
Computational Screening for the Anticancer Potential of Seed-Derived Antioxidant Peptides: A Cheminformatic Approach

 ,
,  , , and
, , and
Abstract
:1. Introduction
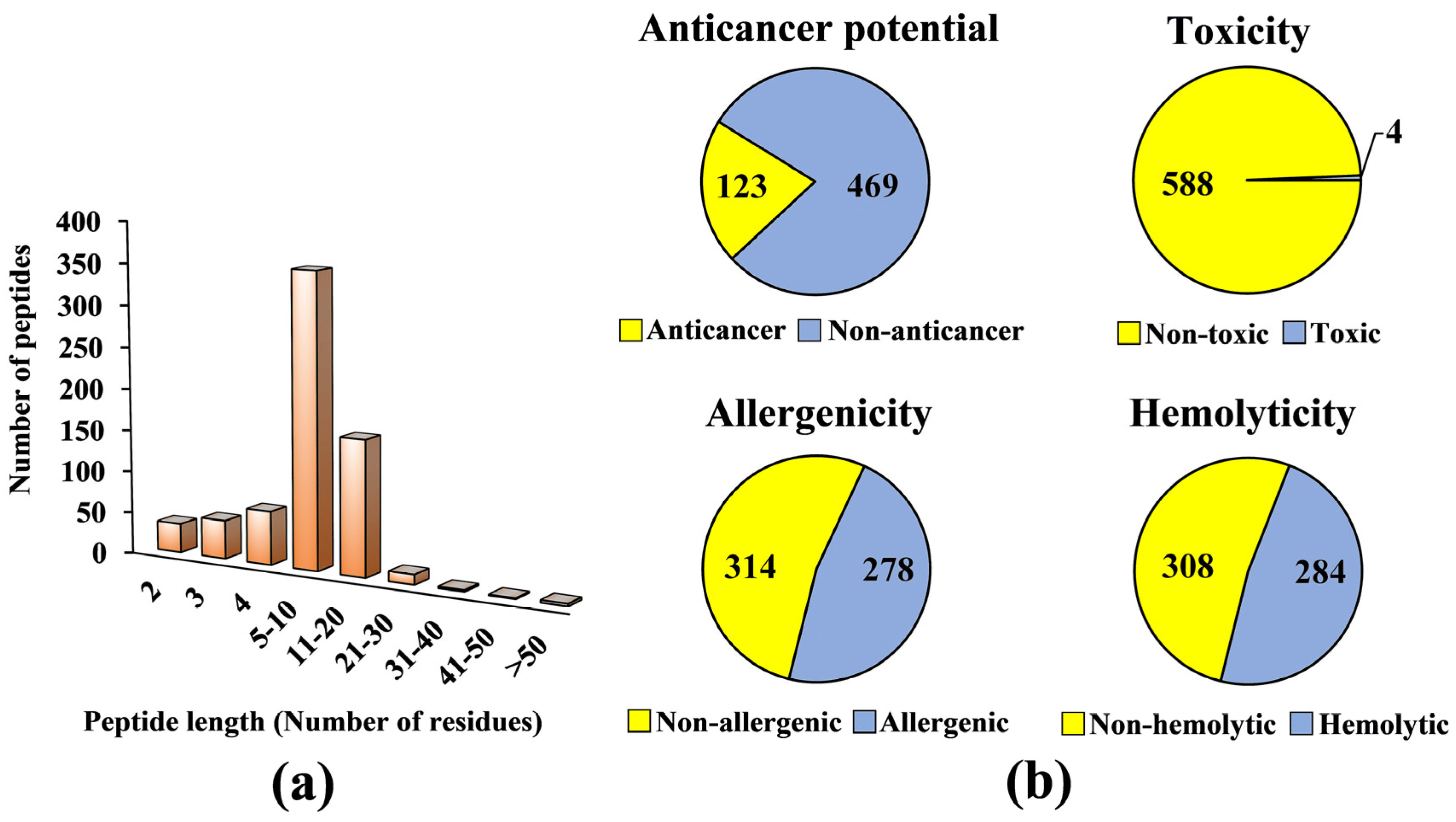
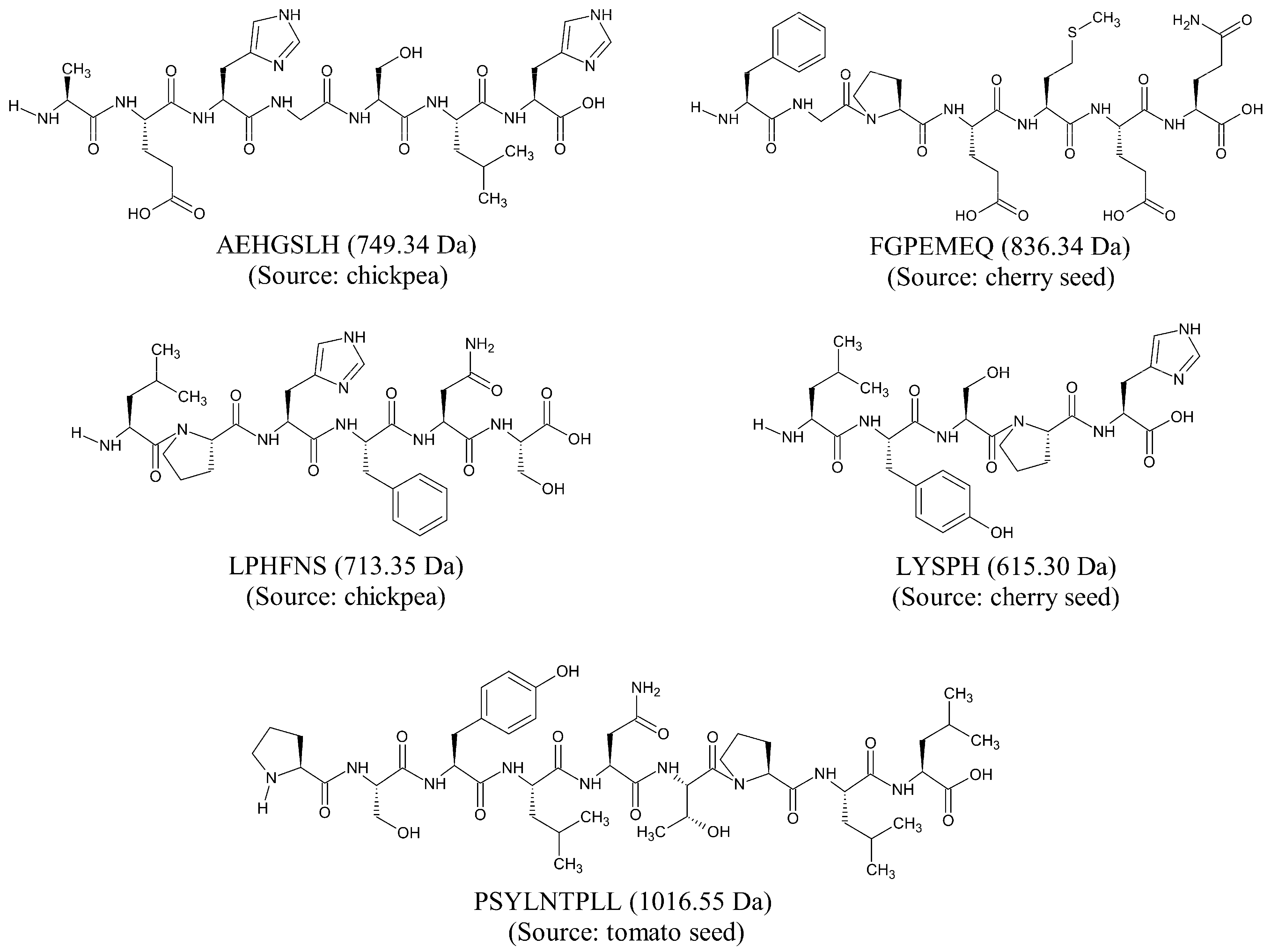
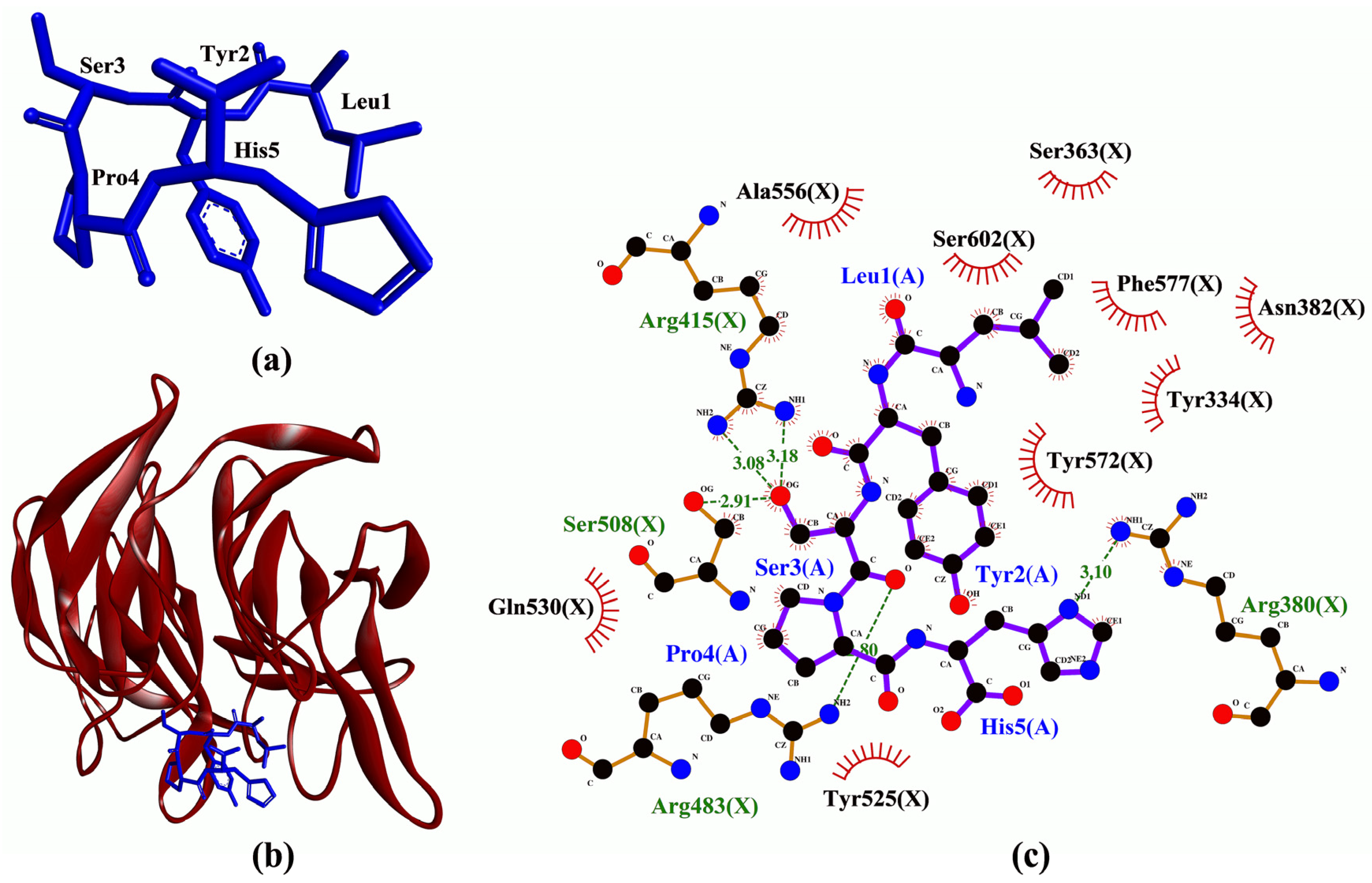
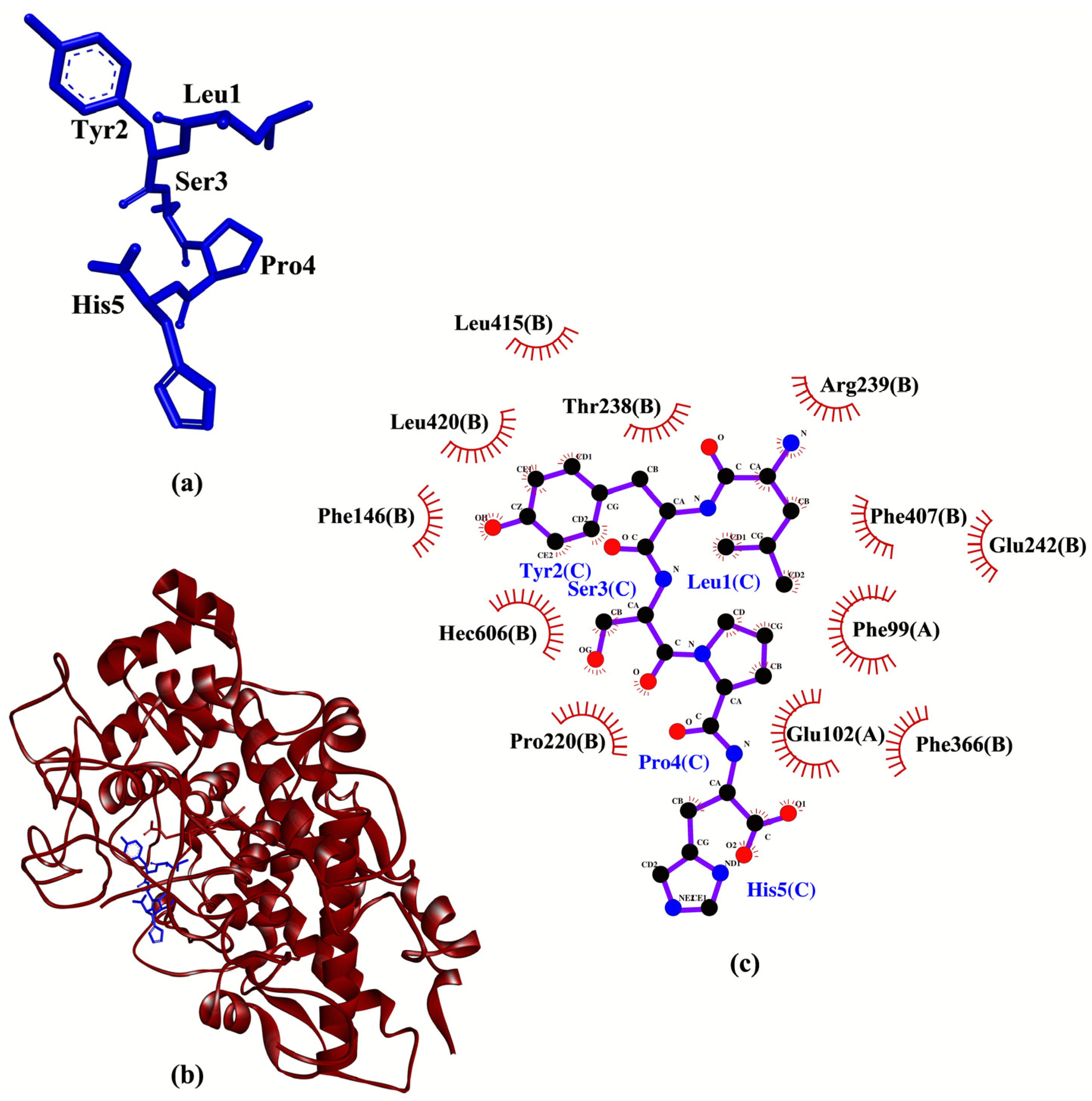
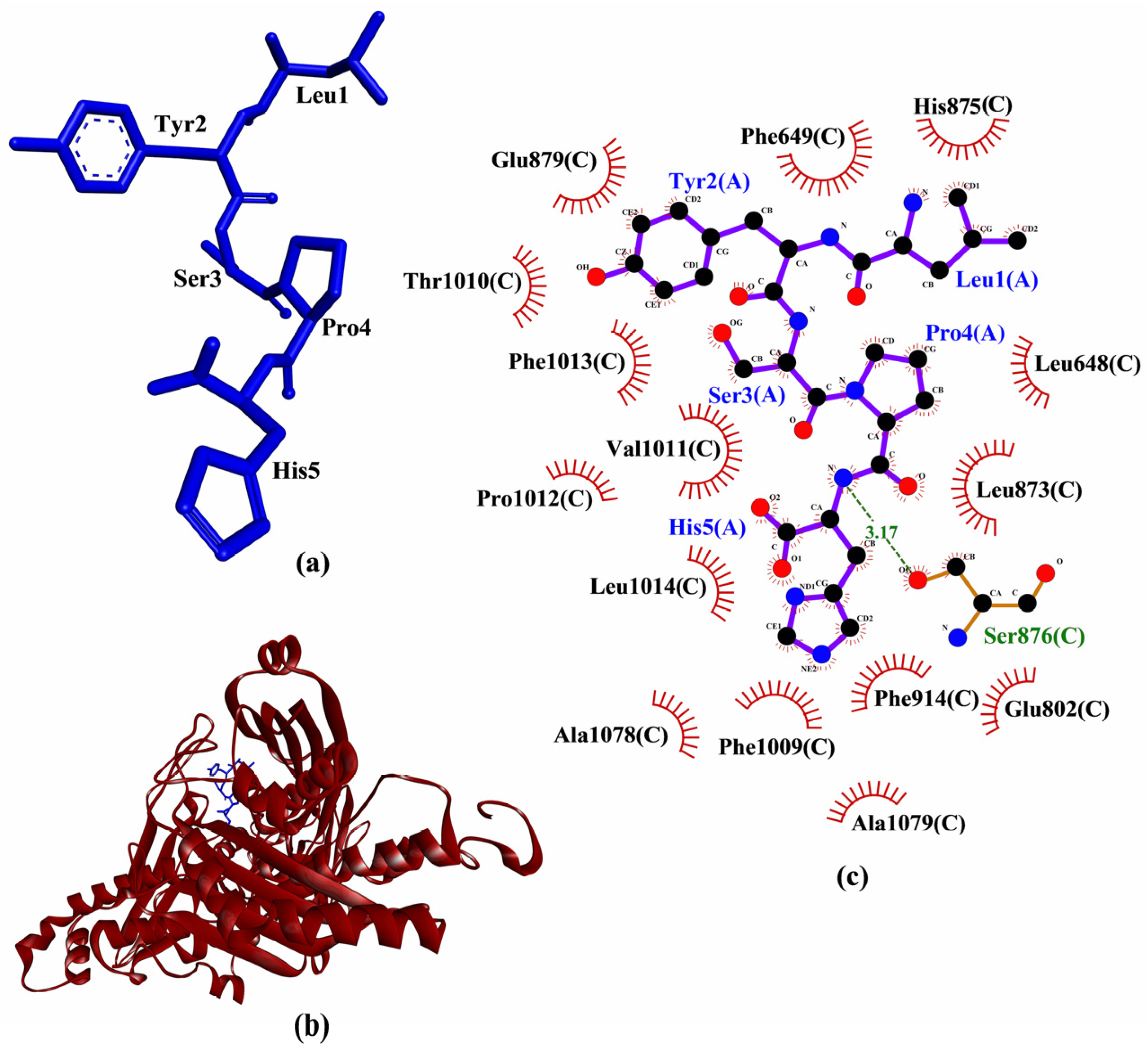
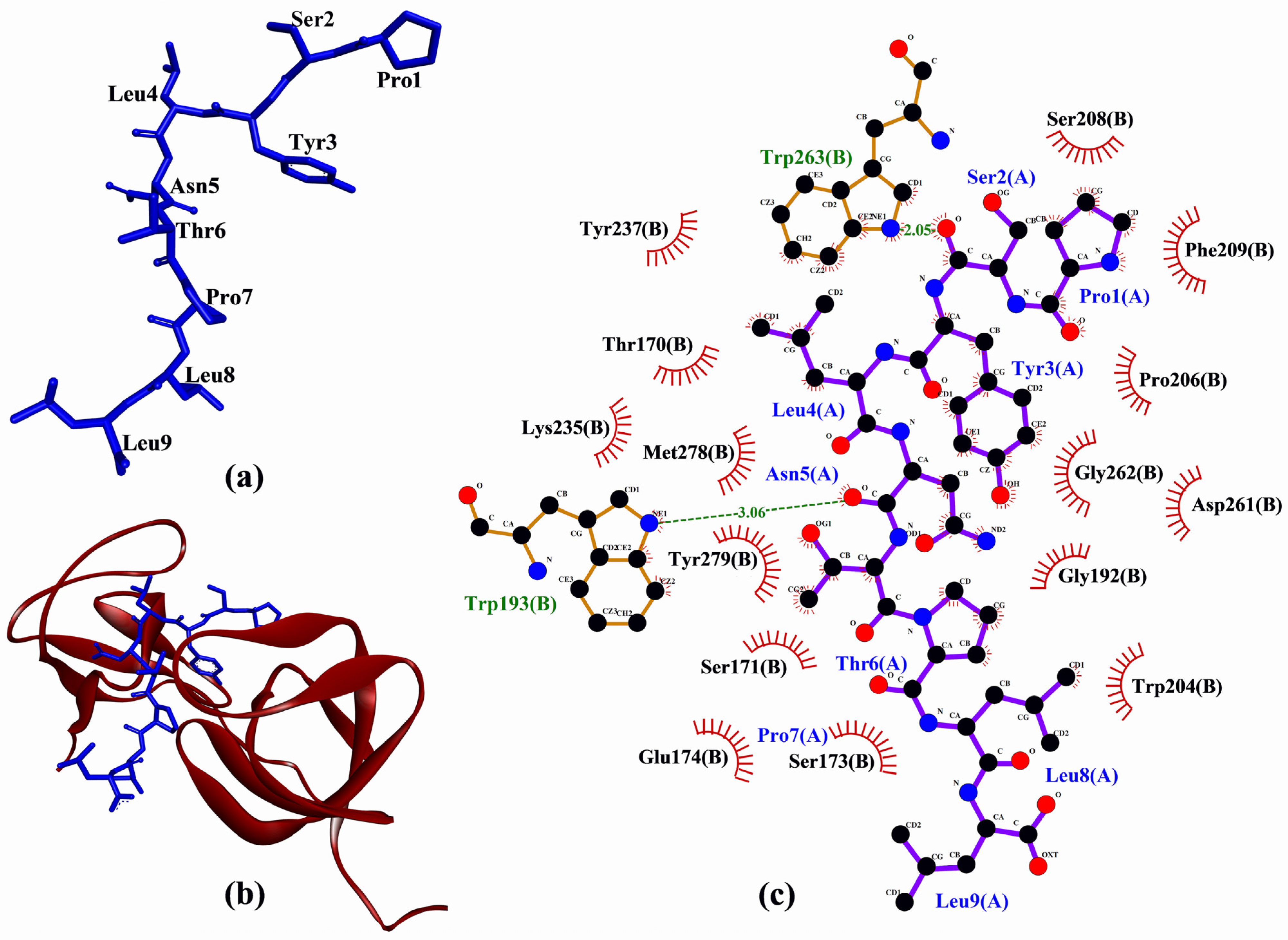
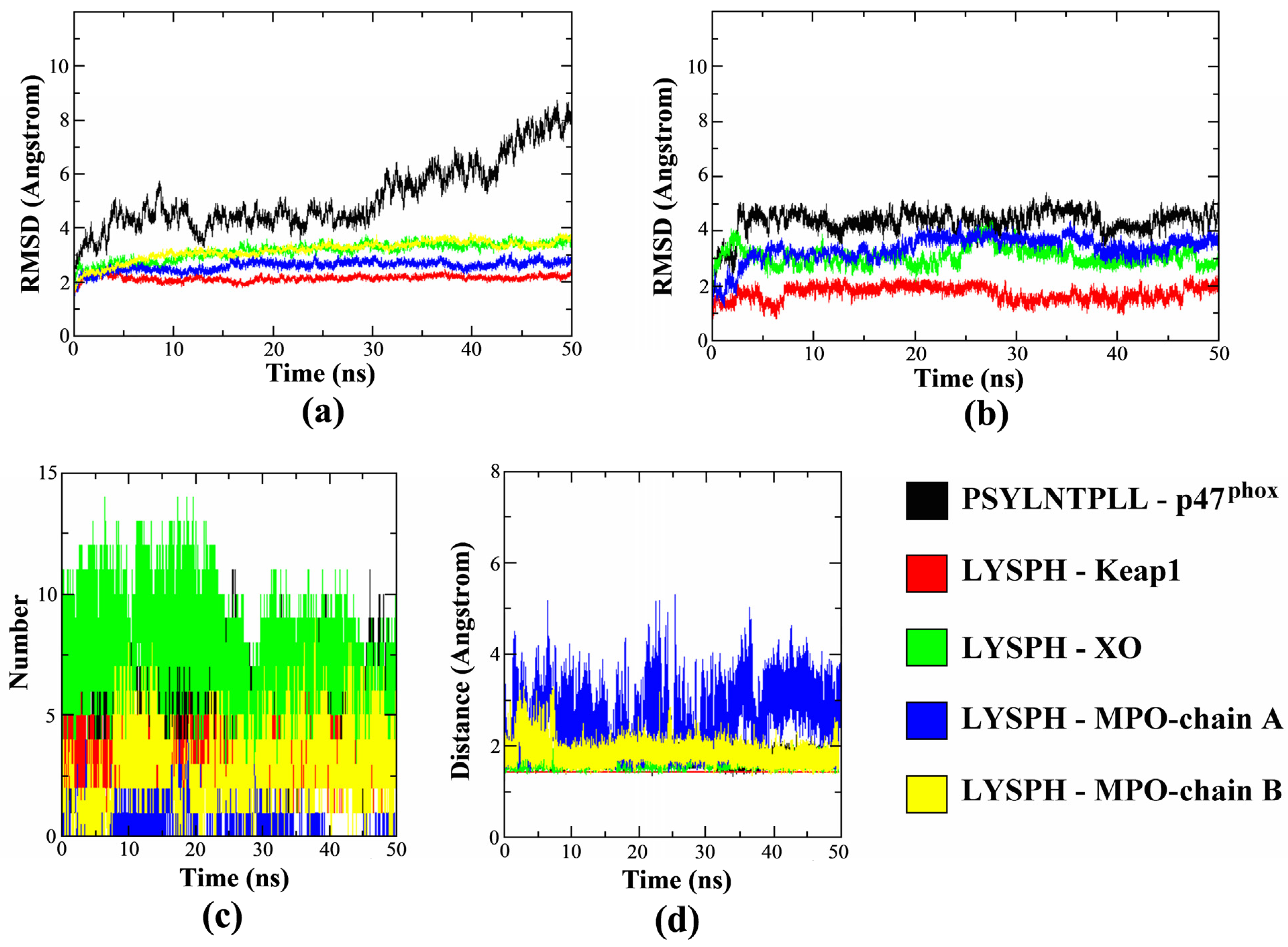
2. Results and Discussion
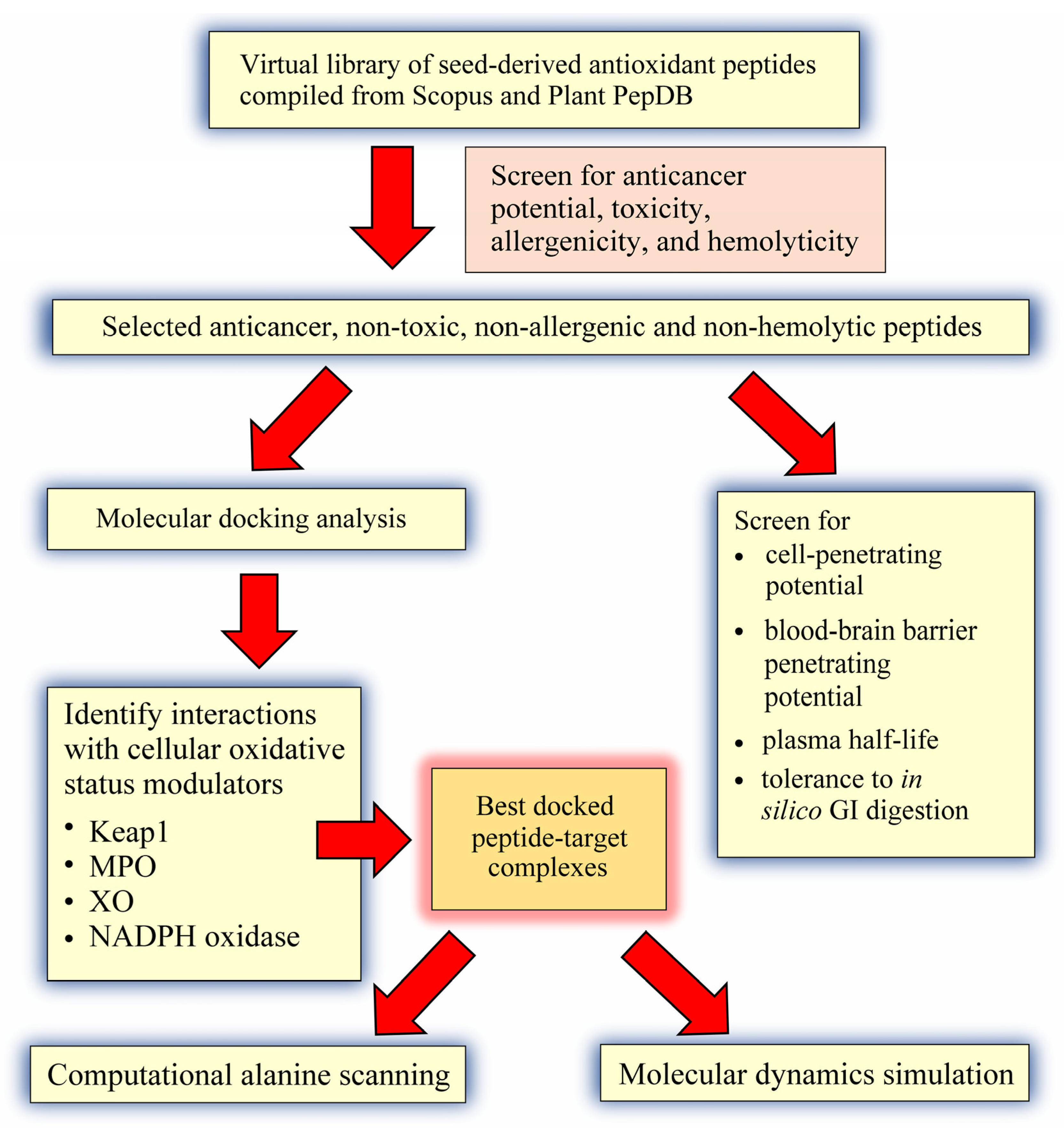
3. Materials and Methods
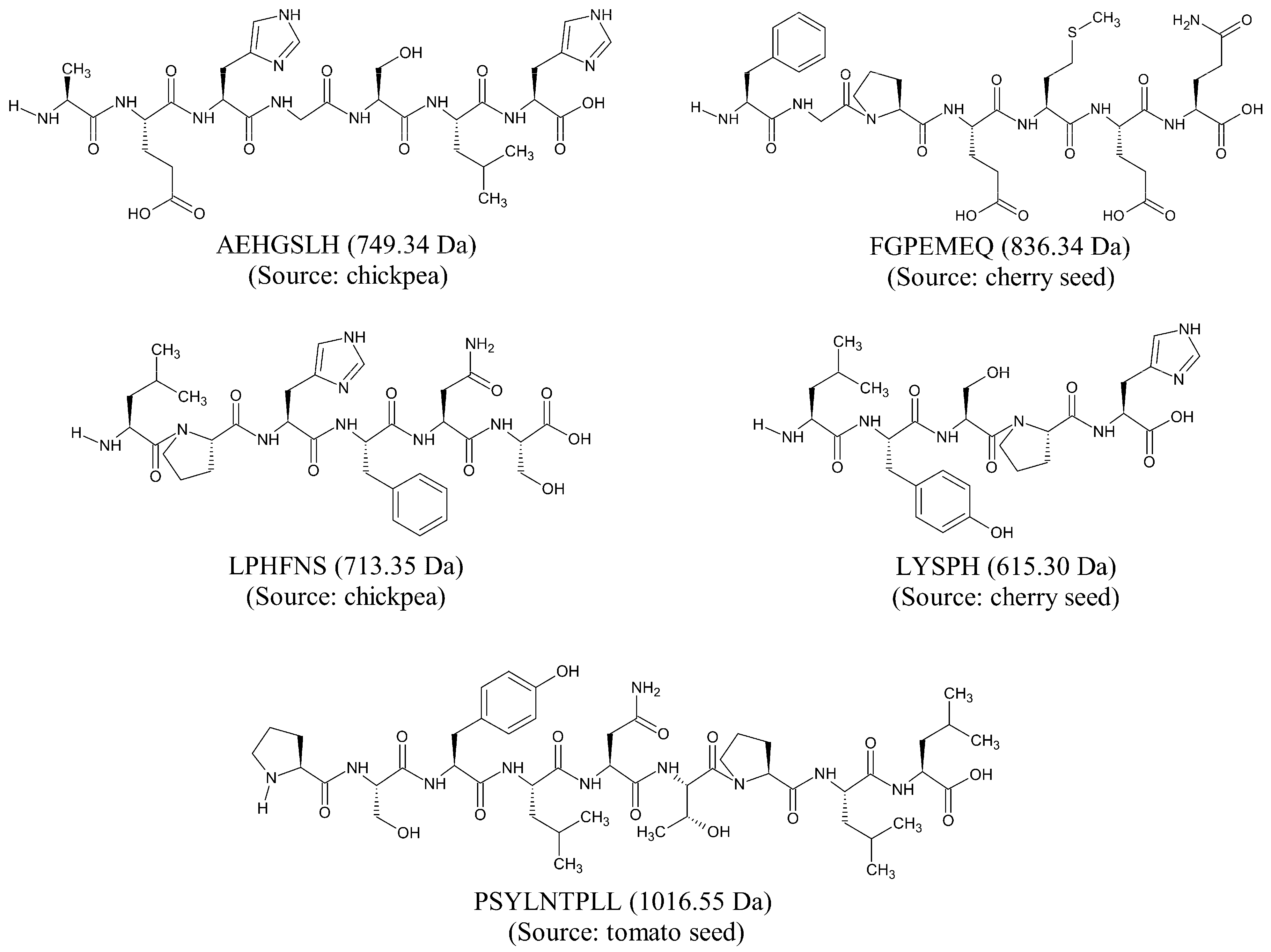
3.1. Compilation of a Virtual Library of Seed-Derived Antioxidant Peptides
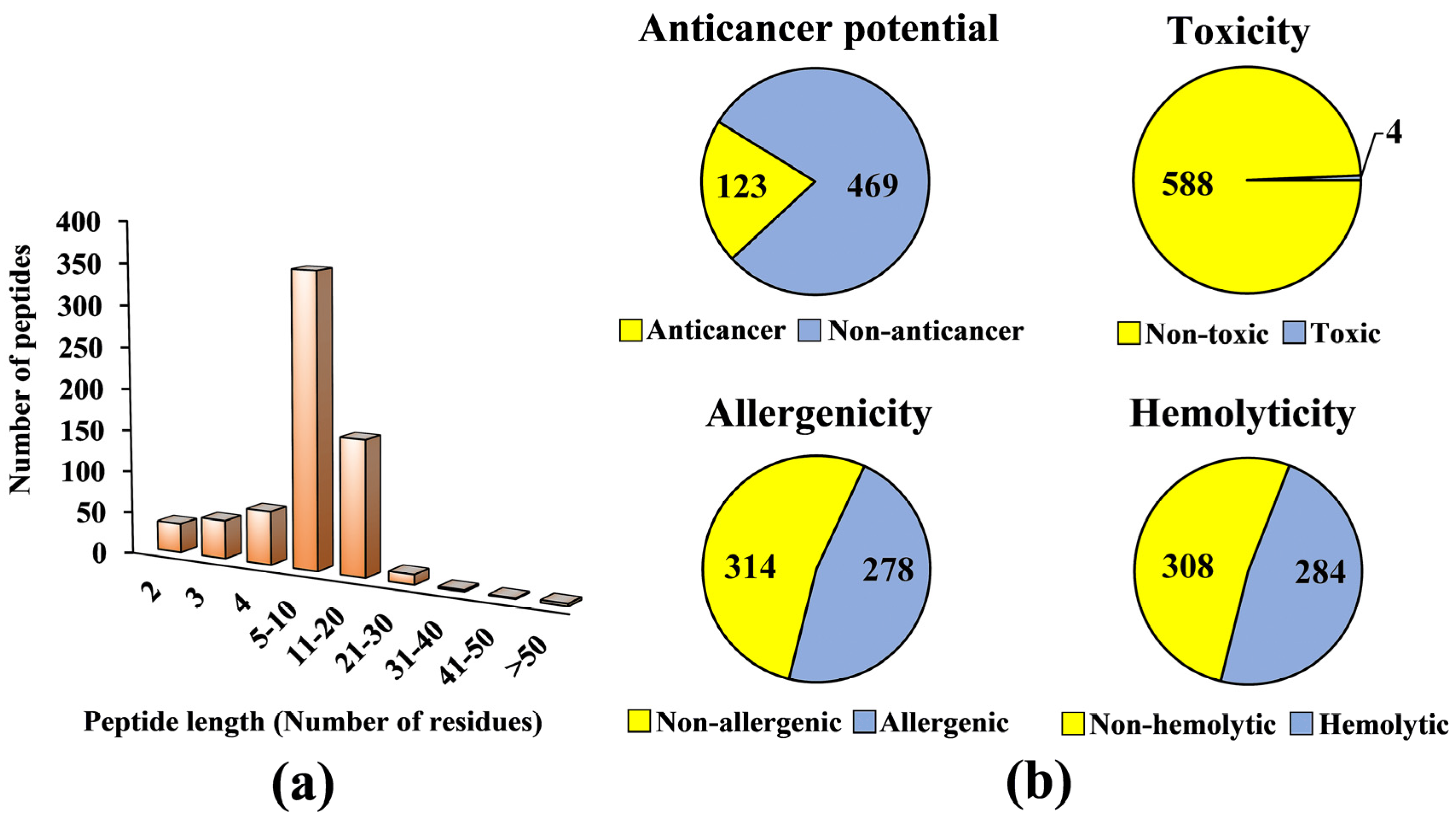
3.2. Virtual Screening for Anticancer Potential, Toxicity, Allergenicity and Hemolyticity
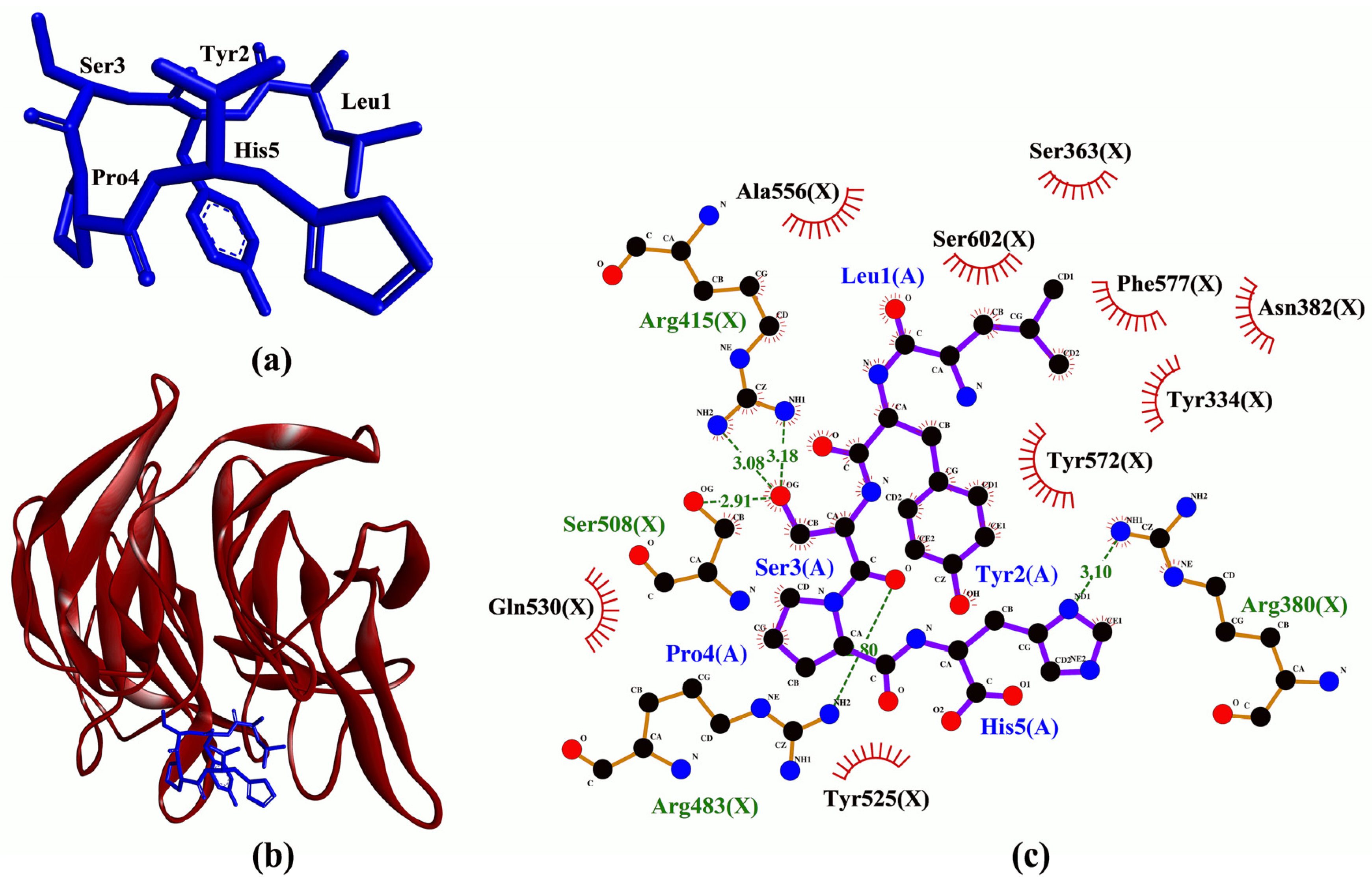
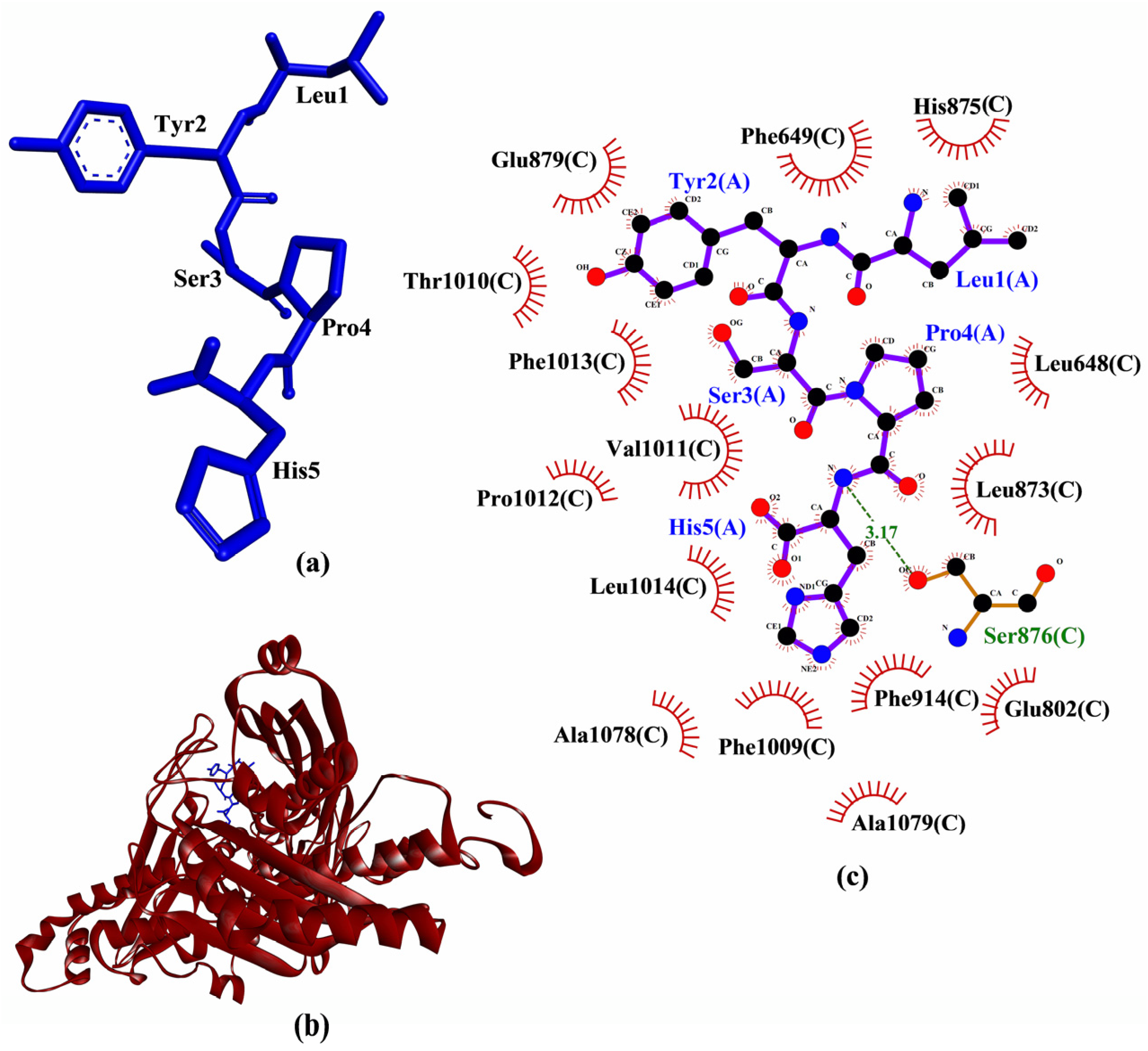
3.3. Molecular Docking Analysis
3.4. Prediction of Cell-Penetrating Potential, Blood-Brain Barrier Penetrating Potential, Plasma Half-Life, and Tolerance to In Silico GI Digestion
3.5. Computational Alanine Scanning Mutagenesis
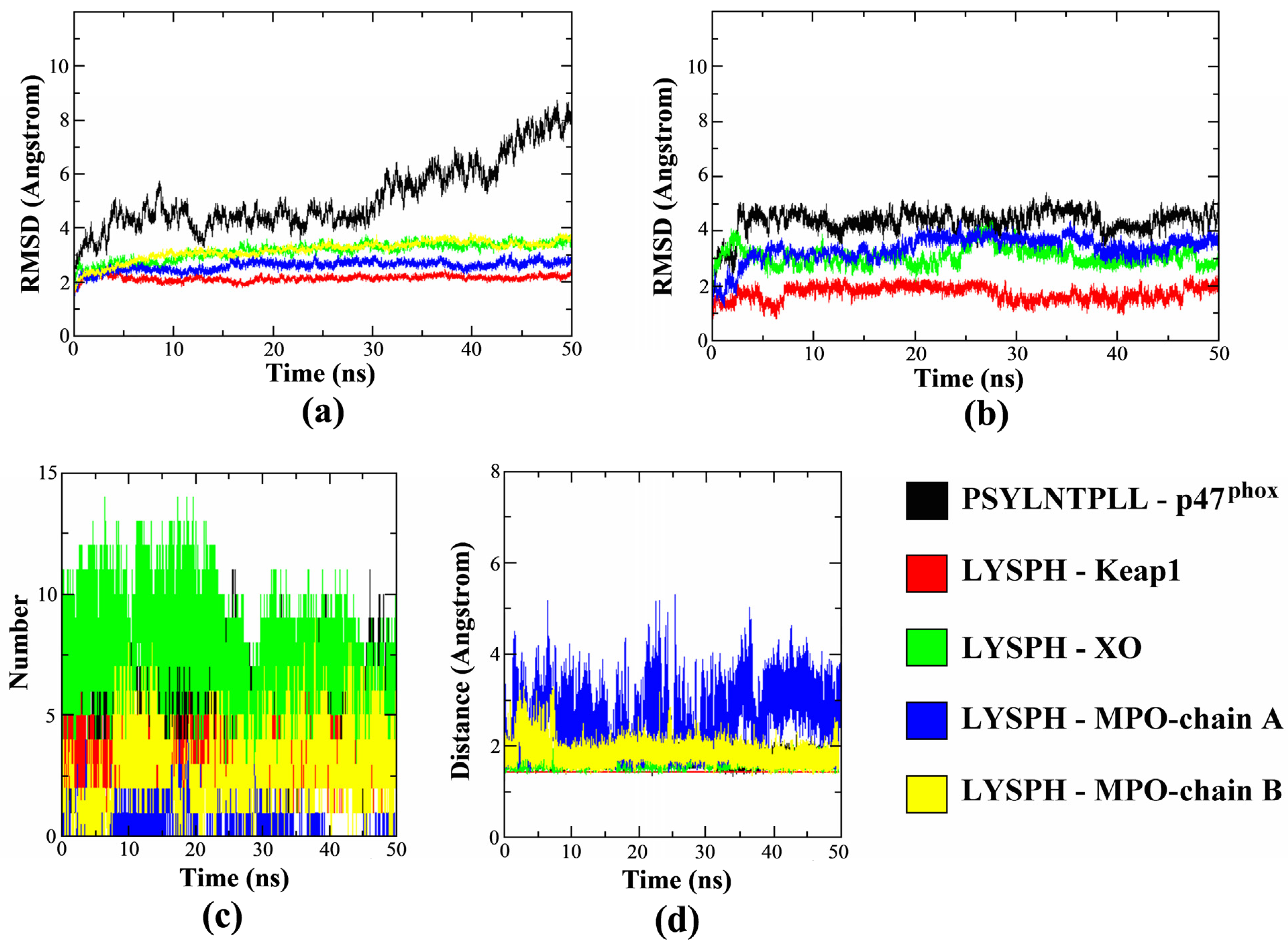
3.6. Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jakubczyk, A.; Karaś, M.; Rybczyńska-Tkaczyk, K.; Zielińska, E.; Zieliński, D. Current trends of bioactive peptides—New sources and therapeutic effect. Foods 2020, 9, 846. [Google Scholar] [CrossRef]
- Wong, F.-C.; Xiao, J.; Wang, S.; Ee, K.-Y.; Chai, T.-T. Advances on the antioxidant peptides from edible plant sources. Trends Food Sci. Technol. 2020, 99, 44–57. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.-T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Lopez, O.P.; Parhiz, H.; et al. A global review on short peptides: Frontiers and perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef]
- Chai, T.-T.; Tan, Y.-N.; Ee, K.-Y.; Xiao, J.; Wong, F.-C. Seeds, fermented foods, and agricultural by-products as sources of plant-derived antibacterial peptides. Crit. Rev. Food Sci. Nutr. 2019, 59, S162–S177. [Google Scholar] [CrossRef]
- Díaz-Gómez, J.L.; Castorena-Torres, F.; Preciado-Ortiz, R.E.; García-Lara, S. Anti-cancer activity of maize bioactive peptides. Front. Chem. 2017, 5, 44. [Google Scholar] [CrossRef]
- Ramkisson, S.; Dwarka, D.; Venter, S.; Mellem, J.J. In vitro anticancer and antioxidant potential of Amaranthus cruentus protein and its hydrolysates. Food Sci. Technol. 2020, 40, 634–639. [Google Scholar] [CrossRef]
- Mika, D.; Guruvayoorappan, C. Myeloperoxidase: The yin and yang in tumour progression. J. Exp. Ther. Oncol. 2011, 9, 93–100. [Google Scholar]
- Kargapolova, Y.; Geißen, S.; Zheng, R.; Baldus, S.; Winkels, H.; Adam, M. The enzymatic and non-enzymatic function of myeloperoxidase (MPO) in inflammatory communication. Antioxidants 2021, 10, 562. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.-L.; Liu, H.-X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef]
- Kazakov, Y.; Tarasov, A.; Alyoshina, L.; Brainina, K. Interplay between antioxidant activity, health and disease. Biointerface Res. Appl. Chem. 2020, 10, 4893–4901. [Google Scholar] [CrossRef]
