
Seriniquinones as Therapeutic Leads for Treatment of BRAF and NRAS Mutant Melanomas

 , , , ,
, , , ,
Abstract
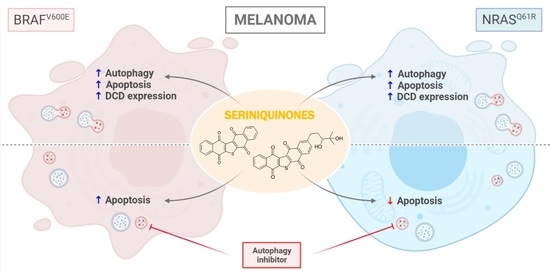
:
1. Introduction
2. Results
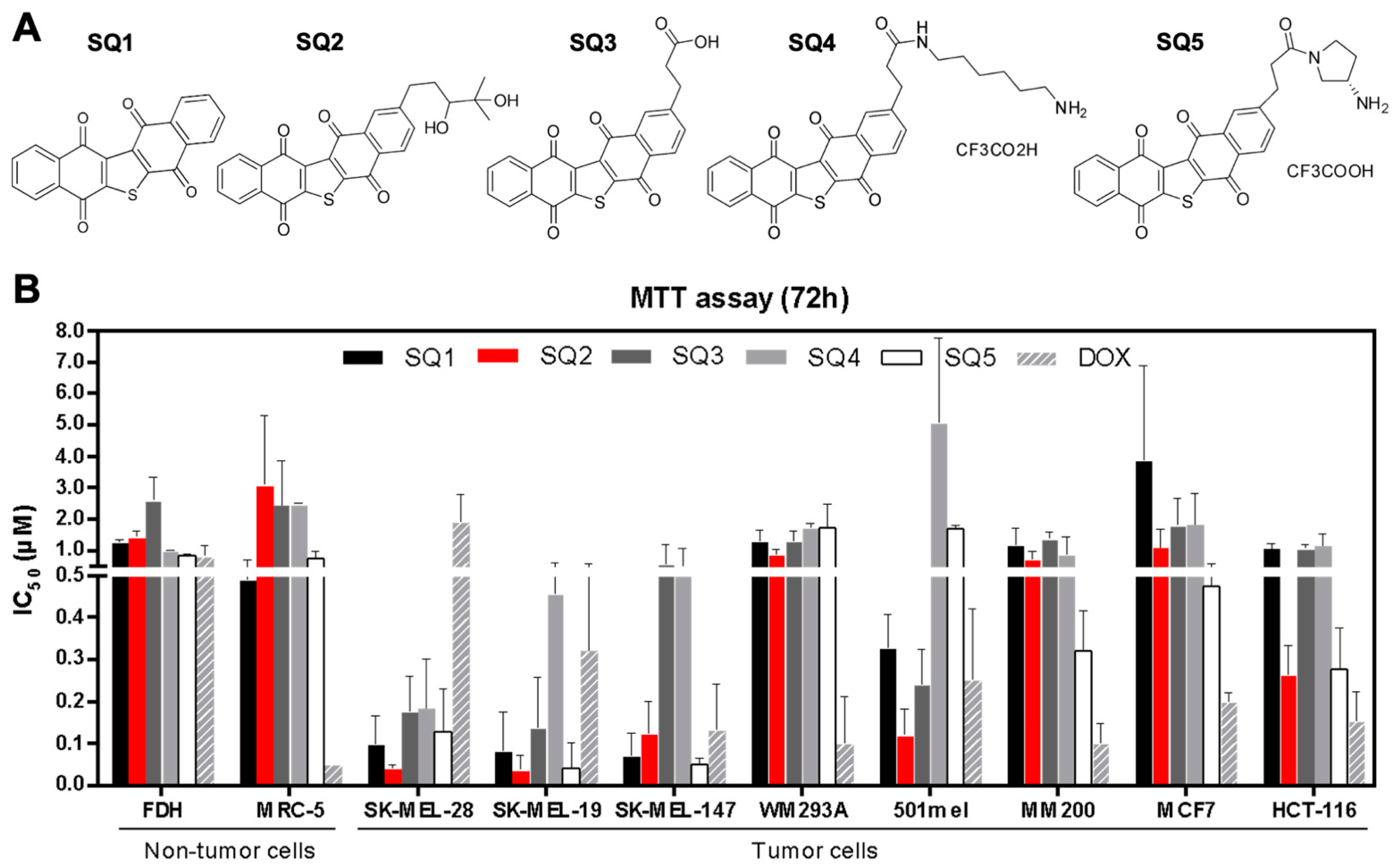
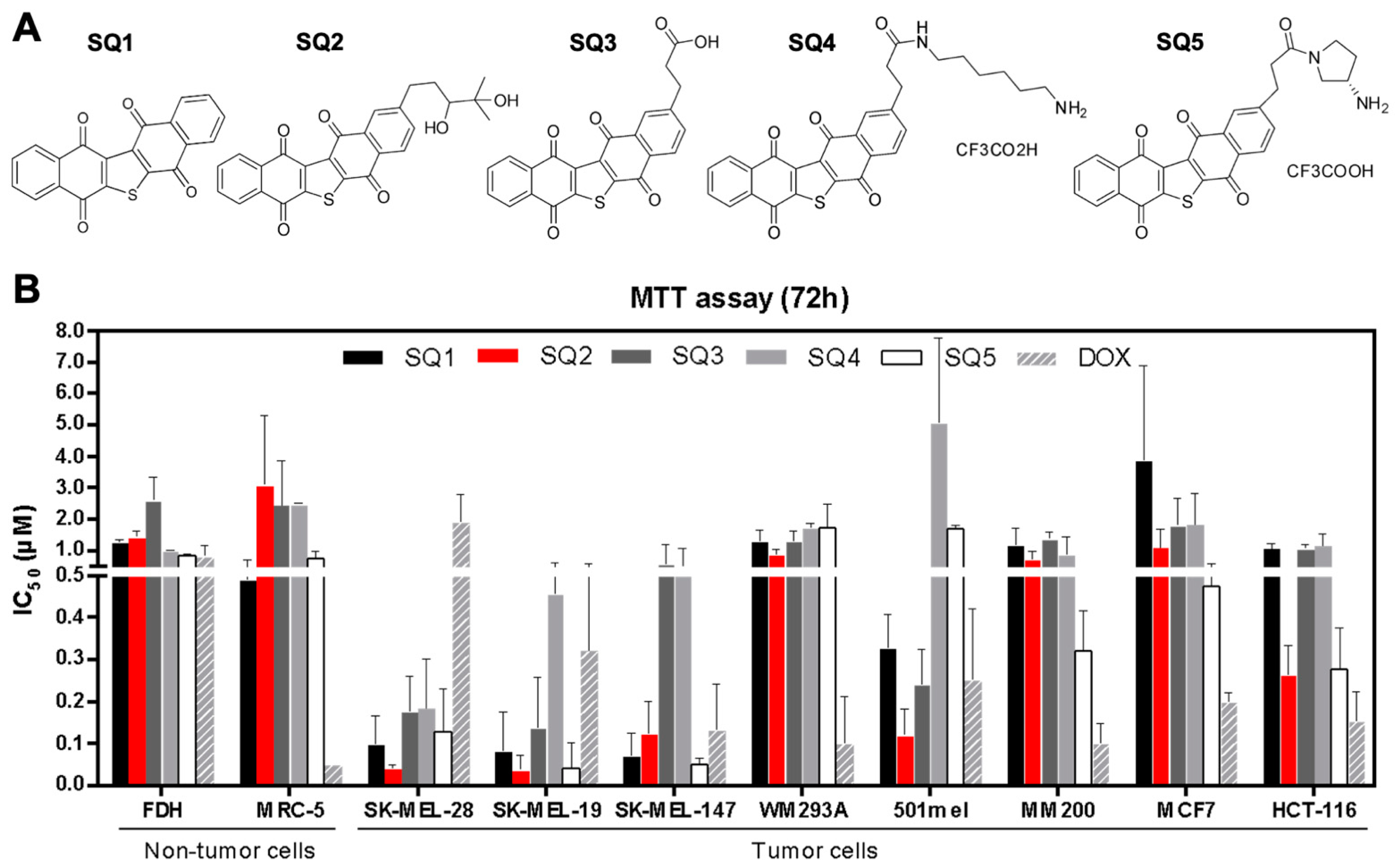
2.1. Seriniquinone Analogues Display Cytotoxic Activity in Cancer Cells
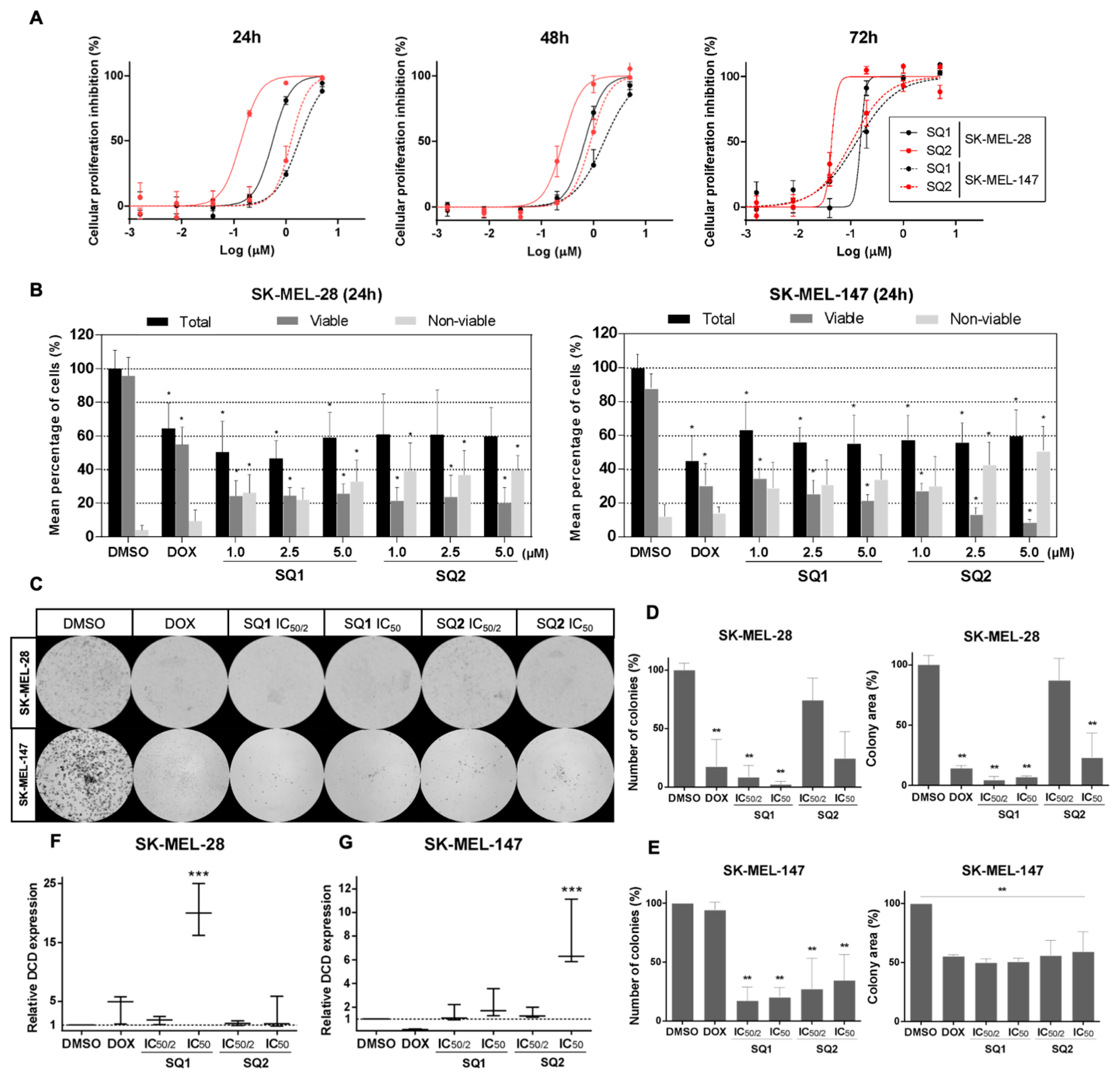
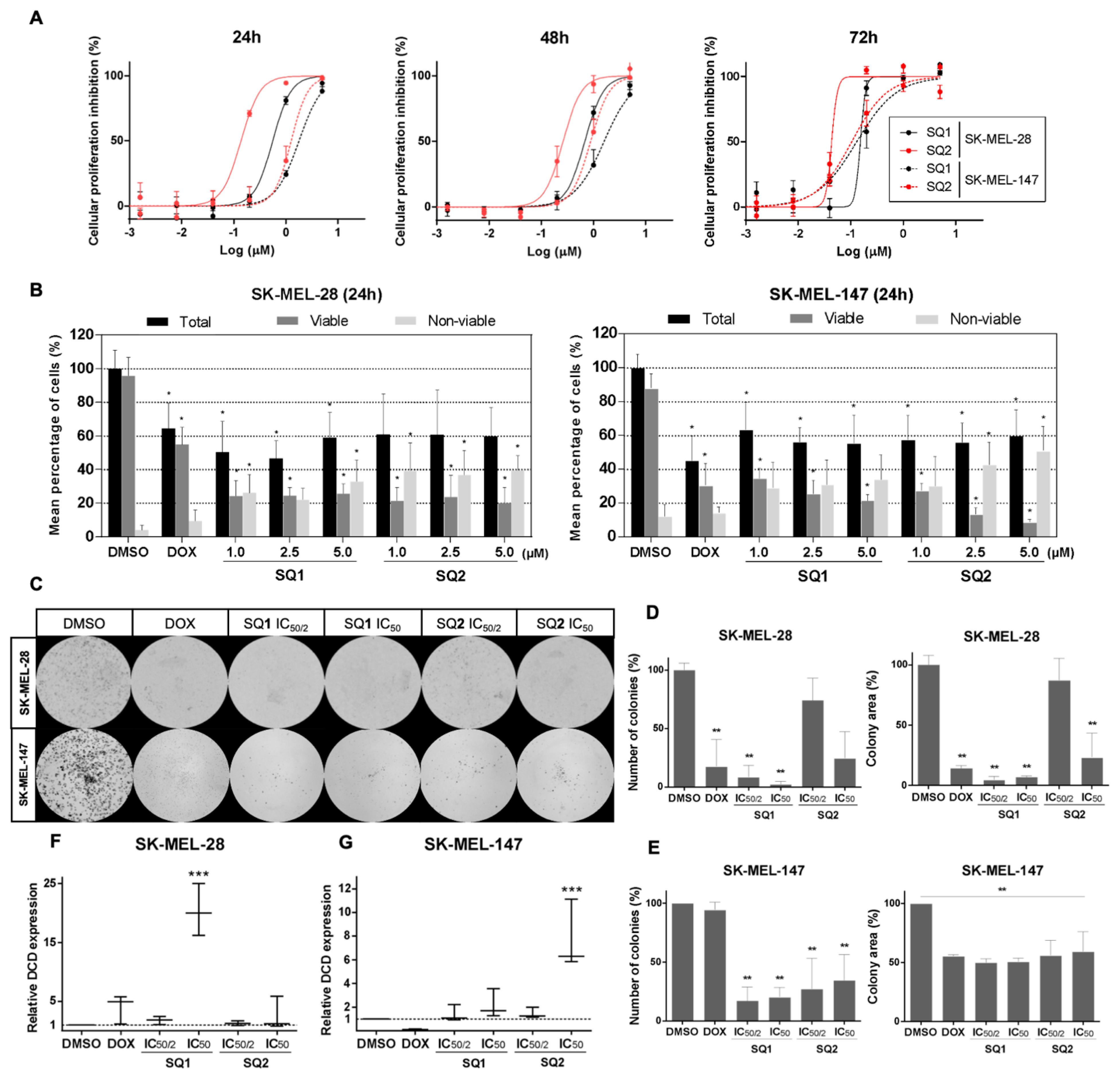
2.2. SQ1 and SQ2 Are Active in BRAF and NRAS Mutant Melanoma Cells
2.3. SQ1 and SQ2 Modulate Dermcidin Expression
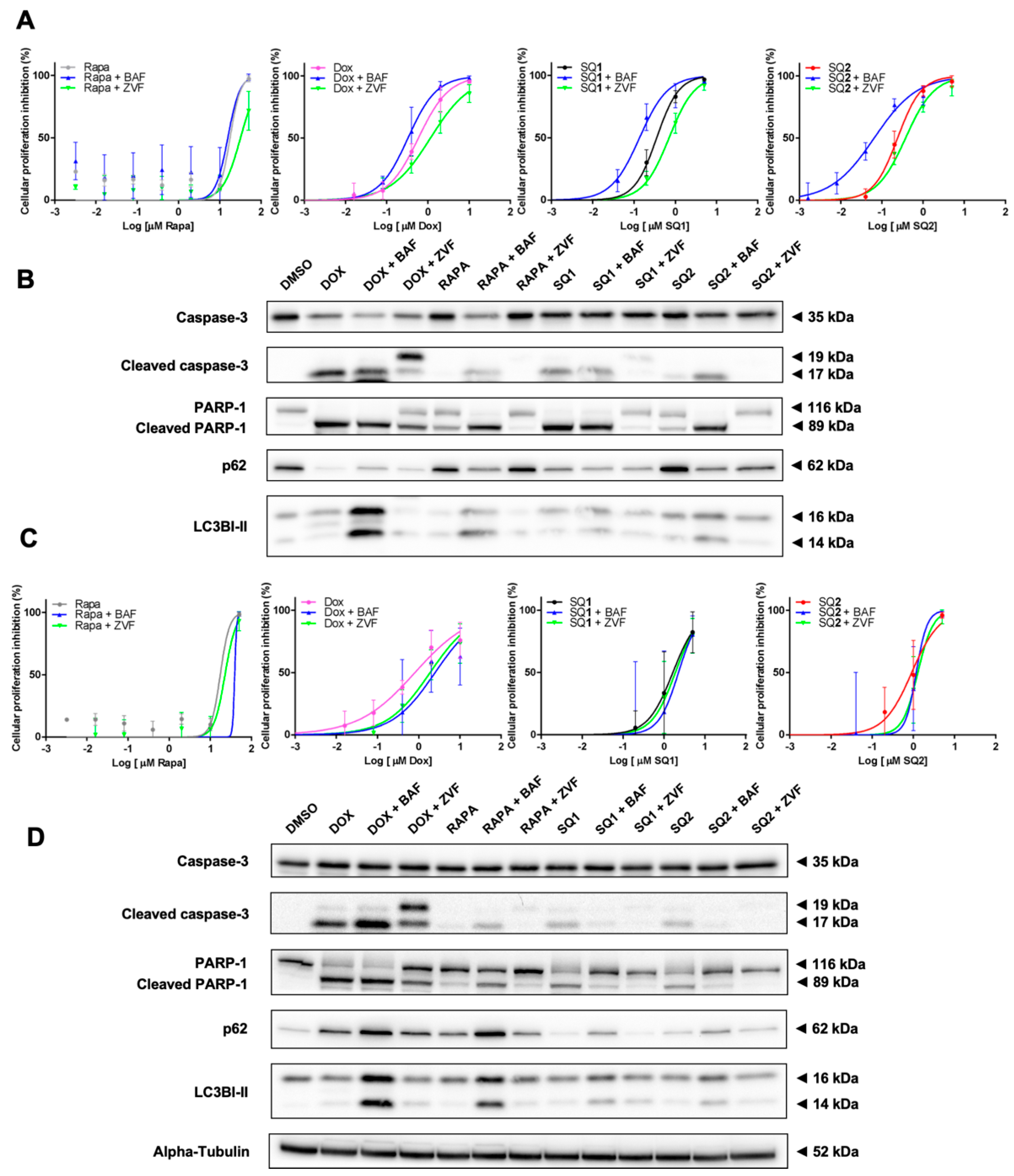
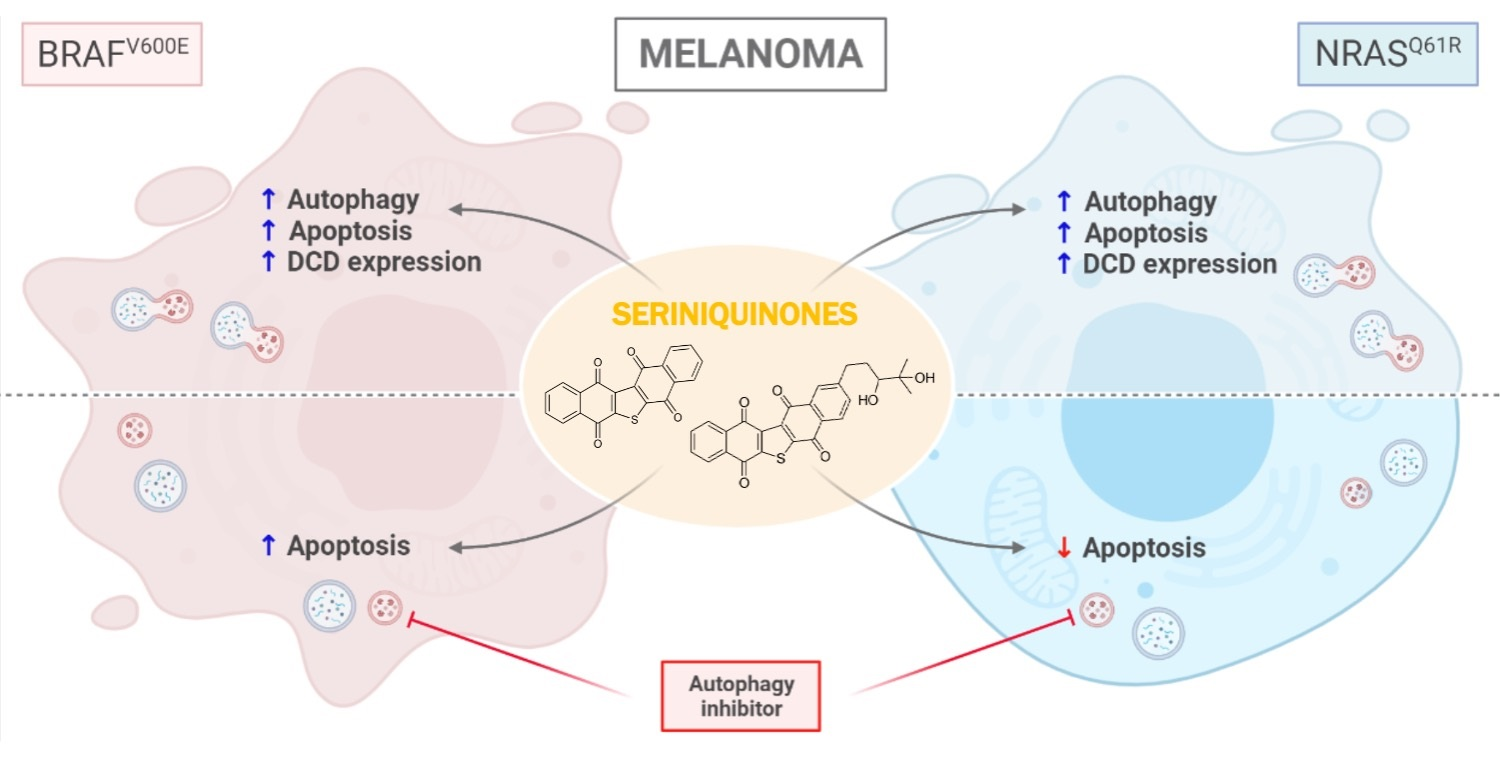
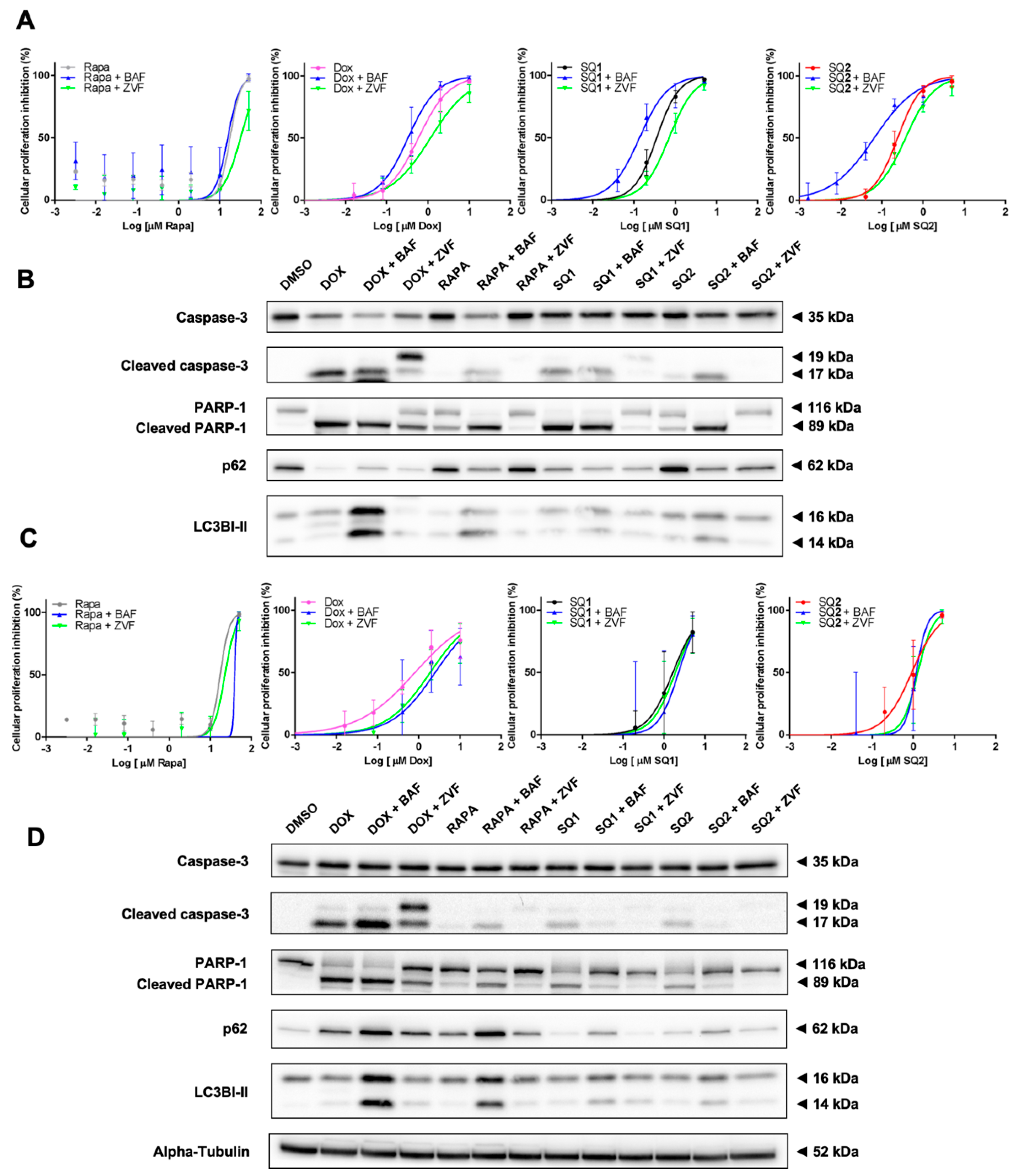
2.4. SQ1 and SQ2 Lead to Cell Death through Autophagy and Apoptosis
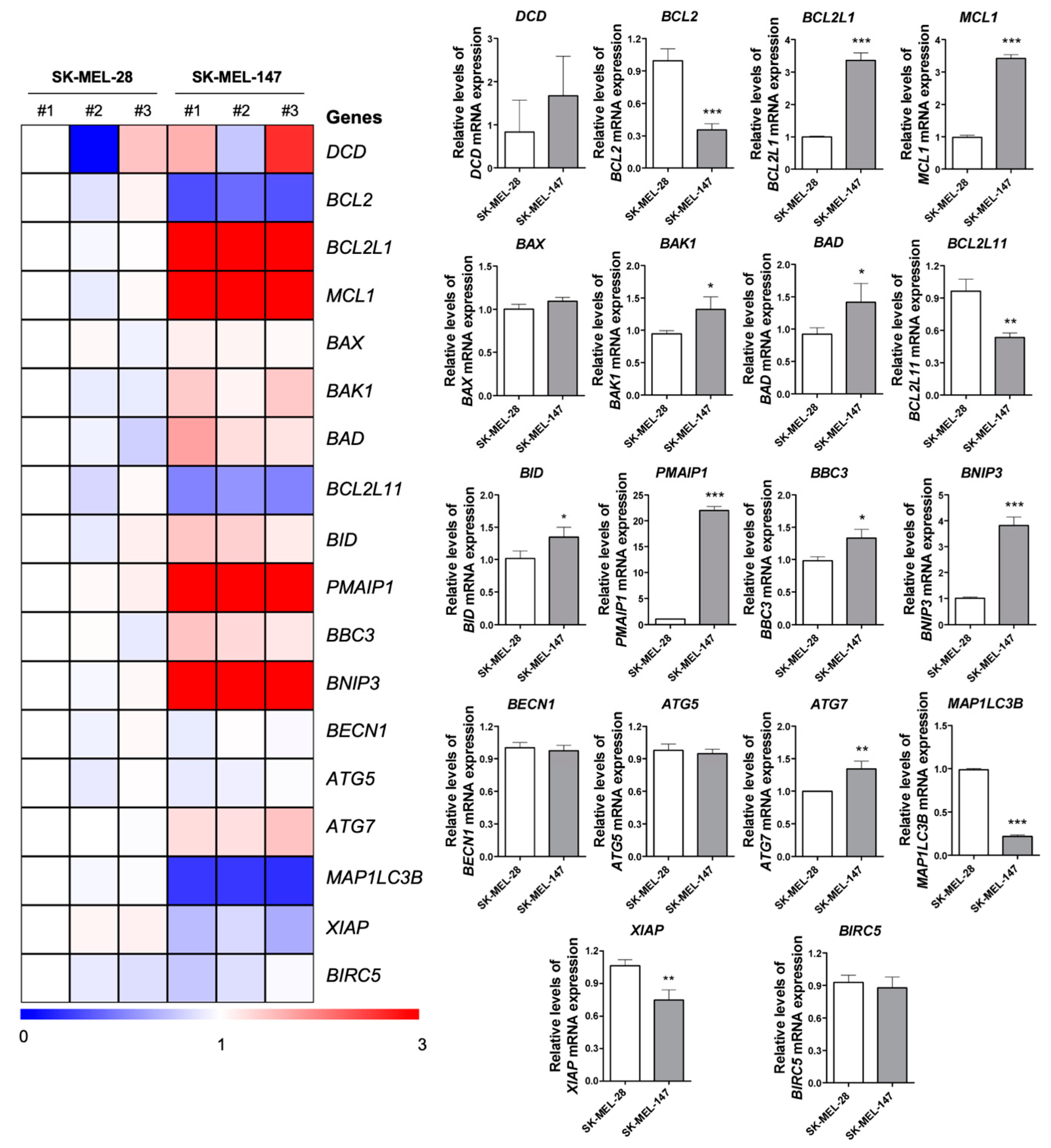
2.5. BRAF and NRAS Mutants Have Different Basal Profiles
2.6. Inhibition of Autophagy Induces Different Responses in NRAS and BRAF Mutated Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Seriniquinone Analogues and Control Agents
4.3. Cytotoxicity Analyses via the MTT Assay
4.4. Selectivity Indices
4.5. Cell Viability Analyses through the Trypan Blue Exclusion Test
4.6. Treatment Scheme
4.7. Clonogenic Assays
4.8. Quantitative PCR Analyses
4.9. Protein Expression Analyses by Western Blotting
