
Micro-Solvated DMABN: Excited State Quantum Dynamics and Dual Fluorescence Spectra



Abstract
:
1. Introduction
2. Methods
2.1. Experimental Section
2.2. Electronic Structure
2.3. LVC Parametrised Potentials
2.4. Quantum Dynamics
3. Results
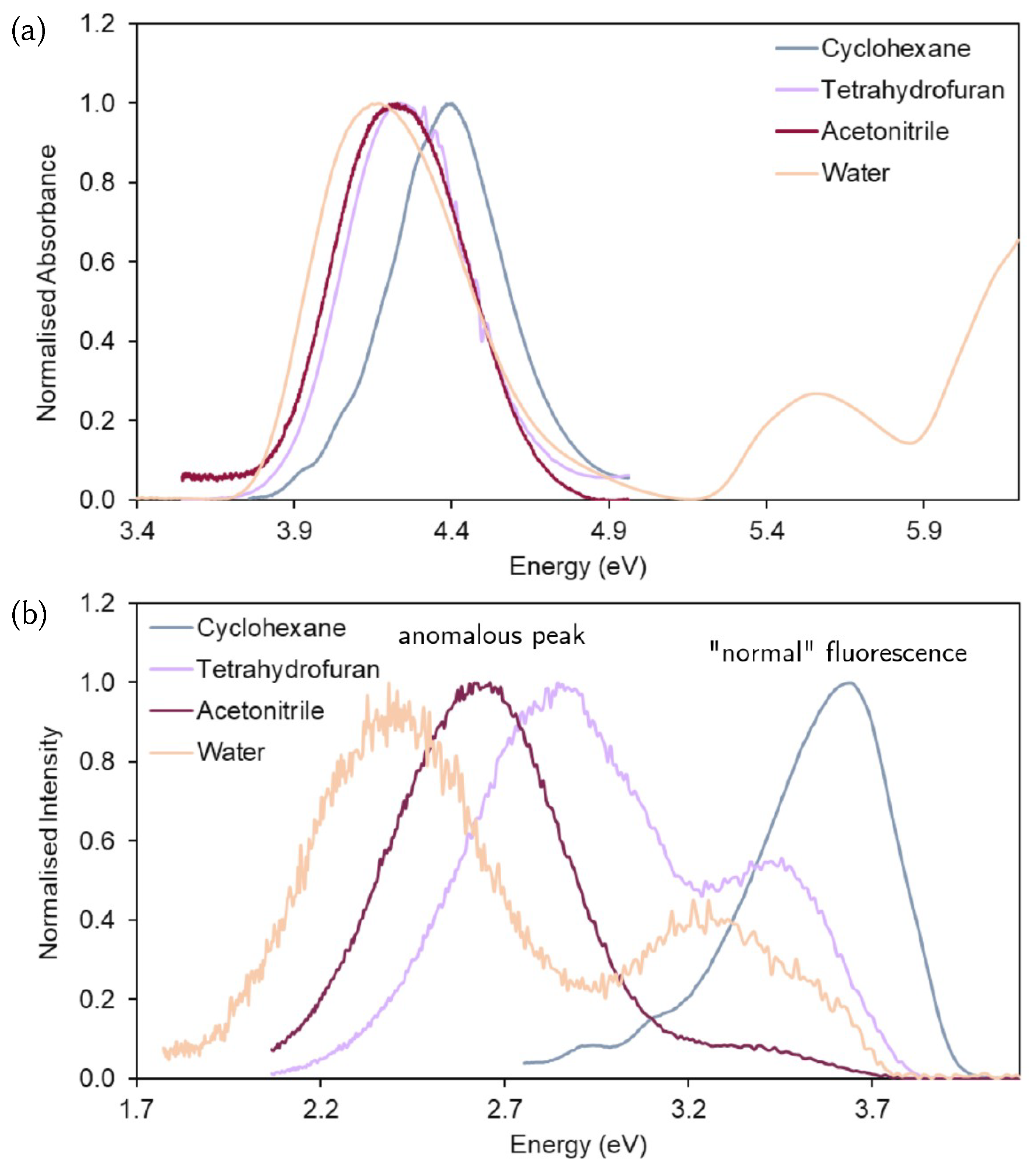
3.1. Analysis of the Experimental Spectra
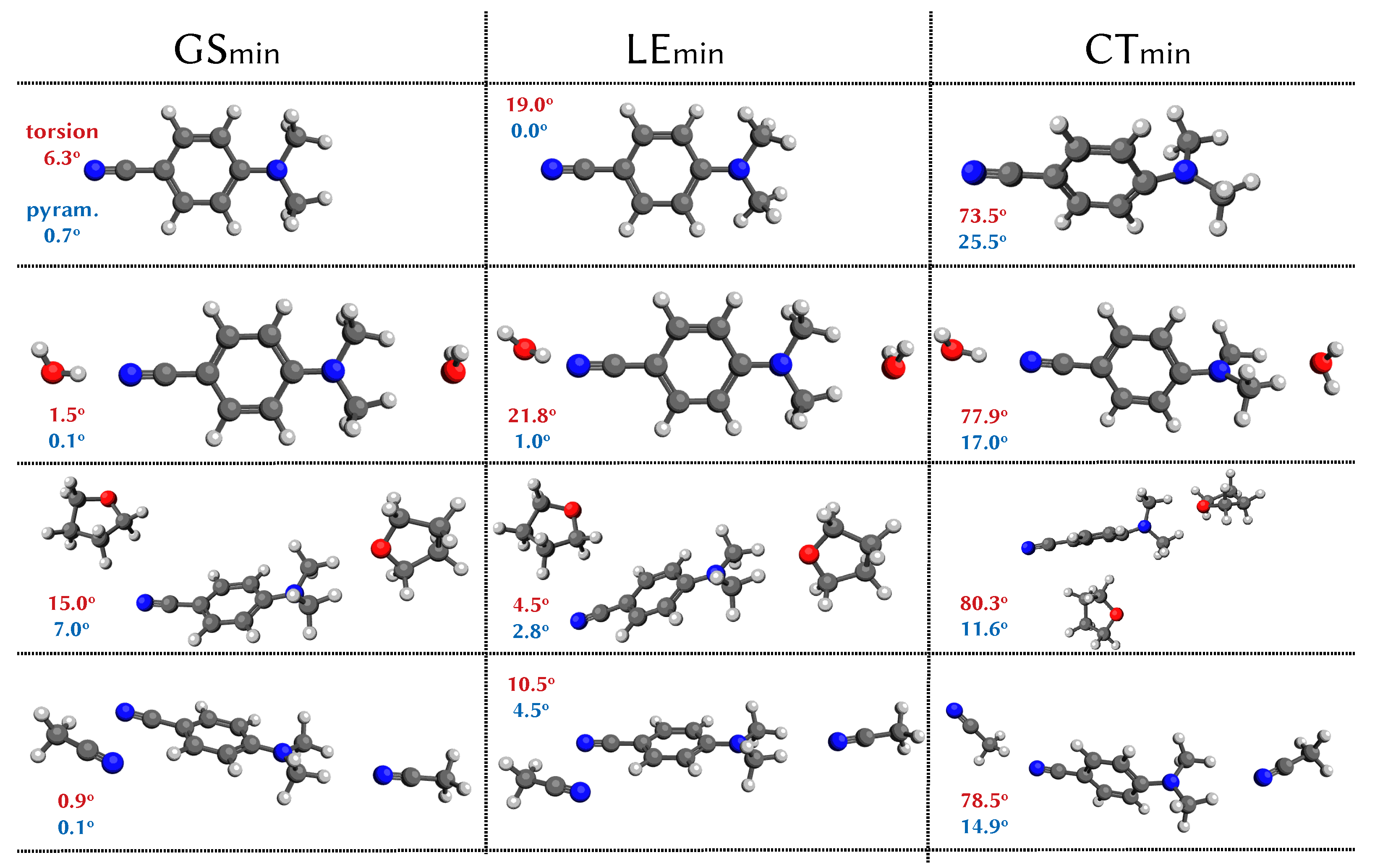
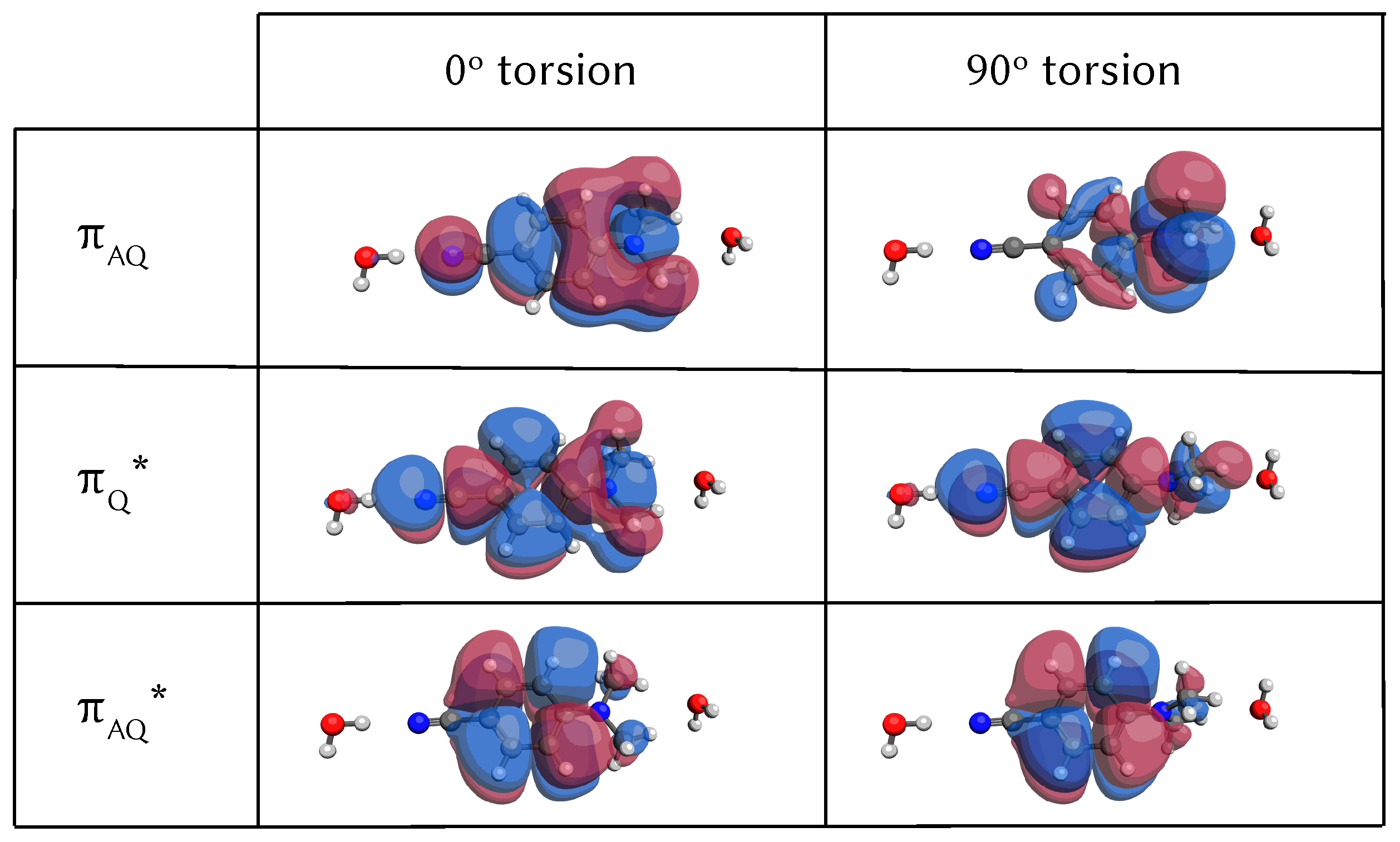
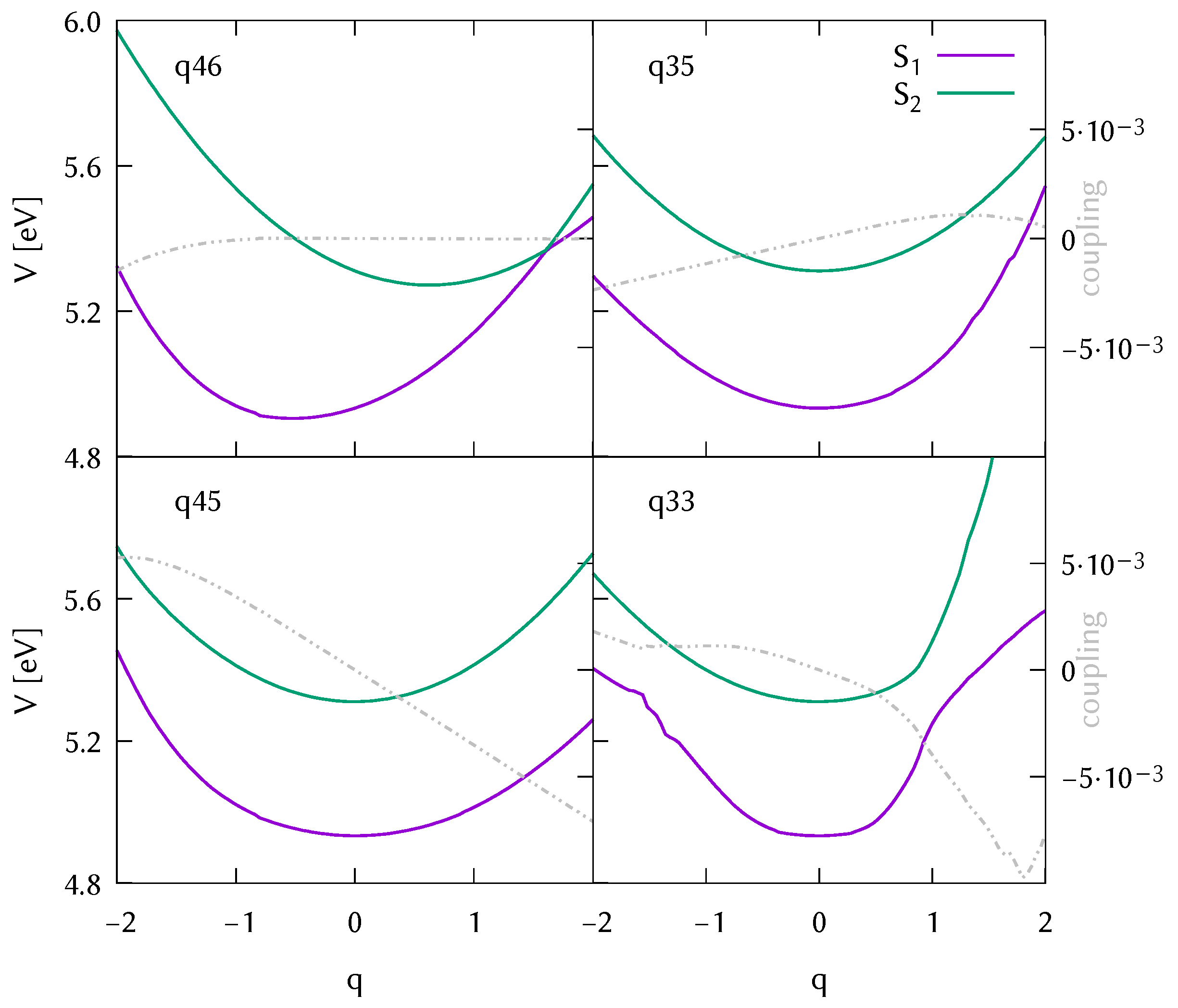
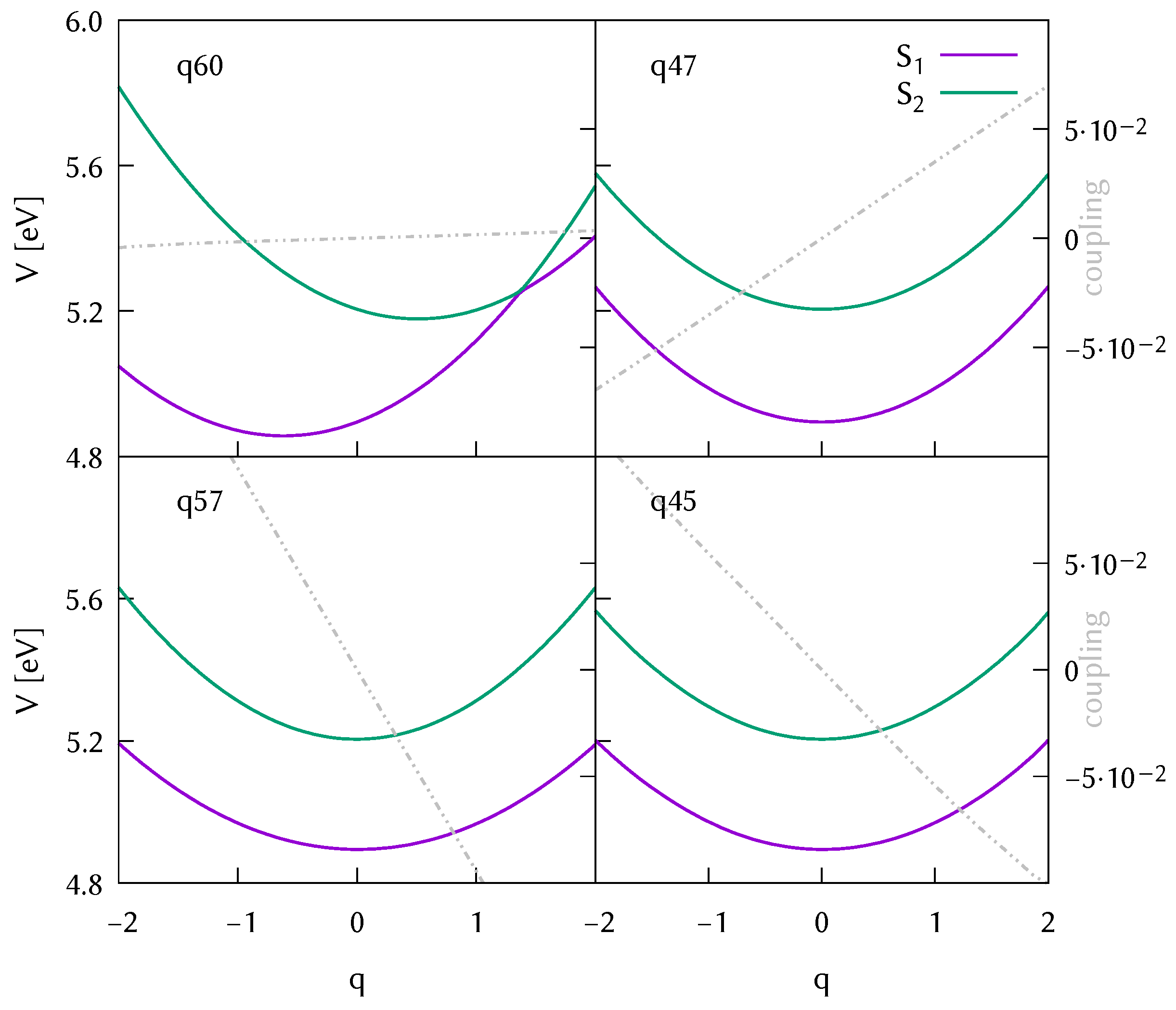
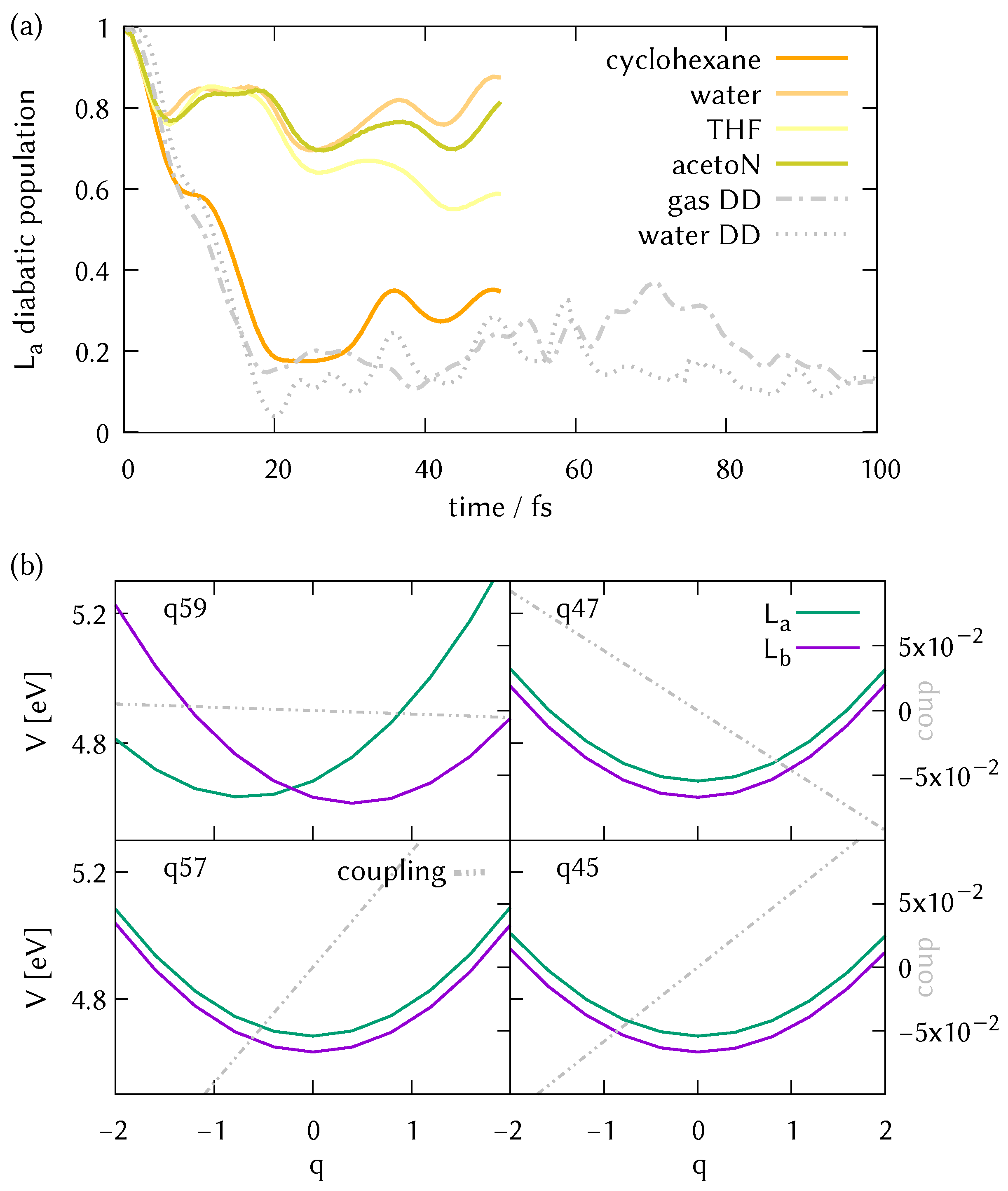
3.2. Analysis of the Potential Energy Surfaces
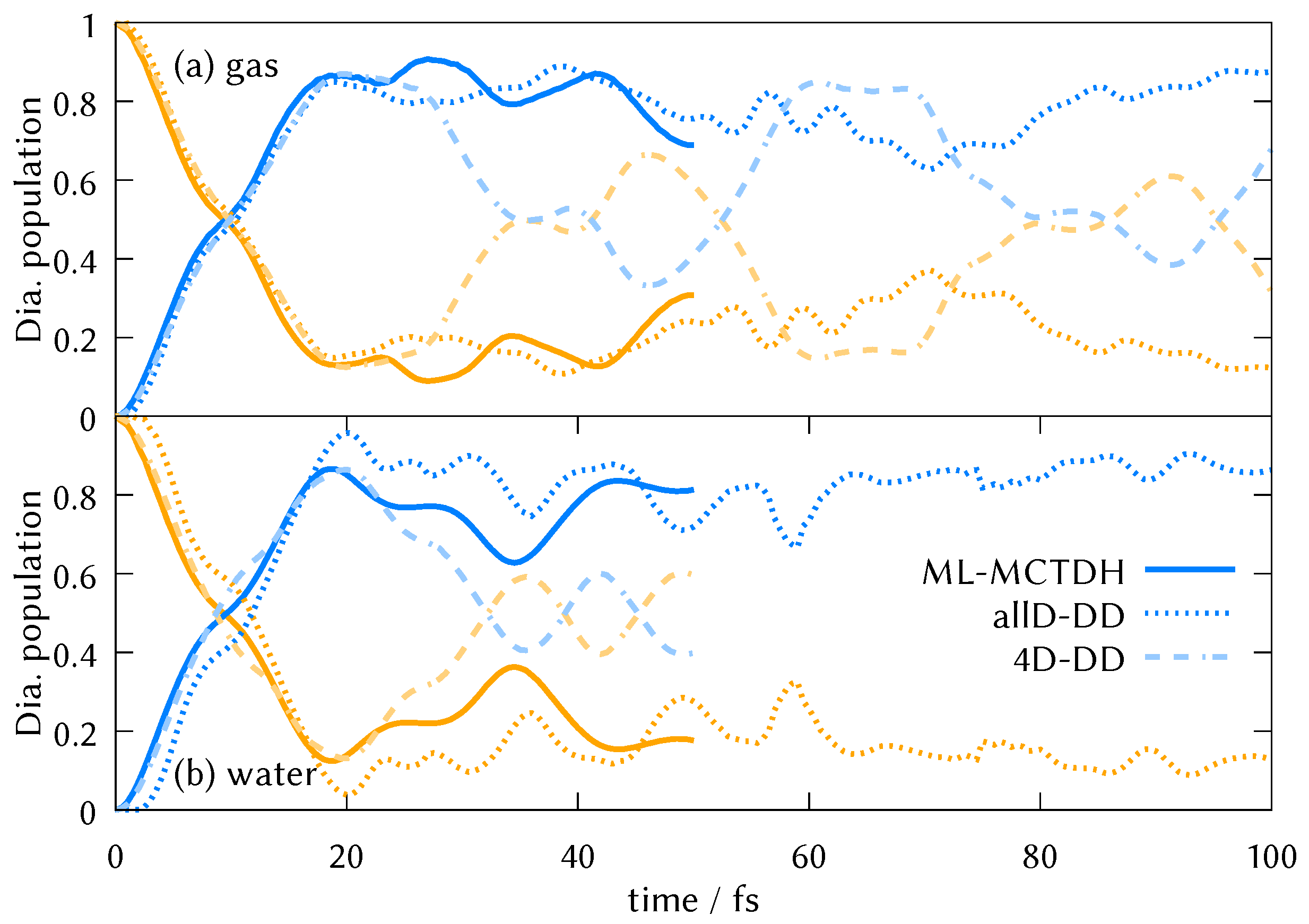
3.3. DD-vMCG Excited State Dynamics
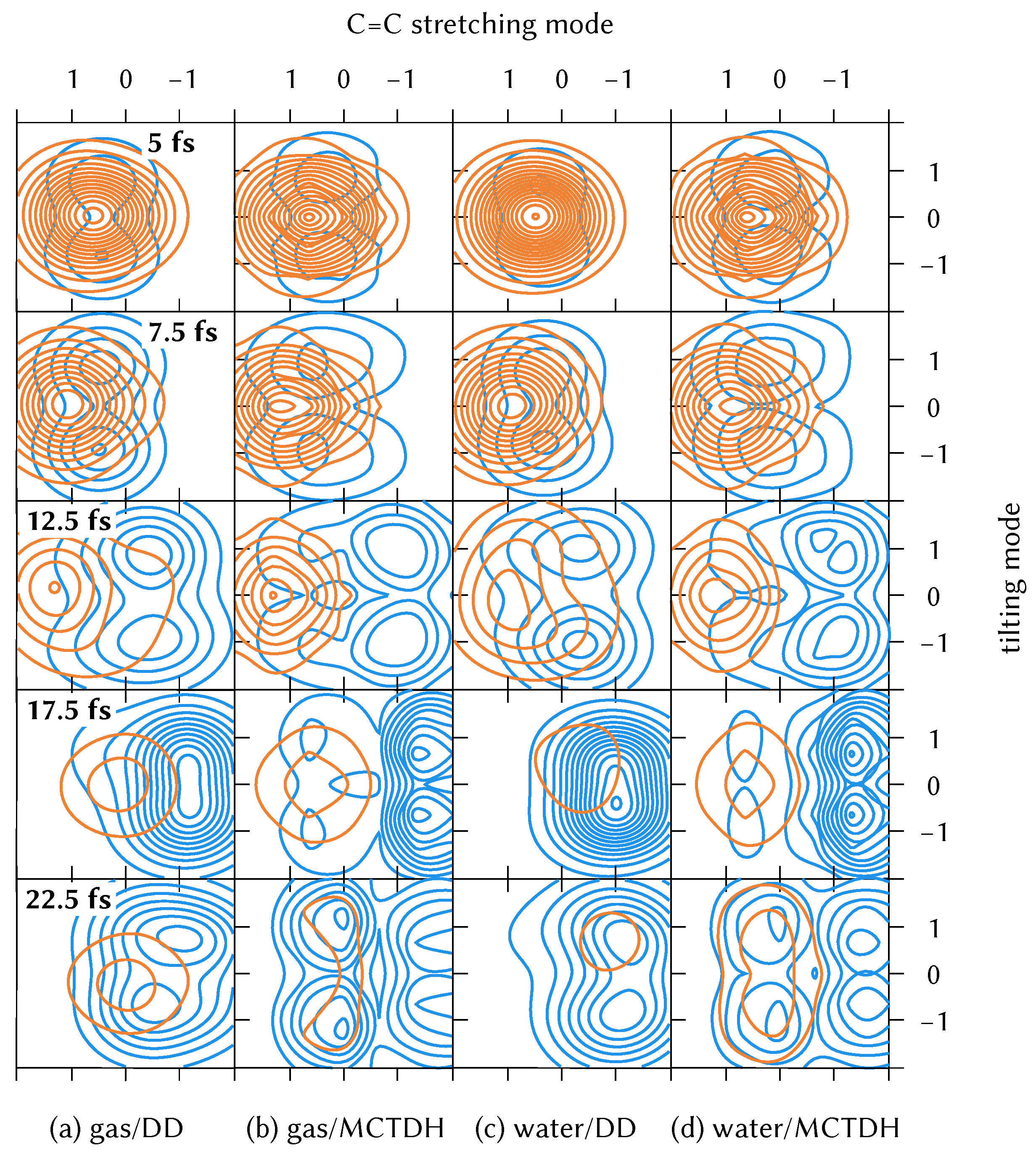
3.4. Normal Mode Analysis of Dynamics
3.5. Solvent Effects: ML-MCTDH Dynamics on Parametrised Hamiltonians
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DMABN | 1,1-dimethylaminobenzonitrile |
| DD-vMCG | Direct dynamics variational multiconfigurational Gaussian |
| MCTDH | Multiconfigurational Time-dependent Hartree |
| CASPT2 | Complete active space perturbation theory to second order |
| TDDFT | Time-dependent density functional theory |
| LVC | Linear Vibronic Coupling |
| PES | Potential Energy Surface |
References
- Lippert, E.; Lüder, W.; Moll, F.; Nägele, W.; Boos, H.; Prigge, H.; Seibold-Blankenstein, I. Umwandlung von Elektronenanregungsenergie. Angew. Chem. 1961, 73, 695–706. [Google Scholar] [CrossRef]
- Khalil, O.S.; Hofeldt, R.; McGlynn, S. Electronic Spectroscopy of Highly-Polar Aromatics. Molecular interactions in the ground and excited states of N,N-Dimethyl-p-cyanoanline. Chem. Phys. Lett. 1972, 17, 479–481. [Google Scholar] [CrossRef]
- Rotkiewicz, K.; Grellmann, K.H.; Grabowski, Z.R. Reinterpretation of the anomalous fluorescense of p-N,N-dimethylamino-benzonitrile. Chem. Phys. Lett. 1973, 19, 315–318. [Google Scholar] [CrossRef]
- Lipiński, J.; Chojnacki, H.; Grabowski, Z.R.; Rotkiewicz, K. Theoretical model for the double fluorescence of p-cyano-N,N-dimethylaniline in polar solvents. Chem. Phys. Lett. 1980, 70, 449–453. [Google Scholar] [CrossRef]
