
The Reversible Carnitine Palmitoyltransferase 1 Inhibitor (Teglicar) Ameliorates the Neurodegenerative Phenotype in a Drosophila Huntington’s Disease Model by Acting on the Expression of Carnitine-Related Genes

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
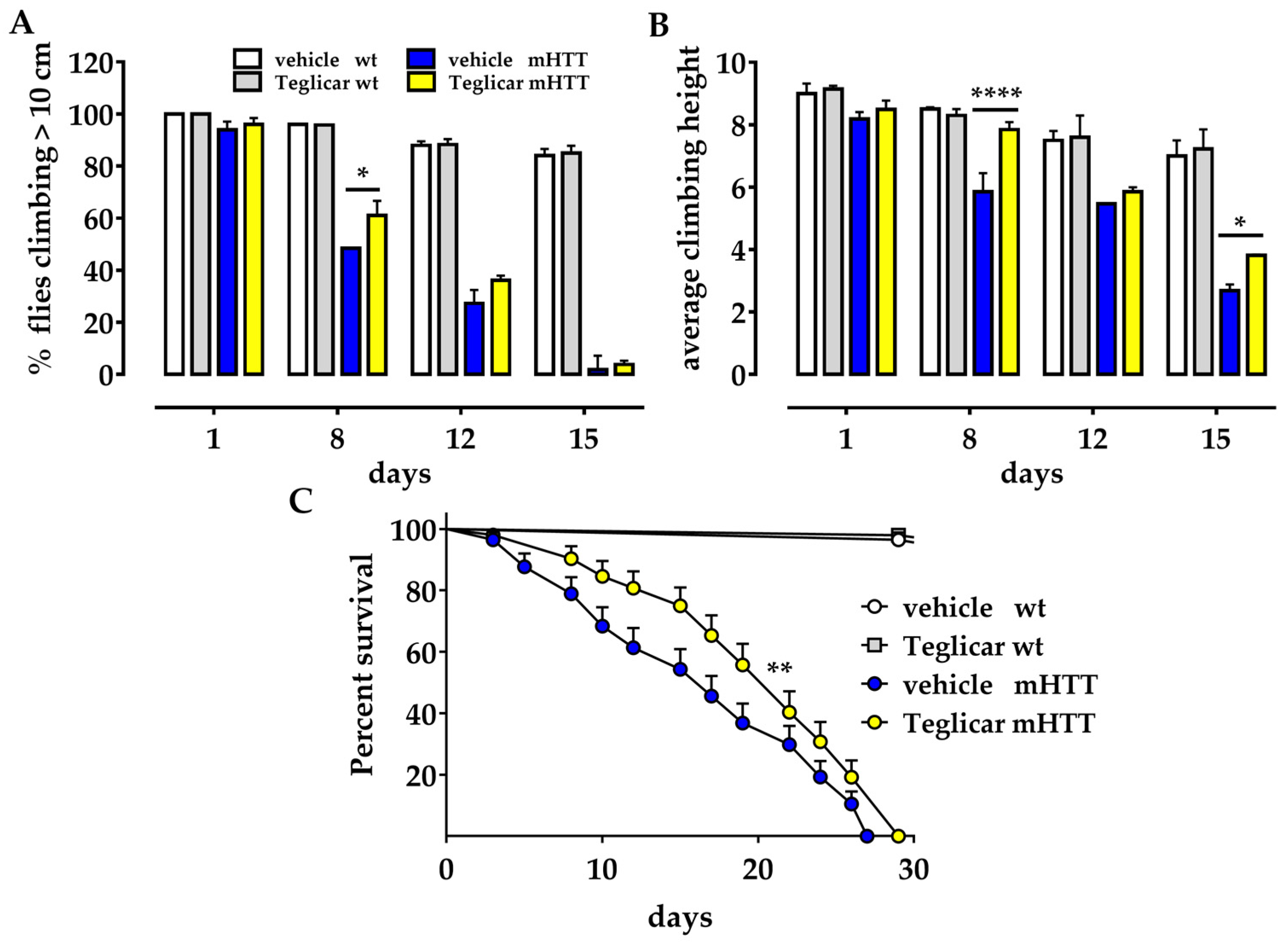
2.1. Pharmacological Inhibition of CPT1 Alleviates Locomotor Defects in the Huntington’s Drosophila Model
2.2. Pharmacological Inhibition of CPT1 Increases Survival in the Huntington’s Drosophila Model
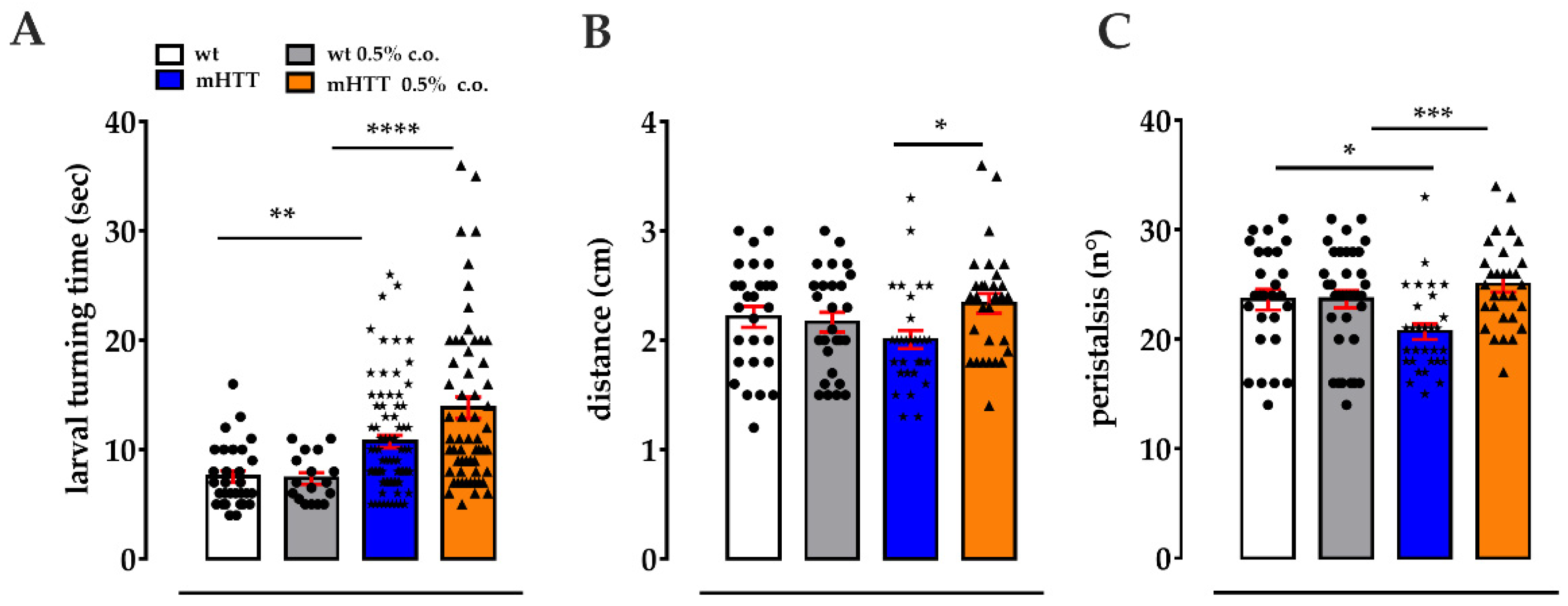
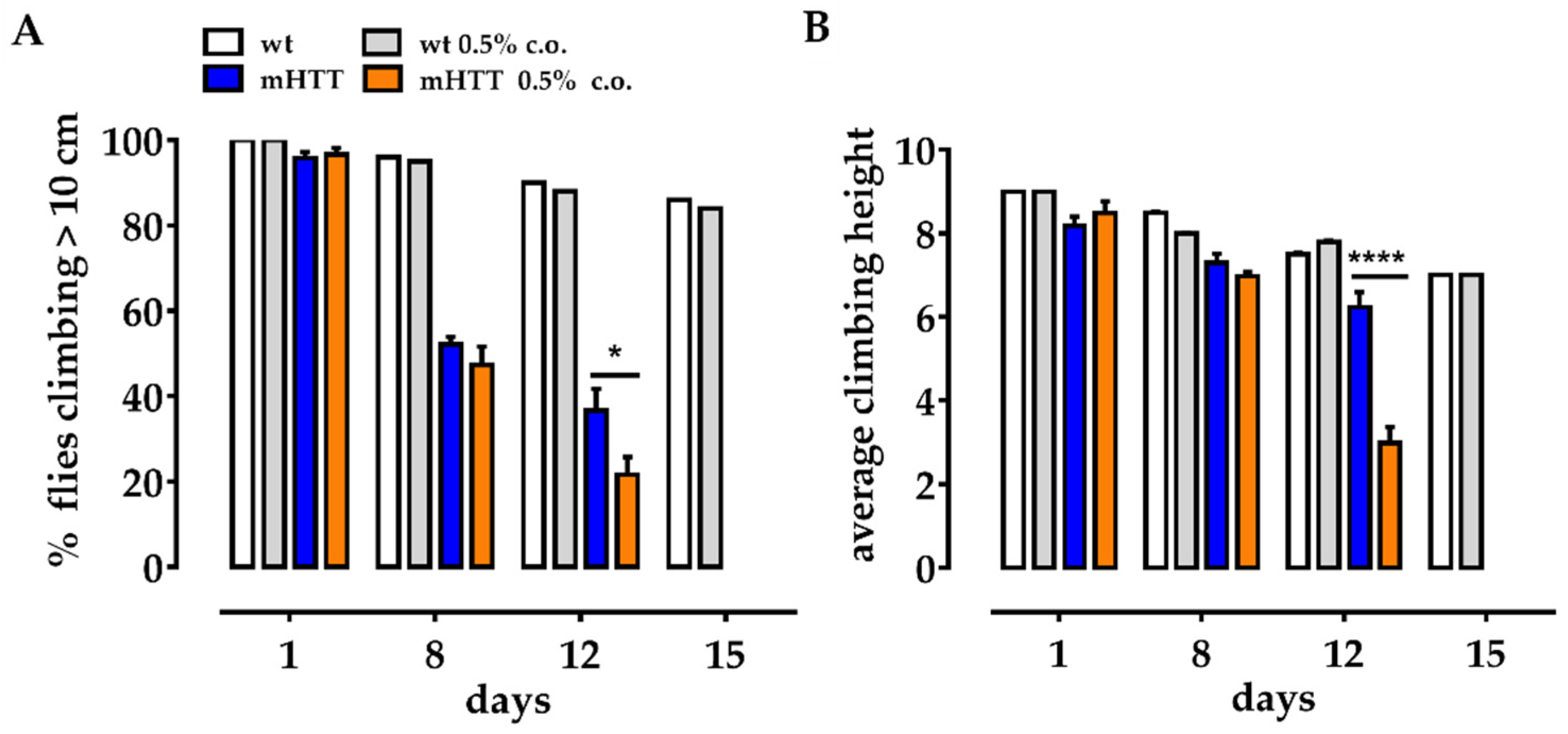
2.3. Dietary Supplementation with Medium-Chain Fatty Acids Affects the Locomotion of Huntington’s Larvae
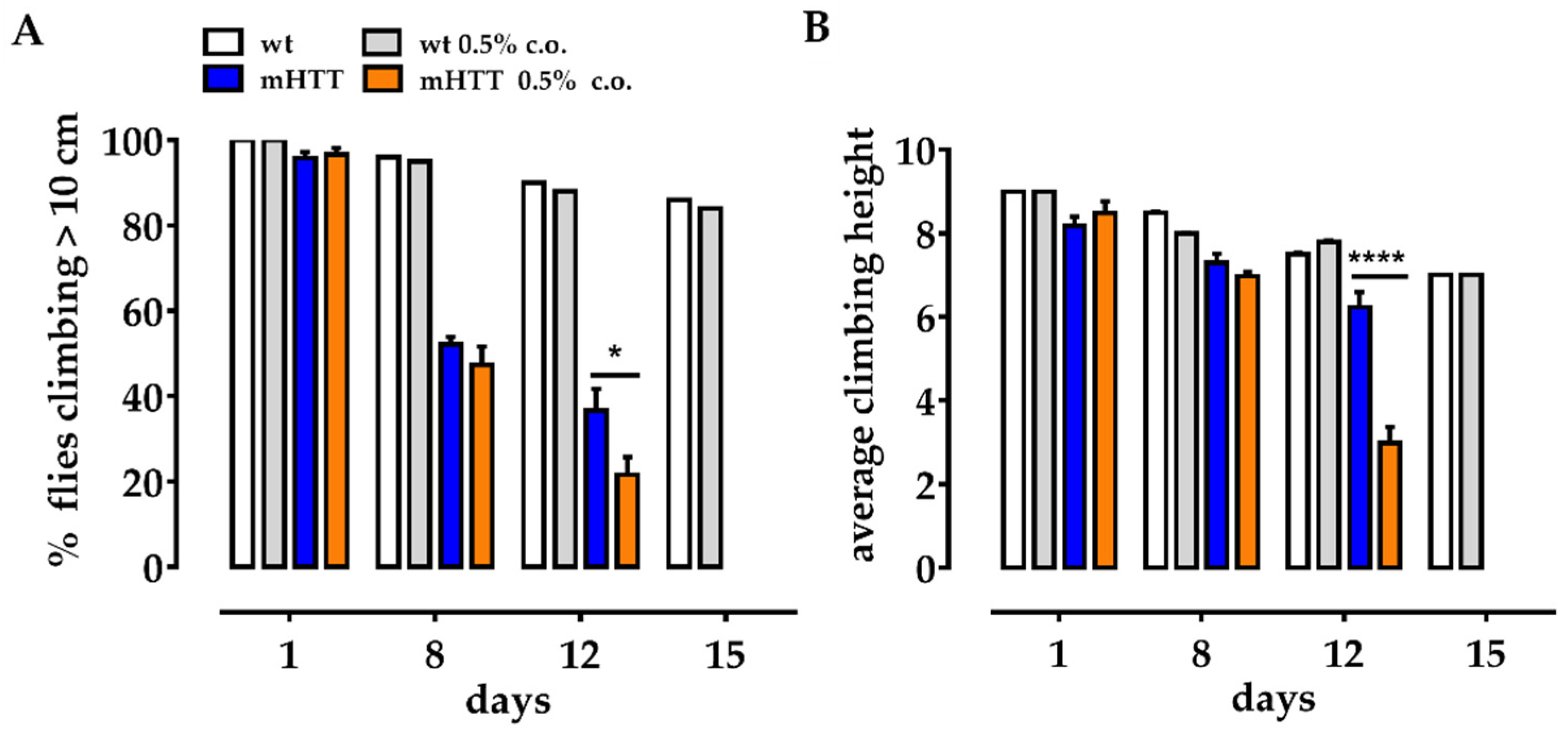
2.4. Dietary Supplementation with Medium-Chain Fatty Acids Worsens the Climbing Ability of mHTT Flies
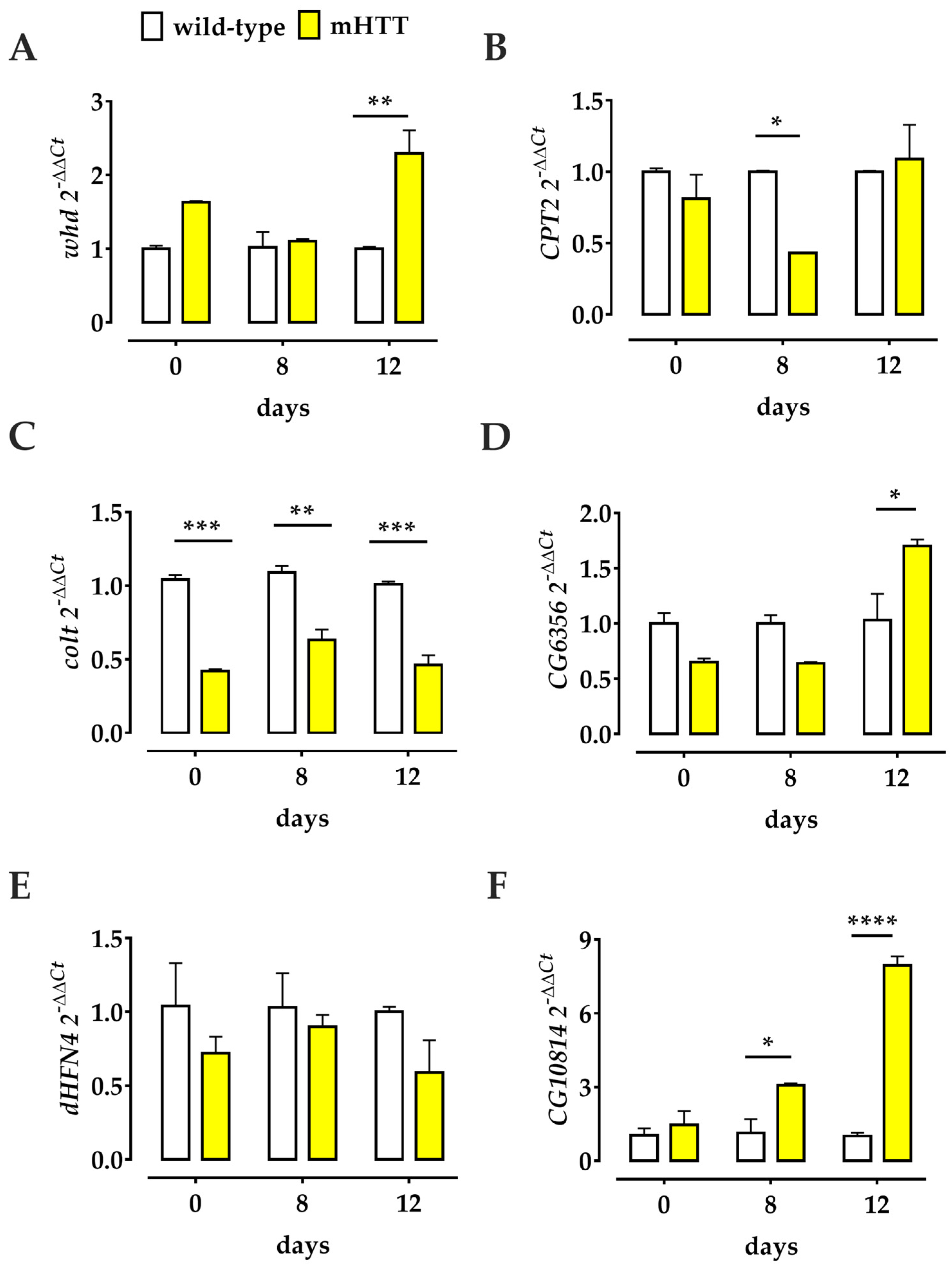
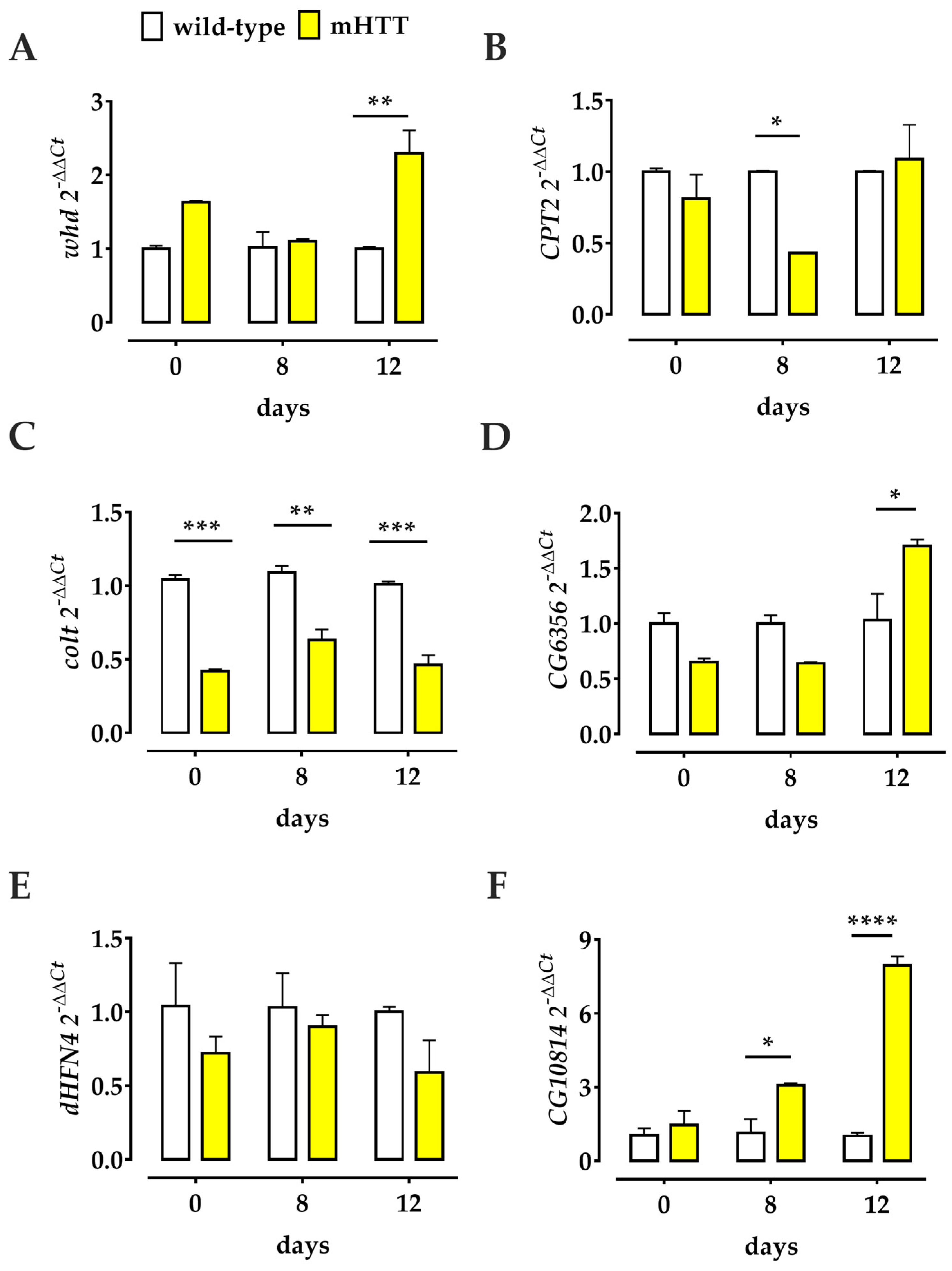
2.5. Transcriptional Profiles of Several Genes Involved in the Drosophila L-Carnitine Cycle Are Deregulated in Q128HD-FL Flies
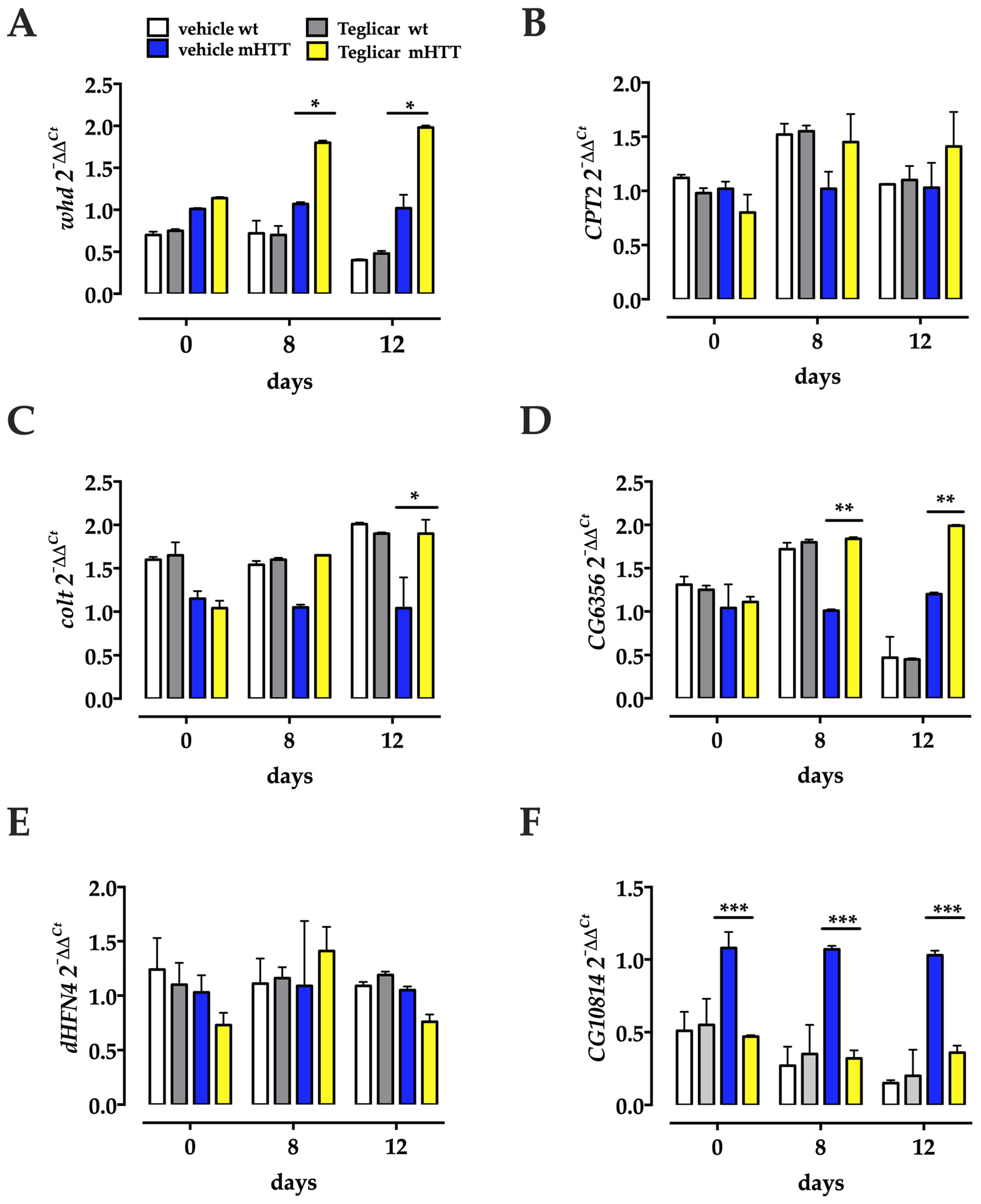
2.6. Pharmacological Inhibition of CPT1 Activity Affects Both Component Genes of the CS and the Transcriptional Profiles of CG6356 and CG10814
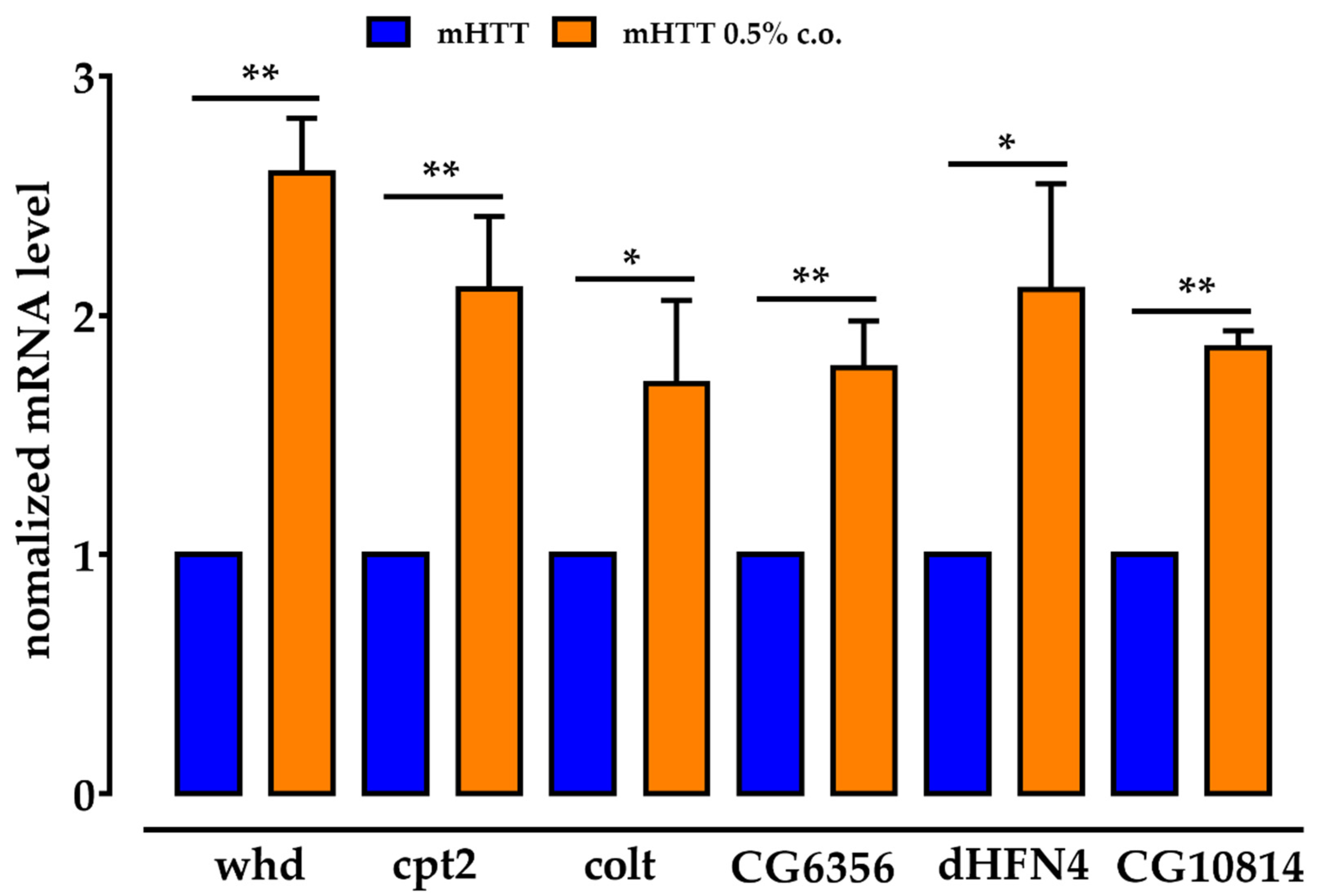
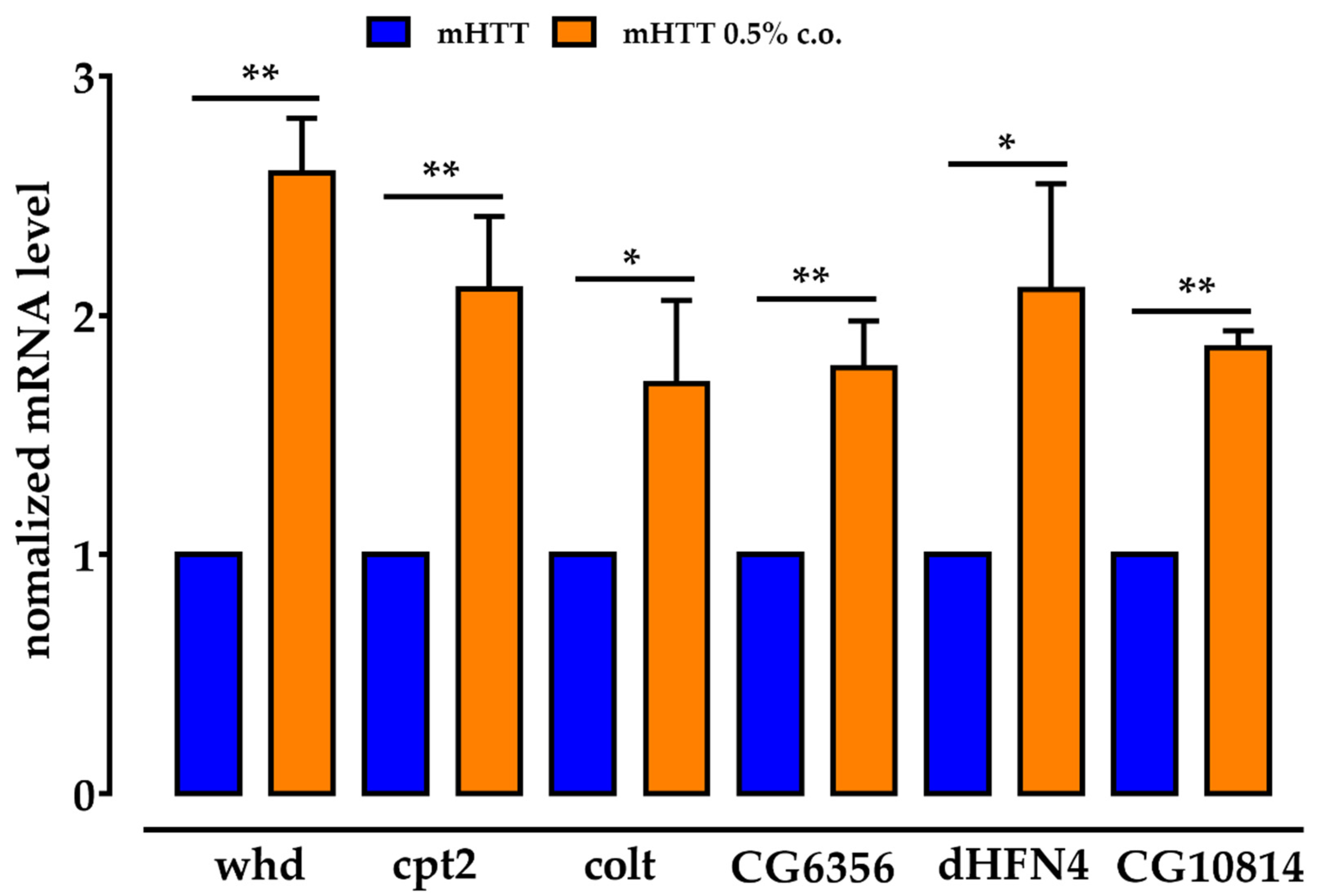
2.7. The Genes for the CS Components Are Upregulated by Dietary Supplementation with Medium-Chain Fatty Acids, as Are CG6356, dHNF4 and CG10814
3. Discussions
4. Materials and Methods
4.1. Drosophila Stocks and Crosses
4.2. Teglicar Treatment
4.3. Climbing Assay
4.4. Lifespan Analysis
4.5. Medium-Chain FAs Fly Food Supplementation
4.6. Larval Turning
4.7. Larval Crawling
4.8. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tabrizi, S.J.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Agerelated Diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Koyuncu, S.; Fatima, A.; Gutierrez-Garcia, R.; Vilchez, D. Proteostasis of huntingtin in health and disease. Int. J. Mol. Sci. 2017, 18, 1568. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Huang, B.; Zhang, B.; Xiang, Y.; Du, Z.; Xu, Q.; Li, Y.; Wang, Q.; Ma, J.; Peng, X.; et al. Resetting Epigenetic Memory by Reprogramming of Histone Modifications in Mammals. Mol. Cell 2016, 63, 1066–1079. [Google Scholar] [CrossRef] [Green Version]
