
E/Z Molecular Photoswitches Activated by Two-Photon Absorption: Comparison between Different Families

 ,
,  , , ,
, , ,  and
and 
Abstract
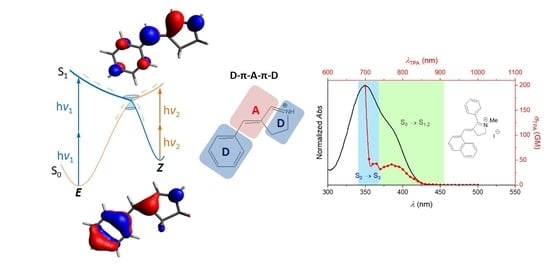
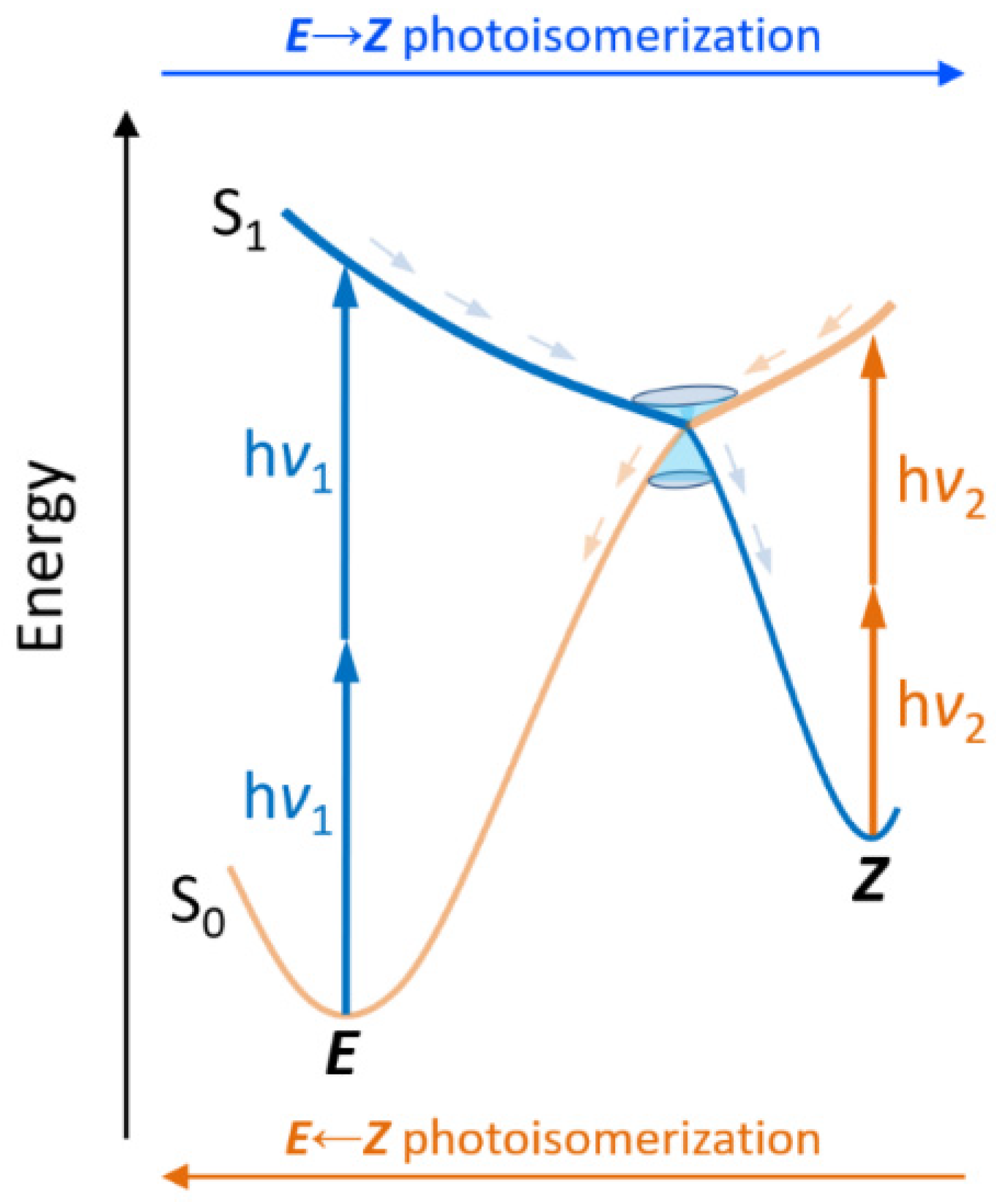
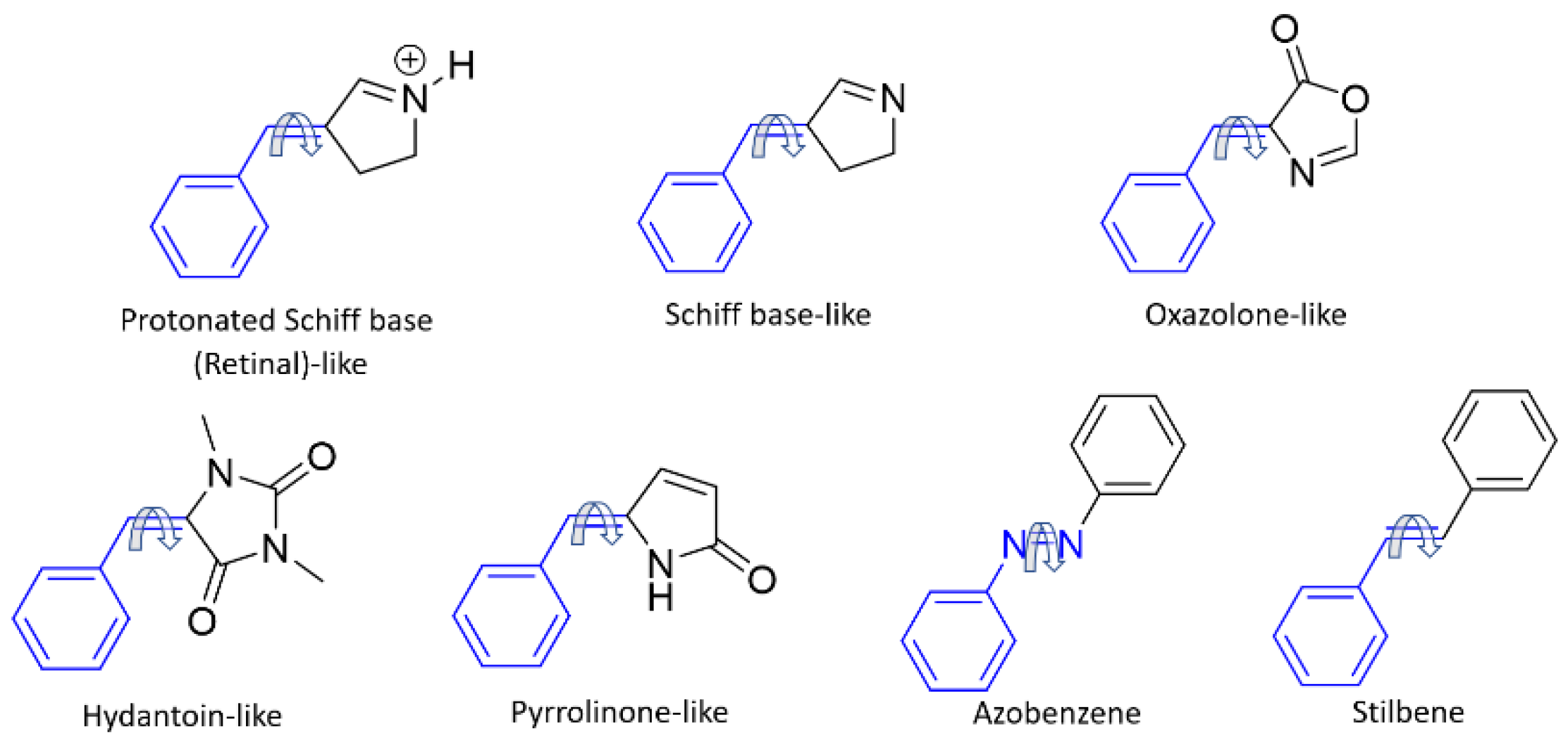
:
1. Introduction
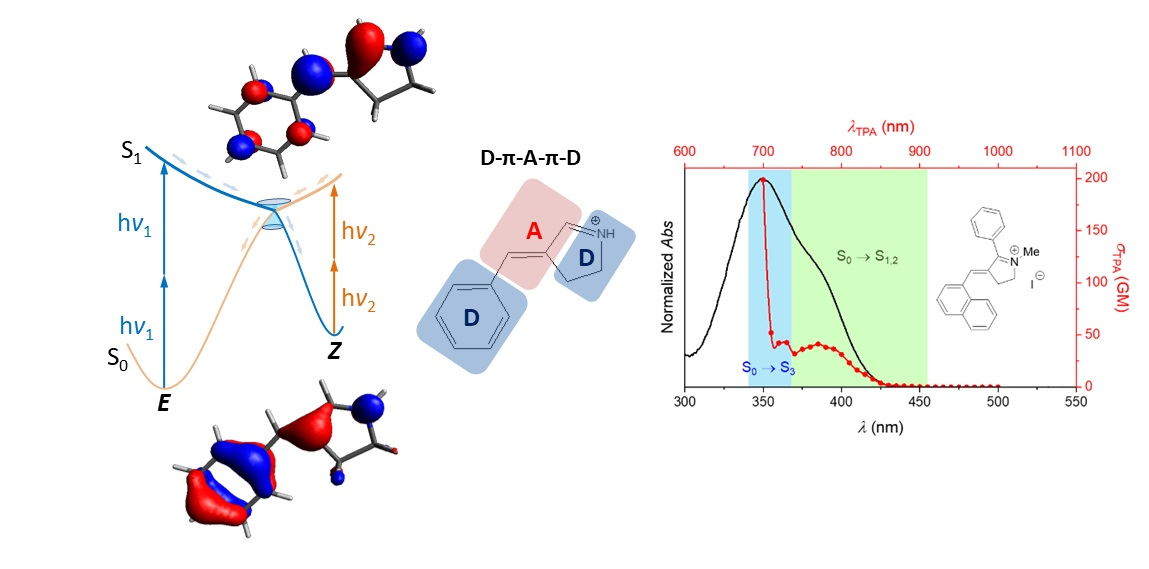
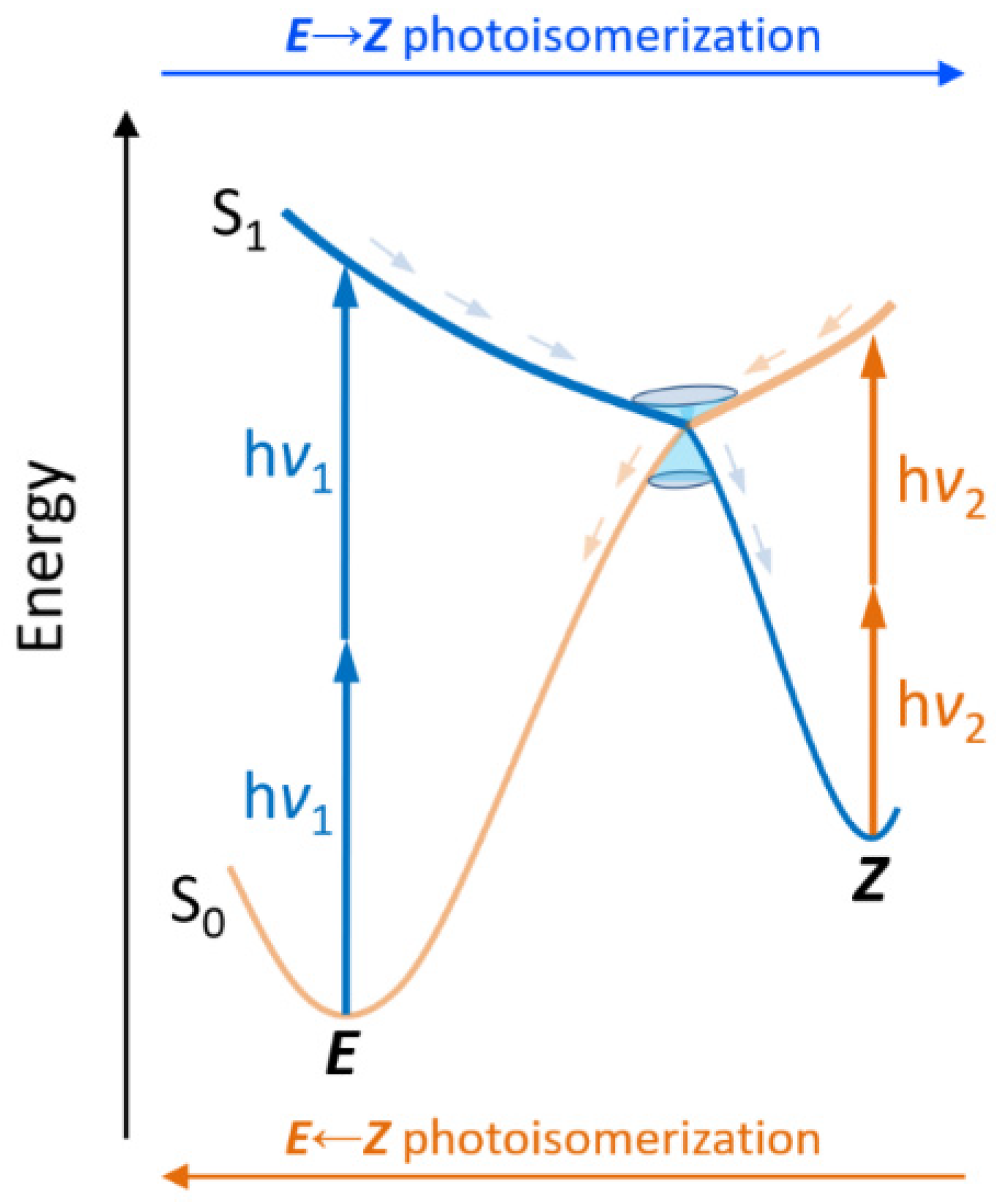
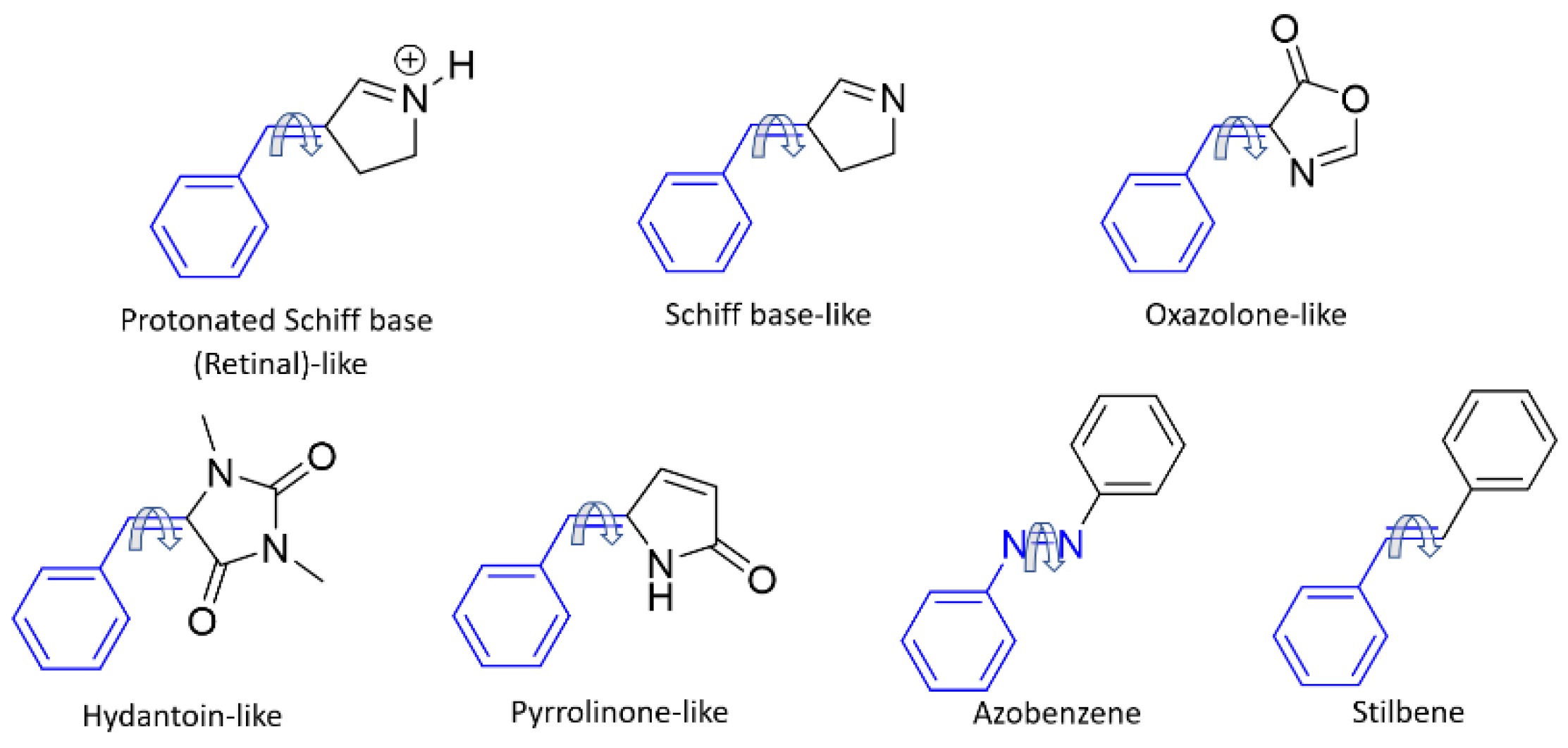
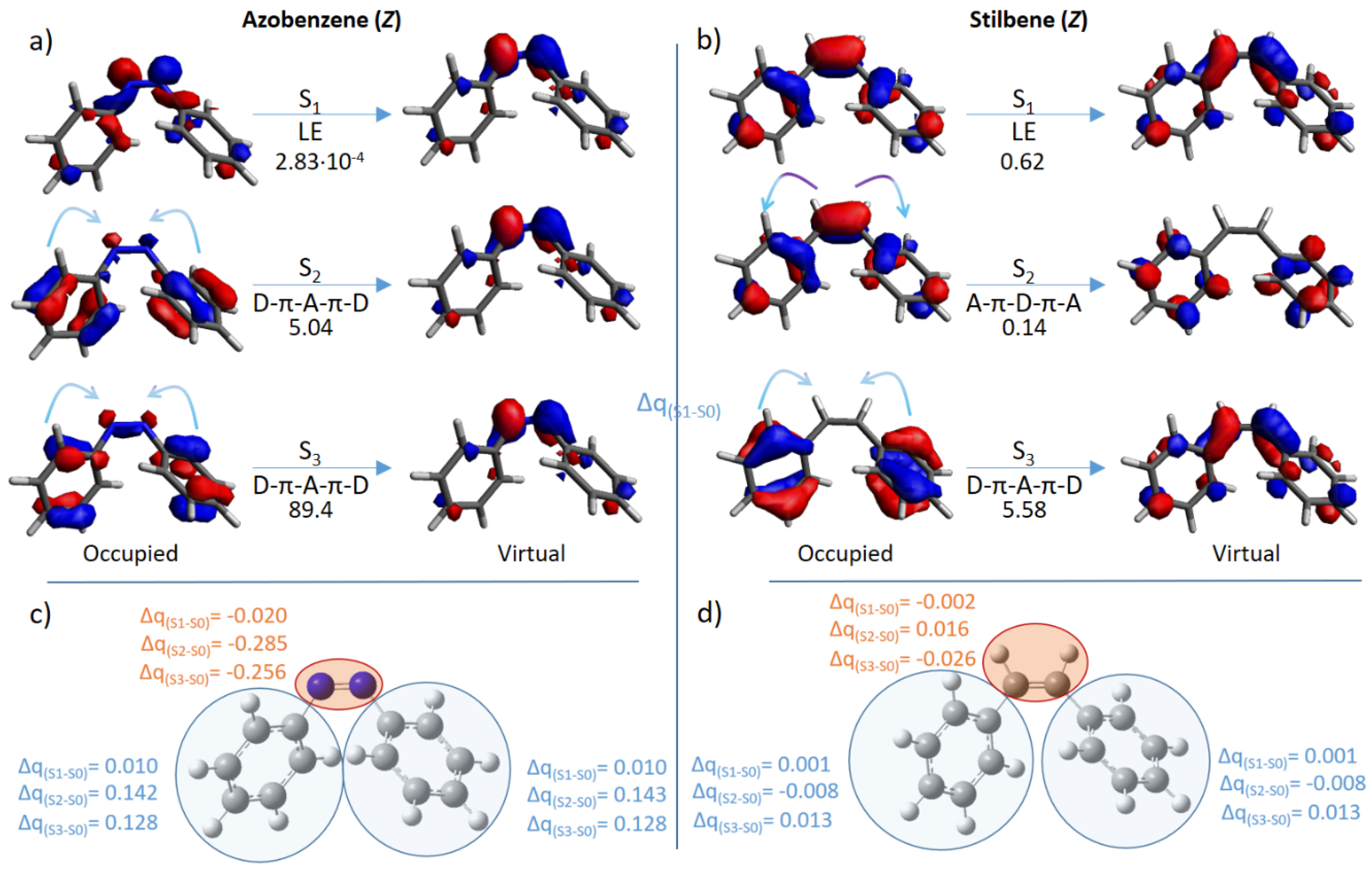
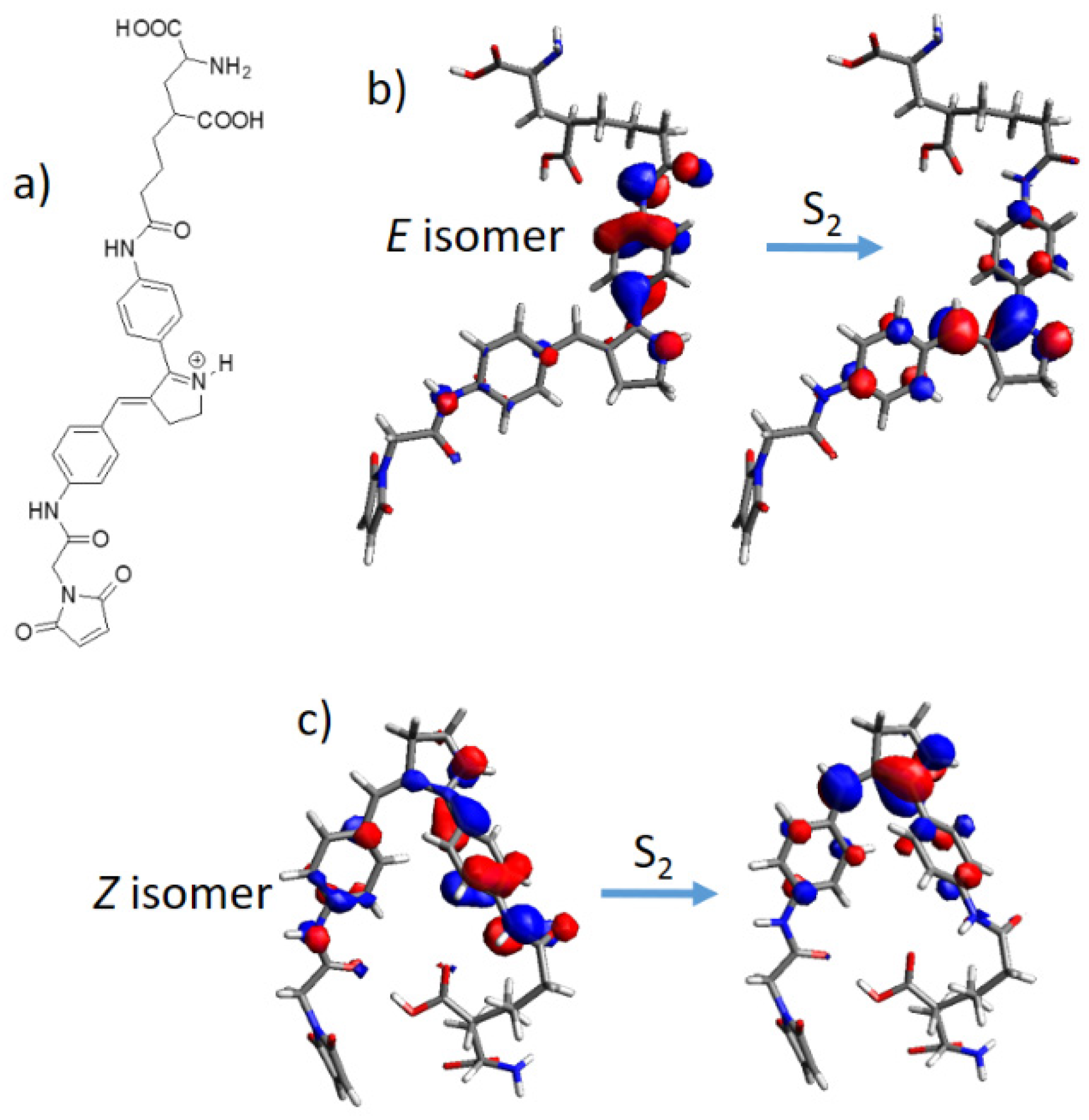
2. Results and Discussion
3. Materials and Methods
3.1. Theoretical Background
3.2. Computational Strategy
3.3. Experimental Section
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Shi, Z.-T.; Zhang, Q.; Tian, H.; Qu, D.-H. Driving Smart Molecular Systems by Artificial Molecular Machines. Adv. Intell. Syst. 2020, 2, 1900169. [Google Scholar] [CrossRef]
- Ariga, K. The evolution of molecular machines through interfacial nanoarchitectonics: From toys to tools. Chem. Sci. 2020, 11, 10594–10604. [Google Scholar] [CrossRef] [PubMed]
- Corra, S.; Curcio, M.; Baroncini, M.; Silvi, S.; Credi, A. Photoactivated Artificial Molecular Machines that Can Perform Tasks. Adv. Mater. 2020, 32, 1906064. [Google Scholar] [CrossRef]
- Baroncini, M.; Canton, M.; Casimiro, L.; Corra, S.; Groppi, J.; La Rosa, M.; Silvi, S.; Credi, A. Photoactive Molecular-Based Devices, Machines and Materials: Recent Advances. Eur. J. Inorg. Chem. 2018, 2018, 4589–4603. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Stoddart, J.F. Photo-driven molecular devices. Chem. Soc. Rev. 2007, 36, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Dattler, D.; Fuks, G.; Heiser, J.; Moulin, E.; Perrot, A.; Yao, X.; Giuseppone, N. Design of Collective Motions from Synthetic Molecular Switches, Rotors, and Motors. Chem. Rev. 2020, 120, 310–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feringa, B.L. The art of building small: From molecular switches to molecular motors. J. Org. Chem. 2007, 72, 6635–6652. [Google Scholar] [CrossRef] [Green Version]
- Roy, P.; Sardjan, A.S.; Cnossen, A.; Browne, W.R.; Feringa, B.L.; Meech, S.R. Excited State Structure Correlates with Efficient Photoconversion in Unidirectional Motors. J. Phys. Chem. Lett. 2021, 12, 3367–3372. [Google Scholar] [CrossRef]
- Kassem, S.; Van Leeuwen, T.; Lubbe, A.S.; Wilson, M.R.; Feringa, B.L.; Leigh, D.A. Artificial molecular motors. Chem. Soc. Rev. 2017, 46, 2592–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asato, R.; Martin, C.J.; Abid, S.; Gisbert, Y.; Asanoma, F.; Nakashima, T.; Kammerer, C.; Kawai, T.; Rapenne, G. Molecular Rotor Functionalized with a Photoresponsive Brake. Inorg. Chem. 2021, 60, 3492–3501. [Google Scholar] [CrossRef]
- García-Iriepa, C.; Marazzi, M.; Frutos, L.M.; Sampedro, D. E/Z Photochemical switches: Syntheses, properties and applications. RSC Adv. 2013, 3, 6241–6266. [Google Scholar] [CrossRef]
