
Human Group IIA Phospholipase A2—Three Decades on from Its Discovery

 , ,
, ,  ,
,
Abstract
1. Introduction
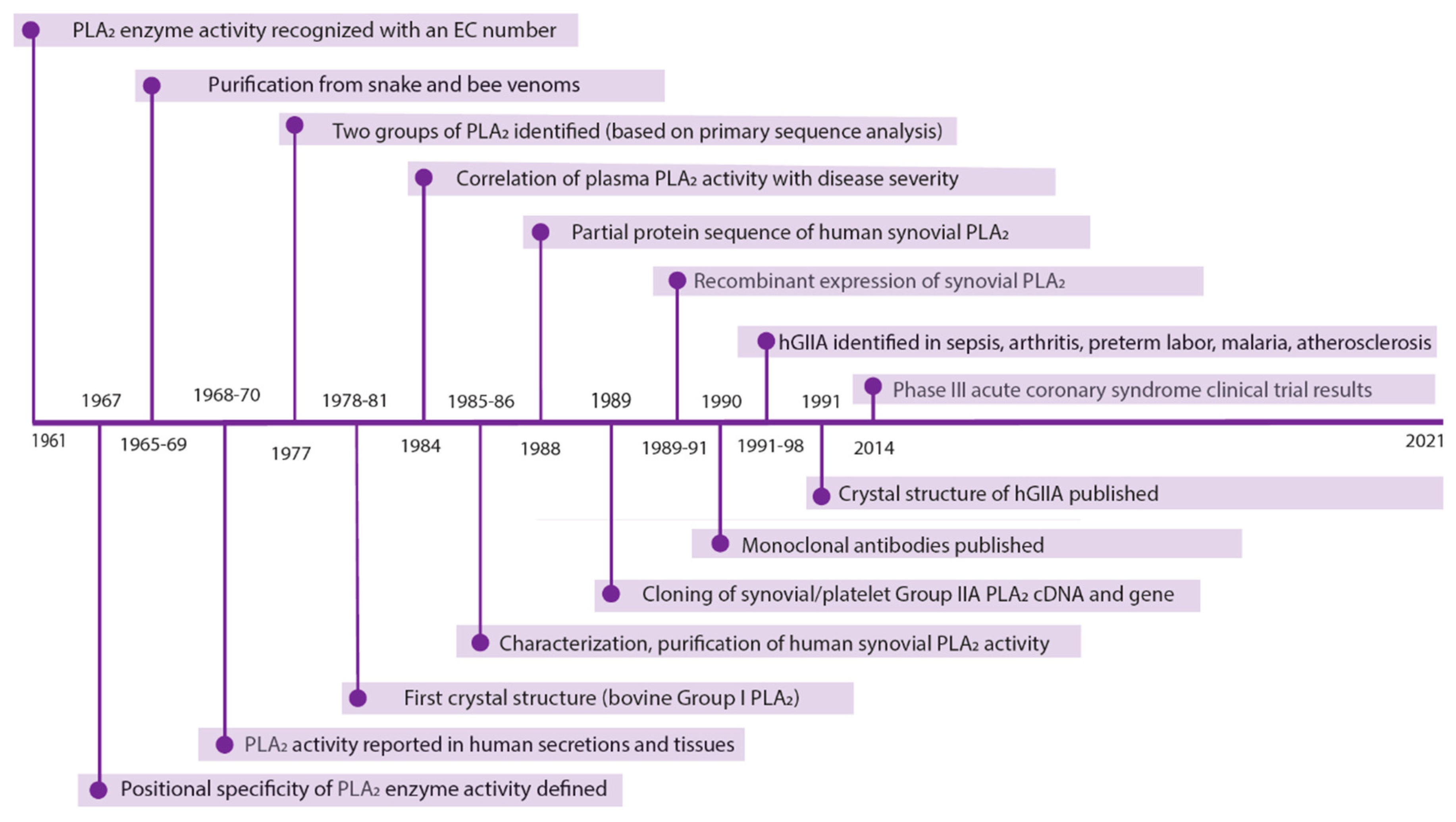
2. Relevant Discoveries, 1991–2014
3. hGIIA Function
3.1. hGIIA Function in Host Defense
3.2. hGIIA Function in Inflammation
3.2.1. hGIIA in Neuroinflammation
3.2.2. hGIIA in Asthma/COVID-19
3.2.3. hGIIA in Atherosclerosis
3.2.4. hGIIA in Sepsis
3.2.5. hGIIA in Rheumatoid Arthritis
3.2.6. hGIIA in Cancer
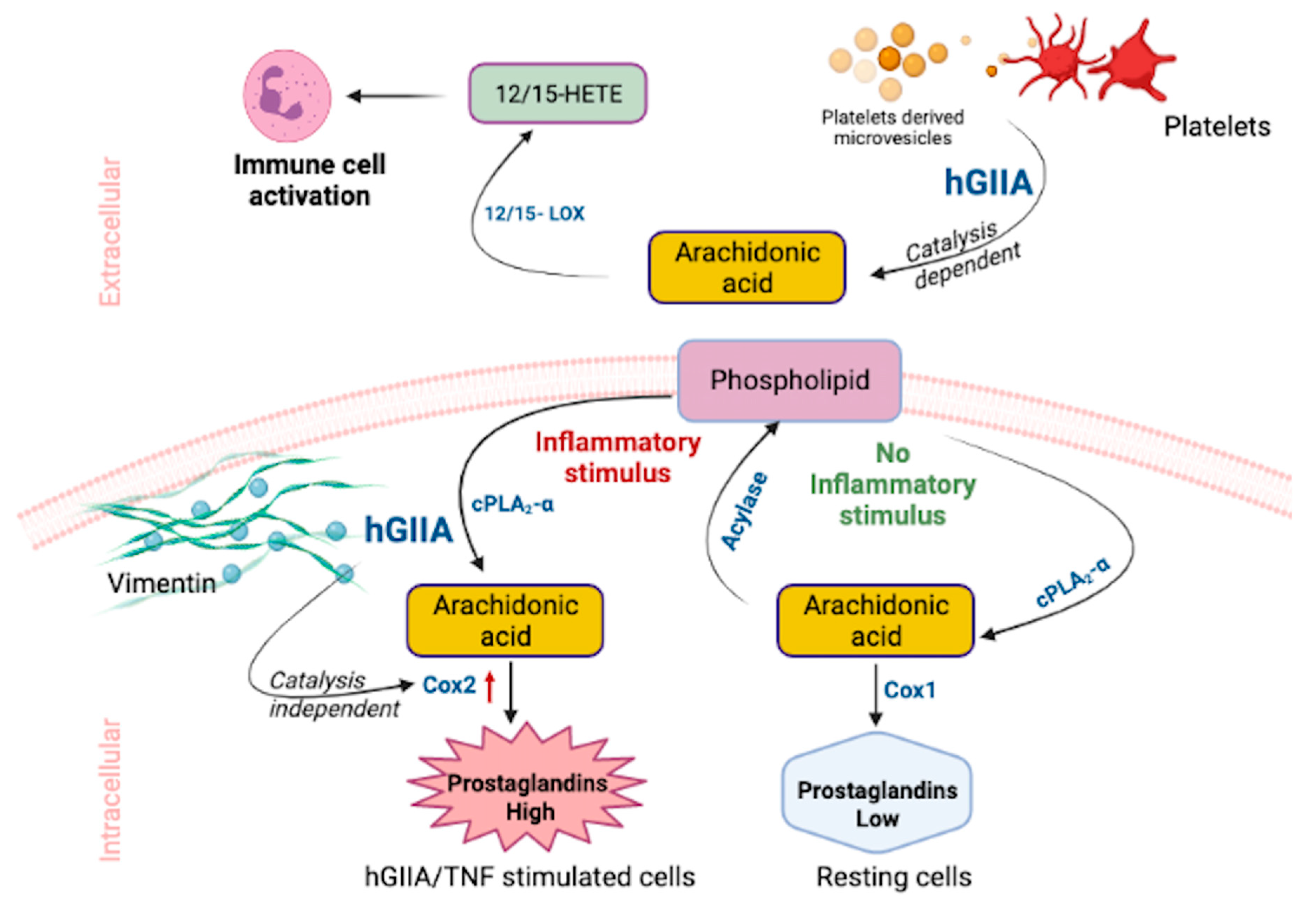
4. hGIIA Mechanism of Action
4.1. Catalysis-Dependent Mechanisms
4.2. Catalysis-Independent Mechanisms
5. Pharmacological Inhibition of hGIIA Function
5.1. Inhibitors of the Catalysis-Dependent Mechanism
5.2. Inhibitors of the Catalysis-Independent Mechanism
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- KEGG Database. Available online: https://www.genome.jp/dbget-bin/www_bget?ec:3.1.1.4 (accessed on 19 October 2021).