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Saudou, F.; Humbert, S. Review The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Dickey, A.S.; Spada, A.R. Therapy development in Huntington disease: From current strategies to emerging opportunities. Am. J. Med. Genet. A 2019, 176, 842–861. [Google Scholar] [CrossRef]
- Chen, J.Y.; Tran, C.; Hwang, L.; Deng, G.; Jung, M.E.; Faull, K.F.; Levine, M.S.; Cepeda, C. Partial Amelioration of Peripheral and Central Symptoms of Huntington’s Disease via Modulation of Lipid Metabolism. J. Huntingt. Dis. 2016, 5, 65–81. [Google Scholar] [CrossRef] [Green Version]
- Block, R.C.; Dorsey, E.R.; Beck, C.A.; Thomas, J.; Shoulson, I. Altered cholesterol and fatty acid metabolism in Huntington disease. J. Clin. Lipidol. 2011, 4, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Nambron, R.; Silajd, E.; Kalliolia, E.; Ottolenghi, C.; Hindmarsh, P.; Hill, N.R.; Costelloe, S.J.; Martin, N.G.; Positano, V.; Watt, H.C.; et al. A Metabolic Study of Huntington’s Disease. PLoS ONE 2016, 11, e0146480. [Google Scholar] [CrossRef] [Green Version]
- Liot, G.; Valette, J.; Pépin, J.; Flament, J.; Brouillet, E. Energy defects in Huntington’s disease: Why “in vivo” evidence matters. Biochem. Biophys. Res. Commun. 2017, 483, 1084–1095. [Google Scholar] [CrossRef]
- Mochel, F.; Haller, R.G. Energy deficit in Huntington disease: Why it matters. J. Clin. Investig. 2011, 121, 493–499. [Google Scholar] [CrossRef]
- Di Cristo, F.; Finicelli, M.; Digilio, F.A.; Paladino, S.; Valentino, A.; Scialò, F.; D’Apolito, M.; Saturnino, C.; Galderisi, U.; Giordano, A.; et al. Meldonium improves Huntington’s disease mitochondrial dysfunction by restoring peroxisome proliferator-activated receptor γ coactivator 1α expression. J. Cell. Physiol. 2019, 234, 9233–9246. [Google Scholar] [CrossRef]
