
The Effects of Streptozotocin-Induced Diabetes and Insulin Treatment on Carnitine Biosynthesis and Renal Excretion


Abstract
:1. Introduction
2. Results
2.1. The Effects of STZ-Induced Diabetes on Physical Characteristics
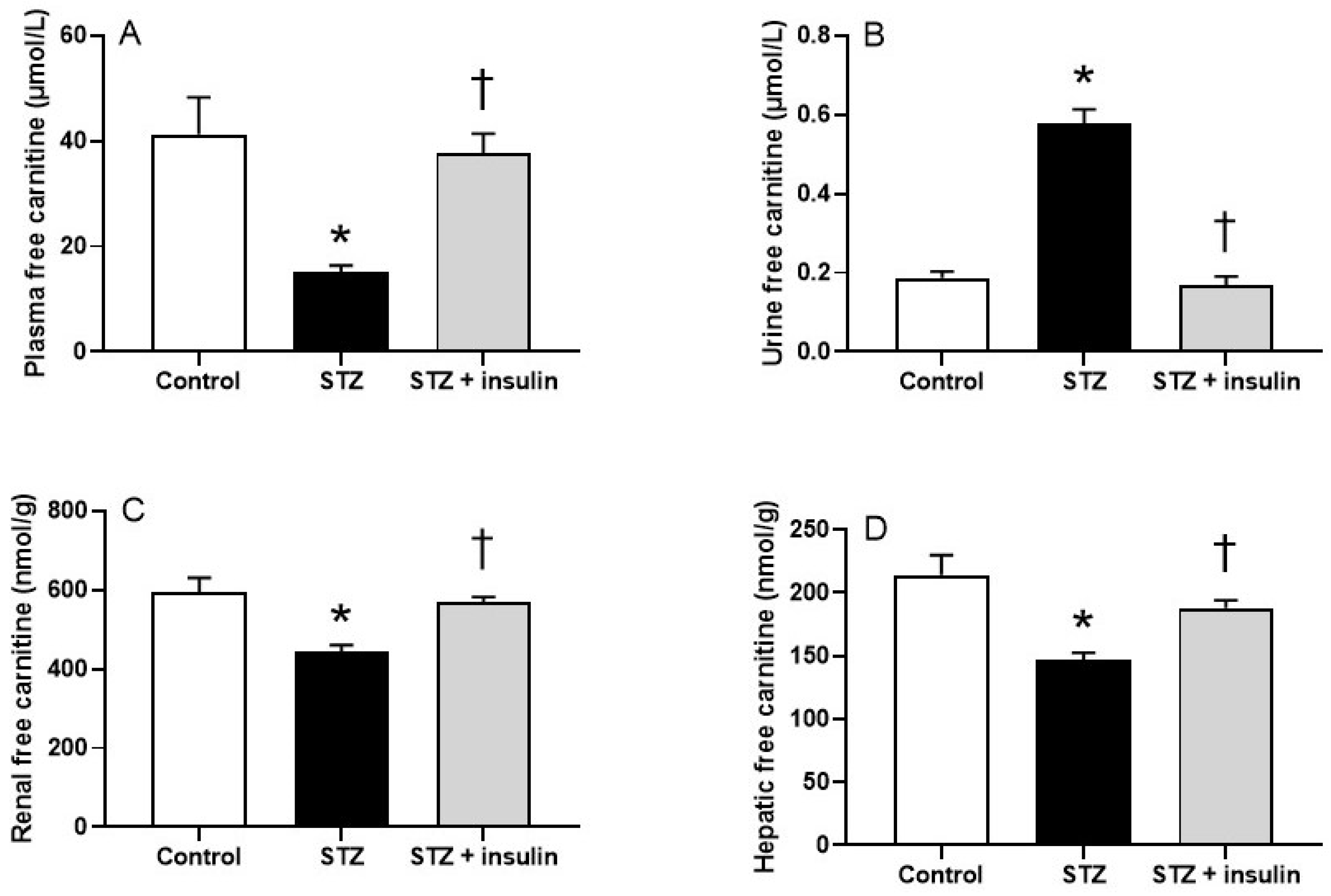
2.2. Plasma, Renal, and Hepatic Free Carnitine Levels in Diabetic Rats
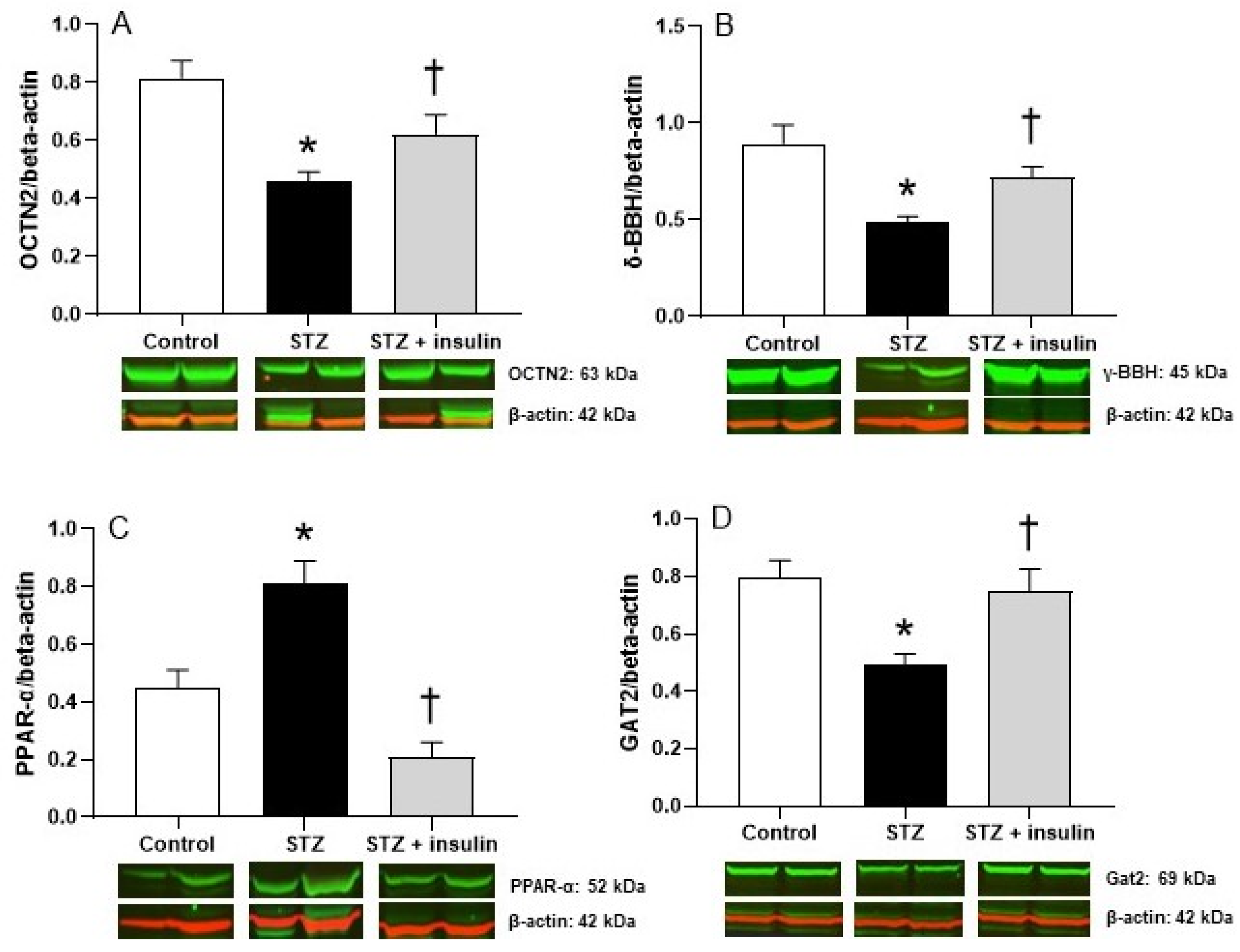
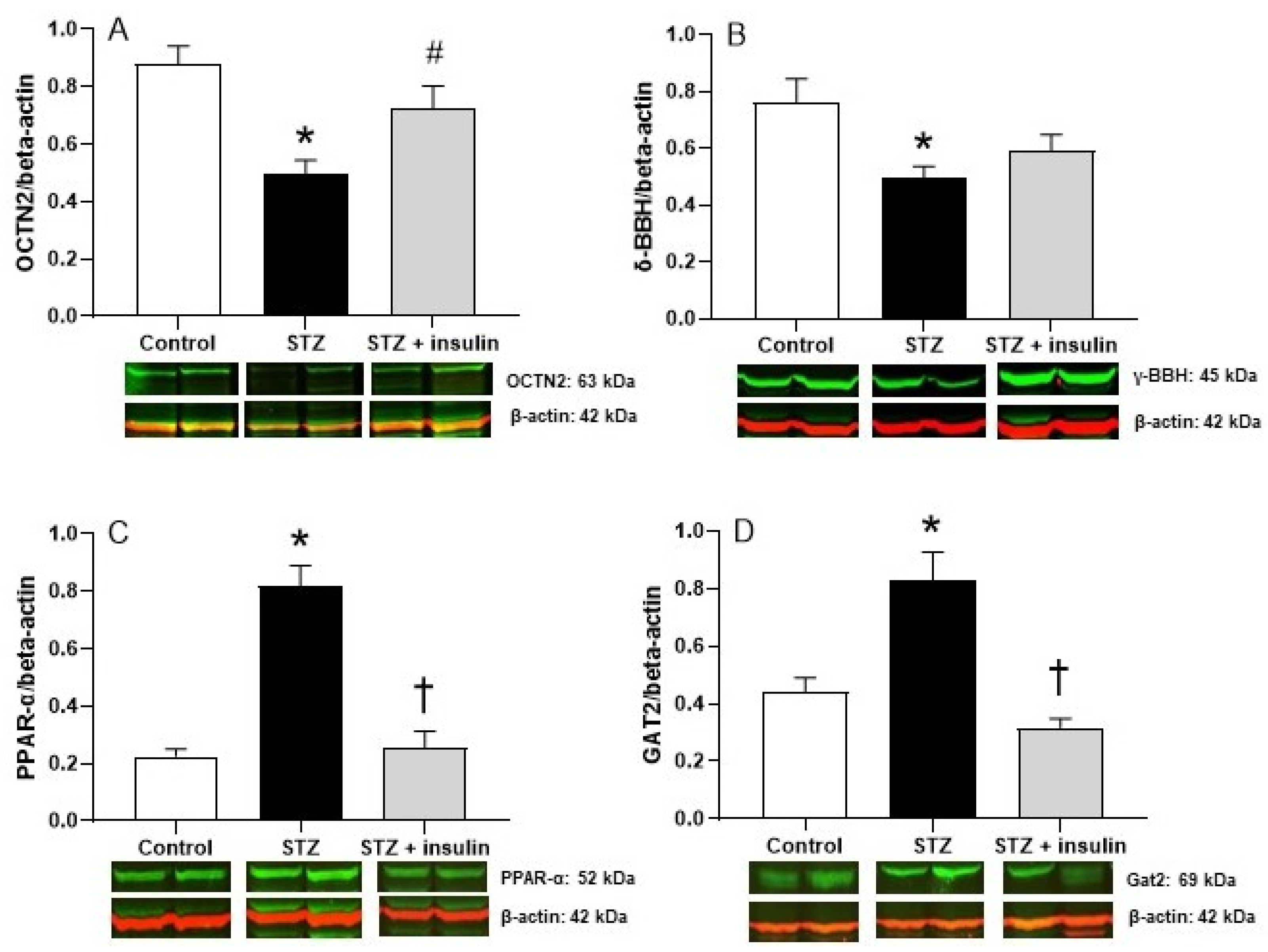
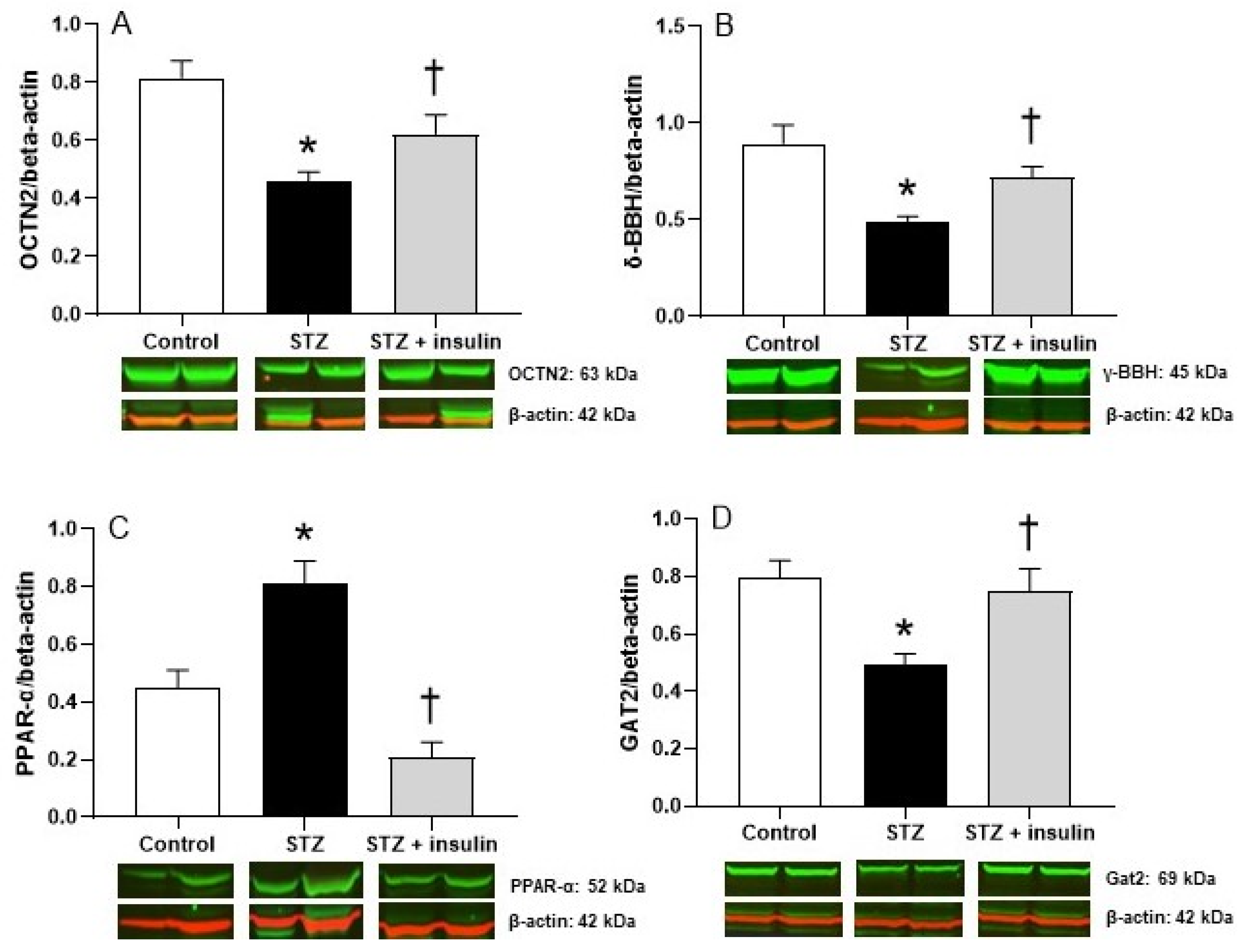
2.3. Expression of Proteins Relating to Carnitine Biosynthesis and Uptake in Kidney of Diabetic Rats
2.4. Expression of Proteins Relating to Carnitine Biosynthesis and Uptake in Liver of Diabetic Rats
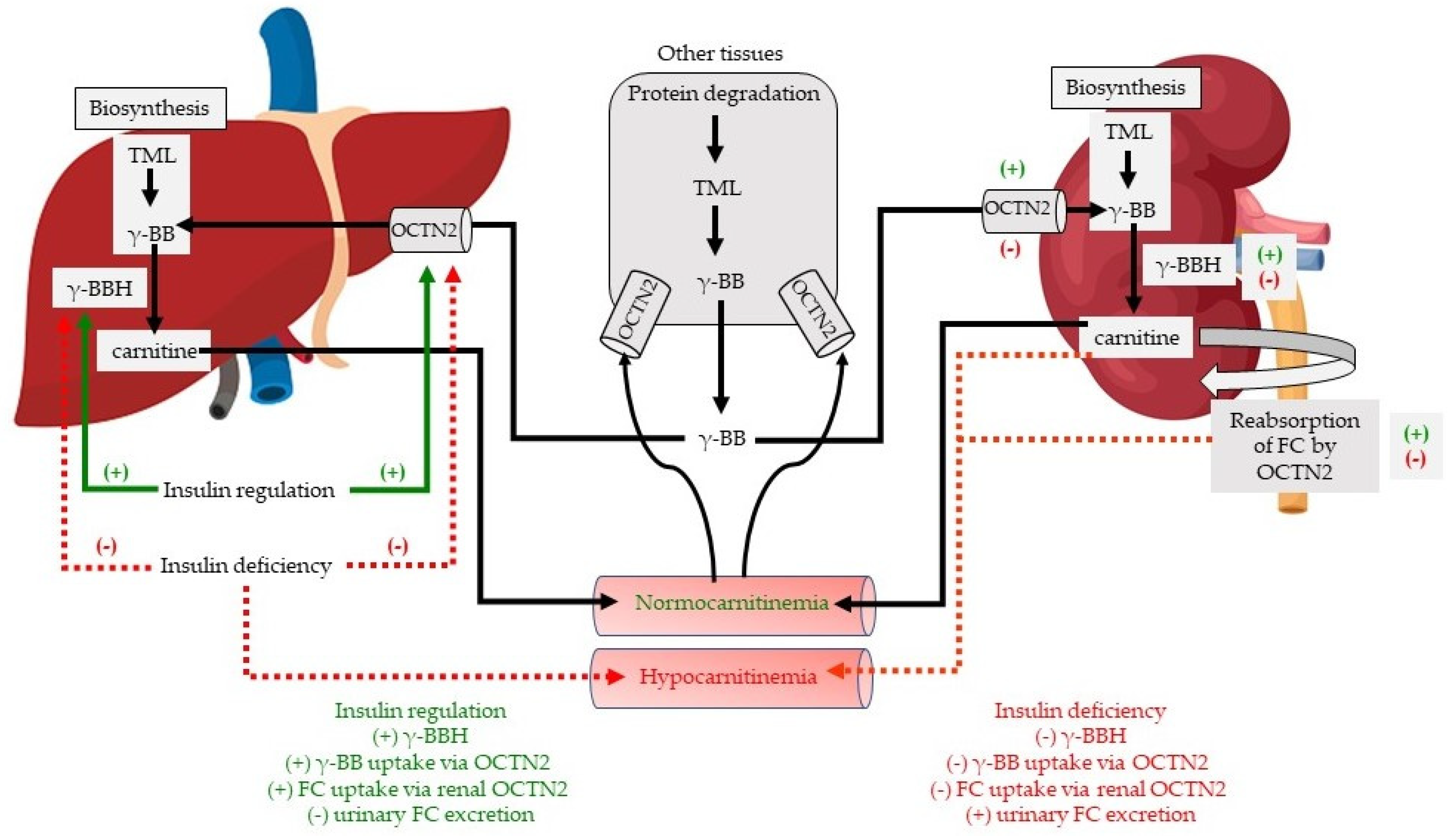
3. Discussion
4. Materials and Methods
4.1. Rat Model of Diabetes
4.2. Food and Water Intake and Urine Collection
4.3. Blood Sampling and Tissue Collection
4.4. Western Blot Analysis
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bremer, J. Carnitine metabolism and functions. Physiol. Rev. 1983, 63, 1420–1480. [Google Scholar] [CrossRef]
- Lysiak, W.; Toth, P.P.; Suelter, C.H.; Bieber, L.L. Quantitation of the efflux of acylcarnitines from rat heart, brain, and liver mitochondria. J. Biol. Chem. 1986, 261, 13698–13703. [Google Scholar] [CrossRef]
- Stanley, C.A. Carnitine Deficiency Disorders in Children. Ann. N. Y. Acad. Sci. 2004, 1033, 42–51. [Google Scholar] [CrossRef]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Frigeni, M.; Balakrishnan, B.; Yin, X.; Calderon, F.R.; Mao, R.; Pasquali, M.; Longo, N. Functional and molecular studies in primary carnitine deficiency. Hum. Mutat. 2017, 38, 1684–1699. [Google Scholar] [CrossRef]