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Trzoss, L.; Fukuda, T.; Costa-Lotufo, L.V.; Jimenez, P.; La Clair, J.J.; Fenical, W. Seriniquinone, a selective anticancer agent, induces cell death by autophagocytosis, targeting the cancer-protective protein dermcidin. Proc. Natl. Acad. Sci. USA 2014, 111, 14687–14692. [Google Scholar] [CrossRef] [Green Version]
- Hammons, J.C.; Trzoss, L.; Jimenez, P.C.; Hirata, A.S.; Costa-Lotufo, L.V.; La Clair, J.J.; Fenical, W. Advance of Seriniquinone Analogues as Melanoma Agents. ACS Med. Chem. Lett. 2019, 10, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Ayala, B.; Kwong, L.N.; et al. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Manzano, J.L.; Layos, L.; Bugés, C.; de Los Llanos Gil, M.; Vila, L.; Martínez-Balibrea, E.; Martinez-Cardus, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lev, D.C.; Onn, A.; Melinkova, V.O.; Miller, C.; Stone, V.; Ruiz, M.; McGary, E.C.; Ananthaswamy, H.N.; Price, J.E.; Bar-Eli, M. Exposure of Melanoma Cells to Dacarbazine Results in Enhanced Tumor Growth and Metastasis In Vivo. J. Clin. Oncol. 2004, 22, 2092–2100. [Google Scholar] [CrossRef]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta Bioenergy 2019, 1871, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schittek, B. The Multiple Facets of Dermcidin in Cell Survival and Host Defense. J. Innate Immun. 2012, 4, 349–360. [Google Scholar] [CrossRef]