- Mehrotra, A.; Sood, A.; Sandhir, R. Mitochondrial modulators improve lipid composition and attenuate memory deficits in experimental model of Huntington’s disease. Mol. Cell. Biochem. 2015, 410, 281–292. [Google Scholar] [CrossRef]
- Kreilaus, F.; Mclean, C.A.; Garner, B.; Jenner, A.M. Evidence for altered cholesterol metabolism in Huntington’s disease post mortem brain tissue. Neuropathol. Appl. Neurobiol. 2016, 42, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Console, L.; Giangregorio, N.; Indiveri, C.; Tonazzi, A. Carnitine/acylcarnitine translocase and carnitine palmitoyltransferase 2 form a complex in the inner mitochondrial membrane. Mol. Cell. Biochem. 2014, 394, 307–314. [Google Scholar] [CrossRef]
- Carillo, M.R.; Bertapelle, C.; Scialò, F.; Siervo, M.; Spagnuolo, G.; Simeone, M.; Peluso, G.; Digilio, F.A. L-Carnitine in Drosophila: A Review. Antioxidants 2020, 9, 1310. [Google Scholar] [CrossRef]
- Longo, N.; Frigeni, M.; Pasquali, M.; Biophys, B.; Author, A. Carnitine transport and fatty acid oxidation hhs public access author manuscript. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- El-Gharbawy, A.; Vockley, J. Inborn Errors of Metabolism with Myopathy: Defects of Fatty Acid Oxidation and the Carnitine Shuttle System. Pediatric Clin. N. Am. 2018, 65, 317–335. [Google Scholar] [CrossRef]
- Pacilli, A.; Calienni, M.; Margarucci, S.; D’Apolito, M.; Petillo, O.; Rocchi, L.; Pasquinelli, G.; Nicolai, R.; Koverech, A.; Calvani, M.; et al. Carnitine-acyltransferase system inhibition, cancer cell death, and prevention of myc-induced lymphomagenesis. J. Natl. Cancer Inst. 2013, 105, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, M.R.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foà, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the leukemia cell metabolism by the CPT1a inhibition: Functional preclinical effects in leukemias. Blood 2015, 126, 1925–1929. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Wang, G.; Wang, Y.; Cai, L.; Qian, K.; Ju, L.; Liu, X.; Xiao, Y.; Wang, X. Fatty acid oxidation inhibitor etomoxir suppresses tumor progression and induces cell cycle arrest via PPARγ-mediated pathway in bladder cancer. Clin. Sci. 2019, 133, 1745–1758. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Cui, F.; Meng, L.; Chen, G.; Li, Z.; Xu, Y.; Ma, Z.; Woldegiorgis, G. Carnitine Palmitoyltransferase Inhibitor in Diabetes. J. Mol. Genet. Med. 2016, 10, 238. [Google Scholar] [CrossRef]
