
Influence of the Substituents on the Opening of Silylepoxy Alcohols: 5-exo-Cyclization towards Tetrahydrofurans vs. Unexpected Side Reaction Leading to Tetrahydropyrans



Abstract
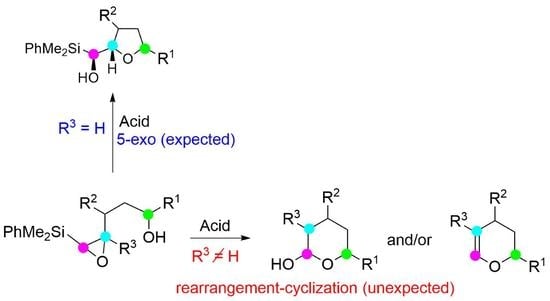
:
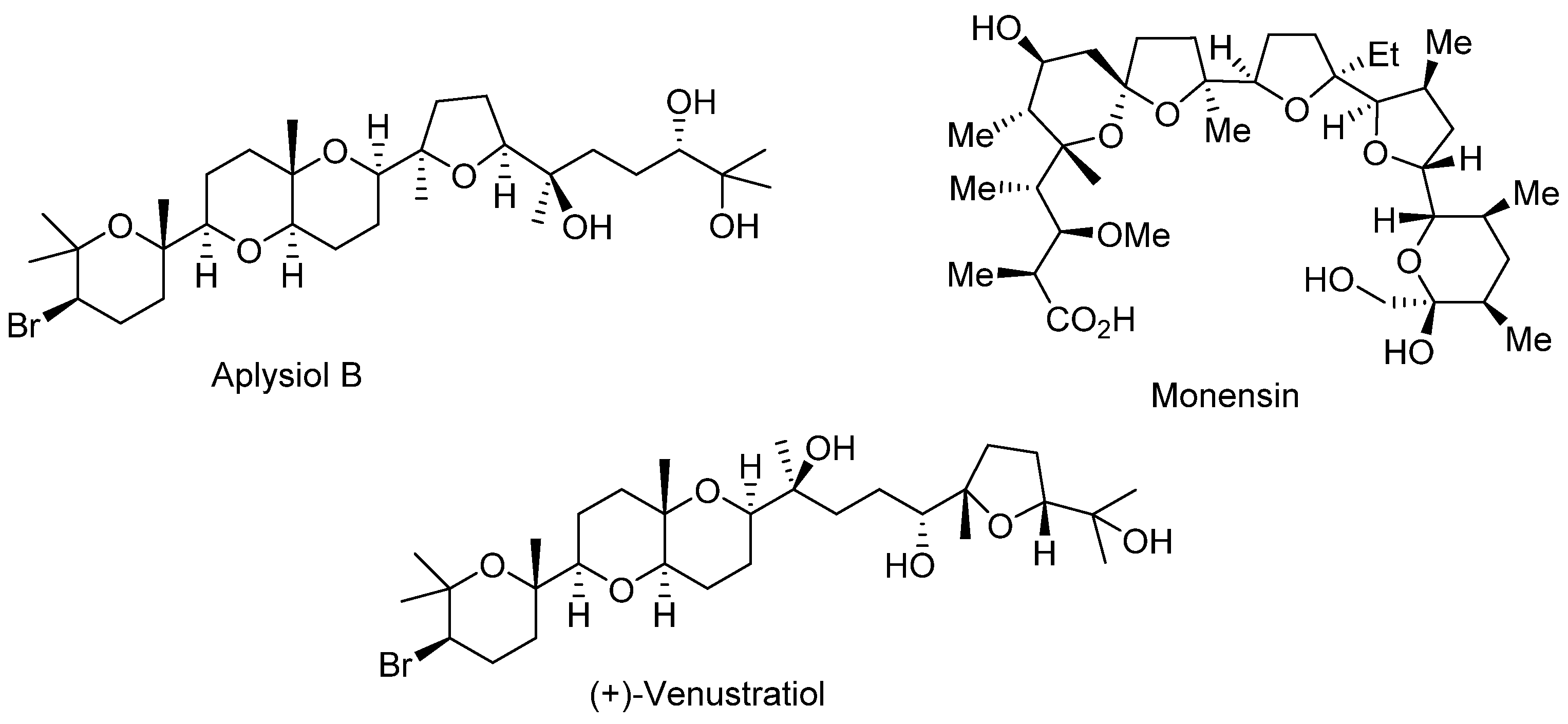
1. Introduction
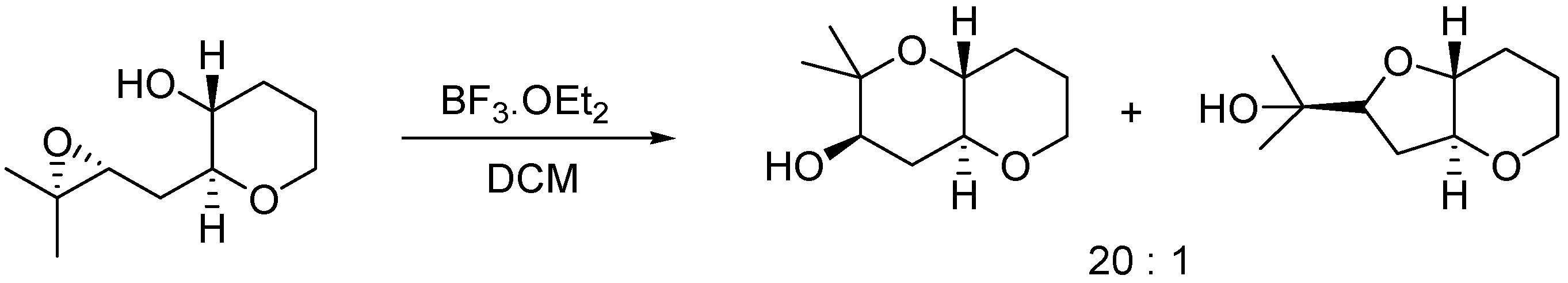
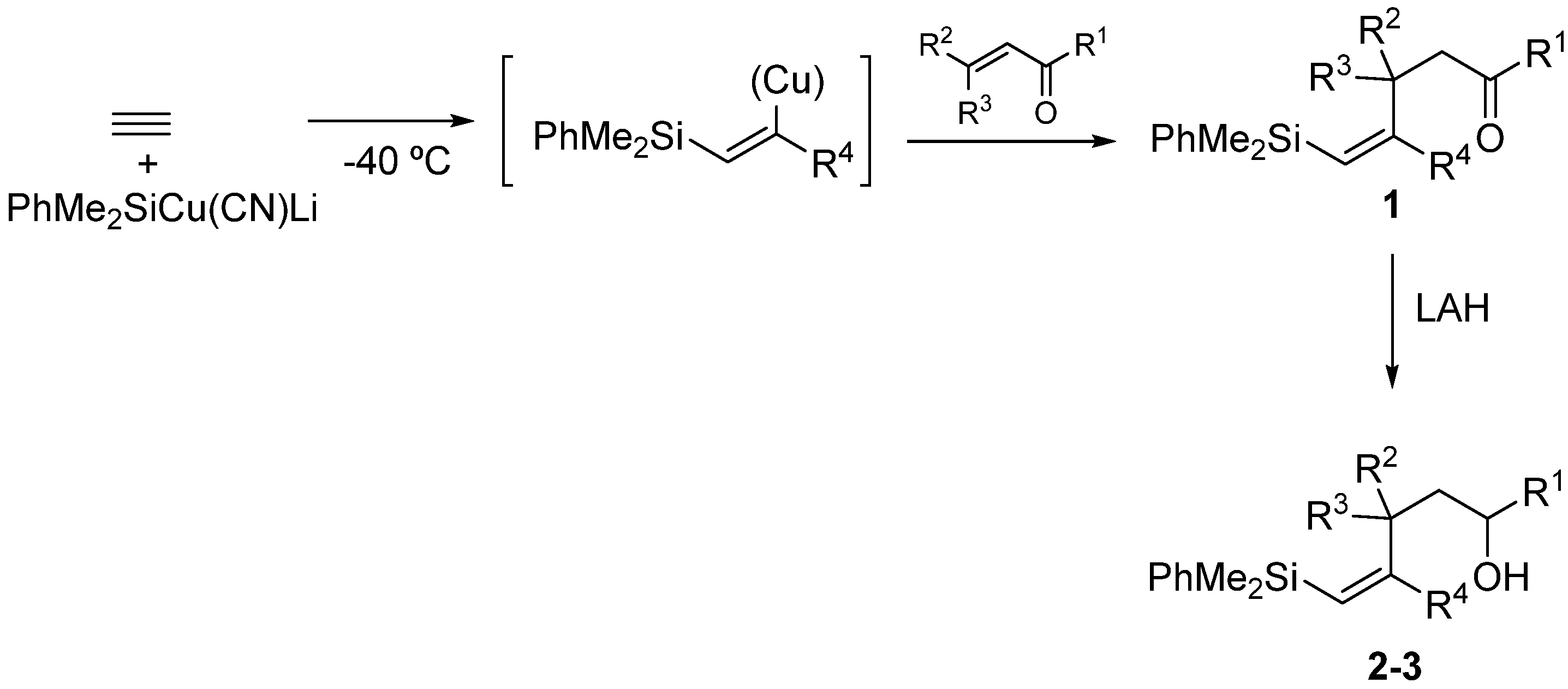
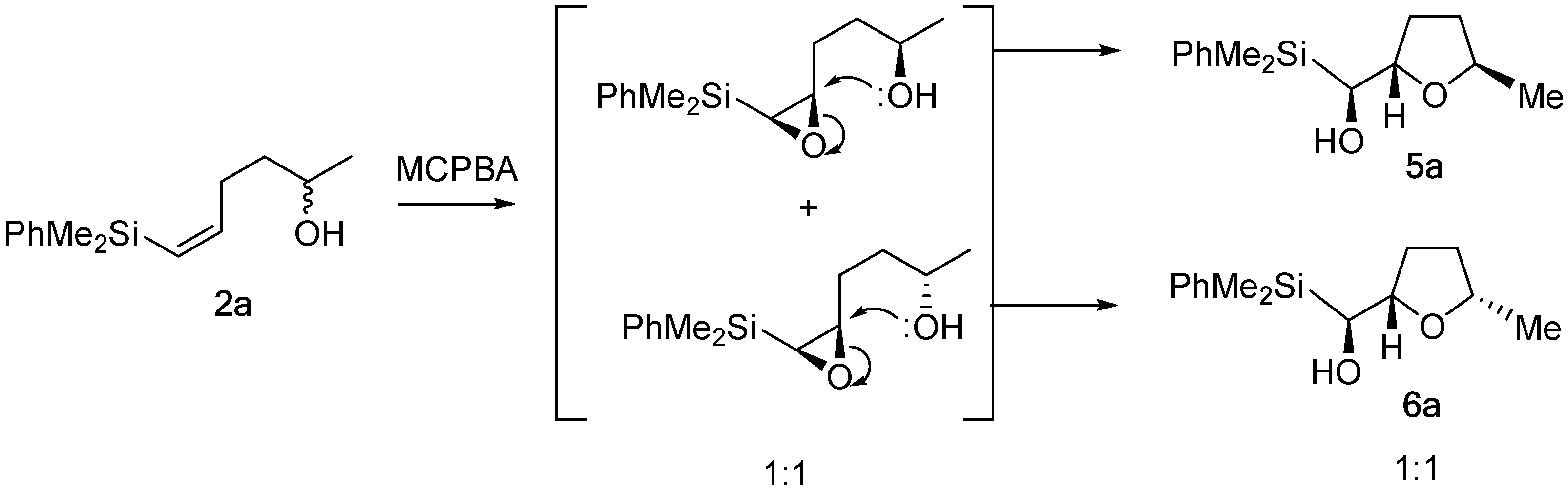
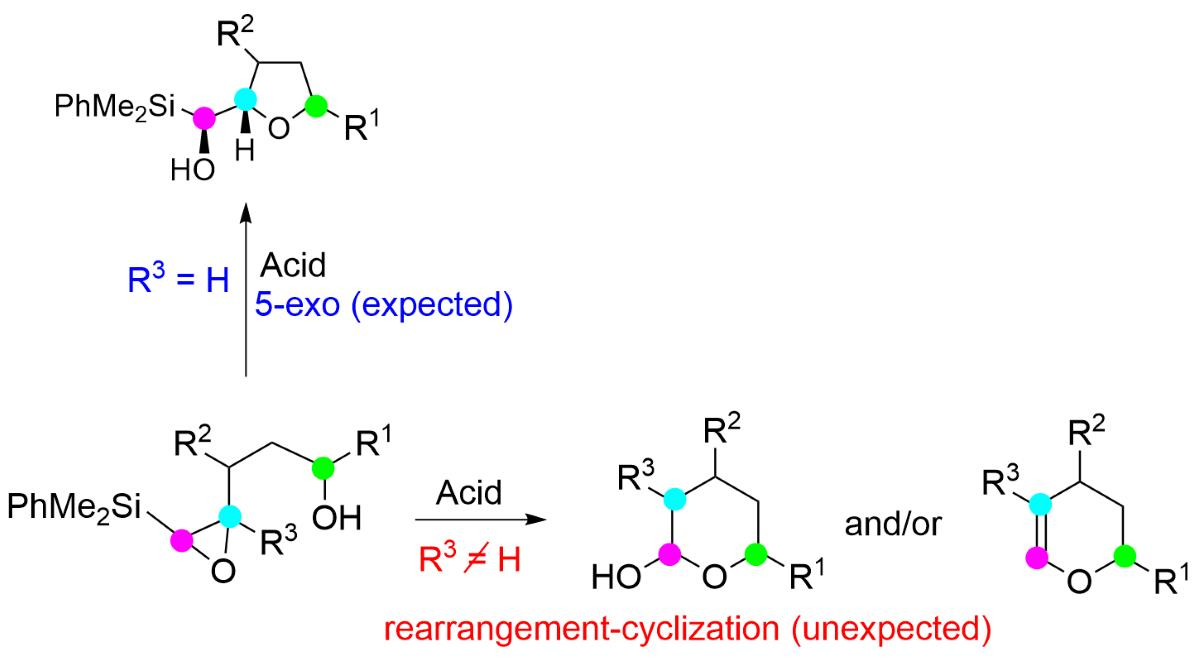
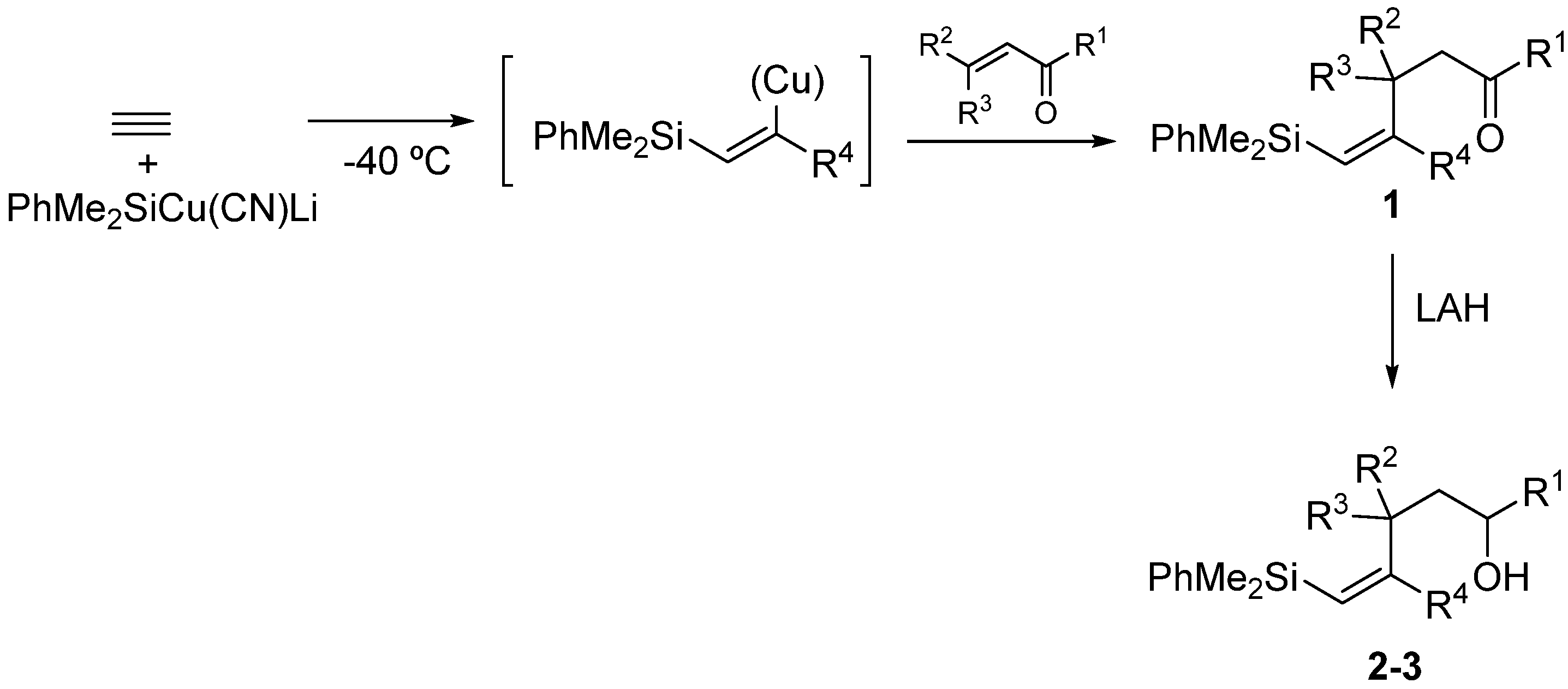
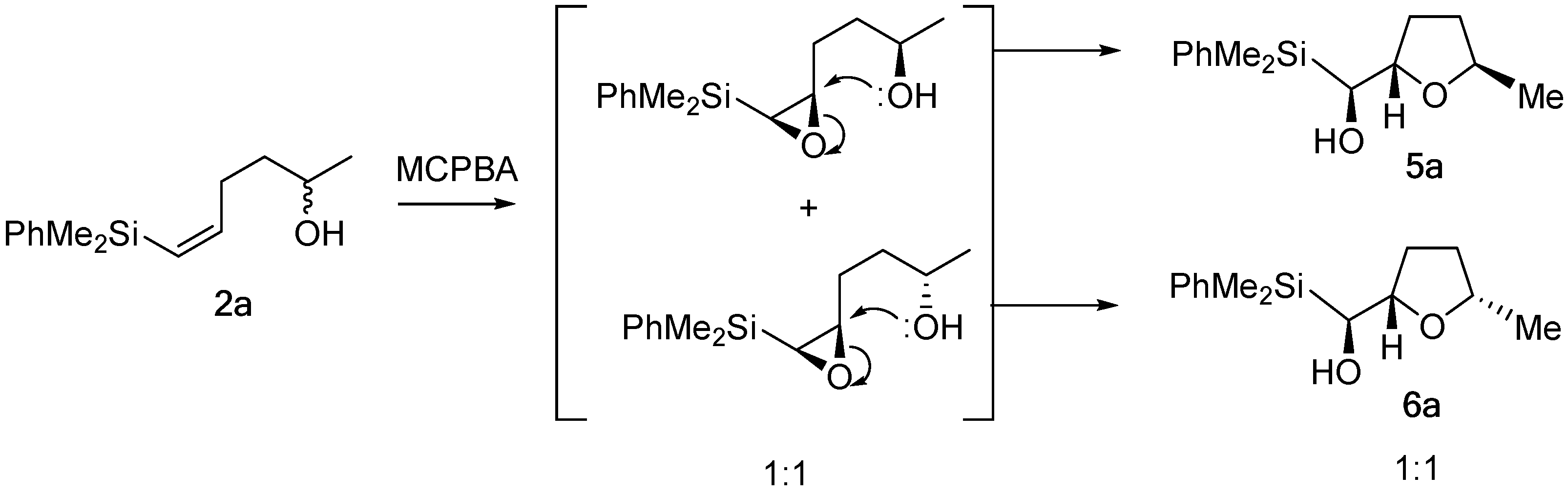
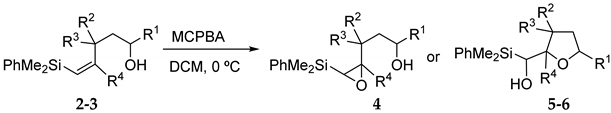
2. Results and Discussion
3. Materials and Methods
3.1. General Procedures
3.2. Preparation of Key Compounds
3.2.1. Epoxidation of Vinylsilyl Alcohols 2-3 with MCPBA