- Lippert, E.; Lüder, W.; Boos, H. Advances in Molecular Spectroscopy; Pergamon Press: Oxford, UK, 1962. [Google Scholar]
- Platt, R.J. Classification of Spectra of Cata-Condensed Hydrocarbons. J. Chem. Phys. 1949, 17, 484–495. [Google Scholar] [CrossRef]
- Kosower, E.M.; Dodiuk, H. Multiple fluorescences. II. A new scheme for 4-(N,N-dimethylamino)benzonitrile including proton transfer. J. Am. Chem. Soc. 1976, 98, 924–929. [Google Scholar] [CrossRef]
- Chandross, E.A. Complexes of Dipolar Excited States and Small Polar Molecules, in The Exciplex; Elsevier Science: New York, NY, USA, 1975; p. 187. [Google Scholar]
- Zachariasse, K.A.; Grobys, M.; Von Der Haar, T.; Hebecker, A.; Il’ichev, V.Y.; Jiang, Y.B.; Morawski, O.; Kühnle, W. Intramolecular charge transfer in the excited state. Kinetics and configurational changes. J. Photochem. Photobiol. A Chem. 1996, 102, 59–70. [Google Scholar] [CrossRef]
- Zachariasse, K.A.; Grobys, M.; Von Der Haar, T.; Hebecker, A.; Il’ichev, V.Y.; Morawski, O.; Rückert, I.; Kühnle, W. Photoinduced intramolecular charge transfer and internal conversion in molecules with a small energy gap between S1 and S2. Dynamics and structure. J. Photochem. Photobiol. A Chem. 1997, 105, 373–383. [Google Scholar] [CrossRef]
- Lewis, F.D.; Holman, B. Singlet states of benzonitrile and p-dimethylaminobenzonitrile. J. Phys. Chem. 1980, 84, 2326–2328. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W. Promotion of intramolecular charge transfer in dimethylamino derivatives: Twisting versus acceptor-group rehybridization. Chem. Phys. Lett. 1996, 259, 119–127. [Google Scholar] [CrossRef]
- Coto, P.B.; Serrano-Andrés, L.; Gustavsson, T.; Fujiwara, T.; Lim, E.C. Intramolecular charge transfer and dual fluorescence of 4-(dimethylamino)benzonitrile: Ultrafast branching followed by a two-fold decay mechanism. Phys. Chem. Chem. Phys. 2011, 13, 15182–15188. [Google Scholar] [CrossRef] [PubMed]
- Zachariasse, K.A. Comment on “pseudo-Jahn-Teller and TICT-models: A photophysical comparison of meta-and para-DMABN derivatives” [Chem. Phys. Lett. 305 (1999) 8]: The PICT model for dual fluorescence of aminobenzonitriles. Chem. Phys. Lett. 2000, 320, 8–13. [Google Scholar] [CrossRef]