- Cox, R.A.; Hoppel, C.L. Carnitine and trimethylaminobutyrate synthesis in rat tissues. Biochem. J. 1974, 142, 699–701. [Google Scholar] [CrossRef]
- Rebouche, C.J.; Lehman, L.J.; Olson, L. Epsilon-N-trimethyllysine availability regulates the rate of carnitine biosynthesis in the growing rat. J. Nutr. 1986, 116, 751–759. [Google Scholar] [CrossRef] [Green Version]
- Vaz, F.M.; Wanders, R.J.A. Carnitine biosynthesis in mammals. Biochem. J. 2012, 361, 417–429. [Google Scholar] [CrossRef]
- Strijbis, K.; Vaz, F.M.; Distel, B. Enzymology of the carnitine biosynthesis pathway. IUBMB Life 2010, 62, 357–362. [Google Scholar] [CrossRef]
- Cederblad, G.; Bylund, A.C.; Holm, J.; Scherstén, T. Carnitine concentration in relation to enzyme activities and substrate utilization in human skeletal muscles. Scand. J. Clin. Lab. Investig. 1976, 36, 547–552. [Google Scholar] [CrossRef]
- Hoppel, C.L.; Davis, A.T. Inter-tissue relationships in the synthesis and distribution of carnitine. Biochem. Soc. Trans. 1986, 14, 673–674. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, T.; Fukuyama, Y.; Fujita, M.; Shirasaka, Y.; Tamai, I. Carnitine precursor γ-butyrobetaine is a novel substrate of the Na(+)- and Cl(-)-dependent GABA transporter Gat2. Drug Metab. Pharmacokinet. 2011, 26, 632–636. [Google Scholar] [CrossRef] [Green Version]
- Tamai, I.; Ohashi, R.; Nezu, J.I.; Sai, Y.; Kobayashi, D.; Oku, A.; Tsuji, A. Molecular and functional characterization of organic cation/carnitine transporter family in mice. J. Biol. Chem. 2000, 275, 40064–40072. [Google Scholar] [CrossRef] [Green Version]