- Altoè, P.; Bernardi, F.; Conti, I.; Garavelli, M.; Negri, F.; Orlandi, G. Light driven molecular switches: Exploring and tuning their photophysical and photochemical properties. Theor. Chem. Acc. 2007, 117, 1041–1059. [Google Scholar] [CrossRef]
- Bléger, D.; Hecht, S. Visible-Light-Activated Molecular Switches. Angew. Chem. Int. Ed. 2015, 54, 11338–11349. [Google Scholar] [CrossRef] [PubMed]
- Beharry, A.A.; Woolley, G.A. Azobenzene photoswitches for biomolecules. Chem. Soc. Rev. 2011, 40, 4422–4437. [Google Scholar] [CrossRef] [PubMed]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Waldeck, D.H. Photoisomerization dynamics of stilbenes. Chem. Rev. 1991, 91, 415–436. [Google Scholar] [CrossRef]
- García-Iriepa, C.; Sampedro, D.; Mendicuti, F.; Léonard, J.; Frutos, L.M. Photoreactivity Control Mediated by Molecular Force Probes in Stilbene. J. Phys. Chem. Lett. 2019, 10, 1063–1067. [Google Scholar] [CrossRef]
- Klajn, R. Spiropyran-based dynamic materials. Chem. Soc. Rev. 2014, 43, 148–184. [Google Scholar] [CrossRef] [Green Version]
- Kortekaas, L.; Browne, W.R. The evolution of spiropyran: Fundamentals and progress of an extraordinarily versatile photochrome. Chem. Soc. Rev. 2019, 48, 3406–3424. [Google Scholar] [CrossRef] [Green Version]
- Wiedbrauk, S.; Dube, H. Hemithioindigo-An emerging photoswitch. Tetrahedron Lett. 2015, 56, 4266–4274. [Google Scholar] [CrossRef]
- Petermayer, C.; Dube, H. Indigoid Photoswitches: Visible Light Responsive Molecular Tools. Acc. Chem. Res. 2018, 51, 1153–1163. [Google Scholar] [CrossRef]
- Blanco-Lomas, M.; Campos, P.J.; Sampedro, D. Synthesis and photoisomerization of rhodopsin-based molecular switches. Eur. J. Org. Chem. 2012, 6328–6334. [Google Scholar] [CrossRef]
- Sampedro, D.; Migani, A.; Pepi, A.; Busi, E.; Basosi, R.; Latterini, L.; Elisei, F.; Fusi, S.; Ponticelli, F.; Zanirato, V.; et al. Design and photochemical characterization of a biomimetic light-driven Z/E switcher. J. Am. Chem. Soc. 2004, 126, 9349–9359. [Google Scholar] [CrossRef]
- Valentini, A.; Rivero, D.; Zapata, F.; García-Iriepa, C.; Marazzi, M.; Palmeiro, R.; Fdez. Galván, I.; Sampedro, D.; Olivucci, M.; Frutos, L.M. Optomechanical Control of Quantum Yield in Trans–Cis Ultrafast Photoisomerization of a Retinal Chromophore Model. Angew. Chem. Int. Ed. 2017, 56, 3842–3846. [Google Scholar] [CrossRef]
- García-Iriepa, C.; Gueye, M.; Léonard, J.; Martínez-López, D.; Campos, P.J.; Frutos, L.M.; Sampedro, D.; Marazzi, M. A biomimetic molecular switch at work: Coupling photoisomerization dynamics to peptide structural rearrangement. Phys. Chem. Chem. Phys. 2016, 18, 6742–6753. [Google Scholar] [CrossRef] [Green Version]
- Rivado-Casas, L.; Blanco-Lomas, M.; Campos, P.J.; Sampedro, D. Photochemical characterization of biomimetic molecular switches. Tetrahedron 2011, 67, 7570–7574. [Google Scholar] [CrossRef]