- Vamos, E.; Voros, K.; Vecsei, L.; Klivenyi, P. Neuroprotective effects of L-carnitine in a transgenic animal model of Huntington’s disease. Biomed. Pharmacother. 2010, 64, 282–286. [Google Scholar] [CrossRef]
- Lee, J.; Kosaras, B.; Del Signore, S.J.; Cormier, K.; McKee, A.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 2011, 121, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Dubinsky, J.M. Towards an Understanding of Energy Impairment in Huntington’s Disease Brain. J. Huntingt. Dis. 2017, 6, 267–302. [Google Scholar] [CrossRef] [Green Version]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Mcgurk, L.; Berson, A.; Bonini, N.M. Drosophila as an In Vivo Model for Human Neurodegenerative Disease. Genetics 2015, 201, 377–402. [Google Scholar] [CrossRef] [Green Version]
- Aditi, K.; Shakarad, M.N.; Agrawal, N. Altered lipid metabolism in Drosophila model of Huntington’s disease. Sci. Rep. 2016, 6, 31411. [Google Scholar] [CrossRef]
- Singh, A.; Agrawal, N. Deciphering the key mechanisms leading to alteration of lipid metabolism in Drosophila model of Huntington’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2021, 1867, 166127. [Google Scholar] [CrossRef] [PubMed]
- Strub, B.R.; Parkes, T.L.; Mukai, S.T.; Bahadorani, S.; Coulthard, A.B.; Hall, N.; Phillips, J.P.; Hilliker, A.J. Mutations of the withered (whd) gene in Drosophila melanogaster confer hypersensitivity to oxidative stress and are lesions of the carnitine palmitoyltransferase I (CPT I) gene. Genome 2008, 51, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.G.; Laranjeira, A.; Van Huffel, L.; Gärtner, A.; Vilain, S.; Bastianen, J.; Van Veldhoven, P.P.; Dotti, C.G. Glial β-Oxidation regulates drosophila energy metabolism. Sci. Rep. 2015, 5, 7805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oey, N.A.; IJlst, L.; Van Roermund, C.W.T.; Wijburg, F.A.; Wanders, R.J.A. dif-1 and colt, both implicated in early embryonic development, encode carnitine acylcarnitine translocase. Mol. Genet. Metab. 