- Zachariasse, K.A.; Yoshihara, T.; Druzhinin, S.I. Picosecond and nanosecond fluorescence decays of 4-(dimethylamino)phenylacetylene in comparison with those of 4-(dimethylamino)benzonitrile. No evidence for intramolecular charge transfer and a nonfluorescing intramolecular charge-transfer state. J. Phys. Chem. A 2002, 106, 6325–6333. [Google Scholar] [CrossRef]
- Zachariasse, K.A.; Druzhinin, S.I.; Bosch, W.; Machinek, R.; Chemie, O. Intramolecular Charge Transfer with the Planarized 4-Aminobenzonitrile 1-tert-Butyl-6-cyano-1,2,3,4-tetrahydroquinoline (NTC6). J. Am. Chem. Soc. 2004, 126, 1705–1715. [Google Scholar] [CrossRef]
- Georgieva, I.; Aquino, A.J.A.; Plasser, F.; Trendafilova, N.; Köhn, A.; Lischka, H. Intramolecular Charge-Transfer Excited-State Processes in 4-(N,N-Dimethylamino)benzonitrile: The Role of Twisting and the πσ* State. J. Phys. Chem. A 2015, 119, 6232–6243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowski, Z.R.; Rotkiewicz, K.; Rettig, W. Structural Changes Accompanying Intramolecular Electron Transfer: Focus on Twisted Intramolecular Charge-Transfer States and Structures. Chem. Rev. 2003, 103, 3899–4032. [Google Scholar] [CrossRef]
- Shang, Q.; Bernstein, E.R. Solvation effects on the electronic structure of 4-N, N-dimethylaminobenzonitrile: Mixing of the local ππ* and charge-transfer states. J. Chem. Phys. 1992, 97, 60–68. [Google Scholar] [CrossRef]
- Zhong, C. The driving forces for twisted or planar intramolecular charge transfer. Phys. Chem. Chem. Phys. 2015, 17, 9248–9257. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Wolschann, P.; Rotkiewicz, K. Solvent effects on intramolecular charge separation. Proc. Indian Acad. Sci. Chem. Sci. 1992, 104, 197–207. [Google Scholar] [CrossRef]
- Tomin, V.I.; Dubrovkin, J.M.; Włodarkiewicz, A. Spectral broadening and red edge excitation effect in liquid and viscous solutions of DMABN. J. Lumin. 2017, 190, 344–352. [Google Scholar] [CrossRef]