- Oh, S.-H.; Choi, S.-Y.; Choi, H.-J.; Ryu, H.-M.; Kim, Y.-J.; Jung, H.-Y.; Cho, J.-H.; Kim, C.-D.; Park, S.-H.; Kwon, T.-H.; et al. The emerging role of xanthine oxidase inhibition for suppression of breast cancer cell migration and metastasis associated with hypercholesterolemia. FASEB J. 2019, 33, 7301–7314. [Google Scholar] [CrossRef]
- Cao, H.; Pauff, J.M.; Hille, R. X-ray crystal structure of a xanthine oxidase complex with the flavonoid inhibitor quercetin. J. Nat. Prod. 2014, 77, 1693–1699. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH oxidases (NOX): An overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Nobuhisa, I.; Yuzawa, S.; Takeya, R.; Torikai, S.; Saikawa, K.; Sumimoto, H.; Inagaki, F. NMR solution structure of the tandem Src homology 3 domains of p47phox complexed with a p22phox-derived proline-rich peptide. J. Biol. Chem. 2006, 281, 3660–3668. [Google Scholar] [CrossRef] [Green Version]
- Macías Pérez, M.E.; Hernández Rodríguez, M.; Cabrera Pérez, L.C.; Fragoso-Vázquez, M.J.; Correa-Basurto, J.; Padilla-Martínez, I.I.; Méndez Luna, D.; Mera Jiménez, E.; Flores Sandoval, C.; Tamay Cach, F.; et al. Aromatic regions govern the recognition of NADPH oxidase inhibitors as diapocynin and its analogues. Arch. Der Pharm. 2017, 350, 1700041. [Google Scholar] [CrossRef]
- Konaté, M.M.; Antony, S.; Doroshow, J.H. Inhibiting the activity of NADPH oxidase in cancer. Antioxid Redox Signal. 2020, 33, 435–454. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1–Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef]
- Tascioglu Aliyev, A.; Panieri, E.; Stepanić, V.; Gurer-Orhan, H.; Saso, L. Involvement of NRF2 in breast cancer and possible therapeutical role of polyphenols and melatonin. Molecules 2021, 26, 1853. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Zhang, J.; Zhang, H.; Duan, Y.; Ma, H. Study on the structure–activity relationship of watermelon seed antioxidant peptides by using molecular simulations. Food Chem. 2021, 364, 130432. [Google Scholar] [CrossRef]
- Koh, J.-A.; Ong, J.-H.; Abd Manan, F.; Ee, K.-Y.; Wong, F.-C.; Chai, T.-T. Discovery of bifunctional anti-DPP-IV and anti-ACE peptides from housefly larval proteins after in silico gastrointestinal digestion. Biointerface Res. Appl. Chem. 2022, 12, 4929–4944. [Google Scholar] [CrossRef]
- Das, D.; Jaiswal, M.; Khan, F.N.; Ahamad, S.; Kumar, S. PlantPepDB: A manually curated plant peptide database. Sci. Rep. 2020, 10, 2194. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, P.; Bhagat, D.; Mahalwal, M.; Sharma, N.; Raghava, G.P.S. AntiCP 2.0: An updated model for predicting anticancer peptides. Brief. Bioinform. 2020, 22, bbaa153. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Subramaniyam, S.; Shin, T.H.; Kim, M.O.; Lee, G. Machine-learning-based prediction of cell-penetrating peptides and their uptake efficiency with improved accuracy. J. Proteome Res. 2018, 17, 2715–2726. [Google Scholar] [CrossRef]
- Kochnev, Y.; Hellemann, E.; Cassidy, K.C.; Durrant, J.D. Webina: An open-source library and web app that runs AutoDock Vina entirely in the web browser. Bioinformatics 2020, 36, 4513–4515. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Shukla, R.; Tripathi, T. Molecular dynamics simulation of protein and protein-ligand complexes. In Computer-Aided Drug Design; Springer: Singapore, 2020; Chapter 7; pp. 133–161. [Google Scholar]
