
Compression Behavior and Vibrational Properties of New Energetic Material LLM-105 Analyzed Using the Dispersion-Corrected Density Functional Theory


Abstract
:1. Introduction
2. Results and Discussion
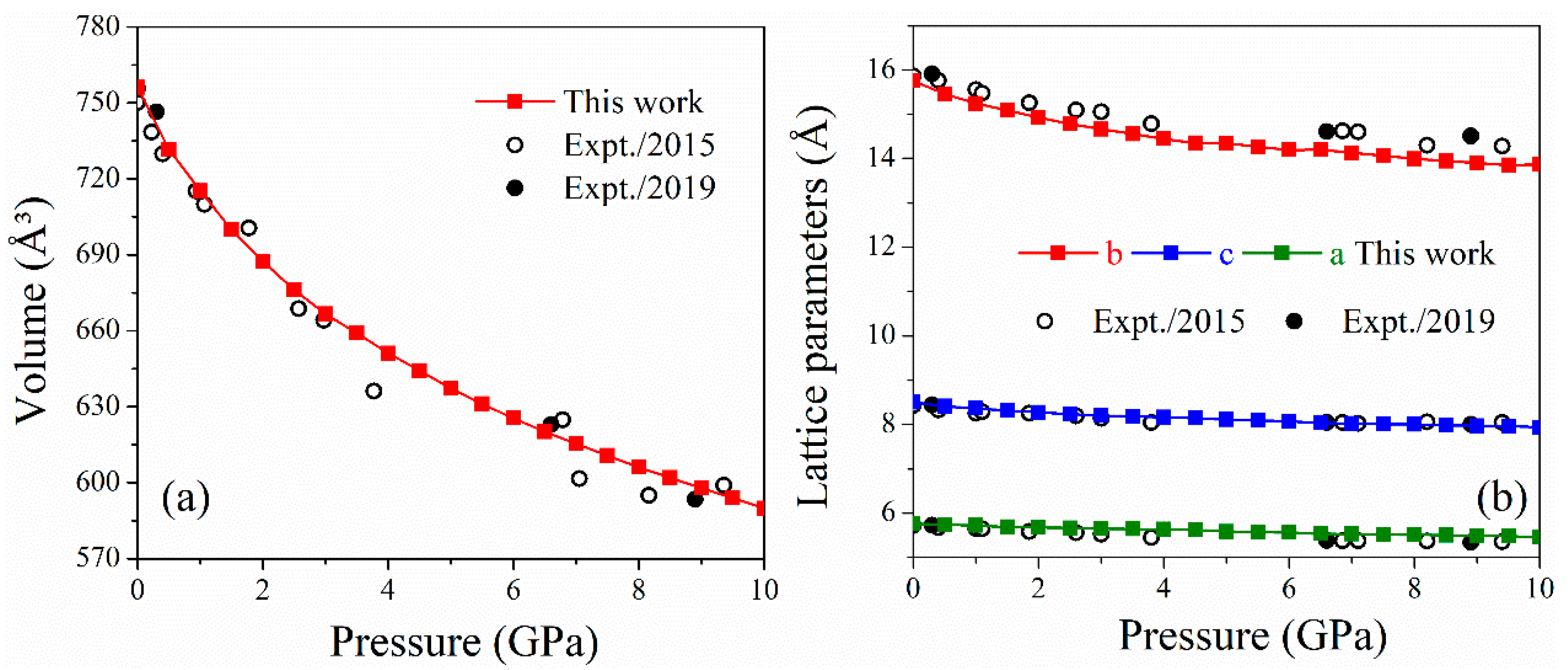
2.1. Lattice Parameters of LLM-105 at Ambient and Pressure Conditions
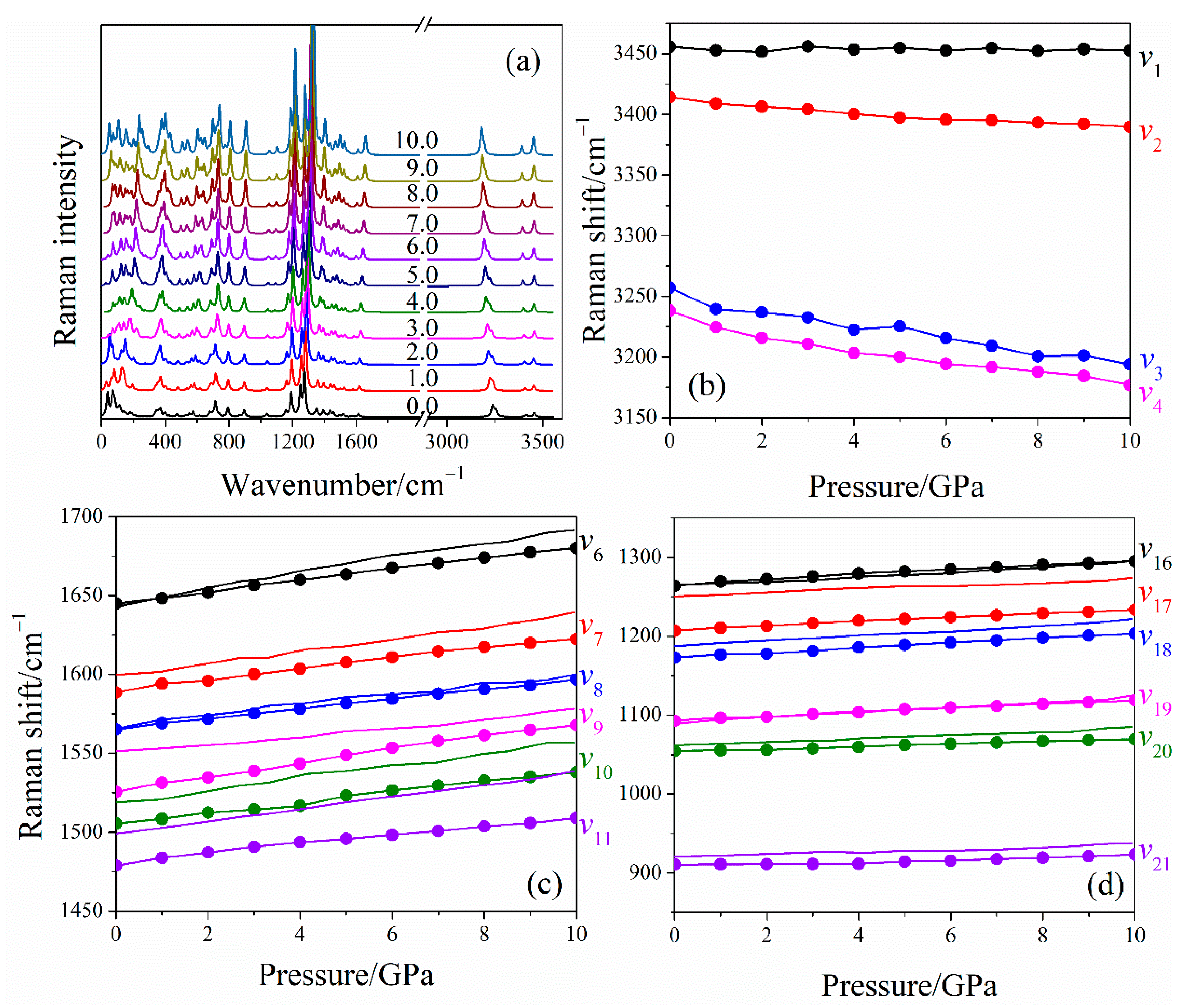
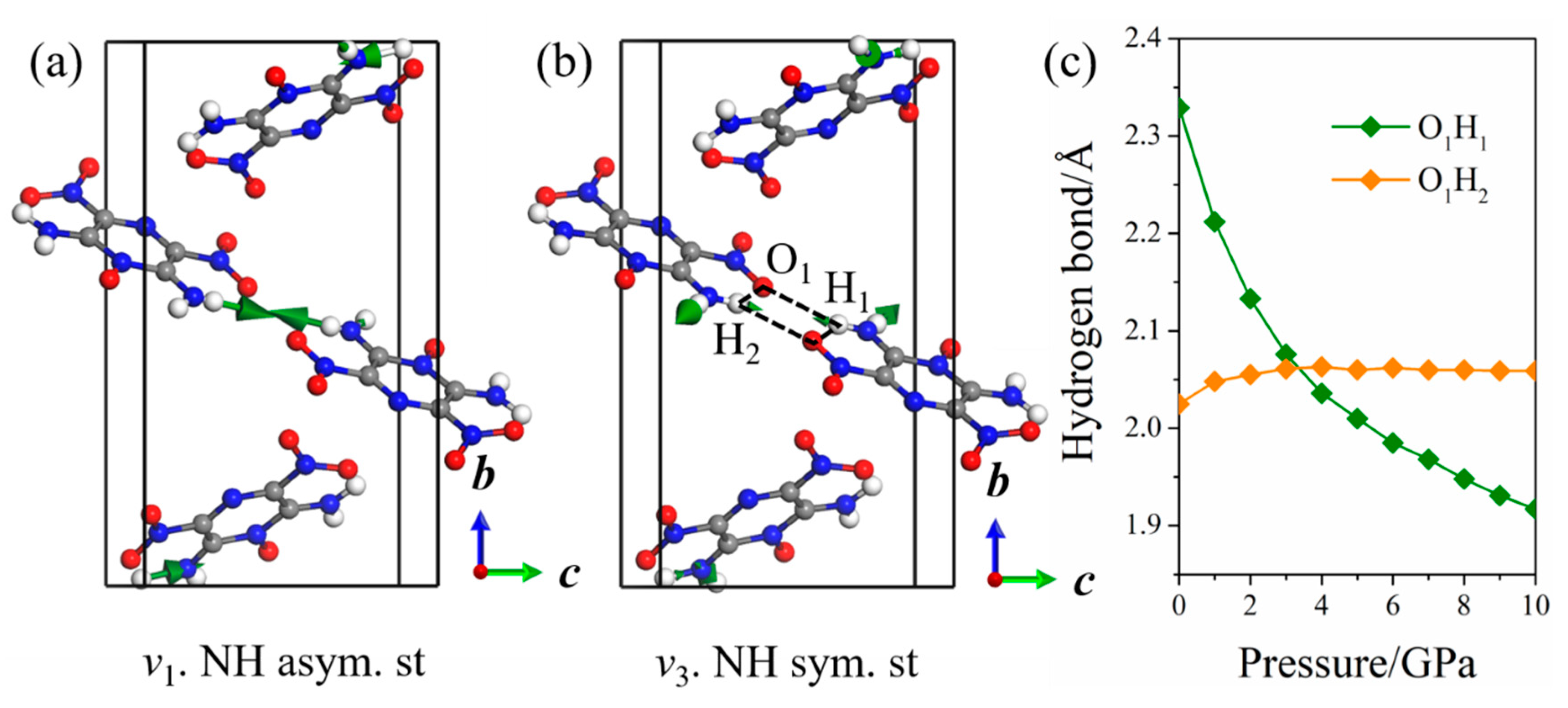
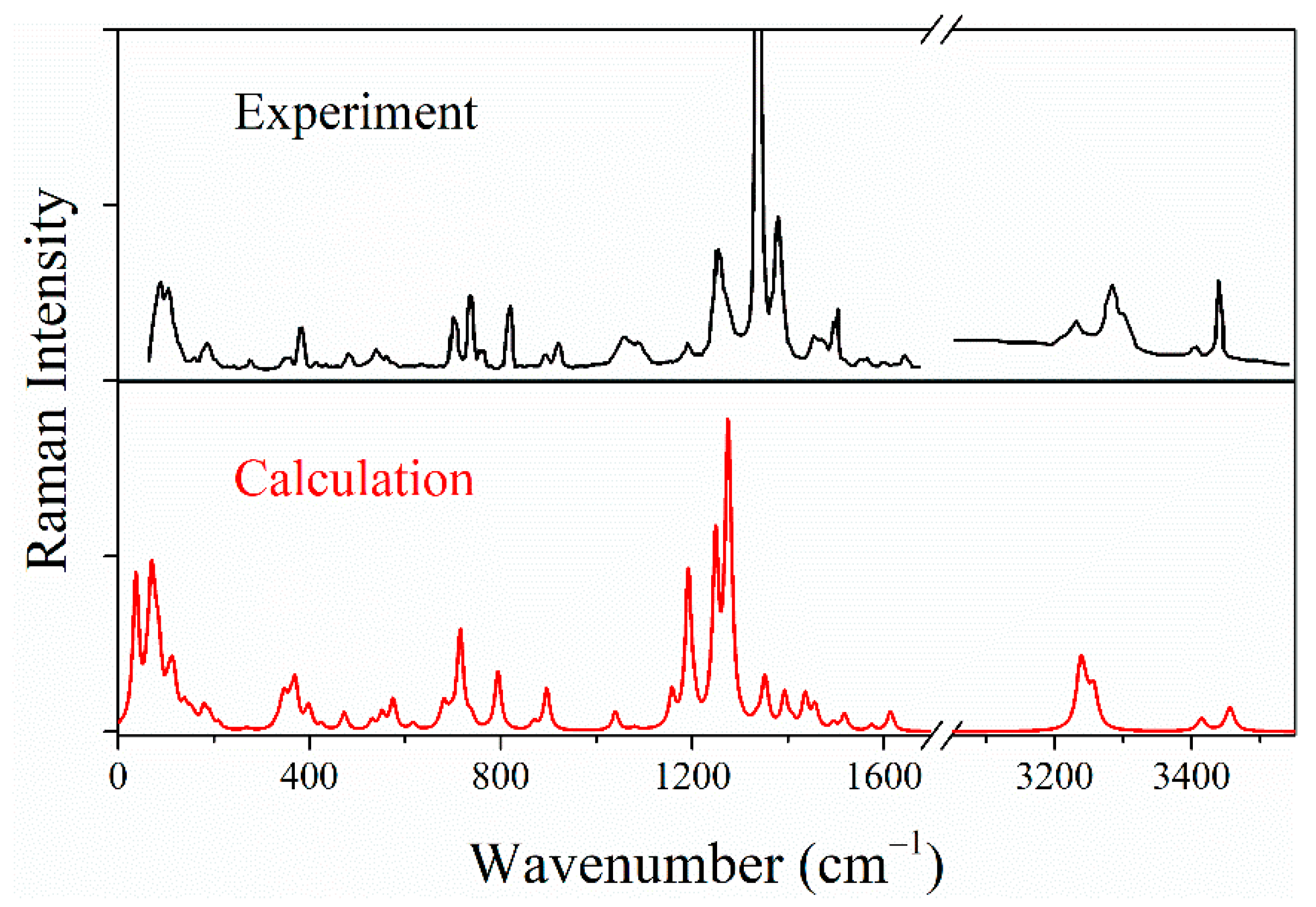
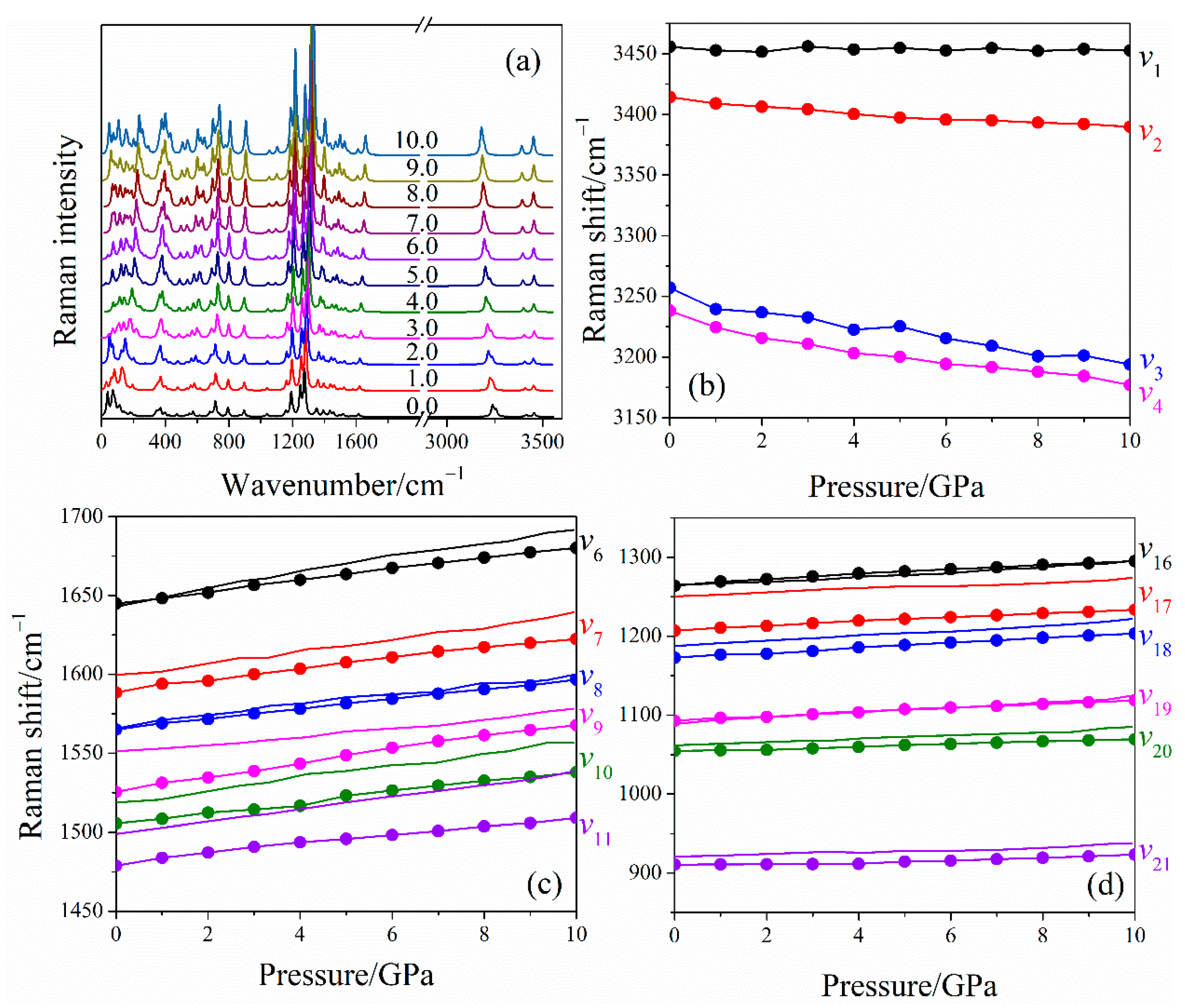
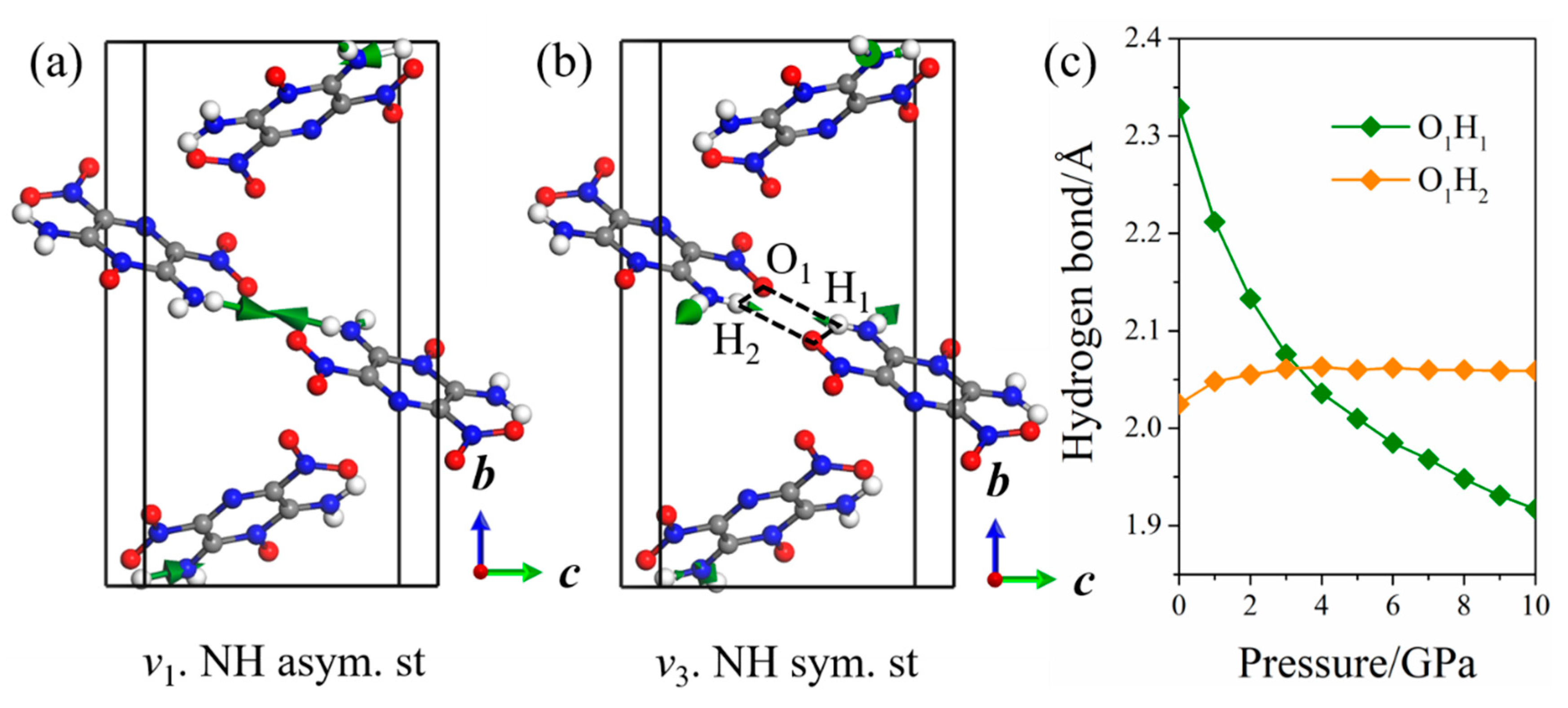
2.2. Raman Spectra of LLM-105 at Ambient and Hydrostatic Conditions
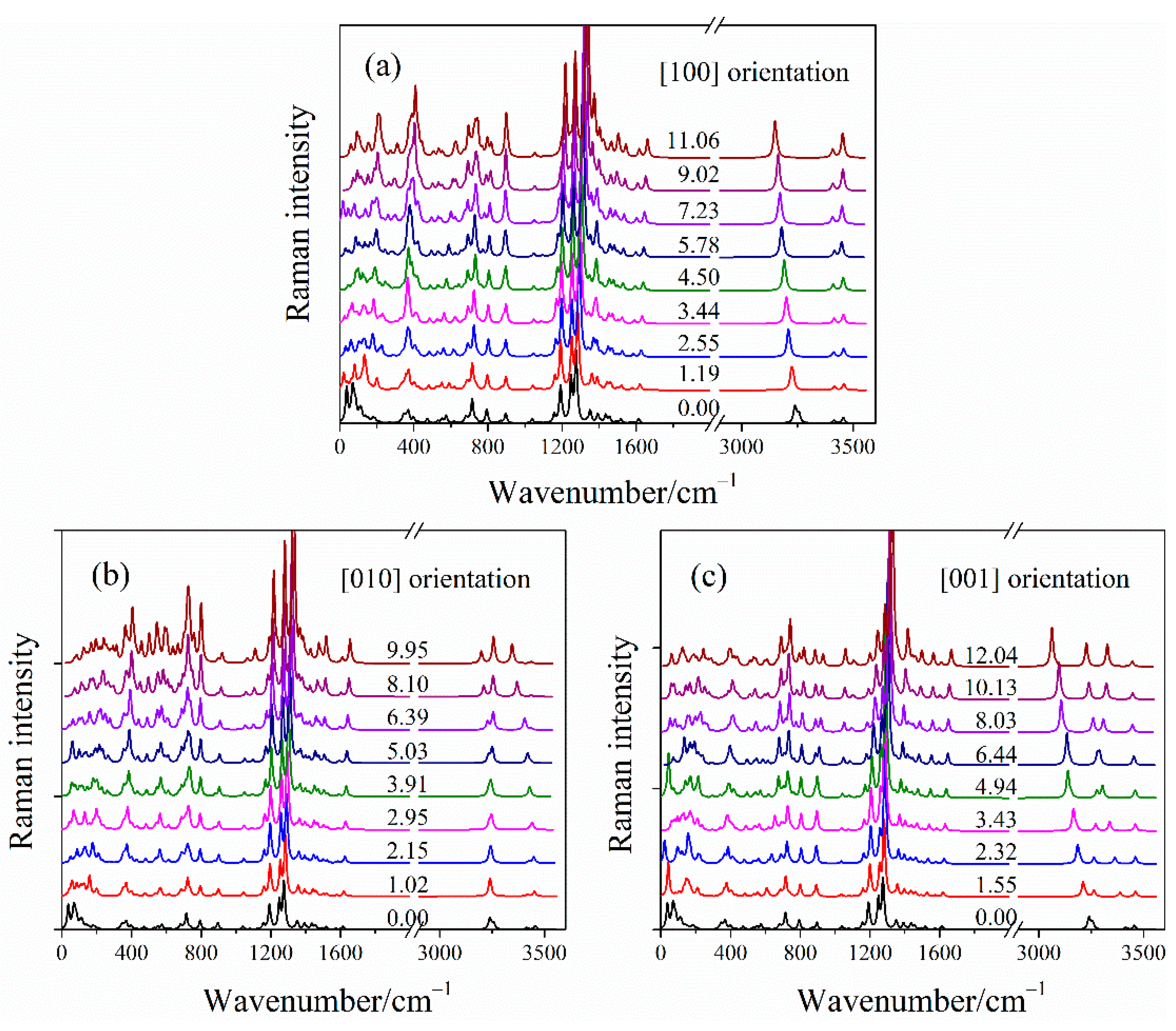
2.3. Raman Spectra of LLM-105 under Uniaxial Compressions
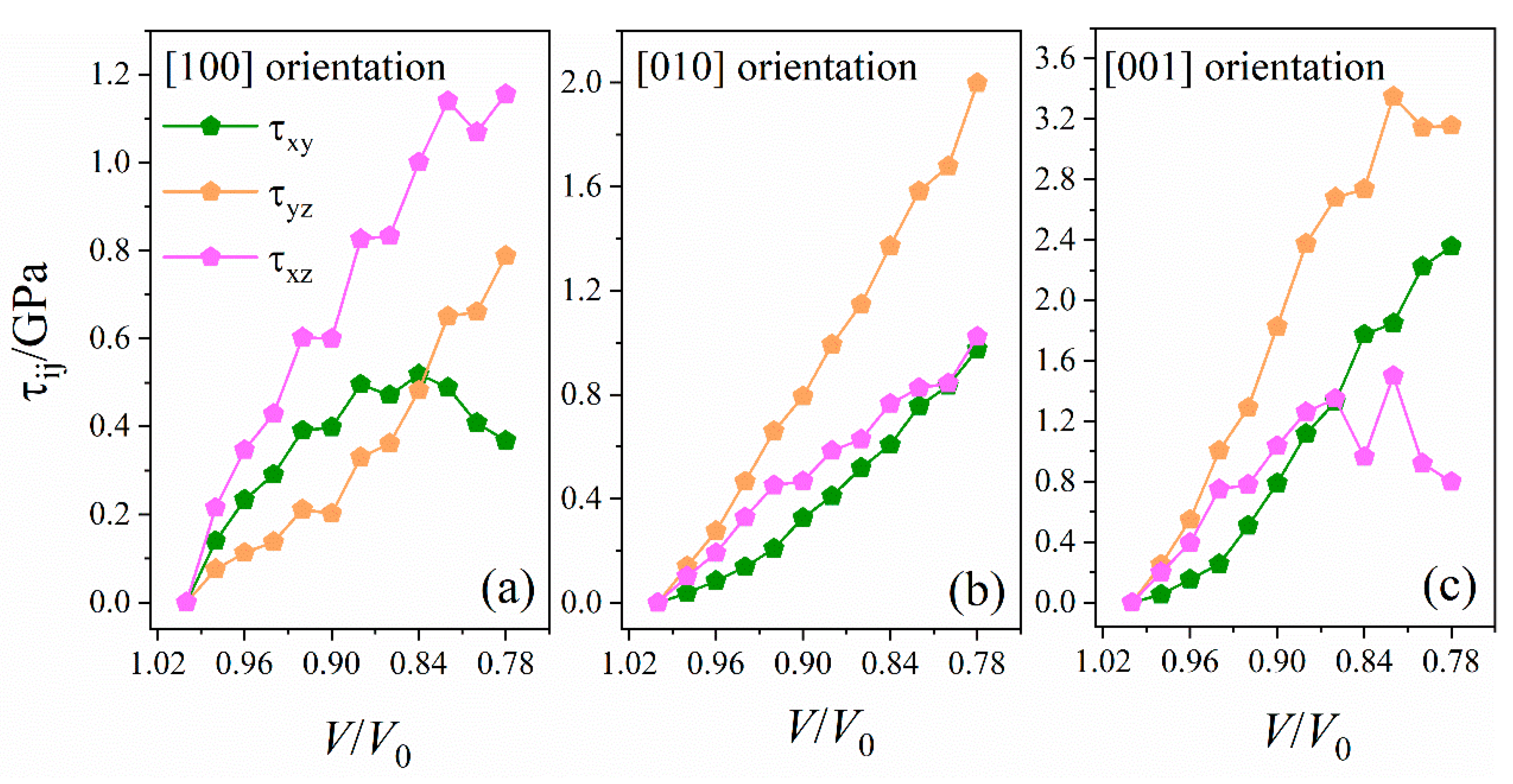
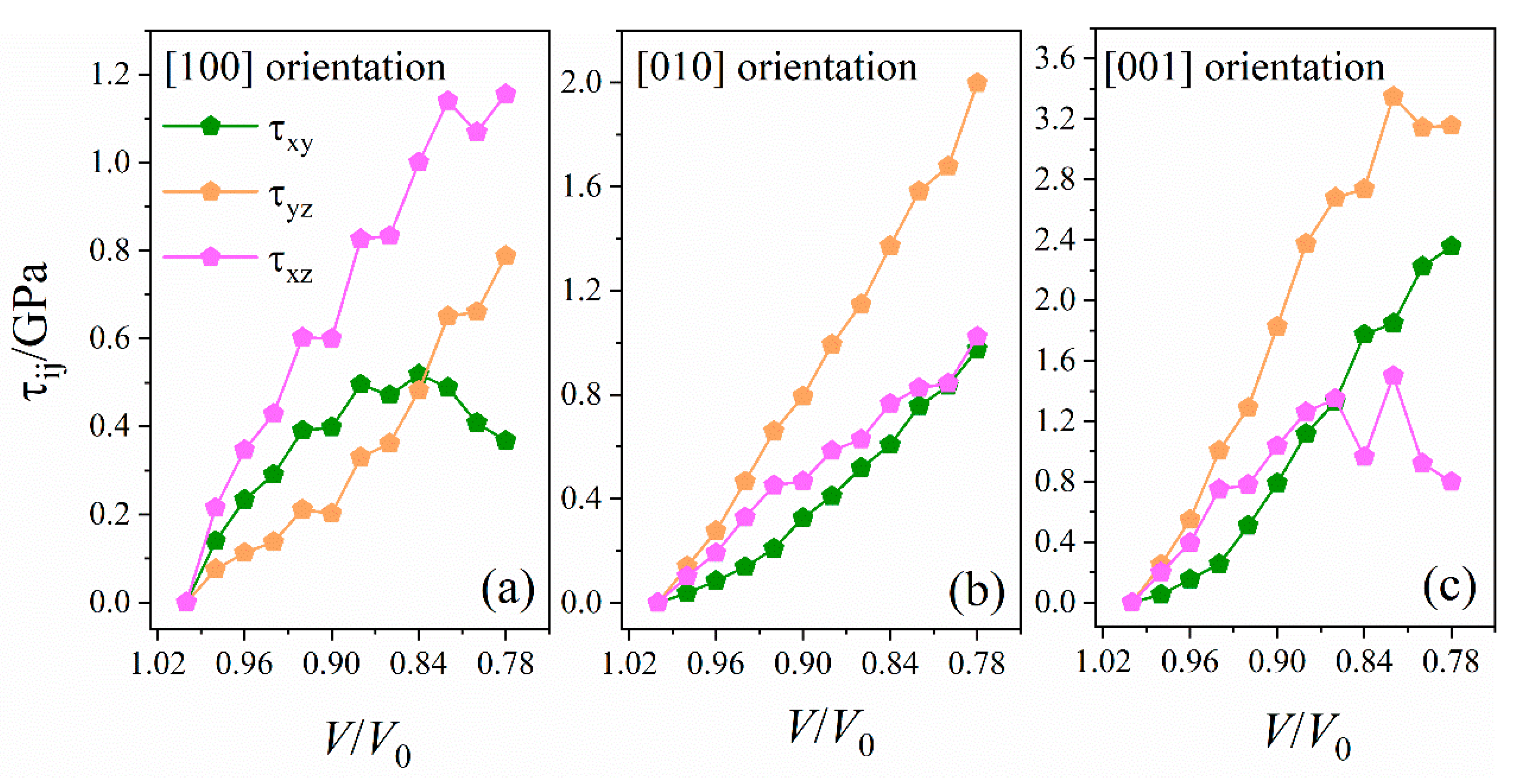
2.3.1. Stress Tensor and Shear Stress
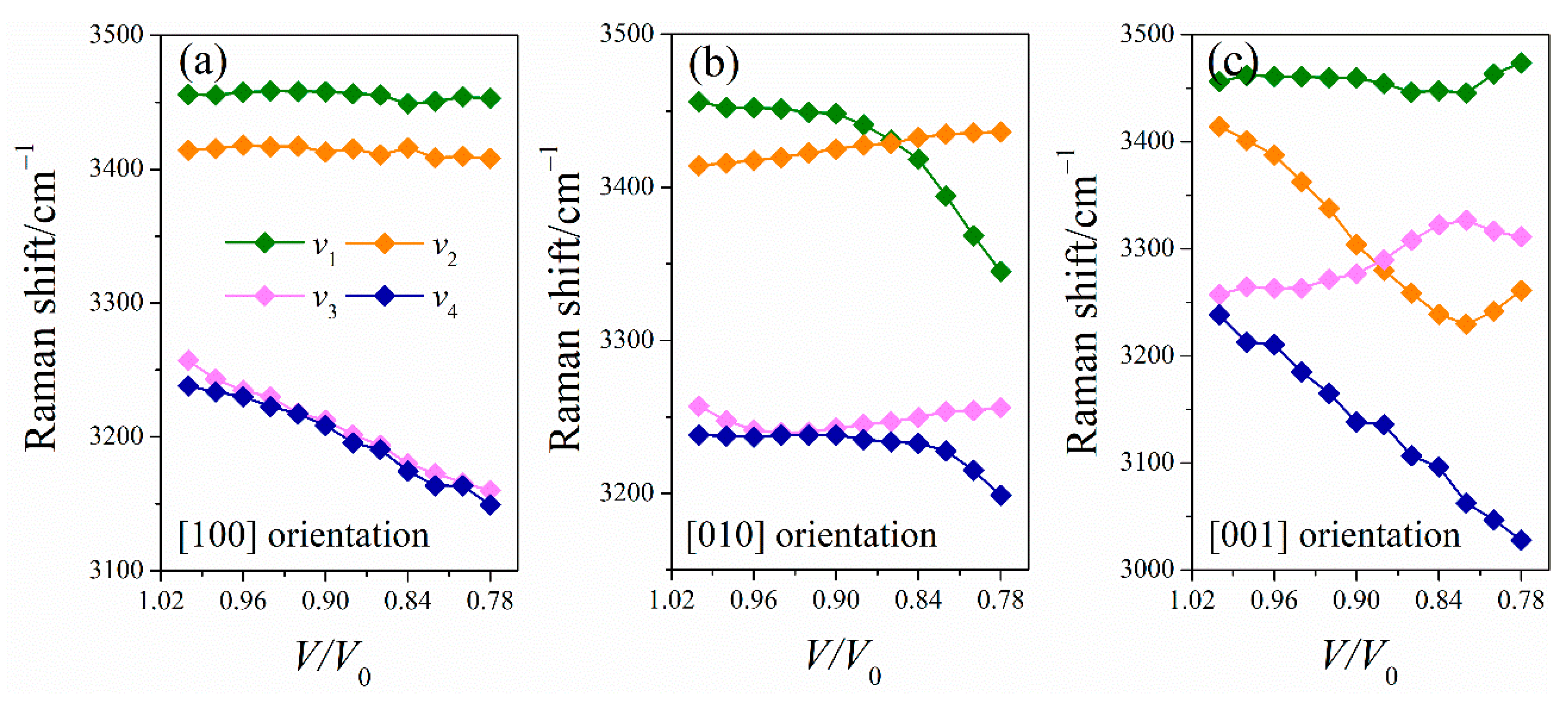
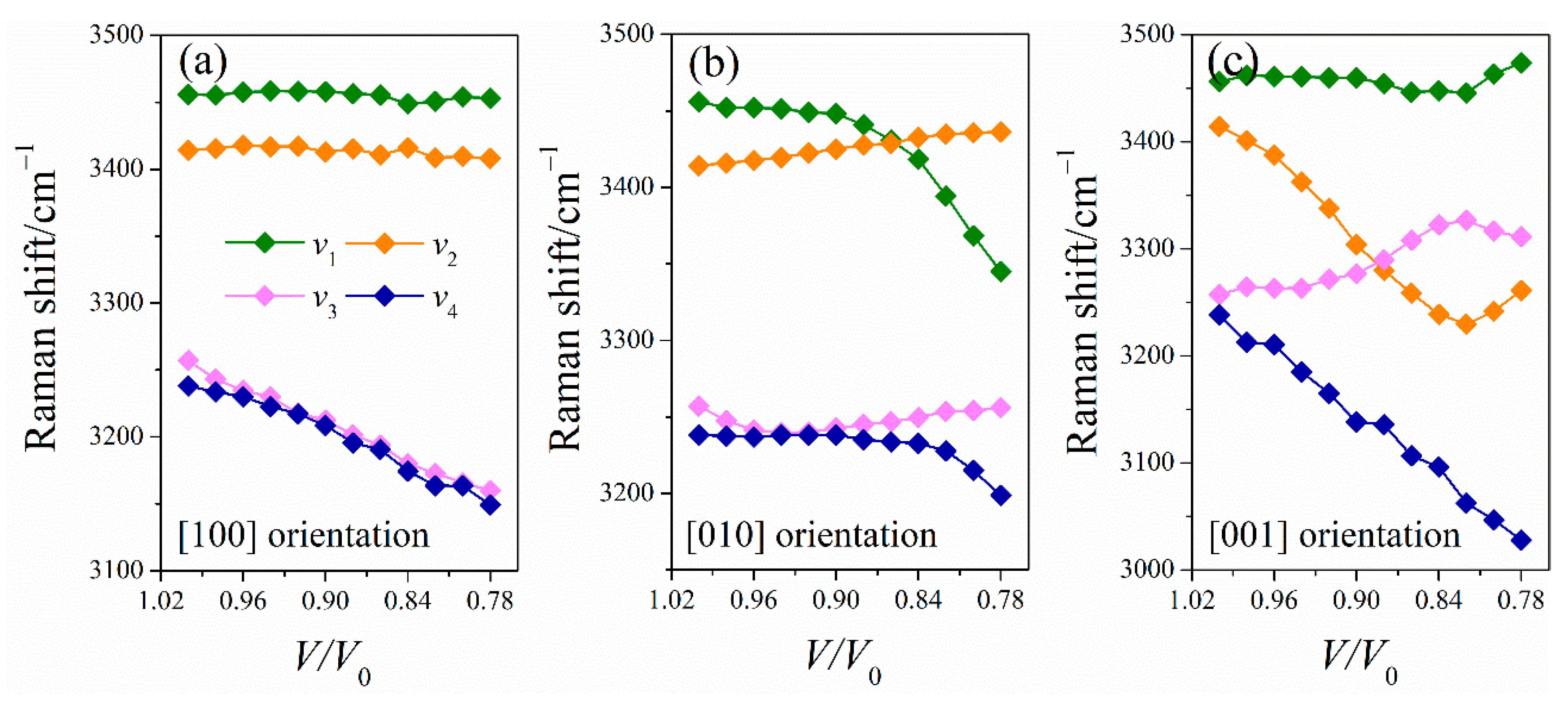
2.3.2. Simulated Raman Spectra of LLM-105 under Uniaxial Compressions
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sikder, A.K.; Sikder, N. A review of advanced high performance, insensitive and thermally stable energetic materials emerging for military and space applications. J. Hazard. Mater. 2004, 112, 1–15. [Google Scholar] [CrossRef]