- Catalán, J. On the dual emission of p-dimethylaminobenzonitrile and its photophysical implications. Phys. Chem. Chem. Phys. 2013, 15, 8811–8820. [Google Scholar] [CrossRef] [PubMed]
- Gómez, I.; Castro, P.J.; Reguero, M. Insight into the Mechanisms of Luminescence of Aminobenzonitrile and Dimethylaminobenzonitrile in Polar Solvents. An ab Initio Study. J. Phys. Chem. A 2015, 119, 1983–1995. [Google Scholar] [CrossRef] [PubMed]
- Fdez Galván, I.; Martín, M.E.; Aguilar, M.A. On the absorption properties of the excited states of DMABN. Chem. Phys. Lett. 2010, 499, 100–102. [Google Scholar] [CrossRef]
- Galván, I.F.; Martín, M.E.; Aguilar, M.A. Theoretical Study of the Dual Fluorescence of 4-(N,N-Dimethylamino)benzonitrile in Solution. J. Chem. Theory Comput. 2010, 6, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- Gómez, I.; Reguero, M.; Boggio-Pasqua, M.; Robb, M.A. Intramolecular charge transfer in 4-aminobenzonitriles does not necessarily need the twist. J. Am. Chem. Soc. 2005, 127, 7119–7129. [Google Scholar] [CrossRef]
- Köhn, A.; Hättig, C. On the nature of the low-lying singlet states of 4-(Dimethyl-amino)benzonitrile. J. Am. Chem. Soc. 2004, 126, 7399–7410. [Google Scholar] [CrossRef]
- Kretz, B.; Egger, D.A. Accurate Molecular Geometries in Complex Excited-State Potential Energy Surfaces from Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2021, 17, 357–366. [Google Scholar] [CrossRef]
- Modesto-Costa, L.; Borges, I. Discrete and continuum modeling of solvent effects in a twisted intramolecular charge transfer system: The 4-N,N-dimethylaminobenzonitrile (DMABN) molecule. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 201, 73–81. [Google Scholar] [CrossRef]
- Nottoli, M.; Mennucci, B.; Lipparini, F. Excited state Born–Oppenheimer molecular dynamics through coupling between time dependent DFT and AMOEBA. Phys. Chem. Chem. Phys. 2020, 22, 19532–19541. [Google Scholar] [CrossRef]
- Du, L.; Lan, Z. An On-the-Fly Surface-Hopping Program JADE for Nonadiabatic Molecular Dynamics of Polyatomic Systems: Implementation and Applications. J. Chem. Theory Comput. 2015, 11, 1360–1374. [Google Scholar] [CrossRef] [Green Version]