- Andresen, M.; Wahl, M.C.; Stiel, A.C.; Gräter, F.; Schäfer, L.V.; Trowitzsch, S.; Weber, G.; Eggeling, C.; Grubmüller, H.; Hell, S.W.; et al. Structure and mechanism of the reversible photoswitch of a fluorescent protein. Proc. Natl. Acad. Sci. USA 2005, 102, 13070–13074. [Google Scholar] [CrossRef] [Green Version]
- Habuchi, S.; Cotlet, M.; Gensch, T.; Bednarz, T.; Haber-Pohlmeier, S.; Rozenski, J.; Dirix, G.; Michiels, J.; Vanderleyden, J.; Heberle, J.; et al. Evidence for the isomerization and decarboxylation in the photoconversion of the red fluorescent protein DsRed. J. Am. Chem. Soc. 2005, 127, 8977–8984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, W.; Helm, V.; Mccammon, J.A.; Langhoff, P.W. Shedding light on the dark and weakly fluorescent states of green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 6177–6182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakhomov, A.A.; Martynov, V.I. GFP Family: Structural Insights into Spectral Tuning. Chem. Biol. 2008, 15, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Q. Light-driven chiral molecular switches or motors in liquid crystals. Adv. Mater. 2012, 24, 1926–1945. [Google Scholar] [CrossRef]
- Shen, Q.; Wang, L.; Liu, S.; Cao, Y.; Gan, L.; Guo, X.; Steigerwald, M.L.; Shuai, Z.; Liu, Z.; Nuckolls, C. Photoactive gate dielectrics. Adv. Mater. 2010, 22, 3282–3287. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, K.; Oh, S.K.; Nakagawa, M. Light-driven motion of liquids on a photoresponsive surface. Science 2000, 288, 1624–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorostiza, P.; Isacoff, E.Y. Optical switches for remote and noninvasive control of cell signaling. Science 2008, 322, 395–399. [Google Scholar] [CrossRef]
- Zhuang, Y.; Ren, X.; Che, X.; Liu, S.; Huang, W.; Zhao, Q. Organic photoresponsive materials for information storage: A review. Adv. Photonics 2020, 3, 014001. [Google Scholar] [CrossRef]
- Jelken, J.; Henkel, C.; Santer, S. Formation of half-period surface relief gratings in azobenzene containing polymer films. Appl. Phys. B Lasers Opt. 2020, 126, 1–14. [Google Scholar] [CrossRef]
- Xie, Q.; Shao, Z.; Zhao, Y.; Yang, L.; Wu, Q.; Xu, W.; Li, K.; Song, Y.; Hou, H. Novel photo-controllable third-order nonlinear optical (NLO) switches based on azobenzene derivatives. Dye. Pigment. 2019, 170, 107599. [Google Scholar] [CrossRef]
- Villarón, D.; Wezenberg, S.J. Stiff-Stilbene Photoswitches: From Fundamental Studies to Emergent Applications. Angew. Chem. Int. Ed. 2020, 59, 13192–13202. [Google Scholar] [CrossRef]
- Pianowski, Z.L. Recent Implementations of Molecular Photoswitches into Smart Materials and Biological Systems. Chem. A Eur. J. 2019, 25, 5128–5144. [Google Scholar] [CrossRef]
- Mayer, G.; Hechel, A. Biologically active molecules with a “light switch”. Angew. Chem. Int. Ed. 2006, 45, 4900–4921. [Google Scholar] [CrossRef]