- OVID Search of Embase Database. Available online: https://www.embase.com/landing?status=grey (accessed on 17 October 2021).
- Dennis, E.A. Diversity of group types, regulation, and function of phospholipase A2. J. Biol. Chem. 1994, 269, 13057–13060. [Google Scholar] [CrossRef]
- Schaloske, R.H.; Dennis, E.A. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta 2006, 1761, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Winkler, H.; Smith, A.D.; Dubois, F.; van den Bosch, H. The positional specificity of lysosomal phospholipase A activities. Biochem. J. 1967, 105, 38C–40C. [Google Scholar] [CrossRef]
- Habermann, E.; Reiz, K.G. On the biochemistry of bee venom peptides, melittin and apamin. Biochem. Z. 1965, 343, 192–203. [Google Scholar] [PubMed]
- De Haas, G.H.; Postema, N.M.; Nieuwenhuizen, W.; van Deenen, L.L. Purification and properties of phospholipase A from porcine pancreas. Biochim. Biophys. Acta 1968, 159, 103–117. [Google Scholar] [CrossRef]
- Wells, M.A.; Hanahan, D.J. Studies on phospholipase A. I. Isolation and characterization of two enzymes from Crotalus adamanteus venom. Biochemistry 1969, 8, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.W.; Tinker, D.O. Phospholipase A from Crotalus atrox venom. I. Purification and some properties. Biochemistry 1969, 8, 1558–1568. [Google Scholar] [CrossRef]
- Dijkstra, B.W.; Drenth, J.; Kalk, K.H.; Vandermaelen, P.J. Three-dimensional structure and disulfide bond connections in bovine pancreatic phospholipase A2. J. Mol. Biol. 1978, 124, 53–60. [Google Scholar] [CrossRef]
- Dijkstra, B.W.; Kalk, K.H.; Hol, W.G.; Drenth, J. Structure of bovine pancreatic phospholipase A2 at 1.7A resolution. J. Mol. Biol. 1981, 147, 97–123. [Google Scholar] [CrossRef]
- Heinrikson, R.L.; Krueger, E.T.; Keim, P.S. Amino acid sequence of phospholipase A2-alpha from the venom of Crotalus adamanteus. A new classification of phospholipases A2 based upon structural determinants. J. Biol. Chem. 1977, 252, 4913–4921. [Google Scholar] [CrossRef]
- Belleville, J.; Clement, J. Trypsin activation of phospholipase A2 from human and rat pancreatic juice. Bull. Soc. Chim. Biol. 1968, 50, 1419–1424. [Google Scholar]
- Illingworth, D.R.; Glover, J. Phospholipases in cerebrospinal fluid and cell fractions of human brain. Biochem. J. 1969, 115, 16P–17P. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.F.; Webster, G.R. The differentiation of phospholipase A1 and A2 in rat and human nervous tissues. J. Neurochem. 1970, 17, 1543–1554. [Google Scholar] [CrossRef]
- Dennis, E.A. Mechanism of phospholipase A2 action toward mixed micelles of detergent and phospholipids. Adv. Exp. Med. Biol. 1978, 101, 165–175. [Google Scholar] [CrossRef]
- Hendrickson, H.S.; Dennis, E.A. Kinetic analysis of the dual phospholipid model for phospholipase A2 action. J. Biol. Chem. 1984, 259, 5734–5739. [Google Scholar] [CrossRef]
- Vadas, P. Elevated plasma phospholipase A2 levels: Correlation with the hemodynamic and pulmonary changes in gram-negative septic shock. J. Lab. Clin. Med. 1984, 104, 873–881. [Google Scholar]
- Vadas, P.; Stefanski, E.; Pruzanski, W. Characterization of extracellular phospholipase A2 in rheumatoid synovial fluid. Life Sci. 1985, 36, 579–587. [Google Scholar] [CrossRef]
- Stefanski, E.; Pruzanski, W.; Sternby, B.; Vadas, P. Purification of a soluble phospholipase A2 from synovial fluid in rheumatoid arthritis. J. Biochem. 1986, 100, 1297–1303. [Google Scholar] [CrossRef]
- Hara, S.; Kudo, I.; Matsuta, K.; Miyamoto, T.; Inoue, K. Amino acid composition and NH2-terminal amino acid sequence of human phospholipase A2 purified from rheumatoid synovial fluid. J. Biochem. 1988, 104, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Kramer, R.M.; Hession, C.; Johansen, B.; Hayes, G.; McGray, P.; Chow, E.P.; Tizard, R.; Pepinsky, R.B. Structure and properties of a human non-pancreatic phospholipase A2. J. Biol. Chem. 1989, 264, 5768–5775. [Google Scholar] [CrossRef]
- Seilhamer, J.J.; Pruzanski, W.; Vadas, P.; Plant, S.; Miller, J.A.; Kloss, J.; Johnson, L.K. Cloning and recombinant expression of phospholipase A2 present in rheumatoid arthritic synovial fluid. J. Biol. Chem. 1989, 264, 5335–5338. [Google Scholar] [CrossRef]
- Tseng, A.; Buchta, R.; Goodman, A.E.; Loughnan, M.; Cairns, D.; Seilhamer, J.; Johnson, L.; Inglis, A.S.; Scott, K.F. A strategy for obtaining active mammalian enzyme from a fusion protein expressed in bacteria using phospholipase A2 as a model. Protein Expr. Purif. 1991, 2, 127–135. [Google Scholar] [CrossRef]
- Johnson, L.K.; Frank, S.; Vades, P.; Pruzanski, W.; Lusis, A.J.; Seilhamer, J.J. Localization and evolution of two human phospholipase A2 genes and two related genetic elements. Adv. Exp. Med. Biol. 1990, 275, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Kudo, I.; Hara, S.; Murakami, M.; Matsuta, K.; Miyamoto, T.; Inoue, K. Monoclonal antibodies against human synovial phospholipase A2. Biochem. Biophys. Res. Commun. 1990, 167, 1309–1315. [Google Scholar] [CrossRef]
- Green, J.A.; Smith, G.M.; Buchta, R.; Lee, R.; Ho, K.Y.; Rajkovic, I.A.; Scott, K.F. Circulating phospholipase A2 activity associated with sepsis and septic shock is indistinguishable from that associated with rheumatoid arthritis. Inflammation 1991, 15, 355–367. [Google Scholar] [CrossRef]
- Smith, G.M.; Ward, R.L.; McGuigan, L.; Rajkovic, I.A.; Scott, K.F. Measurement of human phospholipase A2 in arthritis plasma using a newly developed sandwich ELISA. Br. J. Rheumatol. 1992, 31, 175–178. [Google Scholar] [CrossRef]