- Kochman, M.A.; Tajti, A.; Morrison, C.A.; Miller, R.J.D. Early Events in the Nonadiabatic Relaxation Dynamics of 4-(N,N-Dimethylamino)benzonitrile. J. Chem. Theory Comput. 2015, 11, 1118–1128. [Google Scholar] [CrossRef]
- Kochman, M.A.; Durbeej, B.; Kubas, A. Simulation and Analysis of the Transient Absorption Spectrum of 4-(N,N-Dimethylamino)benzonitrile (DMABN) in Acetonitrile. J. Phys. Chem. A 2021, 125, 8635–8648. [Google Scholar] [CrossRef]
- Medders, G.R.; Alguire, E.C.; Jain, A.; Subotnik, J.E. Ultrafast Electronic Relaxation through a Conical Intersection: Nonadiabatic Dynamics Disentangled through an Oscillator Strength-Based Diabatization Framework. J. Phys. Chem. A 2017, 121, 1425–1434. [Google Scholar] [CrossRef]
- Curchod, B.F.E.; Sisto, A.; Martínez, T.J. Ab Initio Multiple Spawning Photochemical Dynamics of DMABN Using GPUs. J. Phys. Chem. A 2017, 121, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Curchod, B.F.E. Full and Ab Initio Multiple Spawning. In Quantum Chemistry and Dynamics of Excited States; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2020; Chapter 14; pp. 435–467. [Google Scholar] [CrossRef]
- Fdez Galván, I.; Vacher, M.; Alavi, A.; Angeli, C.; Aquilante, F.; Autschbach, J.; Bao, J.J.; Bokarev, S.I.; Bogdanov, N.A.; Carlson, R.K.; et al. OpenMolcas: From Source Code to Insight. J. Chem. Theory Comput. 2019, 15, 5925–5964. [Google Scholar] [CrossRef]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef] [Green Version]
- Mennucci, B. Polarizable continuum model. WIREs Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Mewes, J.M.; You, Z.Q.; Wormit, M.; Kriesche, T.; Herbert, J.M.; Dreuw, A. Experimental Benchmark Data and Systematic Evaluation of Two a Posteriori, Polarizable-Continuum Corrections for Vertical Excitation Energies in Solution. J. Phys. Chem. A 2015, 119, 5446–5464. [Google Scholar] [CrossRef]
- Koppel, H.; Domcke, W.; Cederbaum, L.S. Multimode Molecular Dynamics Beyond the Born-Oppenheimer Approximation. Adv. Chem. Phys. 1984, 57, 59–246. [Google Scholar] [CrossRef]