- Mart, R.J.; Allemann, R.K. Azobenzene photocontrol of peptides and proteins. Chem. Commun. 2016, 52, 12262–12277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, P.; Maclean, M.; MacGregor, S.J.; Anderson, J.G.; Grant, M.H. Cytotoxic responses to 405 nm light exposure in mammalian and bacterial cells: Involvement of reactive oxygen species. Toxicol. In Vitro 2016, 33, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trigo, F.F.; Corrie, J.E.T.; Ogden, D. Laser photolysis of caged compounds at 405 nm: Photochemical advantages, localisation, phototoxicity and methods for calibration. J. Neurosci. Methods 2009, 180, 9–21. [Google Scholar] [CrossRef]
- Sadovski, O.; Beharry, A.A.; Zhang, F.; Woolley, G.A. Spectral tuning of azobenzene photoswitches for biological applications. Angew. Chem. Int. Ed. 2009, 48, 1484–1486. [Google Scholar] [CrossRef]
- Lameijer, L.N.; Budzak, S.; Simeth, N.A.; Hansen, M.J.; Feringa, B.L.; Jacquemin, D.; Szymanski, W. General Principles for the Design of Visible-Light-Responsive Photoswitches: Tetra-ortho-Chloro-Azobenzenes. Angew. Chem. Int. Ed. 2020, 132, 21847–21854. [Google Scholar] [CrossRef]
- Dong, M.; Babalhavaeji, A.; Samanta, S.; Beharry, A.A.; Woolley, G.A. Red-Shifting Azobenzene Photoswitches for in Vivo Use. Acc. Chem. Res. 2015, 48, 2662–2670. [Google Scholar] [CrossRef]
- Göppert-Mayer, M. Über Elementarakte mit zwei Quantensprüngen. Ann. Phys. 1931, 401, 273–294. [Google Scholar] [CrossRef]
- Kaiser, W.; Garrett, C.G.B. Two-photon excitation in CaF2: Eu2+. Phys. Rev. 1961, 7, 229. [Google Scholar] [CrossRef]
- Marazzi, M.; Gattuso, H.; Monari, A.; Assfeld, X. Steady-State Linear and Non-linear Optical Spectroscopy of Organic Chromophores and Bio-macromolecules. Front. Chem. 2018, 6. [Google Scholar] [CrossRef]
- Tsai, C.-L.; Chen, J.-C.; Wang, W.-J. Near-infrared Absorption Property of Biological Soft Tissue Constituents. J. Med. Biol. Eng. 2001, 21, 7–14. [Google Scholar]
- Mojzisova, H.; Vermot, J. When multiphoton microscopy sees near infrared. Curr. Opin. Genet. Dev. 2011, 21, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Rocheleau, J.V.; Piston, D.W. Two-Photon Excitation Microscopy for the Study of Living Cells and Tissues. Curr. Protoc. Cell Biol. 2003, 59, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Mayer, F.; Bunz, U.H.F.; Blasco, E.; Wegener, M. Multi-material multi-photon 3D laser micro- and nanoprinting. Light Adv. Manuf. 2021, 2, 17. [Google Scholar] [CrossRef]
- Antonov, L.; Kamada, K.; Ohta, K.; Kamounah, F.S. A systematic femtosecond study on the two-photon absorbing D-π-A molecules-π-bridge nitrogen insertion and strength of the donor and acceptor groups. Phys. Chem. Chem. Phys. 2003, 5, 1193–1197. [Google Scholar] [CrossRef]
- Derkowska-Zielinska, B.; Matczyszyn, K.; Dudek, M.; Samoc, M.; Czaplicki, R.; Kaczmarek-Kedziera, A.; Smokal, V.; Biitseva, A.; Krupka, O. All-Optical Poling and Two-Photon Absorption in Heterocyclic Azo Dyes with Different Side Groups. J. Phys. Chem. C 2019, 123, 725–734. [Google Scholar] [CrossRef]