- De Palo, E.; Gatti, R.; Sicolo, N.; Padovan, D.; Vettor, R.; Federspil, G. Plasma and urine free L-carnitine in human diabetes mellitus. Acta Diabetol. Lat. 1981, 18, 91–95. [Google Scholar] [CrossRef]
- Cederblad, G.; Hermansson, G.; Ludvigsson, J. Plasma and urine carnitine in children with diabetes mellitus. Clin. Chim. Acta 1982, 125, 207–217. [Google Scholar] [CrossRef]
- Brooks, S.D.; Bahl, J.J.; Bressler, R. Carnitine in the streptozotocin-diabetic rat. J. Nutr. 1985, 115, 1267–1273. [Google Scholar] [CrossRef]
- Winter, S.C.; Simon, M.; Zorn, E.M.; Szabo-Aczel, S.; Vance, W.H.; O’Hara, T.; Higashi, L. Relative carnitine insufficiency in children with type I diabetes mellitus. Am. J. Dis. Child. 1989, 143, 1337–1339. [Google Scholar] [CrossRef]
- Coker, M.; Coker, C.; Darcan, S.; Can, Ş.; Orbak, Z.; Gökşen, D. Carnitine metabolism in diabetes mellitus. J. Pediatr. Endocrinol. Metab. 2002, 15, 841–949. [Google Scholar] [CrossRef]
- Mamoulakis, D.; Galanakis, E.; Dionyssopoulou, E.; Evangeliou, A.; Sbyrakis, S. Carnitine deficiency in children and adolescents with type 1 diabetes. J. Diabetes Complicat. 2004, 18, 271–274. [Google Scholar] [CrossRef]
- Soltész, G.; Melegh, B.; Sándor, A. The relationship between carnitine and ketone body levels in diabetic children. Acta Paediatr. 1983, 72, 511–515. [Google Scholar] [CrossRef]
- Genuth, S.M.; Hoppel, C.L. Plasma and urine carnitine in diabetic ketosis. Diabetes 1979, 28, 1083–1087. [Google Scholar] [CrossRef]