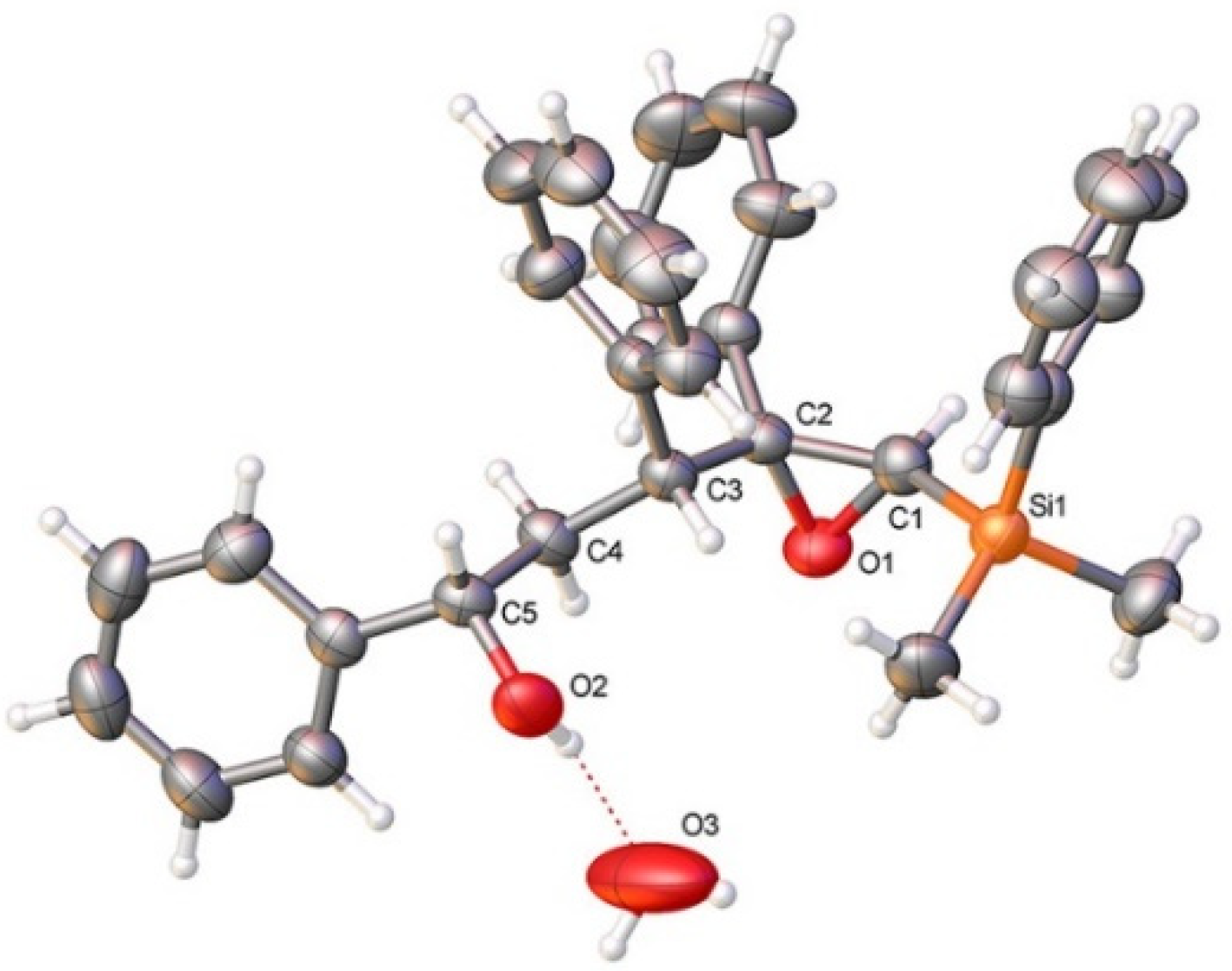
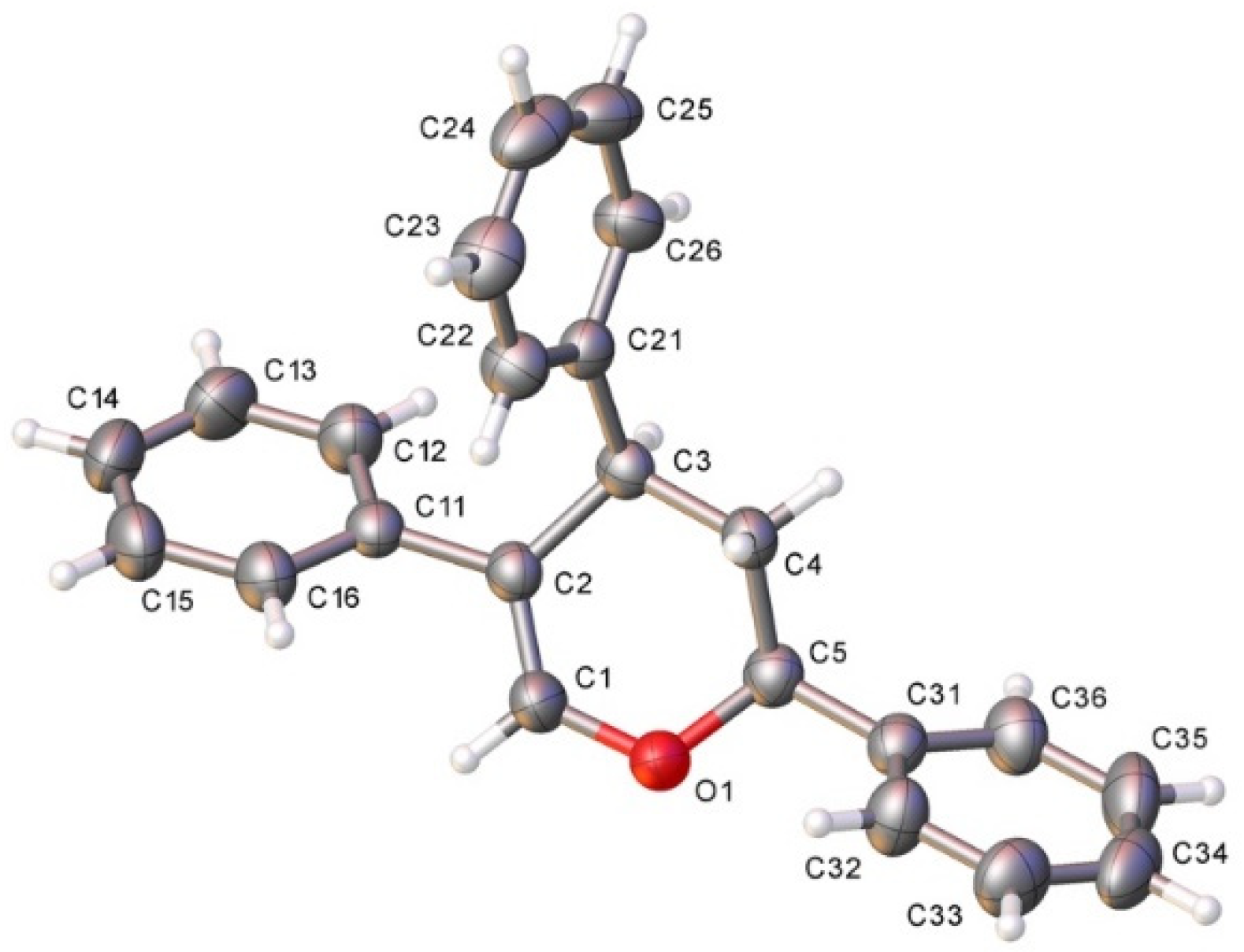
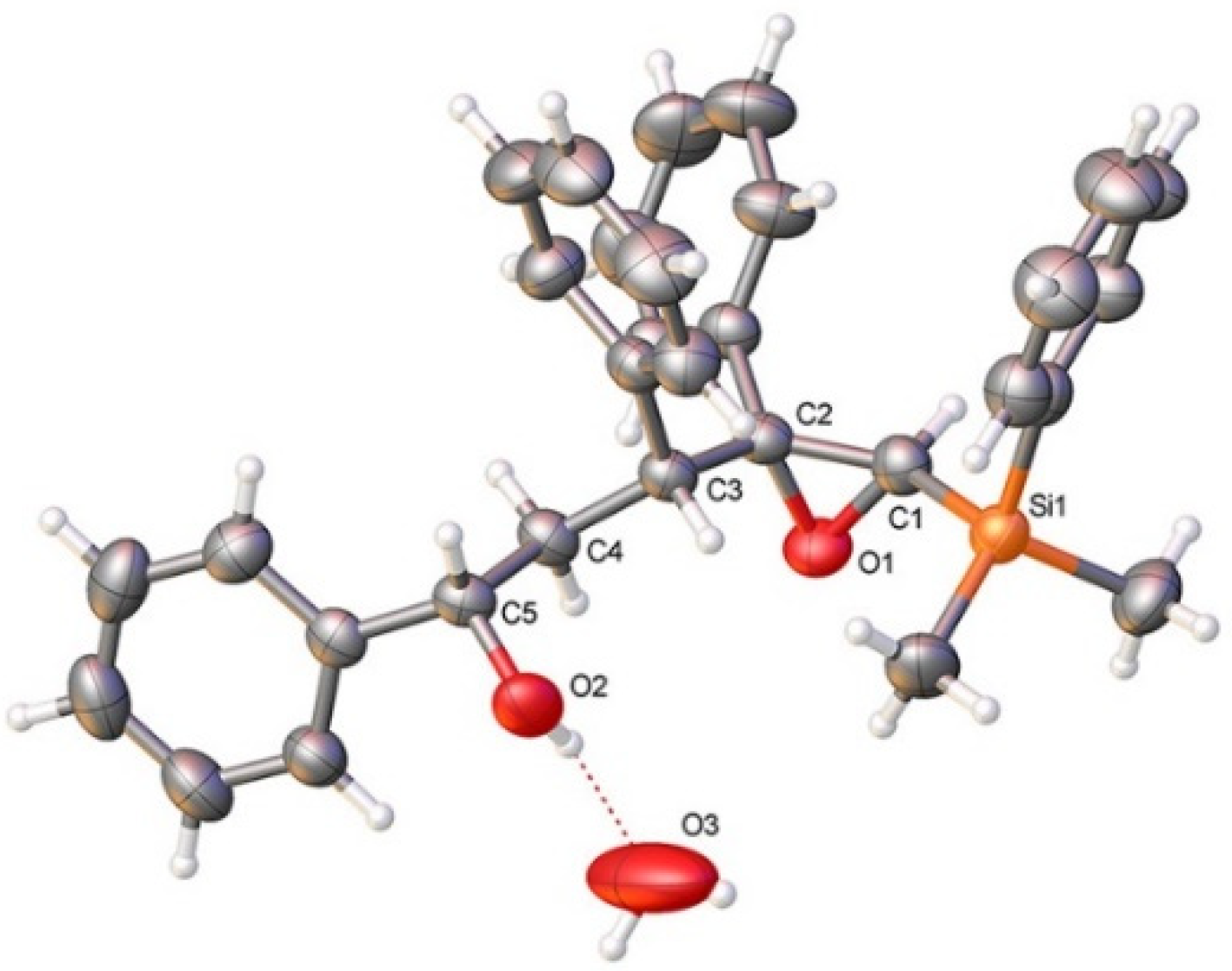
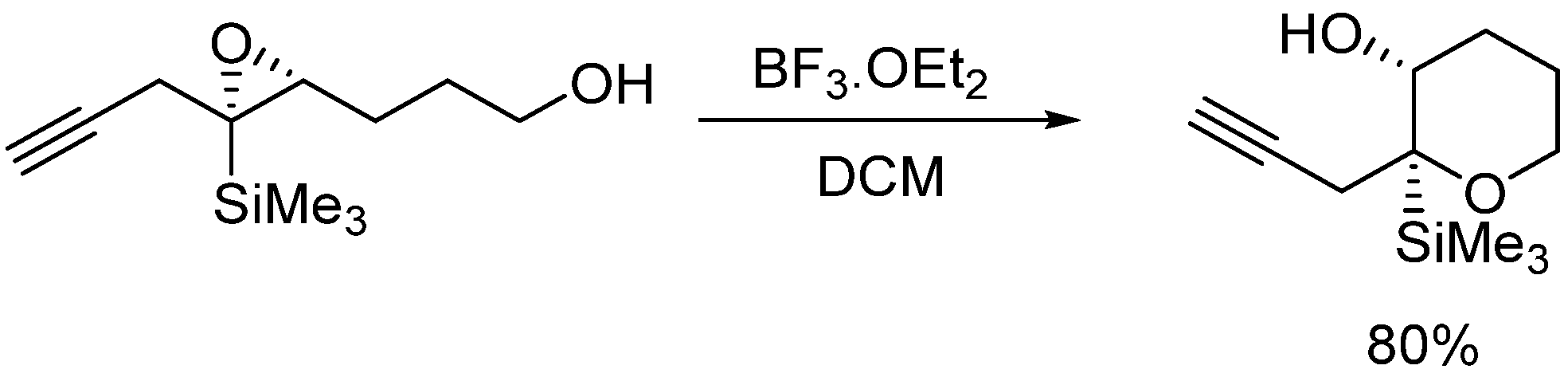
3.2.2. Acid-Catalyzed Intramolecular Cyclization of Epoxysilyl Alcohol 4e
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Agtarap, A.; Chamberlin, J.W.; Pinkerton, M.; Steinrauf, I. Structure of monensic acid, a new biologically active compound. J. Am. Chem. Soc. 1967, 89, 5737–5739. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.E.; Hoehn, M.M. Monensin, a new biologically active compound. I. Discovery and isolation. Antimicrob. Agents Chemother. 1967, 7, 349–352. [Google Scholar] [PubMed]