- Worth, G.A.; Cederbaum, L.S. Beyond Born-Oppenheimer: Conical intersections and their impact on molecular dynamics. Annu. Rev. Phys. Chem. 2004, 55, 127–158. [Google Scholar] [CrossRef] [Green Version]
- Ben-Nun, M.; Quenneville, J.; Martínez, T.J. Ab Initio Multiple Spawning: Photochemistry from First Principles Quantum Molecular Dynamics. J. Phys. Chem. A 2000, 104, 5161–5175. [Google Scholar] [CrossRef]
- Shalashilin, D.V.; Child, M.S. The phase space CCS approach to quantum and semiclassical molecular dynamics for high-dimensional systems. Chem. Phys. 2004, 304, 103–120. [Google Scholar] [CrossRef]
- Richings, G.W.; Polyak, I.; Spinlove, K.E.; Worth, G.A.; Burghardt, I.; Lasorne, B. Quantum Dynamics Simulations using Gaussian Wavepackets: The vMCG Method. Int. Rev. Phys. Chem. 2015, 34, 269–308. [Google Scholar] [CrossRef]
- Spinlove, K.E.; Richings, G.W.; Robb, M.A.; Worth, G.A. Curve Crossing in a Manifold of Coupled Electronic States: Direct Quantum Dynamics Simulations of Formamide. Faraday Discuss. 2018, 212, 191–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christopoulou, G.; Freibert, A.; Worth, G.A. Improved Algorithm for the Direct Dynamics Variational Multi-Configurational Gaussian Method. J. Chem. Phys. 2021, 154, 124127. [Google Scholar] [CrossRef]
- Frenkel, J. Wave Mechanics; Clarendon Press: Oxford, UK, 1934. [Google Scholar]
- Dirac, P.A. Note on Exchange Phenomena in the Thomas Atom. Math. Proc. Camb. Philos. Soc. 1930, 26, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Richings, G.W.; Worth, G.A. Multi-state nonadiabatic direct-dynamics on propagated diabatic potential energy surfaces. Chem. Phys. Lett. 2017, 683, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Frankcombe, T.J.; Collins, M.A.; Worth, G.A. Converged quantum dynamics with modified Shepard interpolation and Gaussian wave packets. Chem. Phys. Lett. 2010, 489, 242–247. [Google Scholar] [CrossRef]
- Worth, G.A. Quantics: A General Purpose Package for Quantum Molecular Dynamics Simulations. Comput. Phys. Commun. 2020, 248, 107040. [Google Scholar] [CrossRef]
- Worth, G.A.; Giri, K.; Richings, G.W.; Beck, M.H.; Jäckle, A.; Meyer, Y.H. The QUANTICS Package, Version 1.1; University of Birmingham: Birmingham, UK, 2015. [Google Scholar]