- Henderson, G.D.; Xue, G.P.; Snoswell, A.M. Carnitine and creatinine content of tissues of normal and alloxan-diabetic sheep and mice. Comp. Biochem. Physiol. B 1983, 76, 295–298. [Google Scholar] [CrossRef]
- Morabito, E.; Corsico, N.; Marzo, A. Serum and urine levels of levocarnitine family components in genetically diabetic rats. Arzneimittel-Forschung 1994, 44, 965–968. [Google Scholar]
- Malone, J.I.; Schocken, D.D.; Morrison, A.D.; Gilbert-Barness, E. Diabetic cardiomyopathy and carnitine deficiency. J. Diabetes Complicat. 1999, 13, 86–90. [Google Scholar] [CrossRef]
- Malone, M.A.; Schocken, D.D.; Hanna, S.K.; Liang, X.; Malone, J.I. Diabetes-induced bradycardia is an intrinsic metabolic defect reversed by carnitine. Metabolism 2007, 56, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Paulson, D.J.; Schmidt, M.J.; Traxler, J.S.; Ramacci, M.T.; Shug, A.L. Improvement of myocardial function in diabetic rats after treatment with L- carnitine. Metabolism 1984, 33, 358–363. [Google Scholar] [CrossRef]
- Broderick, T.L.; Quinney, H.A.; Lopaschuk, G.D. L-carnitine increase glucose metabolism and mechanical function following ischaemia in diabetic rat heart. Cardiovasc. Res. 1995, 29, 373–378. [Google Scholar] [CrossRef]
- Power, R.A.; Hulver, M.W.; Zhang, J.Y.; Dubois, J.; Marchand, R.M.; Ilkayeva, O.; Mynatt, R.L. Carnitine revisited: Potential use as adjunctive treatment in diabetes. Diabetologia 2007, 50, 825–832. [Google Scholar] [CrossRef]