- Sakemi, S.; Higa, T.; Jefford, C.W.; Bernardinelli, G. Venustatriol. A new, anti-viral, triterpene tetracyclic ether from Laurencia venusta. Tetrahedron Lett. 1986, 27, 4287–4290. [Google Scholar] [CrossRef]
- Pereira, R.B.; Andrade, P.B.; Valentão, P. Chemical Diversity and Biological Properties of Secondary Metabolites from Sea Hares of Aplysia Genus. Mar. Drugs 2016, 14, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrosa, I.; Romea, P.; Urpi, F. Synthesis of six-membered oxygenated heterocycles through carbon–oxygen bond-forming reactions. Tetrahedron 2008, 64, 2683–2723. [Google Scholar] [CrossRef]
- Wolfe, J.P.; Hay, M.B. Recent advances in the stereoselective synthesis of tetrahydrofurans. Tetrahedron 2007, 63, 261–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boivin, T.L.B. Synthetic routes to tetrahydrofuran, tetrahydropyran, and spiroketal units of polyether antibiotics and a survey of spiroketals of other natural products. Tetrahedron 1987, 43, 3309–3362. [Google Scholar] [CrossRef]
- Vilotijevic, I.; Jamison, T.F. Synthesis of Marine Polycyclic Polyethers via Endo-Selective Epoxide-Opening Cascades. Mar. Drugs 2010, 8, 763–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morten, C.J.; Jamison, T.F. Water Overcomes Methyl Group Directing Effects in Epoxide-Opening Cascades. J. Am. Chem. Soc. 2009, 131, 6678–6679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heffron, T.P.; Jamison, T.F. SiMe3-Based