- Badgujar, D.M.; Talawar, M.B.; Asthana, S.N.; Mahulikar, P.P. Advances in science and technology of modern energetic materials: An overview. J. Hazard. Mater. 2008, 151, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Xiong, Y.; Li, H.; Zhang, C. Alleviating the energy & safety contradiction to construct new low sensitivity and highly energetic materials through crystal engineering. CrystEngComm 2018, 20, 1757–1768. [Google Scholar]
- Pagoria, P.F.; Lee, G.S.; Mitchell, A.R.; Schmidt, R.D. A review of energetic materials synthesis. Thermochim. Acta 2002, 384, 187–204. [Google Scholar] [CrossRef]
- Gilardi, R.D.; Butcher, R.J. 2,6-Diamino-3,5-dinitro-1,4-pyrazine-1-oxide. Acta Crystallogr. E 2001, 57, o657–o658. [Google Scholar] [CrossRef] [Green Version]
- Averkiev, B.B.; Antipin, M.Y.; Yudin, I.L.; Sheremetev, A.B. X-ray structural study of three derivatives of dinitropyrazine. J. Mol. Struc. 2002, 606, 139–146. [Google Scholar]
- Zhang, C.; Wang, X.; Huang, H. π-stacked interactions in explosive crystals: Buffers against external mechanical stimuli. J. Am. Chem. Soc. 2008, 130, 8359–8365. [Google Scholar] [CrossRef]
- Tarver, C.M.; Urtiew, P.A.; Tran, T.D. Sensitivity of 2,6-Diamino-3,5-Dinitropyrazine-1-Oxide. J. Energ. Mater. 2005, 23, 183–203. [Google Scholar] [CrossRef]