- Noland, R.C.; Koves, T.R.; Seiler, S.E.; Lum, H.; Lust, R.M.; Ilkayeva, O.; Muoio, D.M. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J. Biol. Chem. 2009, 284, 22840–22852. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, Y.; Ueda, E.; Sakurai, H.; Kojima, Y. Anti-diabetes effect of Zn(II)/carnitine complex by oral administration. Chem. Pharm. Bull. (Tokyo) 2003, 51, 230–231. [Google Scholar] [CrossRef] [Green Version]
- Dai, T.; Abou-Rjaily, G.A.; Al-Share, Q.Y.; Yang, Y.; Fernström, M.A.; DeAngelis, A.M.; Najjar, S.M. Interaction between altered insulin and lipid metabolism in CEACAM1- inactive transgenic mice. J. Biol. Chem. 2004, 279, 45155–45161. [Google Scholar] [CrossRef] [Green Version]
- van Vlies, N.; Ferdinandusse, S.; Turkenburg, M.; Wanders, R.J.; Vaz, F.M. PPARα-activation results in enhanced carnitine biosynthesis and OCTN2-medicated hepatic carnitine accumulation. Biochim. Biophys. Acta 2007, 1767, 1134–1142. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Wakasawa, T.; Funabashi, M.; Fukushi, A.; Fujita, M.; Motojima, K.; Tamai, I. Regulation of Octn2 transporter (SLC22A5) by peroxisome proliferator activated receptor alpha. Biol. Pharm. Bull. 2008, 31, 1230–1236. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Nakanishi, T.; Shibue, Y.; Kobayashi, D.; Moseley, R.H.; Shirasaka, Y.; Tamai, I. Hepatic uptake of gamma-butyrobetaine, a precursor of carnitine biosynthesis, in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G681–G686. [Google Scholar] [CrossRef] [PubMed]