- Ong, J.-H.; Liang, C.-E.; Wong, W.-L.; Wong, F.-C.; Chai, T.-T. Multi-target anti-SARS-CoV-2 peptides from mealworm proteins: An in silico study. Malays. J. Biochem. Mol. Biol. 2021, 24, 83–91. [Google Scholar]
- Mohana, D.S.; Chai, T.-T.; Wong, F.-C. Antioxidant and protein protection potentials of fennel seed-derived protein hydrolysates and peptides. Mod. Food Sci. Technol. 2019, 35, 22–29. [Google Scholar] [CrossRef]
- Chai, T.-T.; Soo, Z.-Y.; Hsu, K.-C.; Li, J.-C.; Abd Manan, F.; Wong, F.-C. Antioxidant activity of semen cassiae protein hydrolysate: Thermal and gastrointestinal stability, peptide identification, and in silico analysis. Mod. Food Sci. Technol. 2019, 35, 38–48. [Google Scholar]
- Chai, T.-T.; Xiao, J.; Mohana Dass, S.; Teoh, J.-Y.; Ee, K.-Y.; Ng, W.-J.; Wong, F.-C. Identification of antioxidant peptides derived from tropical jackfruit seed and investigation of the stability profiles. Food Chem. 2021, 340, 127876. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.; Kumar, R.; Singh, S.; Tuknait, A.; Gautam, A.; Mathur, D.; Anand, P.; Varshney, G.C.; Raghava, G.P.S. A web server and mobile app for computing hemolytic potency of peptides. Sci. Rep. 2016, 6, 22843. [Google Scholar] [CrossRef]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Open Source Drug Discovery, C.; Raghava, G.P.S. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Ji, D.; Udenigwe, C.C.; Agyei, D. Antioxidant peptides encrypted in flaxseed proteome: An in silico assessment. Food Sci. Hum. Wellness 2019, 8, 306–314. [Google Scholar] [CrossRef]
- Duan, X.; Zhang, M.; Chen, F. Prediction and analysis of antimicrobial peptides from rapeseed protein using in silico approach. J. Food Biochem. 2021, 45, e13598. [Google Scholar] [CrossRef]
- Taniguchi, M.; Aida, R.; Saito, K.; Kikura, T.; Ochiai, A.; Saitoh, E.; Tanaka, T. Identification and characterization of multifunctional cationic peptides from enzymatic hydrolysates of soybean proteins. J. Biosci. Bioeng. 2020, 129, 59–66. [Google Scholar] [CrossRef]
- Ghribi, A.M.; Sila, A.; Przybylski, R.; Nedjar-Arroume, N.; Makhlouf, I.; Blecker, C.; Attia, H.; Dhulster, P.; Bougatef, A.; Besbes, S. Purification and identification of novel antioxidant peptides from enzymatic hydrolysate of chickpea (Cicer arietinum L.) protein concentrate. J. Funct. Foods 2015, 12, 516–525. [Google Scholar] [CrossRef]
- Chai, T.-T.; Law, Y.-C.; Wong, F.-C.; Kim, S.-K. Enzyme-assisted discovery of antioxidant peptides from edible marine invertebrates: A review. Mar. Drugs 2017, 15, 42. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.-C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liu, J.; Nie, S.; Ding, L.; Wang, L.; Liu, J.; Liu, W.; Zhang, T. Direct inhibition of Keap1–Nrf2 interaction by egg-derived peptides DKK and DDW revealed by molecular docking and fluorescence polarization. RSC Adv. 2017, 7, 34963–34971. [Google Scholar] [CrossRef] [Green Version]