2005, 85, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Engelhart, D.C.; Azad, P.; Ali, S.; Granados, J.C.; Haddad, G.G.; Nigam, S.K. Drosophila SLC22 orthologs related to OATs, OCTs, and OCTNs regulate development and responsiveness to oxidative stress. Int. J. Mol. Sci. 2020, 21, 2002. [Google Scholar] [CrossRef] [Green Version]
- Laranjeira, A.; Schulz, J.; Dotti, C.G. Genes related to fatty acid β-oxidation play a role in the functional decline of the drosophila brain with age. PLoS ONE 2016, 11, e0161143. [Google Scholar] [CrossRef]
- Palanker, L.; Tennessen, J.M.; Lam, G.; Thummel, C.S. Drosophila HNF4 Regulates Lipid Mobilization and β-Oxidation. Cell Metab. 2009, 9, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Heier, C.; Kühnlein, R.P. Triacylglycerol metabolism in drosophila melanogaster. Genetics 2018, 210, 1163–1184. [Google Scholar] [CrossRef] [Green Version]
- Violante, S.; IJlst, L.; Te Brinke, H.; De Almeida, I.T.; Wanders, R.J.A.; Ventura, F.V.; Houten, S.M. Carnitine palmitoyltransferase 2 and carnitine/acylcarnitine translocase are involved in the mitochondrial synthesis and export of acylcarnitines. FASEB J. 2013, 27, 2039–2044. [Google Scholar] [CrossRef]
- Rinaldo, P.; Matern, D.; Bennett, M.J. Fatty acid oxidation disorders. Annu. Rev. Physiol. 2002, 64, 477–502. [Google Scholar] [CrossRef]
- Picao-osorio, J.; Lago-baldaia, I.; Patraquim, P.; Alonso, C.R. Pervasive Behavioral Effects of MicroRNA Regulation in Drosophila. Genetics 2017, 206, 1535–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, M. Fundamental Approaches to Screen Abnormalities in Drosophila (Springer Protocols Handbooks), 1st ed.; Mishra, M., Ed.; Springer: New York, NY, USA, 2020; ISBN 9781493997558. [Google Scholar]