- Couturier, A.; Ringseis, R.; Most, E.; Eder, K. Pharmacological doses of niacin stimulate the expression of genes involved in carnitine uptake and biosynthesis and improve the carnitine status of obese Zucker rats. BMC Pharmacol. Toxicol. 2014, 15, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringseis, R.; Mooren, F.-C.; Keller, J.; Couturier, A.; Wen, G.; Hirche, F.; Krüger, K. Regular endurance exercise improves the diminished hepatic carnitine status in mice fed a high-fat diet. Mol. Nutr. Food Res. 2011, 55, S193–S202. [Google Scholar] [CrossRef]
- Borden, L.A.; Smith, K.E.; Hartig, P.R.; Branchek, T.A.; Weinshank, R.L. Molecular heterogeneity of the gamma-aminobutyric acid (GABA) transport system. Cloning of two novel high affinity GABA transporters from rat brain. J. Biol. Chem. 1992, 267, 21098–21104. [Google Scholar] [CrossRef]
- Stephens, F.B.; Constantin-Teodosiu, D.; Laithwaite, D.; Simpson, E.J.; Greenhaff, P.L. Insulin stimulates L-carnitine accumulation in human skeletal muscle. FASEB J. 2006, 20, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Karwi, Q.G.; Ho, K.L.; Pherwani, S.; Ketema, E.B.; Sun, Q.Y.; Lopaschuk, G.D. Concurrent diabetes and heart failure: Interplay and novel therapeutic approaches. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Luci, S.; Hirche, F.; Eder, K. Fasting and caloric restriction increase mRNA concentrations of novel organic cation transporter-2 and carnitine concentrations in rat tissues. Ann. Nutr. Metab. 2008, 52, 58–67. [Google Scholar] [CrossRef]
- Ringseis, R.; Wege, N.; Wen, G.; Rauer, C.; Hirche, F.; Kluge, H.; Eder, K. Carnitine synthesis and uptake into cells are stimulated by fasting in pigs as a model of nonproliferating species. J. Nutr. Biochem. 2009, 20, 840–847. [Google Scholar] [CrossRef]