- Shaw, S.A.; Vokits, B.P.; Dilger, A.K.; Viet, A.; Clark, C.G.; Abell, L.M.; Locke, G.A.; Duke, G.; Kopcho, L.M.; Dongre, A.; et al. Discovery and structure activity relationships of 7-benzyl triazolopyridines as stable, selective, and reversible inhibitors of myeloperoxidase. Bioorganic Med. Chem. 2020, 28, 115723. [Google Scholar] [CrossRef]
- Maiocchi, S.L.; Ku, J.; Thai, T.; Chan, E.; Rees, M.D.; Thomas, S.R. Myeloperoxidase: A versatile mediator of endothelial dysfunction and therapeutic target during cardiovascular disease. Pharmacol. Ther. 2021, 221, 107711. [Google Scholar] [CrossRef]
- Kang, N.; Kim, E.-A.; Kim, J.; Lee, S.-H.; Heo, S.-J. Identifying potential antioxidant properties from the viscera of sea snails (Turbo cornutus). Mar. Drugs 2021, 19, 567. [Google Scholar] [CrossRef] [PubMed]
- Davey, C.A.; Fenna, R.E. 2.3 Å resolution x-ray crystal structure of the bisubstrate analogue inhibitor salicylhydroxamic acid bound to human myeloperoxidase: A model for a prereaction complex with hydrogen peroxide. Biochemistry 1996, 35, 10967–10973. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhou, Y.; Zhou, S.; Chen, S.; Wu, Y.; Li, L.; Yang, X. Purification and identification of novel xanthine oxidase inhibitory peptides derived from round scad (Decapterus maruadsi) protein hydrolysates. Mar. Drugs 2021, 19, 538. [Google Scholar] [CrossRef]
- Yu, Z.; Kan, R.; Wu, S.; Guo, H.; Zhao, W.; Ding, L.; Zheng, F.; Liu, J. Xanthine oxidase inhibitory peptides derived from tuna protein: Virtual screening, inhibitory activity, and molecular mechanisms. J. Sci. Food Agric. 2021, 101, 1349–1354. [Google Scholar] [CrossRef]
- Nakanishi, A.; Imajoh-Ohmi, S.; Fujinawa, T.; Kikuchi, H.; Kanegasaki, S. Direct evidence for interaction between COOH-terminal regions of cytochrome b558 subunits and cytosolic 47-kDa protein during activation of an O(2−)-generating system in neutrophils. J. Biol. Chem. 1992, 267, 19072–19074. [Google Scholar] [CrossRef]
- Huang, J.; Kleinberg, M.E. Activation of the phagocyte NADPH oxidase protein p47phox: Phosphorylation controls SH3 domain-dependent binding to p22phox *. J. Biol. Chem. 1999, 274, 19731–19737. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Sawney, S.; Saini, V.; Steffi, C.; Tiwari, M.; Saluja, D. Esculetin induces antiproliferative and apoptotic response in pancreatic cancer cells by directly binding to KEAP1. Mol. Cancer 2016, 15, 64. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Jing, X.; Shi, Y.; Xu, H.; Du, J.; Guan, T.; Weihrauch, D.; Jones, D.W.; Wang, W.; Gourlay, D.; et al. N-acetyl lysyltyrosylcysteine amide inhibits myeloperoxidase, a novel tripeptide inhibitor1[S]. J. Lipid Res. 2013, 54, 3016–3029. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Tang, T.; Wang, S.; Cai, T.; Tao, H.; Zhang, Q.; Qi, S.; Qi, Z. Aloin inhibits the proliferation and migration of gastric cancer cells by regulating NOX2-ROS-mediated pro-survival signal pathways. Drug Des. Devel. 