- Ohta, K.; Antonov, L.; Yamada, S.; Kamada, K. Theoretical study of the two-photon absorption properties of several asymmetrically substituted stilbenoid molecules. J. Chem. Phys. 2007, 127, 084504. [Google Scholar] [CrossRef]
- De Boni, L.; Misoguti, L.; Zílio, S.C.; Mendonça, C.R. Degenerate two-photon absorption spectra in azoaromatic compounds. ChemPhysChem 2005, 6, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Carroll, E.C.; Berlin, S.; Levitz, J.; Kienzler, M.A.; Yuan, Z.; Madsen, D.; Larsen, D.S.; Isacoff, E.Y.; Denk, W. Two-photon brightness of azobenzene photoswitches designed for glutamate receptor optogenetics. Proc. Natl. Acad. Sci. USA 2015, 112, E776–E785. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Zhang, J.; Yin, L.; Long, X.; Zhang, W.; Zhang, Q. Recent progress in efficient organic two-photon dyes for fluorescence imaging and photodynamic therapy. J. Mater. Chem. C 2020, 8, 6342–6349. [Google Scholar] [CrossRef]
- De Wergifosse, M.; Houk, A.L.; Krylov, A.I.; Elles, C.G. Two-photon absorption spectroscopy of trans-stilbene, cis-stilbene, and phenanthrene: Theory and experiment. J. Chem. Phys. 2017, 146, 144305. [Google Scholar] [CrossRef] [Green Version]
- Grabarek, D.; Andruniów, T. Assessment of Functionals for TDDFT Calculations of One- and Two-Photon Absorption Properties of Neutral and Anionic Fluorescent Proteins Chromophores. J. Chem. Theory Comput. 2019, 15, 490–508. [Google Scholar] [CrossRef] [PubMed]
- Grabarek, D.; Andruniów, T. Illuminating the origins of two-photon absorption properties in fluorescent protein chromophores. Int. J. Quantum Chem. 2020, 120, e26086. [Google Scholar] [CrossRef]
- Rossano-Tapia, M.; Olsen, J.M.H.; Brown, A. Two-Photon Absorption Cross-Sections in Fluorescent Proteins Containing Non-canonical Chromophores Using Polarizable QM/MM. Front. Mol. Biosci. 2020, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Tahara, T. Two-photon absorption spectrum of all-trans retinal. Chem. Phys. Lett. 2003, 376, 237–243. [Google Scholar] [CrossRef]
- Gholami, S.; Pedraza-González, L.; Yang, X.; Granovsky, A.A.; Ioffe, I.N.; Olivucci, M. Multistate Multiconfiguration Quantum Chemical Computation of the Two-Photon Absorption Spectra of Bovine Rhodopsin. J. Phys. Chem. Lett. 2019, 10, 6293–6300. [Google Scholar] [CrossRef] [PubMed]
- Palczewska, G.; Vinberg, F.; Stremplewski, P.; Bircher, M.P.; Salom, D.; Komar, K.; Zhang, J.; Cascella, M.; Wojtkowski, M.; Kefalov, V.J.; et al. Human infrared Vision is triggered by two-photon chromophore isomerization. Proc. Natl. Acad. Sci. USA 2014, 111, E5445–E5454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neveu, P.; Aujard, I.; Benbrahim, C.; Le Saux, T.; Allemand, J.F.; Vriz, S.; Bensimon, D.; Jullien, L. A caged retinoic acid for one- and two-photon excitation in zebrafish embryos. Angew. Chem. Int. Ed. 2008, 120, 3804–3806. [Google Scholar] [CrossRef]