- Gump, J.C.; Stoltz, C.A.; Mason, B.P.; Freedman, B.G.; Ball, J.R.; Peiris, S.M. Equations of state of 2,6-diamino-3,5-dinitropyrazine-1-oxide. J. Appl. Phys. 2011, 110, 073523. [Google Scholar] [CrossRef]
- Stavrou, E.; Riad Manaa, M.; Zaug, J.M.; Kuo, I.F.; Pagoria, P.F.; Kalkan, B.; Crowhurst, J.C.; Armstrong, M.R. The high pressure structure and equation of state of 2,6-diamino-3,5-dinitropyrazine-1-oxide (LLM-105) up to 20 GPa: X-ray diffraction measurements and first principles molecular dynamics simulations. J. Chem. Phys. 2015, 143, 144506. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Su, H.; Zhou, X.; Wang, X.; Wang, J.; Gao, C.; Sun, X.; Dai, R.; Wang, Z.; Li, H.; et al. Pressure- and Temperature-Dependent Structural Stability of LLM-105 Crystal. J. Phys. Chem. C 2018, 123, 1110–1119. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, Q.; Li, X.; Wang, J.; Wang, X.; Gao, C.; Dai, R.; Wang, Z.; Huang, S.; Liu, Y.; et al. Electronic Structure of LLM-105 Crystal under High Pressure and Low Temperature. J. Phys. Chem. C 2020, 124, 2399–2405. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, C.; Pan, Y.; Xiang, F.; Liu, Z.; Zhu, W.; Xiao, H. First-principles study of the structural transformation, electronic structure, and optical properties of crystalline 2,6-diamino-3,5-dinitropyrazine-1-oxide under high pressure. J. Mol. Model. 2013, 19, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Manaa, M.R.; Kuo, I.F.; Fried, L.E. First-principles high-pressure unreacted equation of state and heat of formation of crystal 2,6-diamino-3, 5-dinitropyrazine-1-oxide (LLM-105). J. Chem. Phys. 2014, 141, 064702. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.H.; Zhang, L.; Zhang, W.B.; Jiang, S.L.; Yu, Y.; Chen, J. Structural, mechanical properties, and vibrational spectra of LLM-105 under high pressures from a first-principles study. J. Mol. Model. 2017, 23, 275. [Google Scholar] [CrossRef]
- Wang, J.; Xiong, Y.; Li, H.; Zhang, C. Reversible Hydrogen Transfer as New Sensitivity Mechanism for Energetic Materials against External Stimuli: A Case of the Insensitive 2,6-Diamino-3,5-dinitropyrazine-1-oxide. J. Phys. Chem. C 2018, 122, 1109–1118. [Google Scholar] [CrossRef]
- Yu, Q.; Liu, Y.; Sui, H.; Sun, J.; Li, J. Kinetic Analysis of Overlapping Multistep Thermal Decomposition of 2,6-Diamino-3,5-dinitropyrazine-1-oxide (LLM-105). J. Phys. Chem. C 2018, 122, 25999–26006. [Google Scholar] [CrossRef]
