression to severe covid-19 and role of variants of concern. Infect. Dis. Ther. 2021, 10, 2479–2488. [Google Scholar] [CrossRef]
- Chau, A.S.; Weber, A.G.; Maria, N.I.; Narain, S.; Liu, A.; Hajizadeh, N.; Malhotra, P.; Bloom, O.; Marder, G.; Kaplan, B. The longitudinal immune response to coronavirus disease 2019: Chasing the cytokine storm. Arthritis Rheumatol. 2021, 73, 23–35. [Google Scholar] [CrossRef]
- Huang, J.; Tao, G.; Liu, J.; Cai, J.; Huang, Z.; Chen, J.-X. Current prevention of COVID-19: Natural products and herbal medicine. Front. Pharmacol. 2020, 11, 588508. [Google Scholar] [CrossRef]
- Boozari, M.; Hosseinzadeh, H. Natural products for COVID-19 prevention and treatment regarding to previous coronavirus infections and novel studies. Phytother. Res. 2021, 35, 864–876. [Google Scholar] [CrossRef]
- Mohammadi Pour, P.; Fakhri, S.; Asgary, S.; Farzaei, M.H.; Echeverria, J. The signaling pathways, and therapeutic targets of antiviral agents: Focusing on the antiviral approaches and clinical perspectives of anthocyanins in the management of viral diseases. Front. Pharmacol. 2019, 10, 1207. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.-J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef]
- Cheng, P.W.; Ng, L.T.; Chiang, L.C.; Lin, C.C. Antiviral effects of saikosaponins on human coronavirus 229E in vitro. Clin. Exp. Pharmacol. Physiol. 2006, 33, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.-Y.; Wu, S.-L.; Chen, J.-C.; Li, C.-C.; Hsiang, C.-Y. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antivir. Res. 2007, 74, 92–101. [Google Scholar] [CrossRef]
- Schwarz, S.; Wang, K.; Yu, W.; Sun, B.; Schwarz, W. Emodin inhibits current through SARS-associated coronavirus 3a protein. Antivir. Res. 2011, 90, 64–69. [Google Scholar] [CrossRef]
- Li, S.-Y.; Chen, C.; Zhang, H.-Q.; Guo, H.-Y.; Wang, H.; Wang, L.; Zhang, X.; Hua, S.-N.; Yu, J.; Xiao, P.-G. Identification of natural compounds with antiviral activities against SARS-associated coronavirus. Antivir. Res. 2005, 67, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Jan, J.-T.; Ma, S.-H.; Kuo, C.-J.; Juan, H.-F.; Cheng, Y.-S.E.; Hsu, H.-H.; Huang, H.-C.; Wu, D.; Brik, A. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 10012–10017. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Jeon, S.; Jang, Y.; Gotina, L.; Won, J.; Ju, Y.H.; Kim, S.; Jang, M.W.; Won, W.; Park, M.G. Platycodin D, a natural component of Platycodon grandiflorum, prevents both lysosome-and TMPRSS2-driven SARS-CoV-2 infection by hindering membrane fusion. Exp. Mol. Med. 2021, 53, 956–972. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Basharat, Z.; Yousuf, M.; Castaldo, G.; Rastrelli, L.; Khan, H. Virtual screening of natural products against type II transmembrane serine protease (TMPRSS2), the priming agent of coronavirus 2 (SARS-CoV-2). Molecules 2020, 25, 2271. [Google Scholar] [CrossRef] [PubMed]
- Chikhale, R.V.; Gupta, V.K.; Eldesoky, G.E.; Wabaidur, S.M.; Patil, S.A.; Islam, M.A. Identification of potential anti-TMPRSS2 natural products through homology modelling, virtual screening and molecular dynamics simulation studies. J. Biomol. Struct. Dyn. 2020, 37, 6660–6675. [Google Scholar] [CrossRef] [PubMed]