- Rice, G.E.; Brennecke, S.P.; Scott, K.F.; Smith, G.M.; Rajkovic, I.A.; Bishop, G.J. Elevated maternal plasma immunoreactive phospholipase A2 in human preterm and term labour. Eicosanoids 1992, 5, 9–12. [Google Scholar]
- Vadas, P.; Keystone, J.; Stefanski, E.; Scott, K.; Pruzanski, W. Induction of circulating group II phospholipase A2 expression in adults with malaria. Infect. Immun. 1992, 60, 3928–3931. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Crozier, J.A.; Lord, R.S.; Tran, D.; Jamal, O.S.; Parsson, H.N.; Scott, K.F. Expression of secretory group II phospholipase A2 by CD1a positive cells-in human atherosclerotic plaques. Atherosclerosis 1996, 127, 283–285. [Google Scholar] [CrossRef]
- Jamal, O.S.; Conaghan, P.G.; Cunningham, A.M.; Brooks, P.M.; Munro, V.F.; Scott, K.F. Increased expression of human type IIa secretory phospholipase A2 antigen in arthritic synovium. Ann. Rheum. Dis. 1998, 57, 550–558. [Google Scholar] [CrossRef]
- Wery, J.P.; Schevitz, R.W.; Clawson, D.K.; Bobbitt, J.L.; Dow, E.R.; Gamboa, G.; Goodson, T., Jr.; Hermann, R.B.; Kramer, R.M.; McClure, D.B.; et al. Structure of recombinant human rheumatoid arthritic synovial fluid phospholipase A2 at 2.2 A resolution. Nature 1991, 352, 79–82. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Kastelein, J.J.; Schwartz, G.G.; Bash, D.; Rosenson, R.S.; Cavender, M.A.; Brennan, D.M.; Koenig, W.; Jukema, J.W.; Nambi, V.; et al. Varespladib and cardiovascular events in patients with an acute coronary syndrome: The VISTA-16 randomized clinical trial. J. Am. Med. Assoc. 2014, 311, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Sato, H.; Miki, Y.; Yamamoto, K.; Taketomi, Y. A new era of secreted phospholipase A2. J. Lipid Res. 2015, 56, 1248–1261. [Google Scholar] [CrossRef]
- Scott, K.F.; Bryant, K.J.; Bidgood, M.J. Functional coupling and differential regulation of the phospholipase A2-cyclooxygenase pathways in inflammation. J. Leukoc. Biol. 1999, 66, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, H.; Fujimoto, C.; Yoda, E.; Shimbara, S.; Nakatani, Y.; Hara, S.; Murakami, M.; Kudo, I. A novel role of group VIB calcium-independent phospholipase A2 (iPLA2gamma) in the inducible expression of group IIA secretory PLA2 in rat fibroblastic cells. J. Biol. Chem. 2007, 282, 20124–20132. [Google Scholar] [CrossRef]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [PubMed]
- Lambeau, G.; Gelb, M.H. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem. 2008, 77, 495–520. [Google Scholar] [CrossRef]
- Boilard, E.; Lai, Y.; Larabee, K.; Balestrieri, B.; Ghomashchi, F.; Fujioka, D.; Gobezie, R.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; et al. A novel anti-inflammatory role for secretory phospholipase A2 in immune complex-mediated arthritis. EMBO Mol. Med. 2010, 2, 172–187. [Google Scholar] [CrossRef]
- Miki, Y.; Yamamoto, K.; Taketomi, Y.; Sata, H.; Shimo, K.; Kobayashi, T.; Ishikawa, Y.; Ishii, T.; Nakanishi, H.; Ikeda, K.; et al. Lymphoid tissue phospholipase A2 group IID resolves contact hyperesensitvty by driving anti-inflammatory lipid mediators. J. Exp. Med. 2013, 210, 1217–1234. [Google Scholar] [CrossRef]
- Smart, B.P.; Oslund, R.C.; Walsh, L.A.; Gelb, M.H. The first potent inhibitor of mammalian group X sectreted phospholipase A2: Elucidation of sites of enhanced binding. J. Med. Chem. 2006, 49, 2858–2860. [Google Scholar] [CrossRef]
- Nevalainen, T.J.; Graham, G.G.; Scott, K.F. Antibacterial actions of secreted phospholipases A2. Review. Biochim. Biophys. Acta 2008, 1781, 1–9. [Google Scholar] [CrossRef]
- Dore, E.; Boilard, E. Roles of secreted phospholipase A2 group IIA in inflammation and host defense. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Snitko, Y.; Yoon, E.T.; Cho, W. High specificity of human secretory class II phospholipase A2 for phosphatidic acid. Biochem. J. 1997, 321, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Snitko, Y.; Koduri, R.S.; Han, S.K.; Othman, R.; Baker, S.F.; Molini, B.J.; Wilton, D.C.; Gelb, M.H.; Cho, W. Mapping the interfacial binding surface of human secretory group IIa phospholipase A2. Biochemistry 1997, 36, 14325–14333. [Google Scholar] [CrossRef]
- Singer, A.G.; Ghomashchi, F.; Le Calvez, C.; Bollinger, J.; Bezzine, S.; Rouault, M.; Sadilek, M.; Nguyen, E.; Lazdunski, M.; Lambeau, G.; et al. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. J. Biol. Chem. 2002, 277, 48535–48549. [Google Scholar] [CrossRef]
- Bezzine, S.; Bollinger, J.G.; Singer, A.G.; Veatch, S.L.; Keller, S.L.; Gelb, M.H. On the binding preference of human groups IIA and X phospholipases A2 for membranes with anionic phospholipids. J. Biol. Chem. 2002, 277, 48523–48534. [Google Scholar] [CrossRef] [PubMed]
- Bezzine, S.; Koduri, R.S.; Valentin, E.; Murakami, M.; Kudo, I.; Ghomashchi, F.; Sadilek, M.; Lambeau, G.; Gelb, M.H. Exogenously added human group X secreted phospholipase A2 but not the group IB, IIA, and V enzymes efficiently release arachidonic acid from adherent mammalian cells. J. Biol. Chem. 2000, 275, 3179–3191. [Google Scholar] [CrossRef]
- Atsumi, G.; Murakami, M.; Tajima, M.; Shimbara, S.; Hara, N.; Kudo, I. The perturbed membrane of cells undergoing apoptosis is susceptible to type II secretory phospholipase A2 to liberate arachidonic acid. Biochim. Biophys. Acta 1997, 1349, 43–54. [Google Scholar] [CrossRef]
- Weiss, J.; Franson, R.C.; Beckerdite, S.; Schmeidler, K.; Elsbach, P. Partial characterization and purification of a rabbit granulocyte factor that increases permeability of Escherichia coli. J. Clin. Investig. 1975, 55, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Elsbach, P.; Weiss, J.; Franson, R.C.; Beckerdite-Quagliata, S.; Schneider, A.; Harris, L. Separation and purification of a potent bactericidal/permeability-increasing protein and a closely associated phospholipase A2 from rabbit polymorphonuclear leukocytes. Observations on their relationship. J. Biol. Chem. 1979, 254, 11000–11009. [Google Scholar] [CrossRef]