- Estes, P.S.; Daniel, S.G.; Mccallum, A.P.; Boehringer, A.V.; Sukhina, A.S.; Zwick, R.A. Motor neurons and glia exhibit specific individualized responses to TDP-43 expression in a Drosophila model of amyotrophic lateral sclerosis. Dis. Models Mech. 2013, 733, 721–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cristo, F.; Calarco, A.; Digilio, F.A.; Sinicropi, M.S.; Rosano, C.; Galderisi, U.; Melone, M.A.B.; Saturnino, C.; Peluso, G. The discovery of highly potent thp derivatives as octn2 inhibitors: From structure-based virtual screening to in vivo biological activity. Int. J. Mol. Sci. 2020, 21, 7431. [Google Scholar] [CrossRef] [PubMed]
- Legan, S.K.; Rebrin, I.; Mockett, R.J.; Radyuk, S.N.; Klichko, V.I.; Sohal, R.S.; Orr, W.C. Overexpression of glucose-6-phosphate dehydrogenase extends the life span of Drosophila melanogaster. J. Biol. Chem. 2008, 283, 32492–32499. [Google Scholar] [CrossRef] [Green Version]
- Besson, M.T.; Alegría, K.; Garrido-Gerter, P.; Barros, L.F.; Liévens, J.C. Enhanced neuronal glucose transporter expression reveals metabolic choice in a HD Drosophila model. PLoS ONE 2015, 10, e0118765. [Google Scholar] [CrossRef] [Green Version]
- Browne, S.E.; Beal, M.F. The Energetics of Huntington’s Disease. Neurochem. Res. 2004, 29, 531–546. [Google Scholar] [CrossRef]
- Manzo, E.; O’Conner, A.G.; Barrows, J.M.; Shreiner, D.D.; Birchak, G.J.; Zarnescu, D.C. Medium-chain fatty acids, beta-hydroxybutyric acid and genetic modulation of the carnitine shuttle are protective in a drosophila model of ALS based on TDP-43. Front. Mol. Neurosci. 2018, 11, 182. [Google Scholar] [CrossRef]
- Wolfgang, W.J.; Miller, T.W.; Webster, J.M.; Huston, J.S.; Thompson, L.M.; Marsh, J.L.; Messer, A. Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2005, 98, 4764–4769. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.; Heidari, R.; Monnier, V.; Tricoire, H. Genetic screen in adult drosophila reveals that dCBP depletion in glial cells mitigates huntington disease pathology through a foxo-dependent pathway. Int. J. Mol. Sci. 2021, 22, 3884. [Google Scholar] [CrossRef]
- Chongtham, A.; Barbaro, B.; Filip, T.; Syed, A.; Huang, W.; Smith, M.R.; Marsh, J.L. Nonmammalian models of Huntington’s disease. Methods Mol. Biol. 2018, 1780, 75–96. [Google Scholar] [CrossRef]