- Gacek, D.A.; Betke, A.; Nowak, J.; Lokstein, H.; Walla, P.J. Two-photon absorption and excitation spectroscopy of carotenoids, chlorophylls and pigment-protein complexes. Phys. Chem. Chem. Phys. 2021, 23, 8731–8738. [Google Scholar] [CrossRef]
- Bort, G.; Gallavardin, T.; Ogden, D.; Dalko, P.I. From One-Photon to Two-Photon Probes: “Caged” Compounds, Actuators, and Photoswitches. Angew. Chem. Int. Ed. 2013, 52, 4526–4537. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Lomas, M.; Martínez-López, D.; Campos, P.J.; Sampedro, D. Tuning of the properties of rhodopsin-based molecular switches. Tetrahedron Lett. 2014, 55, 3361–3364. [Google Scholar] [CrossRef]
- Blanco-Lomas, M.; Funes-Ardoiz, I.; Campos, P.J.; Sampedro, D. Oxazolone-based photoswitches: Synthesis and properties. Eur. J. Org. Chem. 2013, 2013, 6611–6618. [Google Scholar] [CrossRef]
- Blanco-Lomas, M.; Campos, P.J.; Sampedro, D. Benzylidene-oxazolones as molecular photoswitches. Org. Lett. 2012, 14, 4334–4337. [Google Scholar] [CrossRef]
- Funes-Ardoiz, I.; Blanco-Lomas, M.; Campos, P.J.; Sampedro, D. Benzylidene-oxazolones as photoswitches: Photochemistry and theoretical calculations. Tetrahedron 2013, 69, 9766–9771. [Google Scholar] [CrossRef]
- Martínez-López, D.; Yu, M.L.; García-Iriepa, C.; Campos, P.J.; Frutos, L.M.; Golen, J.A.; Rasapalli, S.; Sampedro, D. Hydantoin-based molecular photoswitches. J. Org. Chem. 2015, 80, 3929–3939. [Google Scholar] [CrossRef]
- García-Iriepa, C.; Ernst, H.A.; Liang, Y.; Unterreiner, A.N.; Frutos, L.M.; Sampedro, D. Study of Model Systems for Bilirubin and Bilin Chromophores: Determination and Modification of Thermal and Photochemical Properties. J. Org. Chem. 2016, 81, 6292–6302. [Google Scholar] [CrossRef] [PubMed]
- Kamada, K.; Iwase, Y.; Sakai, K.; Kondo, K.; Ohta, K. Cationic two-photon absorption chromophores with double-and triple-bond cores in symmetric/asymmetric arrangements. J. Phys. Chem. C 2009, 113, 11469–11474. [Google Scholar] [CrossRef]
- Mcclain, W.M. Excited state symmetry assignment through polarized two-photon absorption studies of fluids. J. Chem. Phys. 1971, 55, 2789–2796. [Google Scholar] [CrossRef]
- Arul Murugan, N.; Kongsted, J.; Rinkevicius, Z.; Aidas, K.; Mikkelsen, K.V.; Ågren, H. Hybrid density functional theory/molecular mechanics calculations of two-photon absorption of dimethylamino nitro stilbene in solution. Phys. Chem. Chem. Phys. 2011, 13, 12506–12516. [Google Scholar] [CrossRef]
- Aidas, K.; Angeli, C.; Bak, K.L.; Bakken, V.; Bast, R.; Boman, L.; Christiansen, O.; Cimiraglia, R.; Coriani, S.; Dahle, P.; et al. The Dalton quantum chemistry program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 269–284. [Google Scholar] [CrossRef]
- Beerepoot, M.T.P.; Friese, D.H.; List, N.H.; Kongsted, J.; Ruud, K. Benchmarking two-photon absorption cross sections: Performance of CC2 and CAM-B3LYP. Phys. Chem. Chem. Phys. 2015, 17, 19306–19314. [Google Scholar] [CrossRef] [Green Version]