- Weiss, J.; Beckerdite-Quagliata, S.; Elsbach, P. Determinants of the action of phospholipases A on the envelope phospholipids of Escherichia coli. J. Biol. Chem. 1979, 254, 11010–11014. [Google Scholar] [CrossRef]
- Weinrauch, Y.; Abad, C.; Liang, N.S.; Lowry, S.F.; Weiss, J. Mobilization of potent plasma bactericidal activity during systemic bacterial challenge. Role of group IIA phospholipase A2. J. Clin. Investig. 1998, 102, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Laine, V.J.; Grass, D.S.; Nevalainen, T.J. Protection by group II phospholipase A2 against Staphylococcus aureus. J. Immunol. 1999, 162, 7402–7408. [Google Scholar] [PubMed]
- Brown, S.; Santa Maria, J.P., Jr.; Walker, S. Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336. [Google Scholar] [CrossRef]
- Foreman-Wykert, A.K.; Weinrauch, Y.; Elsbach, P.; Weiss, J. Cell-wall determinants of the bactericidal action of group IIA phospholipase A2 against Gram-positive bacteria. J. Clin. Investig. 1999, 103, 715–721. [Google Scholar] [CrossRef]
- Schneewind, O.; Missiakas, D. Lipoteichoic acids, phosphate-containing polymers in the envelope of gram-positive bacteria. J. Bacteriol. 2014, 196, 1133–1142. [Google Scholar] [CrossRef]
- Koprivnjak, T.; Peschel, A.; Gelb, M.H.; Liang, N.S.; Weiss, J.P. Role of charge properties of bacterial envelope in bactericidal action of human group IIA phospholipase A2 against Staphylococcus aureus. J. Biol. Chem. 2002, 277, 47636–47644. [Google Scholar] [CrossRef]
- Koduri, R.S.; Gronroos, J.O.; Laine, V.J.; Le Calvez, C.; Lambeau, G.; Nevalainen, T.J.; Gelb, M.H. Bactericidal properties of human and murine groups I, II, V, X, and XII secreted phospholipases A2. J. Biol. Chem. 2002, 277, 5849–5857. [Google Scholar] [CrossRef]
- Buckland, A.G.; Wilton, D.C. The antibacterial properties of secreted phospholipases A2. Biochim. Biophys. Acta 2000, 1488, 71–82. [Google Scholar] [CrossRef]
- Wiese, A.; Brandenburg, K.; Lindner, B.; Schromm, A.B.; Carroll, S.F.; Rietschel, E.T.; Seydel, U. Mechanisms of action of the bactericidal/permeability-increasing protein BPI on endotoxin and phospholipid monolayers and aggregates. Biochemistry 1997, 36, 10301–10310. [Google Scholar] [CrossRef]
- Ben Bacha, A.; Abid, I. Secretory phospholipase A2 in dromedary tears: A host defense against staphylococci and other gram-positive bacteria. Appl. Biochem. Biotechnol. 2013, 169, 1858–1869. [Google Scholar] [CrossRef]
- Kim, R.R.; Chen, Z.; Mann, T.J.; Bastard, K.; Scott, K.F.; Church, W.B. Structural and Functional Aspects of Targeting the Secreted Human Group IIA Phospholipase A2. Molecules 2020, 25, 4459. [Google Scholar] [CrossRef]
- Dacheux, M.; Sinou, V.; Payre, C.; Jeammet, L.; Parzy, D.; Grellier, P.; Deregnaucourt, C.; Lambeau, G. Antimalarial Activity of Human Group IIA Secreted Phospholipase A2 in Relation to Enzymatic Hydrolysis of Oxidized Lipoproteins. Infect. Immun. 2019, 87, e00556-19. [Google Scholar] [CrossRef]
- Nevalainen, T.J.; Haapanen, T.J. Distribution of pancreatic (group I) and synovial-type (group II) phospholipases A2 in human tissues. Inflammation 1993, 17, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Triggiani, M.; Granata, F.; Oriente, A.; De Marino, V.; Gentile, M.; Calabrese, C.; Palumbo, C.; Marone, G. Secretory phospholipases A2 induce beta-glucuronidase release and IL-6 production from human lung macrophages. J. Immunol. 2000, 164, 4908–4915. [Google Scholar] [CrossRef] [PubMed]
- Granata, F.; Petraroli, A.; Boilard, E.; Bezzine, S.; Bollinger, J.; Del Vecchio, L.; Gelb, M.H.; Lambeau, G.; Marone, G.; Triggiani, M. Activation of cytokine production by secreted phospholipase A2 in human lung macrophages expressing the M-type receptor. J. Immunol. 2005, 174, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Granata, F.; Frattini, A.; Loffredo, S.; Staiano, R.I.; Petraroli, A.; Ribatti, D.; Oslund, R.; Gelb, M.H.; Lambeau, G.; Marone, G.; et al. Production of vascular endothelial growth factors from human lung macrophages induced by group IIA and group X secreted phospholipases A2. J. Immunol. 2010, 184, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef]
- Dong, Z.; Liu, Y.; Scott, K.F.; Levin, L.; Gaitonde, K.; Bracken, R.B.; Burke, B.; Zhai, Q.J.; Wang, J.; Oleksowicz, L.; et al. Secretory phospholipase A2-IIa is involved in prostate cancer progression and may potentially serve as a biomarker for prostate cancer. Carcinogenesis 2010, 31, 1948–1955. [Google Scholar] [CrossRef]
- Thommesen, L.; Sjursen, W.; Gasvik, K.; Hanssen, W.; Brekke, O.L.; Skattebol, L.; Holmeide, A.K.; Espevik, T.; Johansen, B.; Laegreid, A. Selective inhibitors of cytosolic or secretory phospholipase A2 block TNF-induced activation of transcription factor nuclear factor-kappa B and expression of ICAM-1. J. Immunol. 1998, 161, 3421–3430. [Google Scholar]
- Dong, Z.; Meller, J.; Succop, P.; Wang, J.; Wikenheiser-Brokamp, K.; Starnes, S.; Lu, S. Secretory phospholipase A2-IIa upregulates HER/HER2-elicited signaling in lung cancer cells. Int. J. Oncol. 2014, 45, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Poligone, B.; Baldwin, A.S. Positive and negative regulation of NF-kappaB by COX-2: Roles of different prostaglandins. J. Biol. Chem. 2001, 276, 38658–38664. [Google Scholar] [CrossRef] [PubMed]