Homologation−Epoxidation−Cyclization Strategy for Ladder THP Synthesis. Org. Lett. 2003, 5, 2339–2342. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.L.; Heffron, T.P.; Merino, E.; Jamison, T.F. Ladder Polyether Synthesis via Epoxide-Opening Cascades Using a Disappearing Directing Group. J. Am. Chem. Soc. 2006, 128, 1056–1057. [Google Scholar] [CrossRef] [PubMed]
- Adiwidjaja, G.; Florke, H.; Kirschning, A.; Schaumann, E. Cyclizations of 5-silyl-substituted 4,5-epoxy-1-alkanols: A configuration dependent mode of ring closure. Tetrahedron Lett. 1995, 36, 8771–8774. [Google Scholar] [CrossRef]
- Pulido, F.J.; Barbero, A.; Castreno, P. Seven-Membered Ring Formation from Cyclopropanated Oxo- and Epoxyallylsilanes. J. Org. Chem. 2011, 76, 5850–5855. [Google Scholar] [CrossRef] [PubMed]
- Barbero, A.; Castreno, P.; Fernandez, G.; Pulido, F.J. Intramolecular ene reaction of epoxyallylsilanes: Synthesis of allyl- and vinylsilane-functionalized cyclohexanols. J. Org. Chem. 2005, 70, 10747–10752. [Google Scholar] [CrossRef] [PubMed]
- Barbero, A.; Diez-Varga, A.; Pulido, F.J.; Gonzalez-Ortega, A. Synthesis of Azepane Derivatives by Silyl-aza-Prins Cyclization of Allylsilyl Amines: Influence of the Catalyst in the Outcome of the Reaction. Org. Lett. 2016, 18, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Barbero, A.; Diez-Varga, A.; Herrero, M.; Pulido, F.J. From