- Coonjobeeharry, J.; Spinlove, K.E.; Sanz Sanz, C.; Sapunar, M.; Došlić, N.; Worth, G.A. Mixed-Quantum-Classical or Fully-Quantised Dynamics? A Unified Code to Compare Methods. Philos. Trans. R. Soc. 2021. [Google Scholar] [CrossRef]
- Beck, M.H.; Jäckle, A.; Worth, G.A.; Meyer, H.D. The multiconfiguration time-dependent Hartree (MCTDH) method: A highly efficient algorithm for propagating wavepackets. Phys. Rep. 2000, 324, 1–105. [Google Scholar] [CrossRef]
- Wang, H.; Thoss, M. Multilayer formulation of the multiconfiguration time-dependent Hartree theory. J. Chem. Phys. 2003, 119, 1289–1299. [Google Scholar] [CrossRef]
- Manthe, U. A multilayer multiconfigurational time-dependent Hartree approach for quantum dynamics on general potential energy surfaces. J. Chem. Phys. 2008, 128, 164116. [Google Scholar] [CrossRef] [PubMed]
- Vendrell, O.; Meyer, H.D. Multilayer multiconfiguration time-dependent Hartree method: Implementation and applications to a Henon–Heiles Hamiltonian and to pyrazine. J. Chem. Phys. 2011, 134, 044135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunkenheimer, B.; Köhn, A. Solvent Effects on Electronically Excited States Using the Conductor-Like Screening Model and the Second-Order Correlated Method ADC(2). J. Chem. Theory Comput. 2013, 9, 977–994. [Google Scholar] [CrossRef]
- Fuß, W.; Pushpa, K.K.; Rettig, W.; Schmid, W.E.; Trushin, S.A. Ultrafast charge transfer via a conical intersection in dimethylaminobenzonitrile. Photochem. Photobiol. Sci. 2002, 1, 255–262. [Google Scholar] [CrossRef]
- Kochman, M.A.; Durbeej, B. Simulating the Nonadiabatic Relaxation Dynamics of 4-(N,N-Dimethylamino)benzonitrile (DMABN) in Polar Solution. J. Phys. Chem. A 2020, 124, 2193–2206. [Google Scholar] [CrossRef]
- Ibele, L.M.; Curchod, B.F.E. A molecular perspective on Tully models for nonadiabatic dynamics. Phys. Chem. Chem. Phys. 2020, 22, 15183–15196. [Google Scholar] [CrossRef]