- Hirata, A.S.; Miguel, R.A.; Jimenez, P.C.; Costa-Lotufo, L.V. DCD (dermcidin). Atlas Genet. Cytogenet. Oncol. Haematol. 2021, 25, 73–82. [Google Scholar] [CrossRef]
- Bancovik, J.; Moreira, D.F.; Carrasco, D.; Yao, J.; Porter, D.; Moura, R.; Camargo, A.; Fontes-Oliveira, C.C.; Malpartida, M.G.; Carambula, S.; et al. Dermcidin exerts its oncogenic effects in breast cancer via modulation of ERBB signaling. BMC Cancer 2015, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.C.; Huang, M.S.; Yang, C.J.; Wang, W.Y.; Lai, T.C.; Hsiao, M.; Chen, C.H. Dermcidin identification from exhaled air for lung cancer diagnosis. Eur. Respir. J. 2010, 35, 1182–1185. [Google Scholar] [CrossRef]
- Ortega-Martínez, I.; Gardeazabal, J.; Erramuzpe, A.; Sanchez-Diez, A.; Cortés, J.; García-Vázquez, M.D.; Pérez-Yarza, G.; Izu, R.; Díaz-Ramón, J.L.; de la Fuente, I.M.; et al. Vitronectin and dermcidin serum levels predict the metastatic progression of AJCC I–II early-stage melanoma. Int. J. Cancer 2016, 139, 1598–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, G.M.; Vashisht Gopal, Y.N.; McQuade, J.L.; Peng, W.; DeBerardinis, R.J.; Davies, M.A. Metabolic strategies of melanoma cells: Mechanisms, interactions with the tumor microenvironment, and therapeutic implications. Pigment. Cell Melanoma Res. 2018, 31, 11–30. [Google Scholar] [CrossRef]