- Wang, X.; Zeng, Q.; Li, J.; Yang, M. First-Principles-Based Force Field for 2,6-Diamino-3,5-dinitropyrazine-1-oxide (LLM-105). ACS Omega 2019, 4, 21054–21062. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zhang, Q.; Xu, R.; Chen, D.; Hao, S.; Nie, F.; Li, H. A Novel Spherulitic Self-Assembly Strategy for Organic Explosives: Modifying the Hydrogen Bonds by Polymeric Additives in Emulsion Crystallization. Cryst. Growth Des. 2018, 18, 2417–2423. [Google Scholar] [CrossRef]
- Yang, Z.; Lin, C.; Gong, F.; Zeng, C.; Zhang, J.; Huang, F. Effects of Crystal Quality and Morphology on the Mechanical Performance of LLM-105 Based PBXs. Propell. Explos. Pyrot. 2019, 44, 1219–1225. [Google Scholar] [CrossRef]
- Huang, C.; Liu, J.; Ding, L.; Wang, D.; Yang, Z.; Nie, F. Facile Fabrication of Nanoparticles Stacked 2,6-diamino-3,5-dinitropyrazine-1-oxide (LLM-105) Sub-microspheres via Electrospray Deposition. Propell. Explos. Pyrot. 2018, 43, 188–193. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, J.; Chen, Y.; Wu, Q.; Ju, Z.; Zhang, S. Detonation response mechanism of shocked LLM-105 using ReaxFF-lg and MSST. Mole. Simulat. 2021, 47, 678–687. [Google Scholar] [CrossRef]