- Durai, P.; Batool, M.; Shah, M.; Choi, S. Middle East respiratory syndrome coronavirus: Transmission, virology and therapeutic targeting to aid in outbreak control. Exp. Mol. Med. 2015, 47, e181. [Google Scholar] [CrossRef]
- Ratia, K.; Kilianski, A.; Baez-Santos, Y.M.; Baker, S.C.; Mesecar, A. Structural basis for the ubiquitin-linkage specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog. 2014, 10, e1004113. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-W.; Tsai, F.-J.; Tsai, C.-H.; Lai, C.-C.; Wan, L.; Ho, T.-Y.; Hsieh, C.-C.; Chao, P.-D.L. Anti-SARS coronavirus 3C-like protease effects of Isatis indigotica root and plant-derived phenolic compounds. Antivir. Res. 2005, 68, 36–42. [Google Scholar] [CrossRef]
- Ryu, Y.B.; Park, S.-J.; Kim, Y.M.; Lee, J.-Y.; Seo, W.D.; Chang, J.S.; Park, K.H.; Rho, M.-C.; Lee, W.S. SARS-CoV 3CLpro inhibitory effects of quinone-methide triterpenes from Tripterygium regelii. Bioorg. Med. Chem. Lett. 2010, 20, 1873–1876. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Luo, C.; Liu, H.; Xu, W.; Chen, G.; Liew, O.W.; Zhu, W.; Puah, C.M.; Shen, X. Binding interaction of quercetin-3-β-galactoside and its synthetic derivatives with SARS-CoV 3CLpro: Structure–activity relationship studies reveal salient pharmacophore features. Bioorg. Med. Chem. 2006, 14, 8295–8306. [Google Scholar] [CrossRef] [PubMed]
- Enmozhi, S.K.; Raja, K.; Sebastine, I.; Joseph, J. Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: An in silico approach. J. Biomol. Struct. Dyn. 2021, 39, 3092–3098. [Google Scholar] [CrossRef]
- Park, J.-Y.; Kim, J.H.; Kim, Y.M.; Jeong, H.J.; Kim, D.W.; Park, K.H.; Kwon, H.-J.; Park, S.-J.; Lee, W.S.; Ryu, Y.B. Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorg. Med. Chem. 2012, 20, 5928–5935. [Google Scholar] [CrossRef]
- Park, J.-Y.; Jeong, H.J.; Kim, J.H.; Kim, Y.M.; Park, S.-J.; Kim, D.; Park, K.H.; Lee, W.S.; Ryu, Y.B. Diarylheptanoids from Alnus japonica inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biol. Pharm. Bull. 2012, 35, b12-00623. [Google Scholar] [CrossRef]
- Ganeshpurkar, A.; Gutti, G.; Singh, S. Viral Polymerases: Structures, Functions and Roles as Antiviral Drug Targets; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Allam, A.E.; Amen, Y.; Ashour, A.; Assaf, H.K.; Hassan, H.A.; Abdel-Rahman, I.M.; Sayed, A.M.; Shimizu, K. In silico study of natural compounds from sesame against COVID-19 by targeting M pro, PL pro and RdRp. RSC Adv. 2021, 11, 22398–22408. [Google Scholar] [CrossRef]
- Cho, J.K.; Curtis-Long, M.J.; Lee, K.H.; Kim, D.W.; Ryu, H.W.; Yuk, H.J.; Park, K.H. Geranylated flavonoids displaying SARS-CoV papain-like protease inhibition from the fruits of Paulownia tomentosa. Bioorg. Med. Chem. 2013, 21, 3051–3057. [Google Scholar] [CrossRef]
- Yu, M.-S.; Lee, J.; Lee, J.M.; Kim, Y.; Chin, Y.-W.; Jee, J.-G.; Keum, Y.-S.; Jeong, Y.-J. Identification of myricetin and scutellarein as novel chemical inhibitors of the SARS coronavirus helicase, nsP13. Bioorg. Med. Chem. Lett. 2012, 22, 4049–4054. [Google Scholar] [CrossRef]