- Dorard, C.; Estrada, C.; Barbotin, C.; Larcher, M.; Garancher, A.; Leloup, J.; Beermann, F.; Baccarini, M.; Pouponnot, C.; LaRue, L.; et al. RAF proteins exert both specific and compensatory functions during tumour progression of NRAS-driven melanoma. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; McArthur, G.A. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Albino, A.P.; Le Strange, R.; Oliff, A.I.; Furth, M.E.; Old, L.J. Transforming ras genes from human melanoma: A manifestation of tumour heterogeneity? Nat. Cell Biol. 1984, 308, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Sullivan, R.J. NRAS mutant melanoma: An overview for the clinician for melanoma management. Melanoma Manag. 2016, 3, 47–59. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, G.D.; Lowrie, A.G.; Riddick, A.C.P.; Fearon, K.C.H.; Habib, F.K.; Ross, J.A. Dermcidin expression confers a survival advantage in prostate cancer cells subjected to oxidative stress or hypoxia. Prostate 2007, 67, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Lowrie, A.G.; Wigmore, S.J.; Wright, D.J.; Waddell, I.D.; A Ross, J. Dermcidin expression in hepatic cells improves survival without N-glycosylation, but requires asparagine residues. Br. J. Cancer 2006, 94, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Brauer, H.A.; D’Arcy, M.; Libby, T.; Thompson, H.J.; Yasui, Y.Y.; Hamajima, N.; Li, C.I.; Troester, M.A.; Lampe, P.D. Dermcidin