- Yu, Q.; Zhao, C.; Liao, L.; Li, H.; Sui, H.; Yin, Y.; Li, J. A mechanism for two-step thermal decomposition of 2,6-diamino-3,5-dinitropyrazine-1-oxide (LLM-105). Phys. Chem. Chem. Phys. 2020, 22, 13729–13736. [Google Scholar] [CrossRef]
- Rajan, R.; Ravindran, T.R.; Venkatesan, V.; Chandra, S.; Gupta, M.K.; Mittal, R.; Srihari, V.; Rajaraman, R. Pressure dependent phase transformations of energetic material 2,4−dinitroanisole using Raman spectroscopy, X-ray diffraction and first principles calculations. J. Mol. Struc. 2022, 1247, 131356. [Google Scholar] [CrossRef]
- Rajan, R.; Ravindran, T.R.; Venkatesan, V.; Srihari, V.; Pandey, K.K.; Chandra, S.; Mishra, K.K.; Vargeese, A.A. New High Pressure Phases of Energetic Material TEX: Evidence from Raman Spectroscopy, X-ray Diffraction, and First-Principles Calculations. J. Phys. Chem. A 2018, 122, 6236–6242. [Google Scholar] [CrossRef]
- Birch, F. Finite Elastic Strain of Cubic Crystals. Phys. Rev. 1947, 71, 809–824. [Google Scholar] [CrossRef]
- Rousseau, D.L.; Bauman, R.P.; Porto, S.P.S. Normal Mode Determination in Crystals. J. Raman Spectrosc. 1981, 10, 253–290. [Google Scholar] [CrossRef]
- Tramer, A.; Jungen, C.; Lahmani, F. Energy Dissipation in Molecular Systems; Springer: Berlin, Germany, 2005. [Google Scholar]