- Skotland, T.; Ekroos, K.; Kauhanen, D.; Simolin, H.; Seierstad, T.; Berge, V.; Sandvig, K.; Llorente, A. Molecular lipid species in urinary exosomes as potential prostate cancer biomarkers. Eur. J. Cancer 2017, 70, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Duchez, A.C.; Boudreau, L.H.; Naika, G.S.; Bollinger, J.; Belleannee, C.; Cloutier, N.; Laffont, B.; Mendoza-Villarroel, R.E.; Levesque, T.; Rollet-Labelle, E.; et al. Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc. Natl. Acad. Sci. USA 2015, 112, E3564–E3573. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Gaudette, D.; Furui, T.; Mao, M.; Estrella, V.; Eder, A.; Pustilnik, T.; Sasagawa, T.; Lapushin, R.; Yu, S.; et al. Lysophospholipid growth factors in the initiation, progression, metastases, and management of ovarian cancer. Ann. N. Y. Acad. Sci. 2000, 905, 188–208. [Google Scholar] [CrossRef]
- Hajizadeh, S.; DeGroot, J.; TeKoppele, J.M.; Tarkowski, A.; Collins, L.V. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res. Ther. 2003, 5, R234–R240. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Pare, A.; Rousseau, M.; Naika, G.S.; Levesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef]
- Sun, G.Y.; Xu, J.; Jensen, M.D.; Yu, S.; Wood, W.G.; Gonzalez, F.A.; Simonyi, A.; Sun, A.Y.; Weisman, G.A. Phospholipase A2 in astrocytes: Responses to oxidative stress, inflammation, and G protein-coupled receptor agonists. Mol. Neurobiol. 2005, 31, 27–41. [Google Scholar] [CrossRef]
- Yagami, T. Cerebral arachidonate cascade in dementia: Alzheimer’s disease and vascular dementia. Curr. Neuropharmacol. 2006, 4, 87–100. [Google Scholar] [CrossRef]
- Titsworth, W.L.; Liu, N.K.; Xu, X.M. Role of secretory phospholipase a2 in CNS inflammation: Implications in traumatic spinal cord injury. CNS Neurol. Disord. Drug Targets 2008, 7, 254–269. [Google Scholar] [CrossRef]
- Sun, G.Y.; Shelat, P.B.; Jensen, M.B.; He, Y.; Sun, A.Y.; Simonyi, A. Phospholipases A2 and inflammatory responses in the central nervous system. Neuromol. Med. 2010, 12, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.-T.; Nevalainen, T.J.; Yeo, J.-F.; Ong, W.-Y. Expression profile of multiple secretory phospholipase A2 isoforms in the rat CNS: Enriched expression of sPLA2-IIA in brainstem and spinal cord. J. Chem. Neuroanat. 2010, 39, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.; Burillo, S.L.; Crespo, M.S.; Nieto, M.L. Secretory phospholipase A2 activates the cascade of mitogen-activated protein kinases and cytosolic phospholipase A2 in the human astrocytoma cell line 1321N1. J. Biol. Chem. 1998, 273, 606–612. [Google Scholar] [CrossRef]
- Hernandez, M.; Barrero, M.J.; Alvarez, J.; Montero, M.; Sanchez Crespo, M.; Nieto, M.L. Secretory phospholipase A2 induces phospholipase Cgamma-1 activation and Ca2+ mobilization in the human astrocytoma cell line 1321N1 by a mechanism independent of its catalytic activity. Biochem. Biophys. Res. Commun. 1999, 260, 99–104. [Google Scholar] [CrossRef]
- Villanueva, E.B.; Little, J.P.; Lambeau, G.; Klegeris, A. Secreted phospholipase A2 group IIA is a neurotoxin released by stimulated human glial cells. Mol. Cell. Neurosci. 2012, 49, 430–438. [Google Scholar] [CrossRef]
- David, S.; Lopez-Vales, R. Bioactive Lipid Mediators in the Initiation and Resolution of Inflammation after Spinal Cord Injury. Neuroscience 2021, 466, 273–297. [Google Scholar] [CrossRef]
- Kumar, A.; Behl, T.; Jamwal, S.; Kaur, I.; Sood, A.; Kumar, P. Exploring the molecular approach of COX and LOX in Alzheimer’s and Parkinson’s disorder. Mol. Biol. Rep. 2020, 47, 9895–9912. [Google Scholar] [CrossRef]
- Gulla, S.; Lomada, D.; Lade, A.; Pallu, R.; Reddy, M.C. Role of Prostaglandins in Multiple Sclerosis. Curr. Pharm. Des. 2020, 26, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.A.; Austen, K.F.; Drazen, J.M.; Clark, D.A.; Marfat, A.; Corey, E.J. Slow reacting substances of anaphylaxis: Identification of leukotrienes C-1 and D from human and rat sources. Proc. Natl. Acad. Sci. USA 1980, 77, 3710–3714. [Google Scholar] [CrossRef]
- Bowton, D.L.; Seeds, M.C.; Fasano, M.B.; Goldsmith, B.; Bass, D.A. Phospholipase A2 and arachidonate increase in bronchoalveolar lavage fluid after inhaled antigen challenge in asthmatics. Am. J. Respir. Crit. Care Med. 1997, 155, 421–425. [Google Scholar] [CrossRef]
- Hallstrand, T.S.; Lai, Y.; Ni, Z.; Oslund, R.C.; Henderson, W.R., Jr.; Gelb, M.H.; Wenzel, S.E. Relationship between levels of secreted phospholipase A2 groups IIA and X in the airways and asthma severity. Clin. Exp. Allergy 2011, 41, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Granata, F.; Nardicchi, V.; Loffredo, S.; Frattini, A.; Ilaria Staiano, R.; Agostini, C.; Triggiani, M. Secreted phospholipases A2: A proinflammatory connection between macrophages and mast cells in the human lung. Immunobiology 2009, 214, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Henderson, W.R., Jr.; Chi, E.Y.; Bollinger, J.G.; Tien, Y.T.; Ye, X.; Castelli, L.; Rubtsov, Y.P.; Singer, A.G.; Chiang, G.K.; Nevalainen, T.; et al. Importance of group X-secreted phospholipase A2 in allergen-induced airway inflammation and remodeling in a mouse asthma model. J. Exp. Med. 2007, 204, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Pniewska, E.; Pawliczak, R. The involvement of phospholipases A2 in asthma and chronic obstructive pulmonary disease. Mediat. Inflamm. 2013, 2013, 793505. [Google Scholar] [CrossRef]
- Snider, J.M.; You, J.K.; Wang, X.; Snider, A.J.; Hallmark, B.; Zec, M.M.; Seeds, M.C.; Sergeant, S.; Johnstone, L.; Wang, Q.; et al. Group IIA secreted phospholipase A2 is associated with the pathobiology leading to COVID-19 mortality. J. Clin. Investig. 2021, 131, e149236. [Google Scholar] [CrossRef] [PubMed]