- Broderick, T.L.; El Midaoui, A.; Chiasson, J.L.; Wang, D.; Jankowski, M.; Gutkowska, J. The effects of exercise training on γ-butyrobetaine hydroxylase and novel organic cation transporter-2 gene expression in the rat. Appl. Physiol. Nutr. Metab. 2011, 36, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Makowski, L.; Noland, R.C.; Koves, T.R.; Xing, W.; Ilkayeva, O.R.; Muehlbauer, M.J.; Muoio, D.M. Metabolic profiling of PPARalpha−/− mice reveals defects in carnitine and amino acid homeostasis that are partially reversed by oral carnitine supplementation. FASEB J. 2009, 23, 586–604. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, A.; Al-Nakkash, L.; Broderick, T.L. Effects of exercise training on renal carnitine biosynthesis and uptake in the high fat-high sugar fed mouse. Molecules 2020, 25, 2100. [Google Scholar] [CrossRef] [PubMed]
- Brass, E.P.; Hoppel, C.L. Carnitine metabolism in the fasting rat. J. Biol. Chem. 1978, 253, 2688–2693. [Google Scholar] [CrossRef]
- Frohlich, J.; Seccombe, D.W.; Hahn, P.; Dodek, P.; Hynie, I. Effect of fasting on free and esterified carnitine levels in human serum and urine: Correlation with serum levels of free fatty acids and beta-hydroxybutyrate. Metabolism 1978, 27, 555–561. [Google Scholar] [CrossRef]
- Stearns, S.B. Carnitine content of skeletal muscle from diabetic and insulin-treated diabetic rats. Biochem. Med. 1980, 24, 33–38. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Control | STZ-Induced Diabetes | STZ + Insulin |
|---|---|---|---|