Silylated Trishomoallylic Alcohols to Dioxaspiroundecanes or Oxocanes: Catalyst and Substitution Influence. J. Org. Chem. 2016, 81, 2704–2712. [Google Scholar] [CrossRef] [PubMed]
- Pulido, F.J.; Barbero, A.; Abad, R. One-Pot Synthesis of 2,7-Dioxabicyclo[2.2.1]heptanes from Oxoallylsilanes. Eur. J. Org. Chem. 2011, 2011, 6974–6979. [Google Scholar] [CrossRef]
- CCDC: 2118173 Contains the Supplementary Crystallographic Data for Compound 4e. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 27 October 2021).
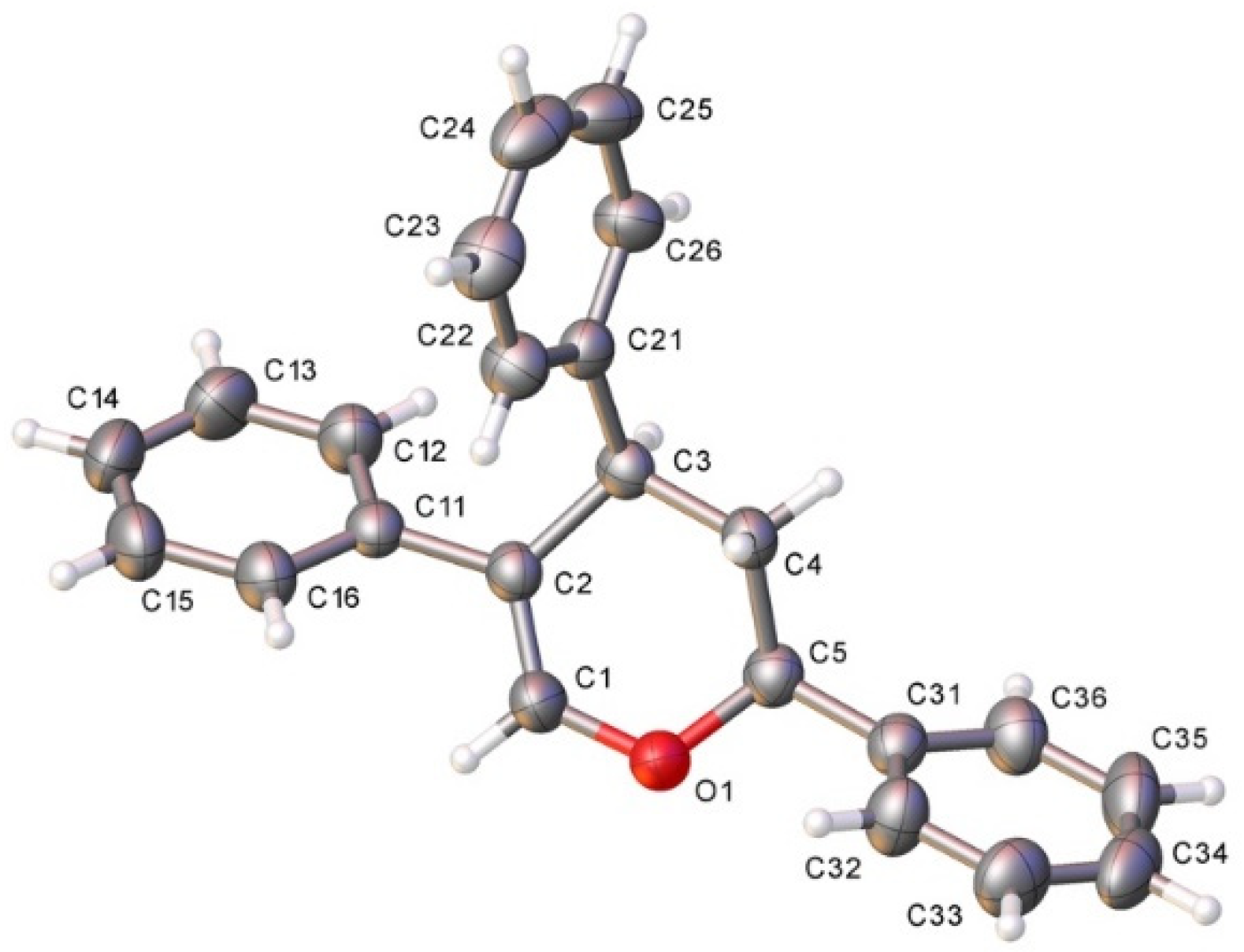
- CCDC: 2118174 Contains the Supplementary Crystallographic Data for Compound 9. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 27 October 2021).