2020, 14, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Rymaszewski, A.L.; Tate, E.; Yimbesalu, J.P.; Gelman, A.E.; Jarzembowski, J.A.; Zhang, H.; Pritchard, K.A., Jr.; Vikis, H.G. The role of neutrophil myeloperoxidase in models of lung tumor development. Cancers 2014, 6, 1111–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quah, Y.; Mohd Ismail, N.I.; Ooi, J.L.S.; Affendi, Y.A.; Abd Manan, F.; Wong, F.-C.; Chai, T.-T. Identification of novel cytotoxic peptide KENPVLSLVNGMF from marine sponge Xestospongia testudinaria, with characterization of stability in human serum. Int. J. Pept. Res. Ther. 2018, 24, 189–199. [Google Scholar] [CrossRef]
- Amigo, L.; Hernández-Ledesma, B. Current evidence on the bioavailability of food bioactive peptides. Molecules 2020, 25, 4479. [Google Scholar] [CrossRef]
- Sharifi, S.; Samani, A.A.; Ahmadian, E.; Eftekhari, A.; Derakhshankhah, H.; Jafari, S.; Mokhtarpour, M.; Vahed, S.Z.; Salatin, S.; Dizaj, S.M. Oral delivery of proteins and peptides by mucoadhesive nanoparticles. Biointerface Res. Appl. Chem. 2019, 9, 3849–3852. [Google Scholar] [CrossRef]
- Wu, Y.; He, H.; Hou, T. Purification, identification, and computational analysis of xanthine oxidase inhibitory peptides from kidney bean. J. Food Sci. 2021, 86, 1081–1088. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 2021, 9, 71. [Google Scholar] [CrossRef]
- Mangold, M.; Gütschow, M.; Stirnberg, M. A short peptide inhibitor as an activity-based probe for matriptase-2. Pharmaceuticals 2018, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- Baig, M.S.; Alagumuthu, M.; Rajpoot, S.; Saqib, U. Identification of a potential peptide inhibitor of SARS-CoV-2 targeting its entry into the host cells. Drugs R D 2020, 20, 161–169. [Google Scholar] [CrossRef]
- Zhou, P.; Miao, Q.; Yan, F.; Li, Z.; Jiang, Q.; Wen, L.; Meng, Y. Is protein context responsible for peptide-mediated interactions? Mol. Omics 2019, 15, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Azizian, H.; Nabati, F.; Sharifi, A.; Siavoshi, F.; Mahdavi, M.; Amanlou, M. Large-scale virtual screening for the identification of new Helicobacter pylori urease inhibitor scaffolds. J. Mol. Model. 2012, 18, 2917–2927. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Ma, B.; Nussinov, R. Hot regions in protein–protein interactions: The organization and contribution of structurally conserved hot spot residues. J. Mol. Biol. 2005, 345, 1281–1294. [Google Scholar] [CrossRef]
- London, N.; Movshovitz-Attias, D.; Schueler-Furman, O. The structural basis of peptide-protein binding strategies. Structure 2010, 18, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Thévenet, P.; Shen, Y.; Maupetit, J.; Guyon, F.; Derreumaux, P.; Tufféry, P. PEP-FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012, 40, W288–W293. [Google Scholar] [CrossRef] [Green Version]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Maupetit, J.; Derreumaux, P.; Tufféry, P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theory Comput. 2014, 10, 4745–4758. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2018, 47, D464–D474. [Google Scholar] [CrossRef] [Green Version]