- Plasser, F.; Gómez, S.; Menger, M.F.S.J.; Mai, S.; González, L. Highly efficient surface hopping dynamics using a linear vibronic coupling model. Phys. Chem. Chem. Phys. 2019, 21, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Dielectric Constant | Refraction Index, n |
|---|---|---|
| Cyclohexane | 2.02 | 1.43 |
| Tetrahydrofuran | 7.58 | 1.41 |
| Acetonitrile | 37.50 | 1.34 |
| Water | 80.10 | 1.33 |
| State Label | Character (symm.) | Solvent | E | E (f) | E (f) |
|---|---|---|---|---|---|
| GS | (A) | gas/ | |||
| L | (B) | cyclohexane | 4.82 (0.00) | 4.84 (0.03) | |
| L | (A) | 4.40 | 5.08 (0.54) | 5.05 (0.68) | |
| LE | (B) | 3.90 | 4.46 (0.04) | 4.24 (0.18) | |
| CT | (A”) | 3.64 | 3.05 (0.00) | 3.06 (0.00) | |
| GS | (A) | water | |||
| L | (B) | 4.90 (0.04) | 4.68 (0.04) | ||
| L | (A) | 4.17 | 5.20 (0.76) | 4.63 (0.81) | |
| LE | (B) | 3.25 | 4.75 (0.75) | 4.28 (1.19) | |
| CT | (A”) | 2.38 | 3.08 (0.00) | 2.95 (0.00) | |
| GS | (A) | THF | |||
| L | (B) | 4.87 (0.03) | 4.70 (0.03) | ||
| L | (A) | 4.24 | 5.18 (0.73) | 4.69 (0.76) | |
| LE | (B) | 3.47 | 4.81 (0.15) | 4.43 (1.11) | |
| CT | (B) | 2.84 | 3.16 (0.00) | 3.09 (0.00) | |
| GS | (A) | acetonitrile | |||
| L | (B) | 4.89 (0.05) | 4.68 (0.04) | ||
| L | (A) | 4.23 | 5.16 (0.74) | 4.66 (0.79) | |
| LE | (B) | 3.38 | 4.83 (0.37) | 4.76 (0.91) | |
| CT | (A”) | 2.66 | 3.11 (0.00) | 3.05 (0.00) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez, S.; Soysal, E.N.; Worth, G.A. Micro-Solvated DMABN: Excited State Quantum Dynamics and Dual Fluorescence Spectra. Molecules 2021, 26, 7247. https://doi.org/10.3390/molecules26237247
Gómez S, Soysal EN, Worth GA. Micro-Solvated DMABN: Excited State Quantum Dynamics and Dual Fluorescence Spectra. Molecules. 2021; 26(23):7247. https://doi.org/10.3390/molecules26237247
Chicago/Turabian StyleGómez, Sandra, Esra N. Soysal, and Graham A. Worth. 2021. "Micro-Solvated DMABN: Excited State Quantum Dynamics and Dual Fluorescence Spectra" Molecules 26, no. 23: 7247. https://doi.org/10.3390/molecules26237247
APA StyleGómez, S., Soysal, E. N., & Worth, G. A. (2021). Micro-Solvated DMABN: Excited State Quantum Dynamics and Dual Fluorescence Spectra. Molecules, 26(23), 7247. https://doi.org/10.3390/molecules26237247