- Beerepoot, M.T.P.; Alam, M.M.; Bednarska, J.; Bartkowiak, W.; Ruud, K.; Zaleśny, R. Benchmarking the Performance of Exchange-Correlation Functionals for Predicting Two-Photon Absorption Strengths. J. Chem. Theory Comput. 2018, 14, 3677–3685. [Google Scholar] [CrossRef] [Green Version]
- Laurent, A.D.; Jacquemin, D. TD-DFT benchmarks: A review. Int. J. Quantum Chem. 2013, 113, 2019–2039. [Google Scholar] [CrossRef]
- Lin, N.; Luo, Y.; Ruud, K.; Zhao, X.; Santoro, F.; Rizzo, A. Differences in two-photon and one-photon absorption profiles induced by vibronic coupling: The case of dioxaborine heterocyclic dye. ChemPhysChem 2011, 12, 3392–3403. [Google Scholar] [CrossRef]
- Cammi, R.; Frediani, L.; Mennucci, B.; Ruud, K. Multiconfigurational self-consistent field linear response for the polarizable continuum model: Theory and application to ground and excited-state polarizabilities of para-nitroaniline in solution. J. Chem. Phys. 2003, 119, 5818–5827. [Google Scholar] [CrossRef]
- Frediani, L.; Ågren, H.; Ferrighi, L.; Ruud, K. Second-harmonic generation of solvated molecules using multiconfigurational self-consistent-field quadratic response theory and the polarizable continuum model. J. Chem. Phys. 2005, 123, 144117. [Google Scholar] [CrossRef] [PubMed]
- Finley, J.; Malmqvist, P.-Å.; Roos, B.O.; Serrano-Andrés, L. The multi-state CASPT2 method. Chem. Phys. Lett. 1998, 288, 299–306. [Google Scholar] [CrossRef]
- Zobel, J.P.; Nogueira, J.J.; González, L. The IPEA dilemma in CASPT2. Chem. Sci. 2017, 8, 1482–1499. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, N.; Malmqvist, P.Å. Multiconfiguration perturbation theory with imaginary level shift. Chem. Phys. Lett. 1997, 274, 196–204. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision A.03; Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Fdez. Galván, I.; Vacher, M.; Alavi, A.; Angeli, C.; Aquilante, F.; Autschbach, J.; Bao, J.J.; Bokarev, S.I.; Bogdanov, N.A.; Carlson, R.K.; et al. OpenMolcas: From Source Code to Insight. J. Chem. Theory Comput. 2019, 15, 5925. [Google Scholar] [CrossRef]
- Brouwer, A.M. Standards for photoluminescence quantum yield measurements in solution (IUPAC technical report). Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar] [CrossRef] [Green Version]
- Rumi, M.; Perry, J.W. Two-photon absorption: An overview of measurements and principles. Adv. Opt. Photonics 2010, 2, 451–518. [Google Scholar] [CrossRef]
- Terenziani, F.; Katan, C.; Badaeva, E.; Tretiak, S.; Blanchara-Desce, M. Enhanced two-photon absorption of organic chromophores: Theoretical and experimental assessments. Adv. Mater. 2008, 20, 4641–4678. [Google Scholar] [CrossRef] [Green Version]
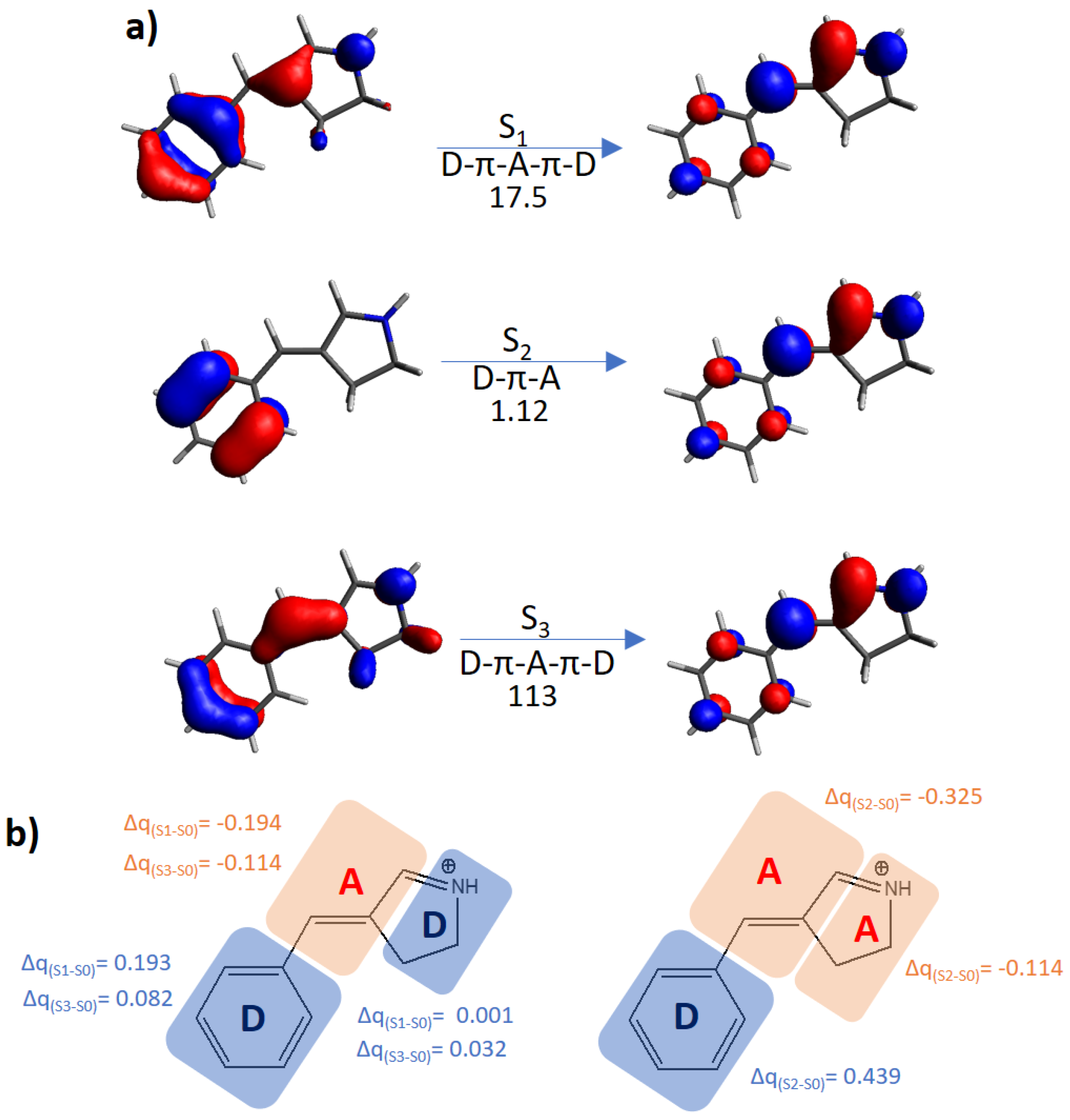

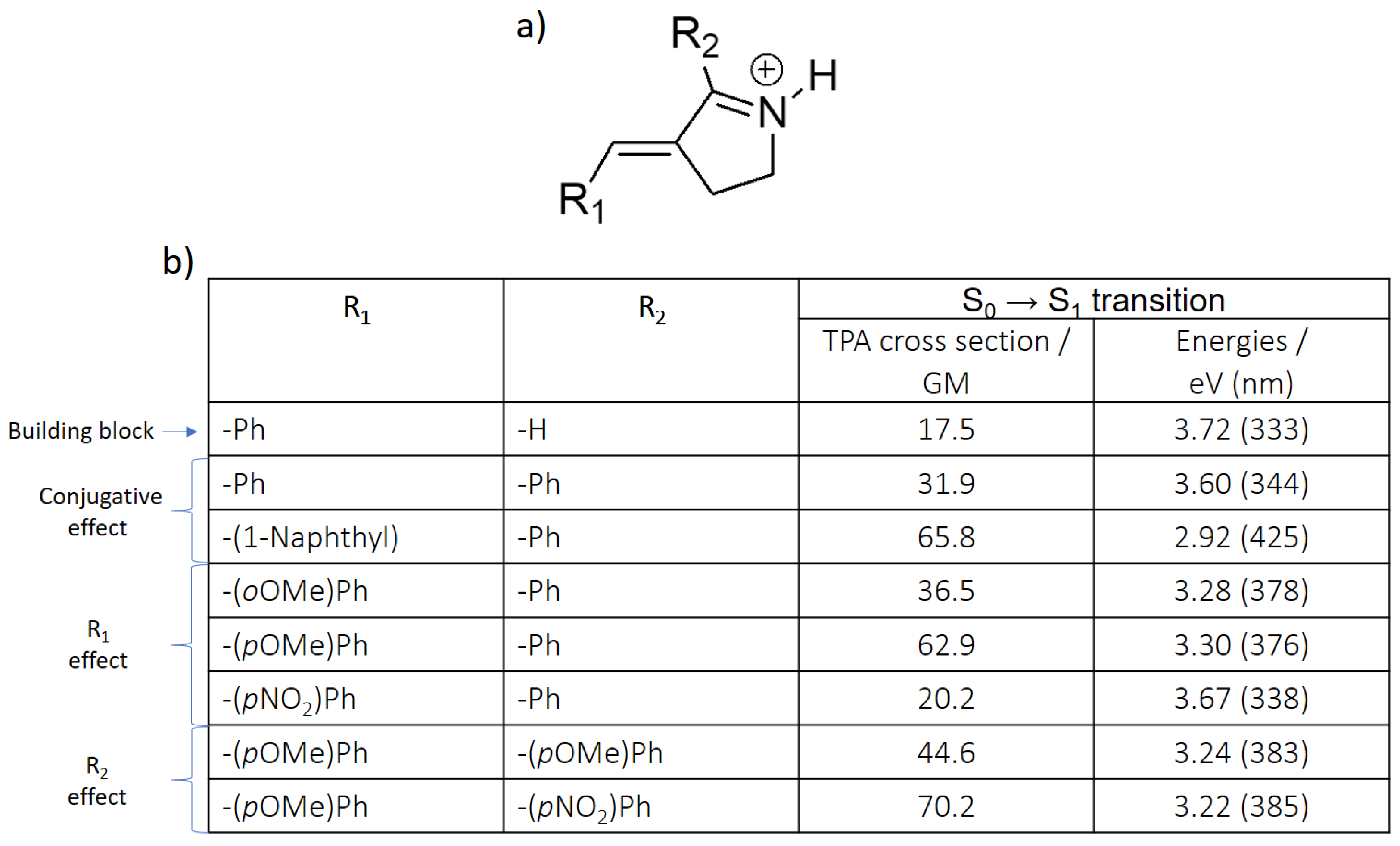
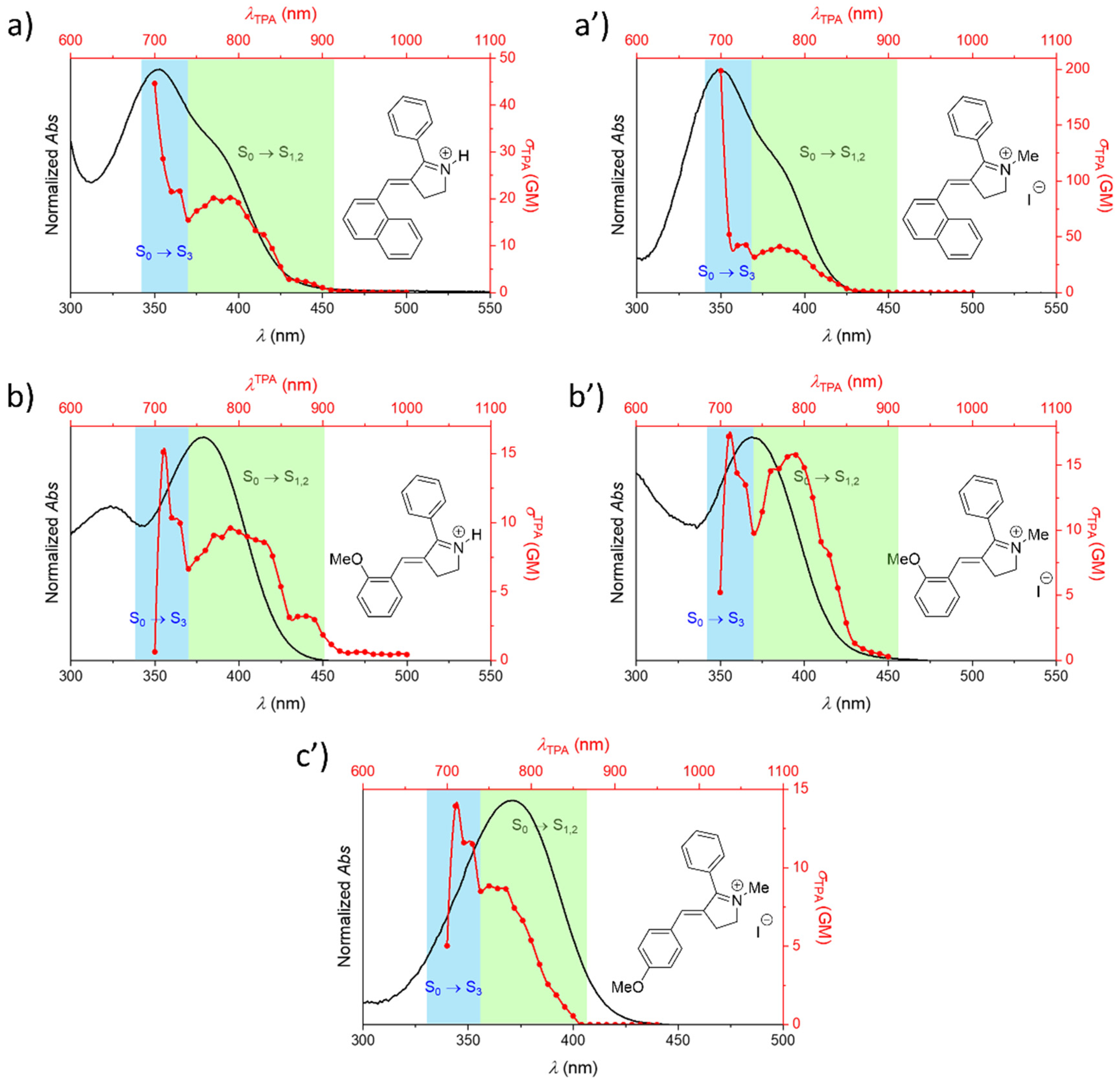
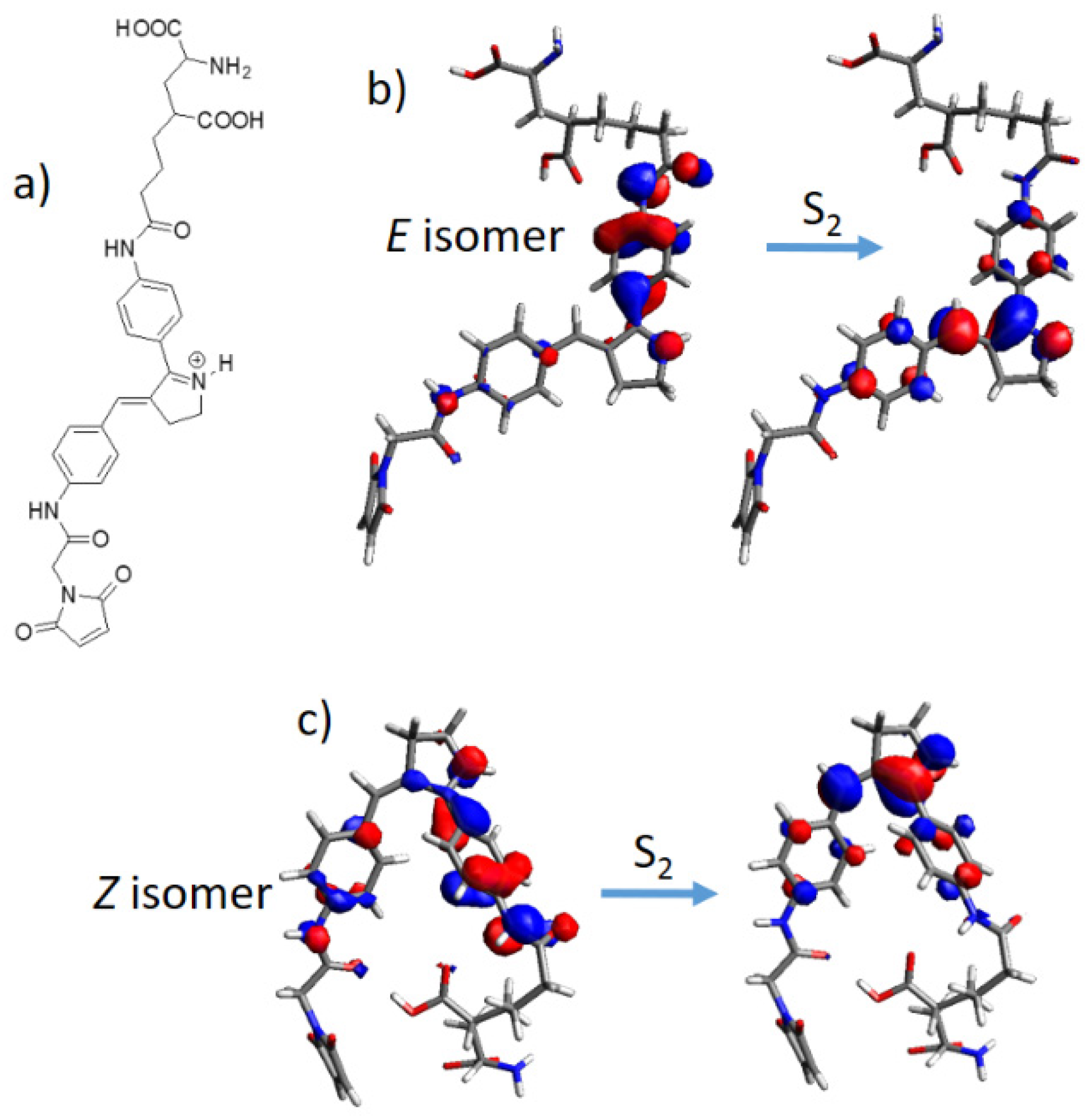

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
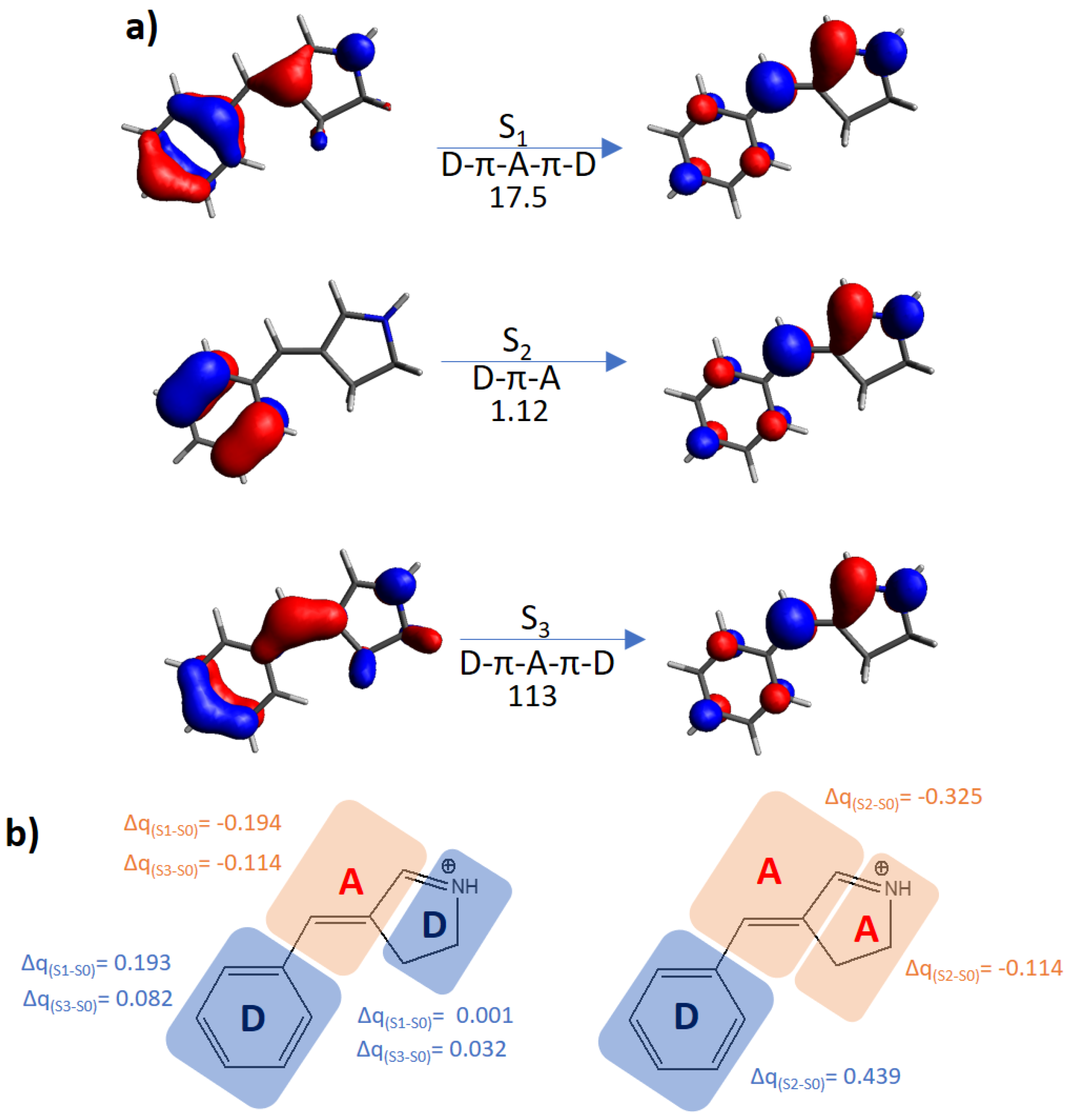
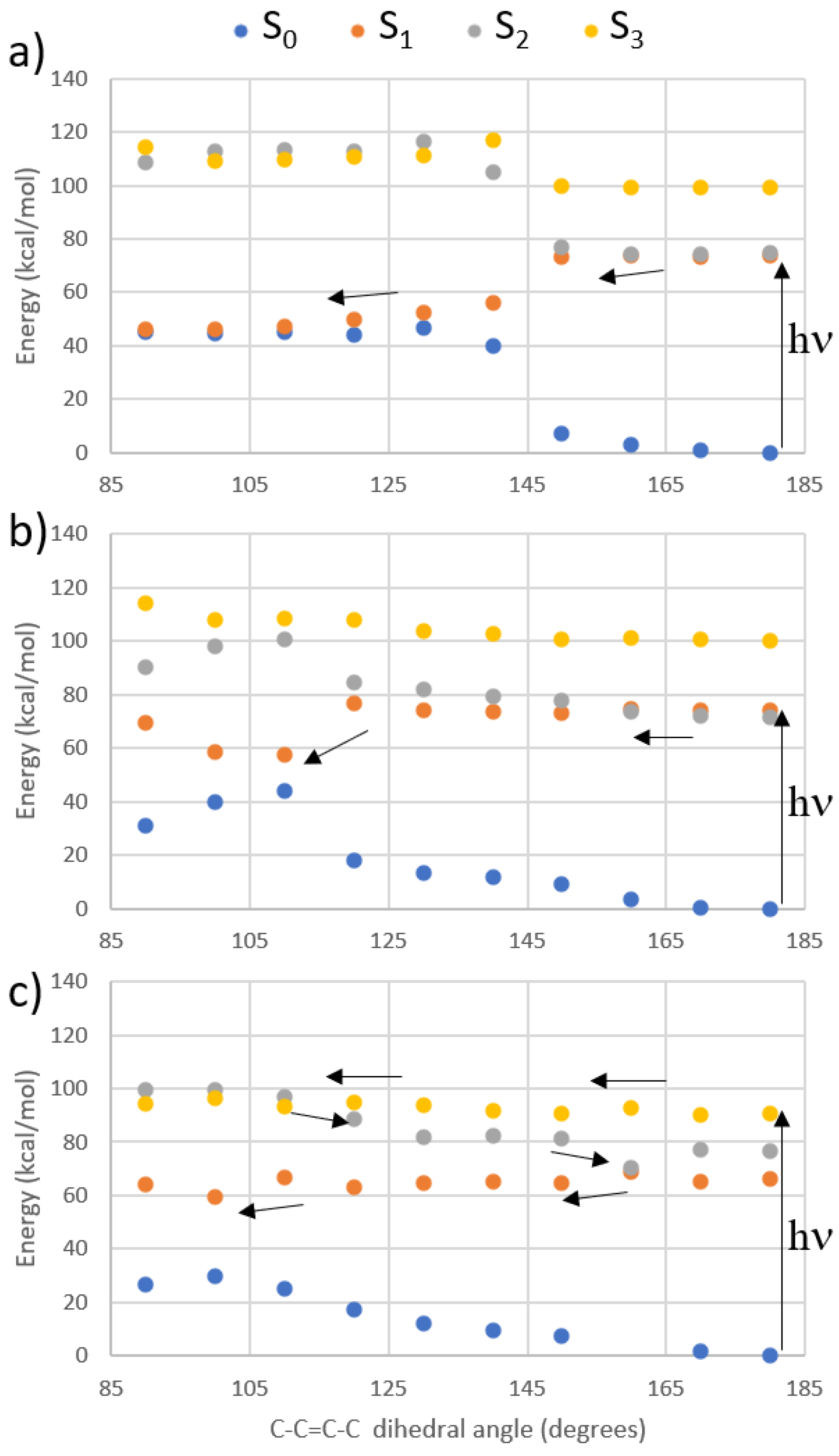
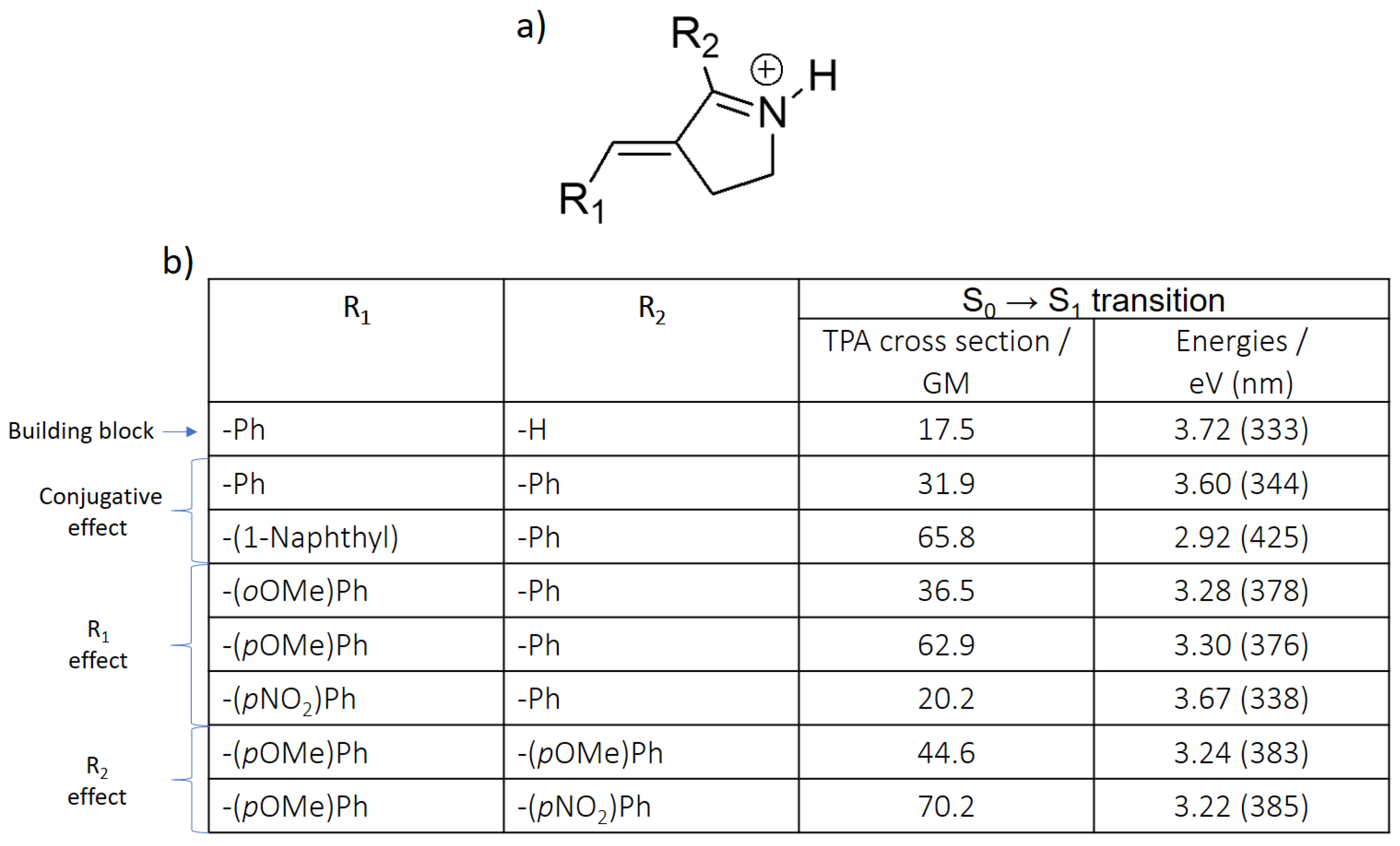