- Jin, Y.-H.; Min, J.S.; Jeon, S.; Lee, J.; Kim, S.; Park, T.; Park, D.; Jang, M.S.; Park, C.M.; Song, J.H. Lycorine, a non-nucleoside RNA dependent RNA polymerase inhibitor, as potential treatment for emerging coronavirus infections. Phytomedicine 2021, 86, 153440. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, A.; Dwivedi, A.; Du Plessis, J. Sinigrin and its therapeutic benefits. Molecules 2016, 21, 416. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-J.; Chao, T.-L.; Kao, H.-C.; Tsai, Y.-M.; Liu, Y.-K.; Wang, L.H.-C.; Hsieh, M.-C.; Chang, S.-Y.; Liang, P.-H. Kinetic characterization and inhibitor screening for the proteases leading to identification of drugs against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65, e02577-20. [Google Scholar] [CrossRef]
- Fan, H.-H.; Wang, L.-Q.; Liu, W.-L.; An, X.-P.; Liu, Z.-D.; He, X.-Q.; Song, L.-H.; Tong, Y.-G. Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus-related coronavirus model. Chin. Med. J. 2020, 133, 1051. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, H.; Watashi, K.; Saso, W.; Shionoya, K.; Iwanami, S.; Hirokawa, T.; Shirai, T.; Kanaya, S.; Ito, Y.; Kim, K.S. Potential anti-COVID-19 agents, cepharanthine and nelfinavir, and their usage for combination treatment. Iscience 2021, 24, 102367. [Google Scholar] [CrossRef]
- Dejani, N.N.; Elshabrawy, H.A.; Bezerra Filho, C.D.S.M.; de Sousa, D.P. Anticoronavirus and Immunomodulatory Phenolic Compounds: Opportunities and Pharmacotherapeutic Perspectives. Biomolecules 2021, 11, 1254. [Google Scholar] [CrossRef]
- Guler, H.I.; Fulya, A.; Zehra, C.; Yakup, K.; Belduz, A.O.; Canakci, S.; Kolayli, S. Targeting CoV-2 Spike RBD and ACE-2 Interaction with Flavonoids of Anatolian Propolis by in silico and in vitro Studies in terms of possible COVID-19 therapeutics. Turk J. Biol. 2021, 45, 530–548. [Google Scholar] [CrossRef]
- Ryu, Y.B.; Jeong, H.J.; Kim, J.H.; Kim, Y.M.; Park, J.-Y.; Kim, D.; Naguyen, T.T.H.; Park, S.-J.; Chang, J.S.; Park, K.H. Biflavonoids from Torreya nucifera displaying SARS-CoV 3CLpro inhibition. Bioorg. Med. Chem. 2010, 18, 7940–7947. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, S.; Shin, D.H.; Kim, M.-S. Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzym. Inhib. Med. Chem. 2020, 35, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-H. Anti–SARS-CoV-2 Natural Products as Potentially Therapeutic Agents. Front. Pharmacol. 2021, 12, 590509. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ye, F.; Sun, Q.; Liang, H.; Li, C.; Li, S.; Lu, R.; Huang, B.; Tan, W.; Lai, L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J. Enzym. Inhib. Med. Chem. 2021, 36, 497–503. [Google Scholar] [CrossRef]
- Chen, C.-N.; Lin, C.P.; Huang, K.-K.; Chen, W.-C.; Hsieh, H.-P.; Liang, P.-H.; Hsu, J.T.-A. Inhibition of SARS-CoV 3C-like protease activity by theaflavin-3, 3’-digallate (TF3). Evid.-Based Complement. Altern. Med. 2005, 2, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Chen, Y.; Wang, Y.-C.; Wang, W.-J.; Yang, C.-S.; Tsai, C.-L.; Hou, M.-H.; Chen, H.-F.; Shen, Y.-C.; Hung, M.-C. Tannic acid suppresses SARS-CoV-2 as a dual inhibitor of the viral main protease and the cellular TMPRSS2 protease. Am. J. Cancer Res. 2020, 10, 4538. [Google Scholar]