- Lindan, P.J.D. First-Principles Simulation: Ideas, Illustrations and the CASTEP Code. J. Phys. Condens. Mat. 2002, 14, 2717–2744. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S. Density functional theory with London dispersion corrections. WIREs. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brilliouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Fischer, T.H.; Almlof, J. General methods for geometry and wave function optimization. J. Phys. Chem. 1992, 96, 9768–9774. [Google Scholar] [CrossRef]
- Refson, K. Variational density-functional perturbation theory for dielectrics and lattice dynamics. Phys. Rev. B 2006, 73, 155114. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| V/Å [3] | a/Å | b/Å | c/Å | β/° | |
|---|---|---|---|---|---|
| Expt. [5] | 748.16 | 5.716 | 15.850 | 8.414 | 101.041 |
| Expt. [9] | 747.3 | 5.72 | 15.82 | 8.42 | 101.15 |
| Expt. [10] | 750.08 | 5.723 | 15.870 | 8.424 | - |
| GGA/PW91 [13] | 939.23 | 6.008 | 18.279 | 8.706 | 100.75 |
| LDA/CA-PZ [13] | 737.24 | 5.837 | 15.844 | 8.416 | 99.51 |
| PBE (HASEM) [15] | 747.38 | 5.64 | 15.96 | 8.46 | 100.93 |
| Force field [18] | 711.00 | 5.64 | 15.55 | 8.24 | 100.96 |
| This work | 756.09 | 5.758 | 15.750 | 8.500 | 101.238 |