| Body weight (g) Fat weight (g) Kidney weight (g) Liver weight (g) Plasma insulin (ng/mL) Plasma glucose (nmol/L) Plasma NEFA (µmol/L) Food intake (g) Water intake (mL) Urine output (mL) | 423 ± 12 12.36 ± 1.03 3.08 ± 0.09 12.51 ± 0.65 1.42 ± 0.18 5.45 ± 0.75 433 ± 11 28.6 ± 2.1 42.9 ± 2.5 12.5 ± 3.3 | 250 ± 14 * 2.61 ± 0.70 * 4.90 ± 1.07 12.60 ± 1.07 0.42 ± 0.03 * 9.37 ± 0.78 * 481 ± 16 * 52.8 ± 3.9 * 257.2 ± 20.8 * 71.9 ± 2.1 * | 410 ± 17 † 10.82 ± 1.50 † 3.54 ± 1.49 15.76 ± 1.73 0.72 ± 0.05 *,† 5.20 ± 0.25 † 445 ± 9 † 37.2 ± 5.3 † 111.7 ± 19.5 † 12.8 ± 2.2 † |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Upadhyay, A.; Boyle, K.E.; Broderick, T.L. The Effects of Streptozotocin-Induced Diabetes and Insulin Treatment on Carnitine Biosynthesis and Renal Excretion. Molecules 2021, 26, 6872. https://doi.org/10.3390/molecules26226872
Upadhyay A, Boyle KE, Broderick TL. The Effects of Streptozotocin-Induced Diabetes and Insulin Treatment on Carnitine Biosynthesis and Renal Excretion. Molecules. 2021; 26(22):6872. https://doi.org/10.3390/molecules26226872
Chicago/Turabian StyleUpadhyay, Aman, Kate E. Boyle, and Tom L. Broderick. 2021. "The Effects of Streptozotocin-Induced Diabetes and Insulin Treatment on Carnitine Biosynthesis and Renal Excretion" Molecules 26, no. 22: 6872. https://doi.org/10.3390/molecules26226872
APA StyleUpadhyay, A., Boyle, K. E., & Broderick, T. L. (2021). The Effects of Streptozotocin-Induced Diabetes and Insulin Treatment on Carnitine Biosynthesis and Renal Excretion. Molecules, 26(22), 6872. https://doi.org/10.3390/molecules26226872