- Jang, M.; Park, Y.-I.; Cha, Y.-E.; Park, R.; Namkoong, S.; Lee, J.I.; Park, J. Tea polyphenols EGCG and theaflavin inhibit the activity of SARS-CoV-2 3CL-protease in vitro. Evid.-Based Complement. Altern. Med. 2020, 2020, 5630838. [Google Scholar] [CrossRef]
- Paraiso, I.L.; Revel, J.S.; Stevens, J.F. Potential use of polyphenols in the battle against COVID-19. Curr. Opin. Food Sci. 2020, 32, 149–155. [Google Scholar] [CrossRef]
- Park, J.Y.; Yuk, H.J.; Ryu, H.W.; Lim, S.H.; Kim, K.S.; Park, K. Evaluation of polyphenols from Broussonetia papyrifera as coronavirus protease inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 504–512. [Google Scholar] [CrossRef]
- Park, J.-Y.; Ko, J.-A.; Kim, D.W.; Kim, Y.M.; Kwon, H.-J.; Jeong, H.J.; Kim, C.Y.; Park, K.H.; Lee, W.S.; Ryu, Y.B. Chalcones isolated from Angelica keiskei inhibit cysteine proteases of SARS-CoV. J. Enzym. Inhib. Med. Chem. 2016, 31, 23–30. [Google Scholar] [CrossRef]
- Van de Sand, L.; Bormann, M.; Alt, M.; Schipper, L.; Heilingloh, C.S.; Steinmann, E.; Todt, D.; Dittmer, U.; Elsner, C.; Witzke, O. Glycyrrhizin Effectively Inhibits SARS-CoV-2 Replication by Inhibiting the Viral Main Protease. Viruses 2021, 13, 609. [Google Scholar] [CrossRef]
- Wen, C.-C.; Kuo, Y.-H.; Jan, J.-T.; Liang, P.-H.; Wang, S.-Y.; Liu, H.-G.; Lee, C.-K.; Chang, S.-T.; Kuo, C.-J.; Lee, S.-S. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J. Med. Chem. 2007, 50, 4087–4095. [Google Scholar] [CrossRef]
- Alhadrami, H.A.; Sayed, A.M.; Sharif, A.M.; Azhar, E.I.; Rateb, M.E. Olive-Derived Triterpenes Suppress SARS COV-2 Main Protease: A Promising Scaffold for Future Therapeutics. Molecules 2021, 26, 2654. [Google Scholar] [CrossRef]
- Sa-Ngiamsuntorn, K.; Suksatu, A.; Pewkliang, Y.; Thongsri, P.; Kanjanasirirat, P.; Manopwisedjaroen, S.; Charoensutthivarakul, S.; Wongtrakoongate, P.; Pitiporn, S.; Chaopreecha, J. Anti-SARS-CoV-2 activity of Andrographis paniculata extract and its major component Andrographolide in human lung epithelial cells and cytotoxicity evaluation in major organ cell representatives. J. Nat. Prod. 2021, 84, 1261–1270. [Google Scholar] [CrossRef]
- Kanjanasirirat, P.; Suksatu, A.; Manopwisedjaroen, S.; Munyoo, B.; Tuchinda, P.; Jearawuttanakul, K.; Seemakhan, S.; Charoensutthivarakul, S.; Wongtrakoongate, P.; Rangkasenee, N. High-content screening of Thai medicinal plants reveals Boesenbergia rotunda extract and its component Panduratin A as anti-SARS-CoV-2 agents. Sci. Rep. 2020, 10, 19963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.W.; Yap, Y.L. Old drugs as lead compounds for a new disease? Binding analysis of SARS coronavirus main proteinase with HIV, psychotic and parasite drugs. Bioorg. Med. Chem. 2004, 12, 2517–2521. [Google Scholar] [CrossRef]
- Guy, R.K.; DiPaola, R.S.; Romanelli, F.; Dutch, R.E. Rapid repurposing of drugs for COVID-19. Science 2020, 368, 829–830. [Google Scholar] [CrossRef] [PubMed]