expression is associated with disease progression and survival among breast cancer patients. Breast Cancer Res. Treat. 2014, 144, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.; Weremowicz, S.; Chin, K.; Seth, P.; Keshaviah, A.; Lahti-Domenici, J.; Bae, Y.K.; Monitto, C.L.; Merlos-Suarez, A.; Chan, J.; et al. A neural survival factor is a candidate oncogene in breast cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 10931–10936. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndoye, A.; Weeraratna, A.T. Autophagy-An emerging target for melanoma therapy. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, E.N.; Bristol, M.L.; Di, X.; Maltese, W.A.; Koterba, K.; Beckman, M.J.; Gewirtz, D.A. A Switch between Cytoprotective and Cytotoxic Autophagy in the Radiosensitization of Breast Tumor Cells by Chloroquine and Vitamin D. Horm. Cancer 2011, 2, 272–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sophia, J.; Kowshik, J.; Dwivedi, A.; Bhutia, S.K.; Manavathi, B.; Mishra, R.; Nagini, S. Nimbolide, a neem limonoid inhibits cytoprotective autophagy to activate apoptosis via modulation of the PI3K/Akt/GSK-3β signalling pathway in oral cancer. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohecker, A.M.; White, E. Targeting Mitochondrial Metabolism by Inhibiting Autophagy in BRAF-Driven Cancers. Cancer Discov. 2014, 4, 766–772. [Google Scholar] [CrossRef] [Green Version]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy Sustains Mitochondrial Glutamine Metabolism and Growth of BrafV600E–Driven Lung Tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Koh, J.Y.; Price, S.; White, E.; Mehnert, J.M. Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discov. 2015, 5, 410–423. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2015, 2, e1054549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.-Y.; Li, J.; Li, F.; Zhu, Y.; Cui, K.; Wong, S.T.; Chang, E.C.; Liao, Y.-H. Induction of N-Ras degradation by flunarizine-mediated autophagy. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Lu, A.; Tebar, F.; Alvarez-Moya, B.; López-Alcalá, C.; Calvo, M.; Enrich, C.; Agell, N.; Nakamura, T.; Matsuda, M.; Bachs, O. A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J. Cell Biol. 2009, 184, 863–879. [Google Scholar] [CrossRef] [Green Version]
- Mattia, G.; Puglisi, R.; Ascione, B.; Malorni, W.; Carè, A.; Matarrese, P. Cell death-based treatments of melanoma:conventional treatments and new therapeutic strategies. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairlie, W.D.; Tran, S.; Lee, E.F. Crosstalk between apoptosis and autophagy signaling pathways. Int. Rev. Cell Mol. Biol. 2020, 352, 115–158. [Google Scholar] [CrossRef]