- Barbero, A.; Barbero, H.; González-Ortega, A.; Pulido, F.J.; Val, P.; Diez-Varga, A.; Morán, J.R. Efficient access to polysubstituted tetrahydrofurans by electrophilic cyclization of vinylsilyl alcohols. RSC Adv. 2015, 5, 49541–49551. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Vinylsilyl Alcohol | Product | Yield (%) |
|---|---|---|---|
| 1 | 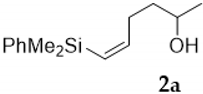 | 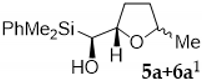 | 60 |
| 2 | 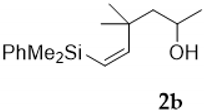 | 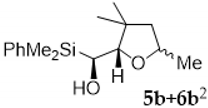 | 61 |
| 3 |  | 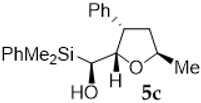 | 59 |
| 4 |  | 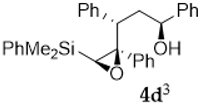 | 69 |
| 5 | 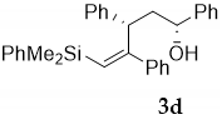 | 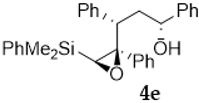 | 65 |
| 6 | 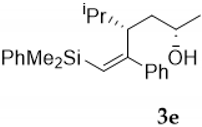 |  | 64 |
| 7 |  | 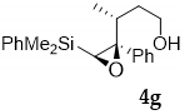 | 62 |
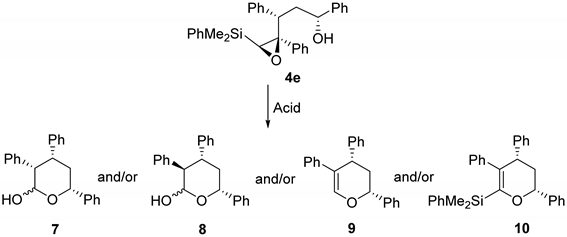
| Entry | Acid | Solvent | Conditions | Product Ratio 7/8/9/10 | Yield (%) |
|---|---|---|---|---|---|
| 1 | pTSA | CH2Cl2 | 0 °C, 2 h | 0/15/80/5 | 88 |
| 2 | pTSA | CH2Cl2 | −40 °C, 4 h | ----- | n.r. 1 |
| 3 | CSA | CH2Cl2 | 0 °C, 4 h | 0/27/73/0 | 78 |
| 4 | H2O | H2O | r.t., 3 days | 0/29/71/0 | 46 |
| 5 | BF3.OEt2 | CH2Cl2 | 0 °C, 30 min | 60/0/40/0 | 83 |
| 6 | BF3.OEt2 | CH2Cl2 | −40 °C, 30 min | 60/40/0/0 | 40 |
| 7 | BF3.OEt2 | CH2Cl2 | −78 °C, 30 min | 30/70/0/0 | 35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díez-Poza, C.; Barbero, A. Influence of the Substituents on the Opening of Silylepoxy Alcohols: 5-exo-Cyclization towards Tetrahydrofurans vs. Unexpected Side Reaction Leading to Tetrahydropyrans. Molecules 2021, 26, 7386. https://doi.org/10.3390/molecules26237386
Díez-Poza C, Barbero A. Influence of the Substituents on the Opening of Silylepoxy Alcohols: 5-exo-Cyclization towards Tetrahydrofurans vs. Unexpected Side Reaction Leading to Tetrahydropyrans. Molecules. 2021; 26(23):7386. https://doi.org/10.3390/molecules26237386
Chicago/Turabian StyleDíez-Poza, Carlos, and Asunción Barbero. 2021. "Influence of the Substituents on the Opening of Silylepoxy Alcohols: 5-exo-Cyclization towards Tetrahydrofurans vs. Unexpected Side Reaction Leading to Tetrahydropyrans" Molecules 26, no. 23: 7386. https://doi.org/10.3390/molecules26237386
APA StyleDíez-Poza, C., & Barbero, A. (2021). Influence of the Substituents on the Opening of Silylepoxy Alcohols: 5-exo-Cyclization towards Tetrahydrofurans vs. Unexpected Side Reaction Leading to Tetrahydropyrans. Molecules, 26(23), 7386. https://doi.org/10.3390/molecules26237386