- Trabjerg, M.S.; Mørkholt, A.S.; Lichota, J.; Oklinski, M.K.E.; Andersen, D.C.; Jønsson, K.; Mørk, K.; Skjønnemand, M.L.N.; Kroese, L.J.; Pritchard, C.E.J.; et al. Dysregulation of metabolic pathways by carnitine palmitoyl-transferase 1 plays a key role in central nervous system disorders: Experimental evidence based on animal models. Sci. Rep. 2020, 10, 15583. [Google Scholar] [CrossRef] [PubMed]
- Mørkholt, A.S.; Oklinski, M.K.; Larsen, A.; Bockermann, I.D.R.; Issazadeh-Navikas, S.; Goller, J.; Nieland, K.; Kwon, T.-H.; Corthals Id, A.; Nielsen, S.; et al. Pharmacological inhibition of carnitine palmitoyl transferase 1 inhibits and reverses experimental autoimmune encephalitis in rodents. PLoS ONE 2020, 15, e0234493. [Google Scholar] [CrossRef] [PubMed]
- Mørkholt, A.S.; Wiborg, O.; Nieland, J.G.K.; Nielsen, S.; Nieland, J.D. Blocking of carnitine palmitoyl transferase 1 potently reduces stress-induced depression in rat highlighting a pivotal role of lipid metabolism. Sci. Rep. 2017, 7, 2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, E.D.; Andrus, P.K.; Oostveen, J.A.; Fleck, T.J.; Gurney, M.E. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. J. Neurosci. Res. 1998, 53, 66–77. [Google Scholar] [CrossRef]
- Oyamada, R.; Hayashi, M.; Katoh, Y.; Tsuchiya, K.; Mizutani, T.; Tominaga, I.; Kashima, H. Neurofibrillary tangles and deposition of oxidative products in the brain in cases of myotonic dystrophy. Neuropathology 2006, 26, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Rosenstock, T.R.; Duarte, A.I.; Cristina Rego, A. Mitochondrial-Associated Metabolic Changes and Neurodegeneration in Huntingtons Disease—From Clinical Features to the Bench. Curr. Drug Targets 2012, 11, 1218–1236. [Google Scholar] [CrossRef]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional regulation of metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef]
- van der Knaap, J.A.; Verrijzer, C.P. Undercover: Gene control by metabolites and metabolic enzymes. Genes Dev. 2016, 30, 2345–2369. [Google Scholar] [CrossRef] [Green Version]