- Arganaraz, G.A.; Palmeira, J.D.F.; Arganaraz, E.R. Phosphatidylserine inside out: A possible underlying mechanism in the inflammation and coagulation abnormalities of COVID-19. Cell Commun. Signal. 2020, 18, 190. [Google Scholar] [CrossRef] [PubMed]
- Kuypers, F.A.; Rostad, C.A.; Anderson, E.J.; Chahroudi, A.; Jaggi, P.; Wrammert, J.; Mantus, G.; Basu, R.; Harris, F.; Hanberry, B.; et al. Secretory phospholipase A2 in SARS-CoV-2 infection and multisystem inflammatory syndrome in children (MIS-C). Exp. Biol. Med. 2021, 1–10. [Google Scholar] [CrossRef]
- O’Donoghue, M.L. Acute coronary syndromes: Targeting inflammation-what has the VISTA-16 trial taught us? Nat. Rev. Cardiol. 2014, 11, 130–132. [Google Scholar] [CrossRef]
- Tietge, U.J.; Maugeais, C.; Cain, W.; Grass, D.; Glick, J.M.; de Beer, F.C.; Rader, D.J. Overexpression of secretory phospholipase A2 causes rapid catabolism and altered tissue uptake of high density lipoprotein cholesteryl ester and apolipoprotein A-I. J. Biol. Chem. 2000, 275, 10077–10084. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Hurt-Camejo, E. Phospholipase A2 enzymes and the risk of atherosclerosis. Eur. Heart J. 2012, 33, 2899–2909. [Google Scholar] [CrossRef]
- Niessen, H.W.; Krijnen, P.A.; Visser, C.A.; Meijer, C.J.; Erik Hack, C. Type II secretory phospholipase A2 in cardiovascular disease: A mediator in atherosclerosis and ischemic damage to cardiomyocytes? Cardiovasc. Res. 2003, 60, 68–77. [Google Scholar] [CrossRef]
- Kugiyama, K.; Ota, Y.; Takazoe, K.; Moriyama, Y.; Kawano, H.; Miyao, Y.; Sakamoto, T.; Soejima, H.; Ogawa, H.; Doi, H.; et al. Circulating levels of secretory type II phospholipase A2 predict coronary events in patients with coronary artery disease. Circulation 1999, 100, 1280–1284. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.L.; Kang, C.W.; Ooi, K.S.; Tan, S.T.; Ahmad, N.S.; Nasuruddin, D.N.; Ithnin, A.; Tajul Arifin, K.; Heng, L.Y.; Hassan, N.I.; et al. Comparison of sPLA2IIA performance with high-sensitive CRP neutrophil percentage PCT and lactate to identify bacterial infection. Sci. Rep. 2021, 11, 11369. [Google Scholar] [CrossRef] [PubMed]
- Nandi, U.; Jones, A.E.; Puskarich, M.A. Group IIA secretory phospholipase 2 independently predicts mortality and positive blood culture in emergency department sepsis patients. J. Am. Coll. Emerg. Physicians Open 2021, 2, e12460. [Google Scholar] [CrossRef] [PubMed]
- Mearelli, F.; Fiotti, N.; Giansante, C.; Casarsa, C.; Orso, D.; De Helmersen, M.; Altamura, N.; Ruscio, M.; Castello, L.M.; Colonetti, E.; et al. Derivation and Validation of a Biomarker-Based Clinical Algorithm to Rule Out Sepsis From Noninfectious Systemic Inflammatory Response Syndrome at Emergency Department Admission: A Multicenter Prospective Study. Crit. Care Med. 2018, 46, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Zeiher, B.G.; Steingrub, J.; Laterre, P.F.; Dmitrienko, A.; Fukiishi, Y.; Abraham, E. LY315920NA/S-5920, a selective inhibitor of group IIA secretory phospholipase A2, fails to improve clinical outcome for patients with severe sepsis. Crit. Care Med. 2005, 33, 1741–1748. [Google Scholar] [CrossRef]
- Lin, M.K.; Farewell, V.; Vadas, P.; Bookman, A.A.; Keystone, E.C.; Pruzanski, W. Secretory phospholipase A2 as an index of disease activity in rheumatoid arthritis. Prospective double blind study of 212 patients. J. Rheumatol. 1996, 23, 1162–1166. [Google Scholar]
- Bryant, K.J.; Bidgood, M.J.; Lei, P.W.; Taberner, M.; Salom, C.; Kumar, V.; Lee, L.; Church, W.B.; Courtenay, B.; Smart, B.P.; et al. A bifunctional role for group IIA secreted phospholipase A2 in human rheumatoid fibroblast-like synoviocyte arachidonic acid metabolism. J. Biol. Chem. 2011, 286, 2492–2503. [Google Scholar] [CrossRef]
- Lee, L.K.; Bryant, K.J.; Bouveret, R.; Lei, P.W.; Duff, A.P.; Harrop, S.J.; Huang, E.P.; Harvey, R.P.; Gelb, M.H.; Gray, P.P.; et al. Selective inhibition of human group IIA-secreted phospholipase A2 (hGIIA) signaling reveals arachidonic acid metabolism is associated with colocalization of hGIIA to vimentin in rheumatoid synoviocytes. J. Biol. Chem. 2013, 288, 15269–15279. [Google Scholar] [CrossRef]
- Ha, V.T.; Lainscek, D.; Gesslbauer, B.; Jarc-Jovicic, E.; Hyotylainen, T.; Ilc, N.; Lakota, K.; Tomsic, M.; van de Loo, F.A.J.; Bochkov, V.; et al. Synergy between 15-lipoxygenase and secreted PLA2 promotes inflammation by formation of TLR4 agonists from extracellular vesicles. Proc. Natl. Acad. Sci. USA 2020, 117, 25679–25689. [Google Scholar] [CrossRef]
- Bradley, J.D.; Dmitrienko, A.A.; Kivitz, A.J.; Gluck, O.S.; Weaver, A.L.; Wiesenhutter, C.; Myers, S.L.; Sides, G.D. A randomized, double-blinded, placebo-controlled clinical trial of LY333013, a selective inhibitor of group II secretory phospholipase A2, in the treatment of rheumatoid arthritis. J. Rheumatol. 2005, 32, 417–423. [Google Scholar]
- Ogawa, M.; Yamashita, S.; Sakamoto, K.; Ikei, S. Elevation of serum group II phospholipase A2 in patients with cancers of digestive organs. Res. Commun. Chem. Pathol. Pharmacol. 1991, 74, 241–244. [Google Scholar]
- Yamashita, S.; Yamashita, J.; Ogawa, M. Overexpression of group II phospholipase A2 in human breast cancer tissues is closely associated with their malignant potency. Br. J. Cancer 1994, 69, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Fijneman, R.J.; Cormier, R.T. The roles of sPLA2-IIA (Pla2g2a) in cancer of the small and large intestine. Front. Biosci. 2008, 13, 4144–4174. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.F.; Sajinovic, M.; Hein, J.; Nixdorf, S.; Galettis, P.; Liauw, W.; de Souza, P.; Dong, Q.; Graham, G.G.; Russell, P.J. Emerging roles for phospholipase A2 enzymes in cancer. Biochimie 2010, 92, 601–610. [Google Scholar] [CrossRef]