- Velázquez-Libera, J.L.; Durán-Verdugo, F.; Valdés-Jiménez, A.; Valdés-Jiménez, A.; Núñez-Vivanco, G.; Caballero, J. LigRMSD: A web server for automatic structure matching and RMSD calculations among identical and similar compounds in protein-ligand docking. Bioinformatics 2020, 36, 2912–2914. [Google Scholar] [CrossRef]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Zou, X. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 66, 399–421. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Zou, X. An iterative knowledge-based scoring function for protein–protein recognition. Proteins: Struct. Funct. Bioinform. 2008, 72, 557–579. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, D.; Huang, S.-Y. Efficient conformational ensemble generation of protein-bound peptides. J. Cheminform. 2017, 9, 59. [Google Scholar] [CrossRef]
- Zhou, P.; Li, B.; Yan, Y.; Jin, B.; Wang, L.; Huang, S.Y. Hierarchical flexible peptide docking by conformer generation and ensemble docking of peptides. J. Chem. Inf. Model. 2018, 58, 1292–1302. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patiyal, S.; Dhall, A.; Sharma, N.; Raghava, G.P.S. B3pred: A random-forest-based method for predicting and designing blood–brain barrier penetrating peptides. Pharmaceutics 2021, 13, 1237. [Google Scholar] [CrossRef]
- Mathur, D.; Singh, S.; Mehta, A.; Agrawal, P.; Raghava, G.P.S. In silico approaches for predicting the half-life of natural and modified peptides in blood. PLoS ONE 2018, 13, e0196829. [Google Scholar] [CrossRef] [Green Version]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM database of bioactive peptides: Current opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef] [Green Version]
- Ibarra, A.A.; Bartlett, G.J.; Hegedüs, Z.; Dutt, S.; Hobor, F.; Horner, K.A.; Hetherington, K.; Spence, K.; Nelson, A.; Edwards, T.A.; et al. Predicting and experimentally validating hot-spot residues at protein–protein interfaces. ACS Chem. Biol. 2019, 14, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.W.; Ibarra, A.A.; Bartlett, G.J.; Wilson, A.J.; Woolfson, D.N.; Sessions, R.B. BAlaS: Fast, interactive and accessible computational alanine-scanning using BudeAlaScan. Bioinformatics 2020, 36, 2917–2919. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Huang, W.; Lin, Z.; van Gunsteren, W.F. Validation of the GROMOS 54A7 force field with respect to β-peptide folding. J. Chem. Theory Comput. 2011, 7, 1237–1243. [Google Scholar] [CrossRef]
- Leherte, L.; Vercauteren, D.P. Reduced point charge models of proteins: Effect of protein–water interactions in molecular dynamics simulations of ubiquitin systems. J. Phys. Chem. B 2017, 121, 9771–9784. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Alves, A.; Ormersbach, F.; Wade, R.C. Prediction of the drug–target binding kinetics for flexible proteins by comparative binding energy analysis. J. Chem. Inf. Model. 2021, 61, 3708–3721. [Google Scholar] [CrossRef]
- Sabri, M.Z.; Hamid, A.A.A.; Hitam, S.M.S.; Rahim, M.Z.A. The assessment of three dimensional modelling design for single strand DNA aptamers for computational chemistry application. Biophys. Chem. 2020, 267, 106492. [Google Scholar] [CrossRef] [PubMed]