- Apolinário, A.C.; Hirata, A.S.; Miguel, R.D.A.; Costa-Lotufo, L.V.; Pessoa, A.; La Clair, J.J.; Fenical, W.; Lopes, L.B. Exploring the benefits of nanotechnology for cancer drugs in different stages of the drug development pipeline. Nanomedicine 2020, 15, 2539–2542. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using realtime quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selectivity Index (SI) | ||||||||
|---|---|---|---|---|---|---|---|---|
| SK-MEL-28 | SK-MEL-19 | SK-MEL-147 | WM293A | 501mel | MM200 | MCF7 | HCT-116 | |
| SQ1 | 4.3 | 10.7 | 4.9 | 0.6 | 2.0 | 0.5 | 0.2 | 0.6 |
| SQ2 | 39.3 | 39.3 | 15.7 | 2.1 | 14.3 | 2.1 | 2.2 | 6.8 |
| SQ3 | 6.0 | 13.0 | 5.5 | 1.0 | 7.2 | 1.0 | 0.8 | 1.4 |
| SQ4 | 12.9 | 7.7 | 6.1 | 1.4 | - | 2.1 | 2.5 | 2.4 |
| SQ5 | 3.7 | 14.0 | 8.0 | 0.3 | 0.3 | 1.9 | 1.1 | 2.5 |
| DOX | 0.04 | 0.1 | 0.3 | 0.9 | 0.2 | 0.5 | 0.1 | 0.3 |
| Selectivity Index (SI) | ||||||||
|---|---|---|---|---|---|---|---|---|
| SK-MEL-28 | SK-MEL-19 | SK-MEL-147 | WM293A | 501mel | MM200 | MCF7 | HCT-116 | |
| SQ1 | 7.7 | 19.2 | 8.8 | 1.1 | 3.6 | 0.8 | 0.3 | 1.1 |
| SQ2 | 29.3 | 29.3 | 11.7 | 1.6 | 10.6 | 1.6 | 1.6 | 5.1 |
| SQ3 | 14.6 | 31.8 | 13.5 | 2.3 | 17.5 | 2.4 | 1.8 | 3.4 |
| SQ4 | 5.3 | 3.2 | 2.5 | 0.6 | - | 0.9 | 1.0 | 1.0 |