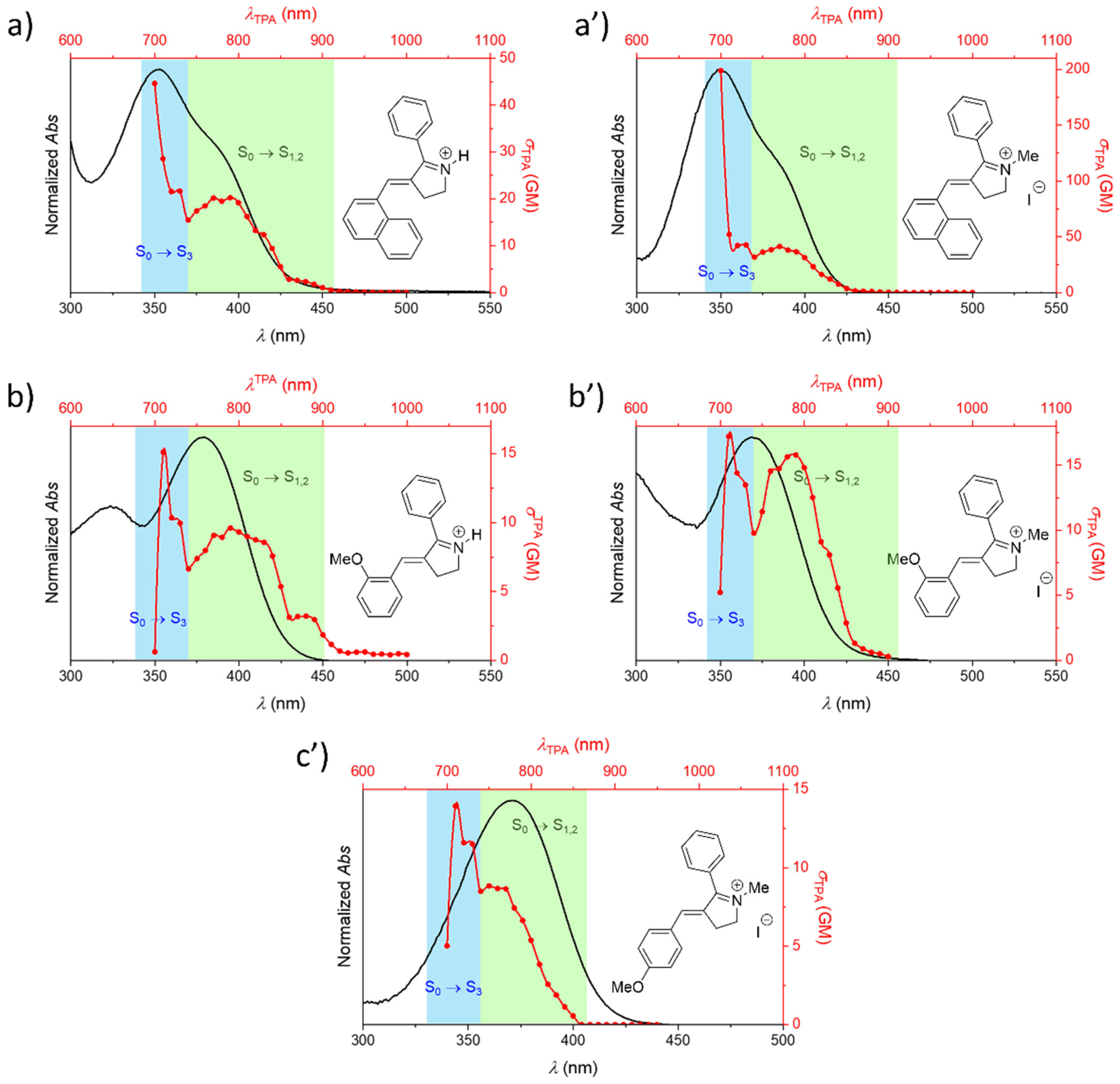
| Building Block | Isomer | S0 → S1 | S0 → S2 | S0 → S3 | |||
|---|---|---|---|---|---|---|---|
| λ/ eV (nm) | σTPA/ GM | λ/ eV (nm) | σTPA/ GM | λ/ eV (nm) | σTPA/ GM | ||
| Protonated Schiff base-like | E | 3.72 (333) | 17.5 | 3.93 (315) | 1.12 | 5.63 (220) | 113 |
| Z | 3.42 (363) | 12.3 | 3.77 (329) | 1.38 | 5.29 (234) | 156 | |
| Schiff base-like | E | 4.51 (275) | 0.72 | 4.83 (257) | 0.07 | 4.95 (250) | 1.34 |
| Z | 4.49 (276) | 1.14 | 4.76 (260) | 0.13 | 5.02 (247) | 1.03 | |
| Oxazolone-like | E | 3.90 (318) | 2.39 | 4.31 (288) | 6.00 × 10−4 | 4.50 (276) | 1.25 |
| Z | 3.96 (313) | 5.42 | 4.33 (286) | 1.00 × 10−4 | 4.57 (271) | 1.57 | |
| Hydantoin-like | E | 3.95 (314) | 2.96 | 4.58 (271) | 5.00 × 10−4 | 4.69 (264) | 1.49 |
| Z | 4.19 (296) | 3.32 | 4.67 (265) | 0.33 | 4.91 (253) | 1.74 | |
| Pyrrolinone-like | E | 3.86 (321) | 4.45 | 4.28 (290) | 0.04 | 4.85 (256) | 1.79 |
| Z | 3.93 (315) | 5.90 | 4.32 (287) | 0.12 | 4.84 (256) | 1.96 | |
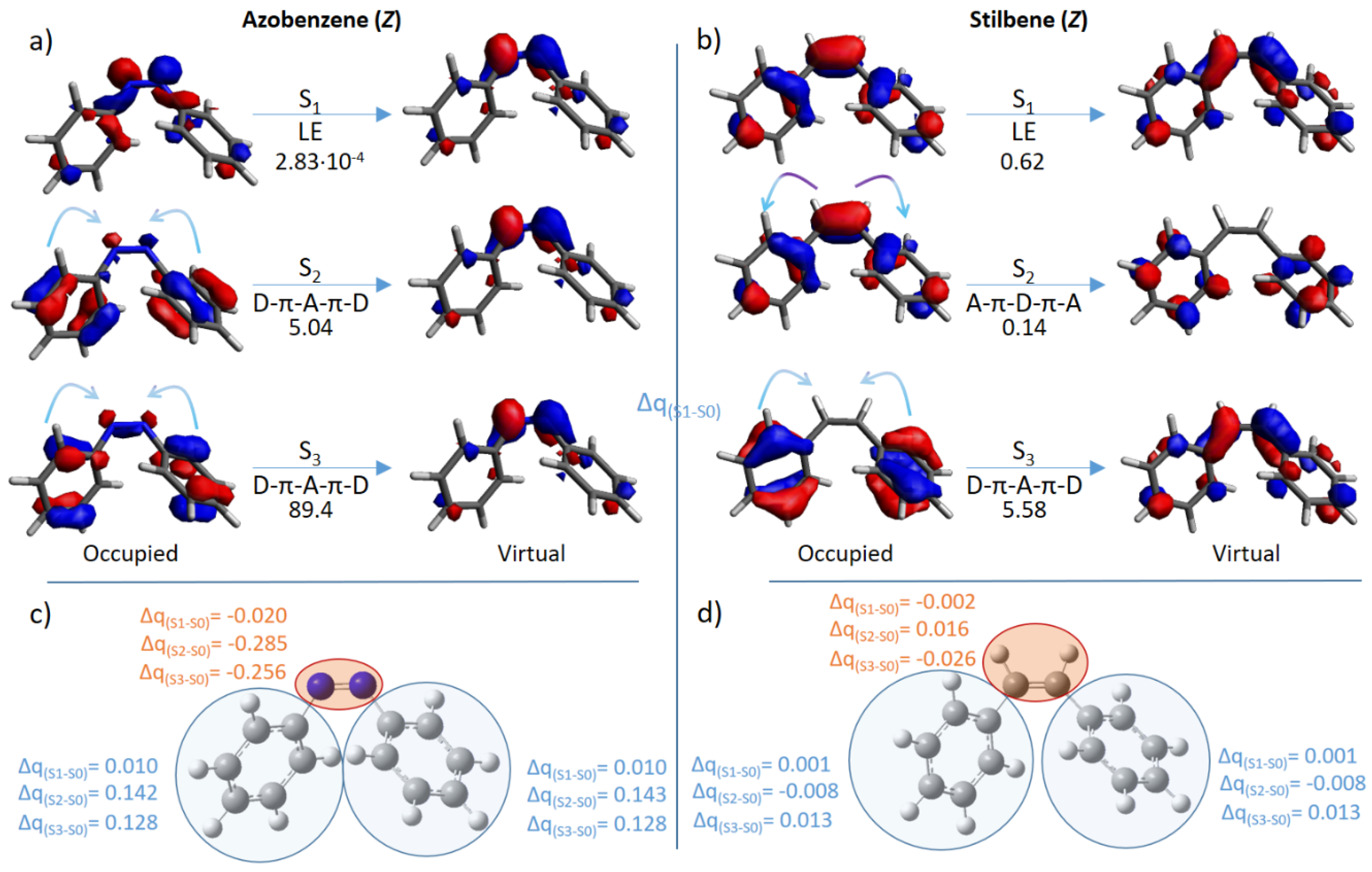
| Azobenzene | E | 2.74 (452) | 4.69 × 10−3 | 3.97 (312) | 3.85 × 10−6 | 4.63 (268) | 1.09 × 10−4 |
| Z | 2.66 (466) | 2.83 × 10−4 | 4.60 (270) | 5.04 | 4.71 (263) | 89.4 | |
| Stilbene | E | 4.13 (300) | 2.83 × 10−7 | 4.90 (253) | 1.25 × 10−5 | 5.70 (218) | 1.78 × 10−2 |
| Z | 4.38 (283) | 0.62 | 4.93 (251) | 0.14 | 5.11 (243) | 5.58 | |
| Photoswitch | Isomer | S0 → S1 | S0 → S2 | ||
|---|---|---|---|---|---|
| λ/ eV (nm) | σTPA/ GM | λ/ eV (nm) | σTPA/ GM | ||
| Protonated Schiff base-like | E | 3.16 (392) | 43.2 | 3.76 (330) | 609.0 |
| Z | 2.86 (434) | 22.0 | 3.62 (343) | 123.0 | |
| Azobenzene | E | 2.80 (443) | 8.04 × 10−3 | 3.54 (350) | 2.07 |
| Z | 2.60 (477) | 4.57 × 10−2 | 3.88 (320) | 4.40 × 10−2 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marazzi, M.; García-Iriepa, C.; Benitez-Martin, C.; Najera, F.; Monari, A.; Sampedro, D. E/Z Molecular Photoswitches Activated by Two-Photon Absorption: Comparison between Different Families. Molecules 2021, 26, 7379. https://doi.org/10.3390/molecules26237379
Marazzi M, García-Iriepa C, Benitez-Martin C, Najera F, Monari A, Sampedro D. E/Z Molecular Photoswitches Activated by Two-Photon Absorption: Comparison between Different Families. Molecules. 2021; 26(23):7379. https://doi.org/10.3390/molecules26237379
Chicago/Turabian StyleMarazzi, Marco, Cristina García-Iriepa, Carlos Benitez-Martin, Francisco Najera, Antonio Monari, and Diego Sampedro. 2021. "E/Z Molecular Photoswitches Activated by Two-Photon Absorption: Comparison between Different Families" Molecules 26, no. 23: 7379. https://doi.org/10.3390/molecules26237379
APA StyleMarazzi, M., García-Iriepa, C., Benitez-Martin, C., Najera, F., Monari, A., & Sampedro, D. (2021). E/Z Molecular Photoswitches Activated by Two-Photon Absorption: Comparison between Different Families. Molecules, 26(23), 7379. https://doi.org/10.3390/molecules26237379