- Sabri, M.Z.; Abdul Hamid, A.A.; Sayed Hitam, S.M.; Abdul Rahim, M.Z. In silico screening of aptamers configuration against hepatitis b surface antigen. Adv. Bioinform. 2019, 2019, 6912914. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Binding Affinity (kcal/mol) | Interaction with Keap1 a | ||
|---|---|---|---|---|
| Hydrogen Bond | Hydrophobic Interaction | Salt Bridge | ||
| LYSPH | −7.6 | Arg380, Arg415(2), Arg483, Ser508 | Tyr334, Ser363, Arg380, Asn382, Arg415, Ser508, Tyr525, Gln530, Ala556, Tyr572, Phe577, Ser602 | - |
| LPHFNS | −7.4 | Ser363, Arg380, Asn414, Arg415(2), Gln530 | Tyr334, Ser363, Arg380, Asn382, Asn414, Arg415, Arg483, Tyr525, Gln530, Ser555, Ala556, Tyr572, Phe577, Ser602 | - |
| AEHGSLH | −7.2 | Tyr334(2), Arg380, Asn382(2), Arg415 | Tyr334, Arg336, Ser363, Arg380, Asn382, Ser383, Pro384, Arg415, Ile461, Arg483, Ser508, Tyr525, Gln530, Ala556, Tyr572, Ser602 | - |
| FGPEMEQ | −7.0 | Ser363, Arg380(2), Asn382(2), Asn387, Asn414 | Tyr334, Ser363, Arg380, Asn382, Asn387, Asp389, Arg415, Gly433, Ile461, Ser555, Ala556, Tyr572, Phe577 | Arg380(2) |
| PSYLNTPLL | −6.4 | Arg380(2), Asn382, Arg415, Arg483, Ser555, Tyr572 | Tyr334, Ser363, Gly364, Arg380, Asn382, Arg415, Arg483, Tyr525, Gln530, Ser555, Ala556, Tyr572, Phe577 | - |
| Peptide | Binding Affinity (kcal/mol) | Interaction with MPO | |
|---|---|---|---|
| Hydrogen Bond | Hydrophobic Interaction | ||
| LYSPH | −7.1 | - | Phe99, Glu102, Phe146, Pro220, Thr238, Arg239, Glu242, Phe366, Phe407, Leu415, Leu420, Hec606 |
| LPHFNS | −6.9 | - | Phe99, Glu102, Glu116, Phe147, Pro220, Thr238, Arg239, Phe366, Phe407, Met411, Hec606 |
| FGPEMEQ | −6.8 | Glu102 | Phe99, Thr100, Glu102, Glu116, Pro145, Phe147, Leu216, Pro220, Thr238, Arg239, Glu242, Phe366, Phe407, Met411, Leu415, Arg424, Hec606 |
| AEHGSLH | −6.3 | - | Phe99, Thr100, Glu102, Glu116, Pro145, Phe146, Phe147, Thr238, Arg239, Glu242, Phe366, Phe407, Val410, Met411, Leu415, Arg424, Hec606 |
| PSYLNTPLL | −3.0 | Thr100 | Phe99, Glu102, Glu116, Pro145, Phe147, Leu216, Pro220, Thr238, Arg239, Phe366, Phe407, Val410, Met411, Arg412, Leu415, Hec606 |
| Peptide | Binding Affinity (kcal/mol) | Interaction with XO | |||
|---|---|---|---|---|---|
| Hydrogen Bond a | Hydrophobic Interaction | Salt Bridge | External Bond | ||
| LYSPH | −6.2 | Ser876 | Leu648, Phe649, Glu802, Leu873, His875, Ser876, Glu879, Phe914, Phe1009, Thr1010, Val1011, Pro1012, Phe1013, Leu1014, Ala1078, Ala1079 | - | - |
| FGPEMEQ | −5.9 | Glu802, Ser876(2), Ala1079 | Leu648, Phe649, Gln767, Phe798, Gly799, Glu802, Thr803, Leu873, His875, Ser876, Glu879, Arg880, Ala910, Phe911, Arg912, Phe914, Phe1009, Thr1010, Val1011, Pro1012, Leu1014, Pro1076, Ala1078, Ala1079, Ser1080, Glu1261 | His875 | - |
| LPHFNS | −4.7 | His875, Ser876 | Leu648, Phe649, Glu802, Leu873, His875, Ser876, Glu879, Arg880, Phe914, Phe1009, Thr1010, Val1011, Pro1012, Phe1013, Leu1014, Ala1078, Ala1079, Glu1261 | - | - |
| AEHGSLH | −3.4 | Glu879 | Leu648, Phe649, Leu712, Glu802, Leu873, His875, Ser876, Glu879, Phe914, Phe1009, Thr1010, Val1011, Pro1012, Phe1013, Leu1014, Pro1076, Tyr1140, Phe1142 | His875 | - |