| Deviation/% | 1.06 | 0.73 | −0.63 | 1.02 | 0.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Fan, J.; Wang, Z.; Qi, H.; Su, Y.; Zhao, J. Compression Behavior and Vibrational Properties of New Energetic Material LLM-105 Analyzed Using the Dispersion-Corrected Density Functional Theory. Molecules 2021, 26, 6831. https://doi.org/10.3390/molecules26226831
Li T, Fan J, Wang Z, Qi H, Su Y, Zhao J. Compression Behavior and Vibrational Properties of New Energetic Material LLM-105 Analyzed Using the Dispersion-Corrected Density Functional Theory. Molecules. 2021; 26(22):6831. https://doi.org/10.3390/molecules26226831
Chicago/Turabian StyleLi, Tianming, Junyu Fan, Zhuoran Wang, Hanhan Qi, Yan Su, and Jijun Zhao. 2021. "Compression Behavior and Vibrational Properties of New Energetic Material LLM-105 Analyzed Using the Dispersion-Corrected Density Functional Theory" Molecules 26, no. 22: 6831. https://doi.org/10.3390/molecules26226831
APA StyleLi, T., Fan, J., Wang, Z., Qi, H., Su, Y., & Zhao, J. (2021). Compression Behavior and Vibrational Properties of New Energetic Material LLM-105 Analyzed Using the Dispersion-Corrected Density Functional Theory. Molecules, 26(22), 6831. https://doi.org/10.3390/molecules26226831