- El Mazouri, S.; Aanniz, T.; Touhtouh, J.; Kandoussi, I.; Hakmi, M.; Belyamani, L.; Ibrahimi, A.; Ouadghiri, M. Anthraquinones: A Promising Multi-target Therapeutic Scaffold To Treat Covid-19. Int. J. Appl. Biol. Pharm. Technol. 2021, 12, 338–355. [Google Scholar] [CrossRef]
- Stewart, C.A.; Gay, C.M.; Ramkumar, K.; Cargill, K.R.; Cardnell, R.J.; Nilsson, M.B.; Heeke, S.; Park, E.M.; Kundu, S.T.; Diao, L. Lung cancer models reveal SARS-CoV-2-induced EMT contributes to COVID-19 pathophysiology. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wang, J.Y.; Zhang, W.; Roehrl, M.W.; Roehrl, V.B.; Roehrl, M.H. An autoantigen profile of human A549 lung cells reveals viral and host etiologic molecular attributes of autoimmunity in COVID-19. J. Autoimmun. 2021, 120, 102644. [Google Scholar] [CrossRef]
- Liu, F.; Ma, J.; Shi, Z.; Zhang, Q.; Wang, H.; Li, D.; Song, Z.; Wang, C.; Jin, J.; Xu, J. Clerodane diterpenoids isolated from the leaves of Casearia graveolens. J. Nat. Prod. 2020, 83, 36–44. [Google Scholar] [CrossRef]
- Luo, W.; Sun, R.; Chen, X.; Li, J.; Jiang, J.; He, Y.; Shi, S.; Wen, H. ERK activation-mediated autophagy induction resists licochalcone A-induced anticancer activities in lung cancer cells in vitro. OncoTargets Ther. 2020, 13, 13437. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wu, Q.; Feng, J.; Yan, L.; Sun, Y.; Liu, S.; Xiang, Y.; Zhang, M.; Pan, T.; Chen, X. Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct. Target. Ther. 2020, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Boonjing, S.; Pothongsrisit, S.; Wattanathamsan, O.; Sritularak, B.; Pongrakhananon, V. Erianthridin induces non-small cell lung cancer cell apoptosis through the suppression of extracellular signal-regulated kinase activity. Planta Med. 2021, 87, 283–293. [Google Scholar] [CrossRef]
- Yang, J.; Cao, L.; Li, Y.; Liu, H.; Zhang, M.; Ma, H.; Wang, B.; Yuan, X.; Liu, Q. Gracillin isolated from Reineckia carnea induces apoptosis of A549 Cells via the mitochondrial pathway. Drug Des. Dev. Ther. 2021, 15, 233. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Zhang, W.; Li, T.; Jiang, L.; Lu, X.; Lin, J. Hispidulin exhibits potent anticancer activity in vitro and in vivo through activating ER stress in non-small-cell lung cancer cells. Oncol. Rep. 2020, 43, 1995–2003. [Google Scholar] [CrossRef]
- Sheng, H.; Lv, W.; Zhu, L.; Wang, L.; Wang, Z.; Han, J.; Hu, J. Liriopesides B induces apoptosis and cell cycle arrest in human non-small cell lung cancer cells. Int. J. Mol. Med. 2020, 46, 1039–1050. [Google Scholar] [CrossRef]




| Proteins | Function | References |
|---|---|---|
| Spike glycoprotein (S protein) | Mediate receptor recognition and membrane fusion for viral entry. | [34] |
| Angiotensin-converting enzyme 2 (ACE2) | Functional cellular receptor on host cell membrane. | [35] |
| Cathepsin L-cysteine peptidase | Facilitate the cleavage of S protein for activating membrane fusion. | [36] |
| Transmembrane protease serine 2 (TMPRSS2) | Cleave C-terminal of ACE2 and activate S-protein. | [37] |
| Nonstructural protein 1 (Nsp1) | Interact with 40S ribosome subunit to induce host mRNA degradation and inhibit type I interferon production. | [38] |