- Brglez, V.; Lambeau, G.; Petan, T. Secreted phospholipases A2 in cancer: Diverse mechanisms of action. Biochimie 2014, 107 Pt A, 114–123. [Google Scholar] [CrossRef]
- Chen, J.; Ye, L.; Sun, Y.; Takada, Y. A Concise Update on the Relevance of Secretory Phospholipase A2 Group IIA and its Inhibitors with Cancer. Med. Chem. 2017, 13, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Chang, Y.; Fan, J.; Ji, W.; Su, C. Phospholipase A2 superfamily in cancer. Cancer Lett. 2021, 497, 165–177. [Google Scholar] [CrossRef]
- Sved, P.; Scott, K.F.; McLeod, D.; King, N.J.; Singh, J.; Tsatralis, T.; Nikolov, B.; Boulas, J.; Nallan, L.; Gelb, M.H.; et al. Oncogenic action of secreted phospholipase A2 in prostate cancer. Cancer Res. 2004, 64, 6934–6940. [Google Scholar] [CrossRef]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Cormier, R.T. PLA2G2A (Phospholipase A2, Group IIA (Platelets, Synovial Fluid). Atlas of Genetics and Cytogen-etics in Oncology and Haematology. 2011. Available online: http://atlasgeneticsoncology.org/Genes/PLA2G2AID41730ch1p36.html (accessed on 22 March 2021).
- Cummings, B.S. Phospholipase A2 as targets for anti-cancer drugs. Biochem. Pharmacol. 2007, 74, 949–959. [Google Scholar] [CrossRef]
- Dong, Q.; Patel, M.; Scott, K.F.; Graham, G.G.; Russell, P.J.; Sved, P. Oncogenic action of phospholipase A2 in prostate cancer. Cancer Lett. 2006, 240, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Dennis, E.A. Critical role of a hydrogen bond in the interaction of phospholipase A2 with transition-state and substrate analogues. Proc. Natl. Acad. Sci. USA 1991, 88, 9325–9329. [Google Scholar] [CrossRef]
- Berg, O.G.; Gelb, M.H.; Tsai, M.D.; Jain, M.K. Interfacial enzymology: The secreted phospholipase A2-paradigm. Chem. Rev. 2001, 101, 2613–2654. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.F.; Othman, R.; Wilton, D.C. Tryptophan-containing mutant of human (group IIa) secreted phospholipase A2 has a dramatically increased ability to hydrolyze phosphatidylcholine vesicles and cell membranes. Biochemistry 1998, 37, 13203–13211. [Google Scholar] [CrossRef]
- Harkewicz, R.; Dennis, E.A. Applications of mass spectrometry to lipids and membranes. Annu. Rev. Biochem. 2011, 80, 301–325. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.V.; Nunez, J.K.; Doudna, J.A. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef]
- Hernandez, M.; Martin, R.; Garcia-Cubillas, M.D.; Maeso-Hernandez, P.; Nieto, M.L. Secreted PLA2 induces proliferation in astrocytoma through the EGF receptor: Another inflammation-cancer link. Neuro Oncol. 2010, 12, 1014–1023. [Google Scholar] [CrossRef]
- Bidgood, M.J.; Jamal, O.S.; Cunningham, A.M.; Brooks, P.M.; Scott, K.F. Type IIA secretory phospholipase A2 up-regulates cyclooxygenase-2 and amplifies cytokine-mediated prostaglandin production in human rheumatoid synoviocytes. J. Immunol. 2000, 165, 2790–2797. [Google Scholar] [CrossRef]
- Lu, S.; Dong, Z. Overexpression of secretory phospholipase A2-IIa supports cancer stem cell phenotype via HER/ERBB-elicited signaling in lung and prostate cancer cells. Int. J. Oncol. 2017, 50, 2113–2122. [Google Scholar] [CrossRef]
- Murakami, M.; Kambe, T.; Shimbara, S.; Yamamoto, S.; Kuwata, H.; Kudo, I. Functional association of type IIA secretory phospholipase A2 with the glycosylphosphatidylinositol-anchored heparan sulfate proteoglycan in the cyclooxygenase-2-mediated delayed prostanoid-biosynthetic pathway. J. Biol. Chem. 1999, 274, 29927–29936. [Google Scholar] [CrossRef] [PubMed]
- Mounier, C.M.; Ghomashchi, F.; Lindsay, M.R.; James, S.; Singer, A.G.; Parton, R.G.; Gelb, M.H. Arachidonic acid release from mammalian cells transfected with human groups IIA and X secreted phospholipase A2 occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A2-alpha. J. Biol. Chem. 2004, 279, 25024–25038. [Google Scholar] [CrossRef]
- Fujita, M.; Zhu, K.; Fujita, C.K.; Zhao, M.; Lam, K.S.; Kurth, M.J.; Takada, Y.K.; Takada, Y. Proinflammatory secreted phospholipase A2 type IIA (sPLA-IIA) induces integrin activation through direct binding to a newly identified binding site (site 2) in integrins alphavbeta3, alpha4beta1, and alpha5beta1. J. Biol. Chem. 2015, 290, 259–271. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Cytoplasmic Intermediate Filaments in Cell Biology. Annu. Rev. Cell Dev. Biol. 2018, 34, 1–28. [Google Scholar] [CrossRef]
- Boilard, E.; Bourgoin, S.G.; Bernatchez, C.; Poubelle, P.E.; Surette, M.E. Interaction of low molecular weight group IIA phospholipase A2 with apoptotic human T cells: Role of heparan sulfate proteoglycans. Fed. Am. Soc. Exp. Biol. J. 2003, 17, 1068–1080. [Google Scholar] [CrossRef]
- Nakatani, Y.; Tanioka, T.; Sunaga, S.; Murakami, M.; Kudo, I. Identification of a cellular protein that functionally interacts with the C2 domain of cytosolic phospholipase A2alpha. J. Biol. Chem. 2000, 275, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Perlson, E.; Michaelevski, I.; Kowalsman, N.; Ben-Yaakov, K.; Shaked, M.; Seger, R.; Eisenstein, M.; Fainzilber, M. Vimentin binding to phosphorylated Erk sterically hinders enzymatic dephosphorylation of the kinase. J. Mol. Biol. 2006, 364, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, A.; Kokotou, M.G.; Vasilakaki, S.; Kokotos, G. Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 941–956. [Google Scholar] [CrossRef]