| PSYLNTPLL | 3.0 | Asn768, Asp872, Ser876(2), Arg880, Thr1010(2) | Leu648, Phe649, Leu712, Asn768, Glu802, Thr803, Arg871, Asp872, Leu873, Ser874, His875, Ser876, Glu879, Arg880, Phe914, Ser1008, Phe1009, Thr1010, Val1011, Pro1012, Phe1013, Leu1014, Pro1076, Ala1079, Tyr1140, Phe1142, Glu1261 | - | Ala1079 |
| Peptide | Docking Score | Interaction with p47phox | |
|---|---|---|---|
| Hydrogen Bond | Hydrophobic Interaction | ||
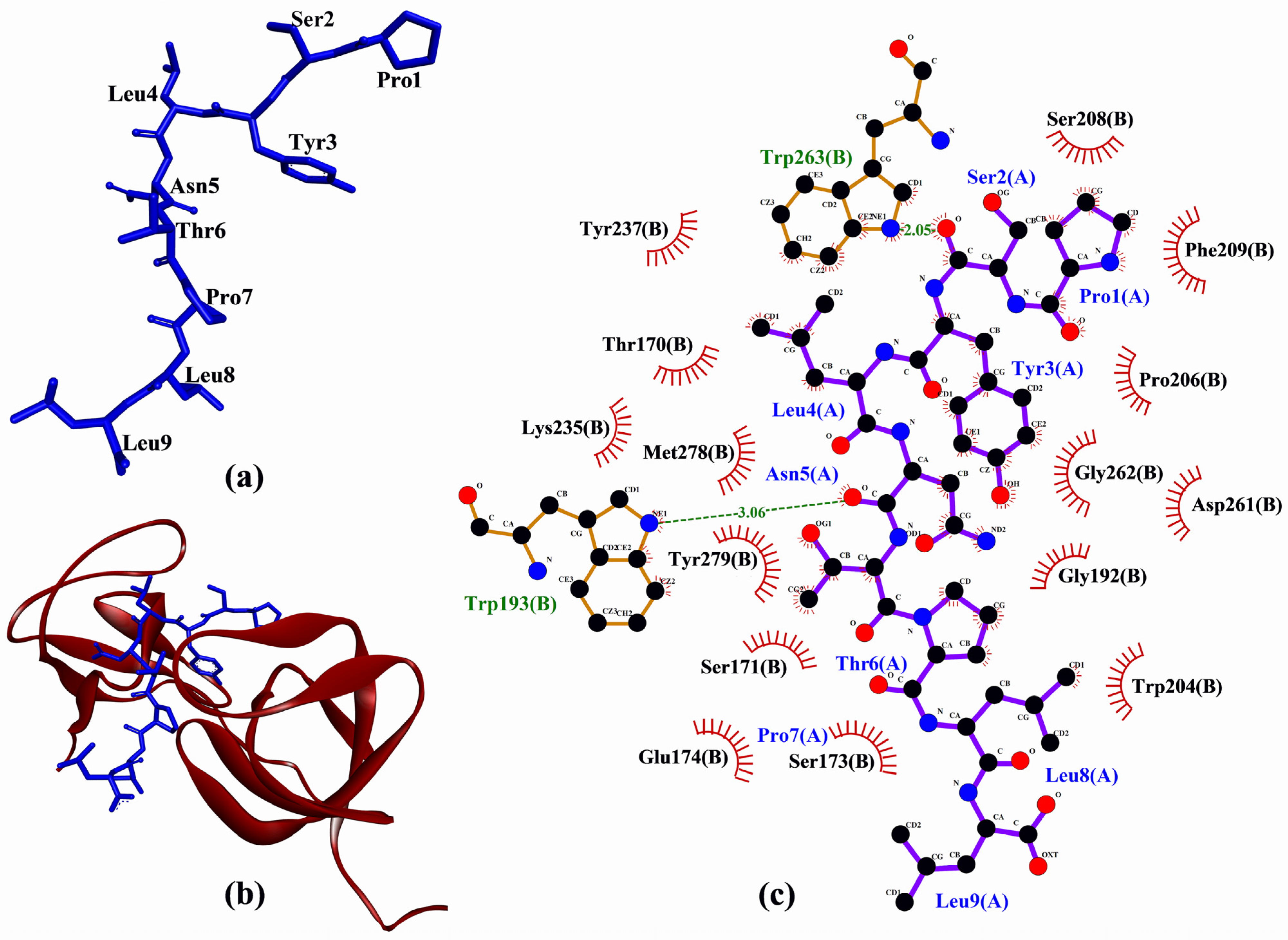
| PSYLNTPLL | −216.493 | Trp193, Trp263 | Thr170, Ser171, Ser173, Glu174, Gly192, Trp193, Trp204, Pro206, Ser208, Phe209, Lys235, Tyr237, Asp261, Gly262, Trp263, Met278, Tyr279 |
| LPHFNS | −195.377 | Tyr279 | Tyr167, Thr170, Ser191, Trp193, Pro206, Tyr237, Asp243, Glu244, Asp261, Gly262, Trp263, Tyr274, Pro276, Tyr279 |
| LYSPH | −185.715 | Thr170, Ser208 | Tyr167, Thr170, Ser171, Ser173, Glu174, Trp193, Trp204, Pro206, Ser208, Phe209, Trp263, Met278 |
| AEHGSLH | −181.729 | Trp263 | Tyr167, Thr170, Ser171, Glu174, Trp193, Glu241, Asp243, Trp263, Tyr274, Pro276, Met278 |
| FGPEMEQ | −175.680 | Thr170, Trp193 | Thr170, Ser173, Glu174, Gly192, Trp193, Trp204, Pro206, Phe209, Asp261, Met278, Tyr279 |
| Peptide | Cell-Penetrating Potential | Blood-Brain Barrier Penetrating Potential | Plasma Half-Life (Seconds) | Tolerance to In Silico GI Digestion |
|---|---|---|---|---|
| AEHGSLH | No | No | 828.91 | No |
| FGPEMEQ | No | Yes | 796.21 | No |
| LPHFNS | Yes | Yes | 823.51 | No |
| LYSPH | Yes | Yes | 832.41 | No |
| PSYLNTPLL | Yes | Yes | 833.41 | No |
| Peptide | Residue | ∆∆G (kJ/mol) | |||
|---|---|---|---|---|---|
| Keap1 | MPO | XO | p47phox | ||
| LYSPH | Leu | 8.4596 | 7.7764 | 6.3704 | - |
| Tyr | 14.3232 | 23.0458 | 16.8951 | - | |
| Ser | 2.5540 | −0.1909 | −0.1396 | - | |
| Pro | 5.0370 | 5.2764 | 6.3965 | - | |
| His | 0.6399 | 3.1014 | 14.2670 | - | |
| PSYLNTPLL | Pro | - | - | - | 0.0856 |
| Ser | - | - | - | 0.5437 | |
| Tyr | - | - | - | 27.1615 | |
| Leu | - | - | - | −0.4367 | |
| Asn | - | - | - | 0.7800 | |
| Thr | - | - | - | 1.1903 | |
| Pro | - | - | - | 2.9660 | |
| Leu | - | - | - | 2.7811 | |
| Leu | - | - | - | 0.4836 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chai, T.-T.; Koh, J.-A.; Wong, C.C.-C.; Sabri, M.Z.; Wong, F.-C. Computational Screening for the Anticancer Potential of Seed-Derived Antioxidant Peptides: A Cheminformatic Approach. Molecules 2021, 26, 7396. https://doi.org/10.3390/molecules26237396
Chai T-T, Koh J-A, Wong CC-C, Sabri MZ, Wong F-C. Computational Screening for the Anticancer Potential of Seed-Derived Antioxidant Peptides: A Cheminformatic Approach. Molecules. 2021; 26(23):7396. https://doi.org/10.3390/molecules26237396
Chicago/Turabian StyleChai, Tsun-Thai, Jiun-An Koh, Clara Chia-Ci Wong, Mohamad Zulkeflee Sabri, and Fai-Chu Wong. 2021. "Computational Screening for the Anticancer Potential of Seed-Derived Antioxidant Peptides: A Cheminformatic Approach" Molecules 26, no. 23: 7396. https://doi.org/10.3390/molecules26237396
APA StyleChai, T.-T., Koh, J.-A., Wong, C. C.-C., Sabri, M. Z., & Wong, F.-C. (2021). Computational Screening for the Anticancer Potential of Seed-Derived Antioxidant Peptides: A Cheminformatic Approach. Molecules, 26(23), 7396. https://doi.org/10.3390/molecules26237396