| Open reading frame 7a (ORF7a) | Block the activity of bone marrow stromal antigen 2 (BST-2) by directly binding and disrupting the glycosylation of BST-2. (BST-2 mediates the restriction of virus-like particle release.) | [39] |
| Replicase polyprotein 1ab | Transcription and replication of viral RNAs. | [40] |
| Papain-like proteinase (PLpro) | Cleave the N-terminal of replicase polyprotein leading to release of Nsp2 and Nsp3, which are in turn involved in viral replication. | [41] |
| Viral main protease (3C-like protease (3CLpro) or Mpro) | Essential for viral replication by controlling the activity of replication complex. | [42] |
| RNA dependent RNA polymerase (RdRp) (Nsp12) | Catalyze the replication of viral RNA from RNA template. | [33] |
| Non-structural protein 13 (Nsp13) helicase | NTPase, duplex RNA/DNA-unwinding and RNA-capping activities. | [43] |
| Phyto-Chemicals | Plants | Target Cell Lines | IC50 | Signaling Pathways | References |
|---|---|---|---|---|---|
| Graveospene A | Casearia graveolens | A549 | 1.9 M | Triggers apoptosis by inducing cell cycle arrest in phase G0/G1. | [225] |
| Licochalcone A | Xinjiang licorice and Glycyrrhiza inflata | A549, H460, SPC-A1, H23, and H1299 | <40 μM | Induces apoptosis by downregulating the expression of anti-apoptotic proteins such as c-IAP1, c-IAP2, XIAP, survivin, and c-FLIPL by inhibiting the activity of phosphorylated extracellular signal-regulated kin (ERK). | [226] |
| Erianin | Dendrobium chrysotoxum Lindl | H460 and H1299 | >100 nM | Induces apoptosis by promoting G2/M-phase arrest. | [227] |
| Erianthridin | Dendrobium formosum | H460 | 150.9 μM | Induces apoptosis by suppressing extracellular signal-regulated kinase activity. | [228] |
| A549 | 161.9 μM | ||||
| Gracillin | Reineckia carnea | A549 | 2.54 mol/L | Induces apoptosis via the mitochondrial pathway. | [229] |
| Hispidulin | Saussurea involucrate | A549 and NCL-H460 | <30 μM | Promotes apoptosis by increasing ROS production and activating ER stress. | [230] |
| Liriopesides B (LPB) | Liriope platyphylla | H460 | 42.62 µM | Induces apoptosis by inhibiting the progression of the cell cycle from G1 to S phase. | [231] |
| H1975 | 32.25 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yapasert, R.; Khaw-on, P.; Banjerdpongchai, R. Coronavirus Infection-Associated Cell Death Signaling and Potential Therapeutic Targets. Molecules 2021, 26, 7459. https://doi.org/10.3390/molecules26247459
Yapasert R, Khaw-on P, Banjerdpongchai R. Coronavirus Infection-Associated Cell Death Signaling and Potential Therapeutic Targets. Molecules. 2021; 26(24):7459. https://doi.org/10.3390/molecules26247459
Chicago/Turabian StyleYapasert, Rittibet, Patompong Khaw-on, and Ratana Banjerdpongchai. 2021. "Coronavirus Infection-Associated Cell Death Signaling and Potential Therapeutic Targets" Molecules 26, no. 24: 7459. https://doi.org/10.3390/molecules26247459
APA StyleYapasert, R., Khaw-on, P., & Banjerdpongchai, R. (2021). Coronavirus Infection-Associated Cell Death Signaling and Potential Therapeutic Targets. Molecules, 26(24), 7459. https://doi.org/10.3390/molecules26247459