- Kyger, E.M.; Franson, R.C. Non-specific inhibition of enzymes by p-bromophenacyl bromide. Inhibition of human phospholipase C and modification of sulfhydryl groups. Biochim. Biophys. Acta 1984, 794, 96–103. [Google Scholar] [CrossRef]
- Yap, W.H.; Ahmed, N.; Lim, Y.M. Inhibition of Human Group IIA-Secreted Phospholipase A2 and THP-1 Monocyte Recruitment by Maslinic Acid. Lipids 2016, 51, 1153–1159. [Google Scholar] [CrossRef]
- Yap, W.H.; Ooi, B.K.; Ahmed, N.; Lim, Y.M. Maslinic acid modulates secreted phospholipase A2-IIA (sPLA2-IIA)-mediated inflammatory effects in macrophage foam cells formation. J. Biosci. 2018, 43, 277–285. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, W.; Kim, J.; Bae, J.S. Inhibitory Effect of Sulforaphane on Secretory Group IIA Phospholipase A2. Int. J. Pharmacol. 2018, 14, 187–193. [Google Scholar] [CrossRef]
- Bae, J.S. Inhibitory Effect of Orientin on Secretory Group IIA Phospholipase A2. Inflammation 2015, 38, 1631–1638. [Google Scholar] [CrossRef]
- Lee, I.-C.; Bae, J.-S. Inhibitory effect of vicenin-2 and scolymoside on secretory group IIA phospholipase A2. Anim. Cells Syst. 2015, 19, 305–311. [Google Scholar] [CrossRef]
- Moneriz, C.; Marin-Garcia, P.; Garcia-Granados, A.; Bautista, J.M.; Diez, A.; Puyet, A. Parasitostatic effect of maslinic acid. I. Growth arrest of Plasmodium falciparum intraerythrocytic stages. Malar. J. 2011, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Tseng, A.; Inglis, A.S.; Scott, K.F. Native peptide inhibition. Specific inhibition of type II phospholipases A2 by synthetic peptides derived from the primary sequence. J. Biol. Chem. 1996, 271, 23992–23998. [Google Scholar] [CrossRef] [PubMed]
- Church, W.B.; Inglis, A.S.; Tseng, A.; Duell, R.; Lei, P.W.; Bryant, K.J.; Scott, K.F. A novel approach to the design of inhibitors of human secreted phospholipase A2 based on native peptide inhibition. J. Biol. Chem. 2001, 276, 33156–33164. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Dickerson, T.; Kaur, H.; Takada, Y.K.; Fujita, M.; Liu, R.; Knapp, J.M.; Lam, K.S.; Schore, N.E.; Kurth, M.J.; et al. Identification of inhibitors against interaction between pro-inflammatory sPLA2-IIA protein and integrin alphavbeta3. Bioorg. Med. Chem. Lett. 2013, 23, 340–345. [Google Scholar] [CrossRef][Green Version]
- Hou, S.; Xu, T.; Xu, J.; Qu, L.; Xu, Y.; Chen, L.; Liu, J. Structural basis for functional selectivity and ligand recognition revealed by crystal structures of human secreted phospholipase A2 group IIE. Sci. Rep. 2017, 7, 10815. [Google Scholar] [CrossRef]
- Hopkins, A.I. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.K.M.; Fatima, S.; Caprano, A.; Wates, S.A.; Vafaee, F. Integrative resource for network-based investigation of COVID-19 combinatorial drug repositioning and mechanism of action. Patterns 2021, 2, 100325. [Google Scholar] [CrossRef] [PubMed]
- Denti, V.; Mahajneh, A.; Capitoli, G.; Clerici, F.; Piga, L.; Pagani, L.; Chinello, C.; Bolognesi, M.M.; Paglia, G.; Galimberti, S.; et al. Lipidomic typing of colorectal cancer tissue containing tumour-infiltrating lymphocytes by MALDI mass spectrometry imaging. Metabolites 2021, 11, 599. [Google Scholar] [CrossRef]
- Lundberg, E.; Borner, G.H.H. Spatial proteomics: A powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Nagle, M.P.; Tam, G.S.; Maltz, E.; Hemminger, Z.; Wolman, R. From cell biology to physiology using in situ single cell technologies. Cell Syst. 2021, 12, 388–400. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cancer Type | Expression | Association with Prognosis | Reference |
|---|---|---|---|
| Prostate | Increased | Shorter Patient Survival | [118] |
| Breast | Increased | Shorter Patient Survival | [118] |
| Gastric | Increased | Leading to Longer Patient Survival and Less Metastasis | [118] |
| Lung | Increased | Shorter Patient Survival | [118] |
| Oesophageal | Increased | Unknown | [118] |
| Colon | Varies from Site | Protective | [119] |
| Liver | High | Shorter Patient Survival | [119] |
| Brain | High | Shorter Patient Survival | [119] |
| Pancreatic | High Early Stage | Better Prognosis | [119] |
| Low Late Stage | Poor Prognosis | [119] | |
| Ovarian | High | Shorter Patient Survival | [119] |
| Decreased | Following Chemotherapy | [124] | |
| Skin | Increased | [125] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scott, K.F.; Mann, T.J.; Fatima, S.; Sajinovic, M.; Razdan, A.; Kim, R.R.; Cooper, A.; Roohullah, A.; Bryant, K.J.; Gamage, K.K.; et al. Human Group IIA Phospholipase A2—Three Decades on from Its Discovery. Molecules 2021, 26, 7267. https://doi.org/10.3390/molecules26237267
Scott KF, Mann TJ, Fatima S, Sajinovic M, Razdan A, Kim RR, Cooper A, Roohullah A, Bryant KJ, Gamage KK, et al. Human Group IIA Phospholipase A2—Three Decades on from Its Discovery. Molecules. 2021; 26(23):7267. https://doi.org/10.3390/molecules26237267
Chicago/Turabian StyleScott, Kieran F., Timothy J. Mann, Shadma Fatima, Mila Sajinovic, Anshuli Razdan, Ryung Rae Kim, Adam Cooper, Aflah Roohullah, Katherine J. Bryant, Kasuni K. Gamage, and et al. 2021. "Human Group IIA Phospholipase A2—Three Decades on from Its Discovery" Molecules 26, no. 23: 7267. https://doi.org/10.3390/molecules26237267
APA StyleScott, K. F., Mann, T. J., Fatima, S., Sajinovic, M., Razdan, A., Kim, R. R., Cooper, A., Roohullah, A., Bryant, K. J., Gamage, K. K., Harman, D. G., Vafaee, F., Graham, G. G., Church, W. B., Russell, P. J., Dong, Q., & de Souza, P. (2021). Human Group IIA Phospholipase A2—Three Decades on from Its Discovery. Molecules, 26(23), 7267. https://doi.org/10.3390/molecules26237267