| SQ5 | 6.2 | 23.3 | 13.3 | 0.6 | 0.5 | 3.1 | 1.9 | 4.2 |
| DOX | 0.6 | 2.0 | 4.3 | 13.0 | 3.0 | 7.8 | 1.4 | 5.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirata, A.S.; Rezende-Teixeira, P.; Machado-Neto, J.A.; Jimenez, P.C.; Clair, J.J.L.; Fenical, W.; Costa-Lotufo, L.V. Seriniquinones as Therapeutic Leads for Treatment of BRAF and NRAS Mutant Melanomas. Molecules 2021, 26, 7362. https://doi.org/10.3390/molecules26237362
Hirata AS, Rezende-Teixeira P, Machado-Neto JA, Jimenez PC, Clair JJL, Fenical W, Costa-Lotufo LV. Seriniquinones as Therapeutic Leads for Treatment of BRAF and NRAS Mutant Melanomas. Molecules. 2021; 26(23):7362. https://doi.org/10.3390/molecules26237362
Chicago/Turabian StyleHirata, Amanda S., Paula Rezende-Teixeira, João Agostinho Machado-Neto, Paula C. Jimenez, James J. La Clair, William Fenical, and Leticia V. Costa-Lotufo. 2021. "Seriniquinones as Therapeutic Leads for Treatment of BRAF and NRAS Mutant Melanomas" Molecules 26, no. 23: 7362. https://doi.org/10.3390/molecules26237362
APA StyleHirata, A. S., Rezende-Teixeira, P., Machado-Neto, J. A., Jimenez, P. C., Clair, J. J. L., Fenical, W., & Costa-Lotufo, L. V. (2021). Seriniquinones as Therapeutic Leads for Treatment of BRAF and NRAS Mutant Melanomas. Molecules, 26(23), 7362. https://doi.org/10.3390/molecules26237362