- Lal, J.J.; Sreeranjit Kumar, C.V.; Indira, M. Coconut Palm. Encycl. Food Sci. Nutr. 2003, 1, 1464–1475. [Google Scholar] [CrossRef]
- Bravo-Ruiz, I.; Medina, M.Á.; Martínez-Poveda, B. From food to genes: Transcriptional regulation of metabolism by lipids and carbohydrates. Nutrients 2021, 13, 1513. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Bovier, T.F.; Cavaliere, D.; Colombo, M.; Peluso, G.; Giordano, E.; Digilio, F.A. Methods to test endocrine disruption in drosophila melanogaster. J. Vis. Exp. 2019, 2019, e59535. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertapelle, C.; Carillo, M.R.; Cacciola, N.A.; Shidlovskii, Y.V.; Peluso, G.; Digilio, F.A. The Reversible Carnitine Palmitoyltransferase 1 Inhibitor (Teglicar) Ameliorates the Neurodegenerative Phenotype in a Drosophila Huntington’s Disease Model by Acting on the Expression of Carnitine-Related Genes. Molecules 2022, 27, 3125. https://doi.org/10.3390/molecules27103125
Bertapelle C, Carillo MR, Cacciola NA, Shidlovskii YV, Peluso G, Digilio FA. The Reversible Carnitine Palmitoyltransferase 1 Inhibitor (Teglicar) Ameliorates the Neurodegenerative Phenotype in a Drosophila Huntington’s Disease Model by Acting on the Expression of Carnitine-Related Genes. Molecules. 2022; 27(10):3125. https://doi.org/10.3390/molecules27103125
Chicago/Turabian StyleBertapelle, Carla, Maria Rosaria Carillo, Nunzio Antonio Cacciola, Yulii V. Shidlovskii, Gianfranco Peluso, and Filomena Anna Digilio. 2022. "The Reversible Carnitine Palmitoyltransferase 1 Inhibitor (Teglicar) Ameliorates the Neurodegenerative Phenotype in a Drosophila Huntington’s Disease Model by Acting on the Expression of Carnitine-Related Genes" Molecules 27, no. 10: 3125. https://doi.org/10.3390/molecules27103125
APA StyleBertapelle, C., Carillo, M. R., Cacciola, N. A., Shidlovskii, Y. V., Peluso, G., & Digilio, F. A. (2022). The Reversible Carnitine Palmitoyltransferase 1 Inhibitor (Teglicar) Ameliorates the Neurodegenerative Phenotype in a Drosophila Huntington’s Disease Model by Acting on the Expression of Carnitine-Related Genes. Molecules, 27(10), 3125. https://doi.org/10.3390/molecules27103125