
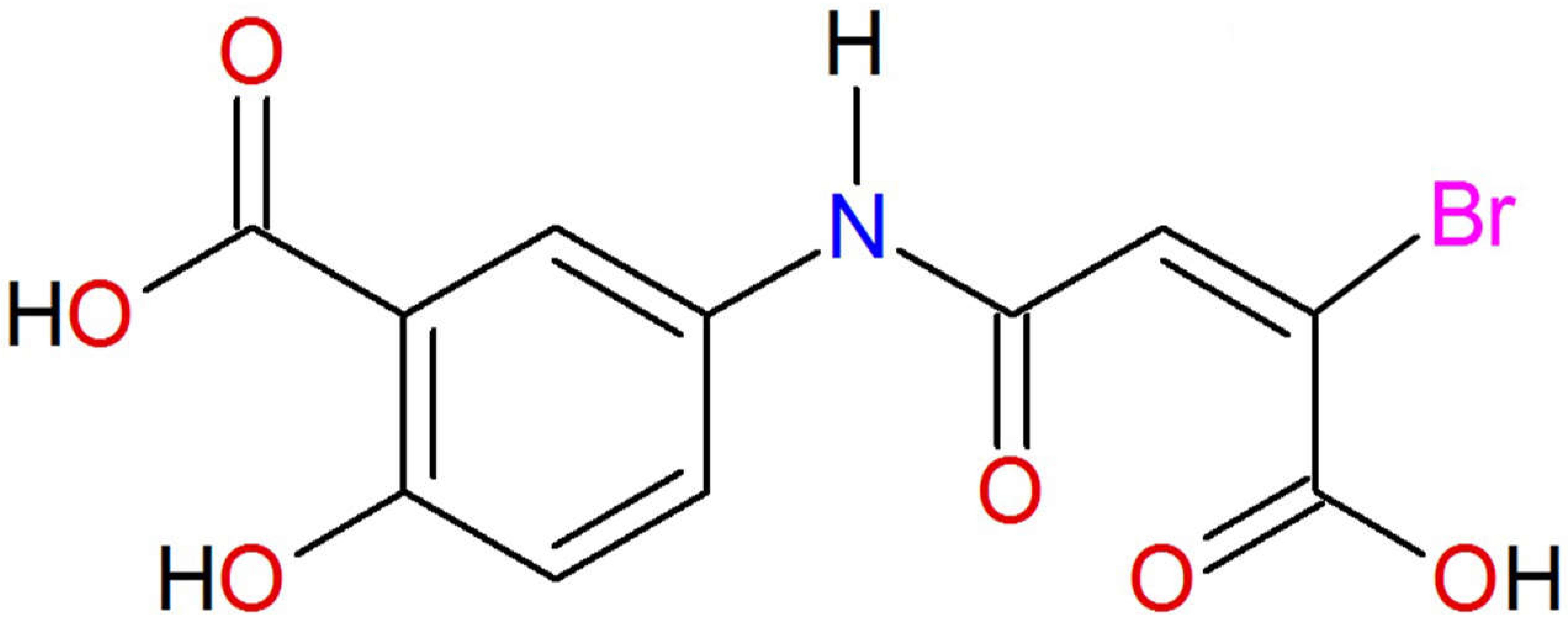
Preclinical Pharmacokinetics and Acute Toxicity in Rats of 5-{[(2E)-3-Bromo-3-carboxyprop-2-enoyl]amino}-2-hydroxybenzoic Acid: A Novel 5-Aminosalicylic Acid Derivative with Potent Anti-Inflammatory Activity

 , , , and
, , , and
Abstract
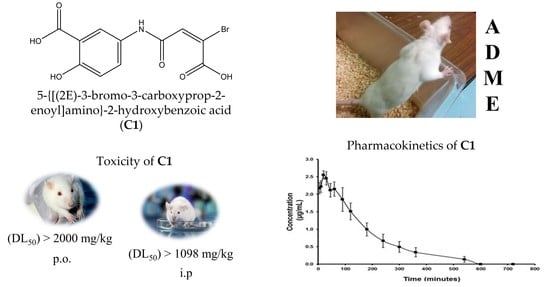
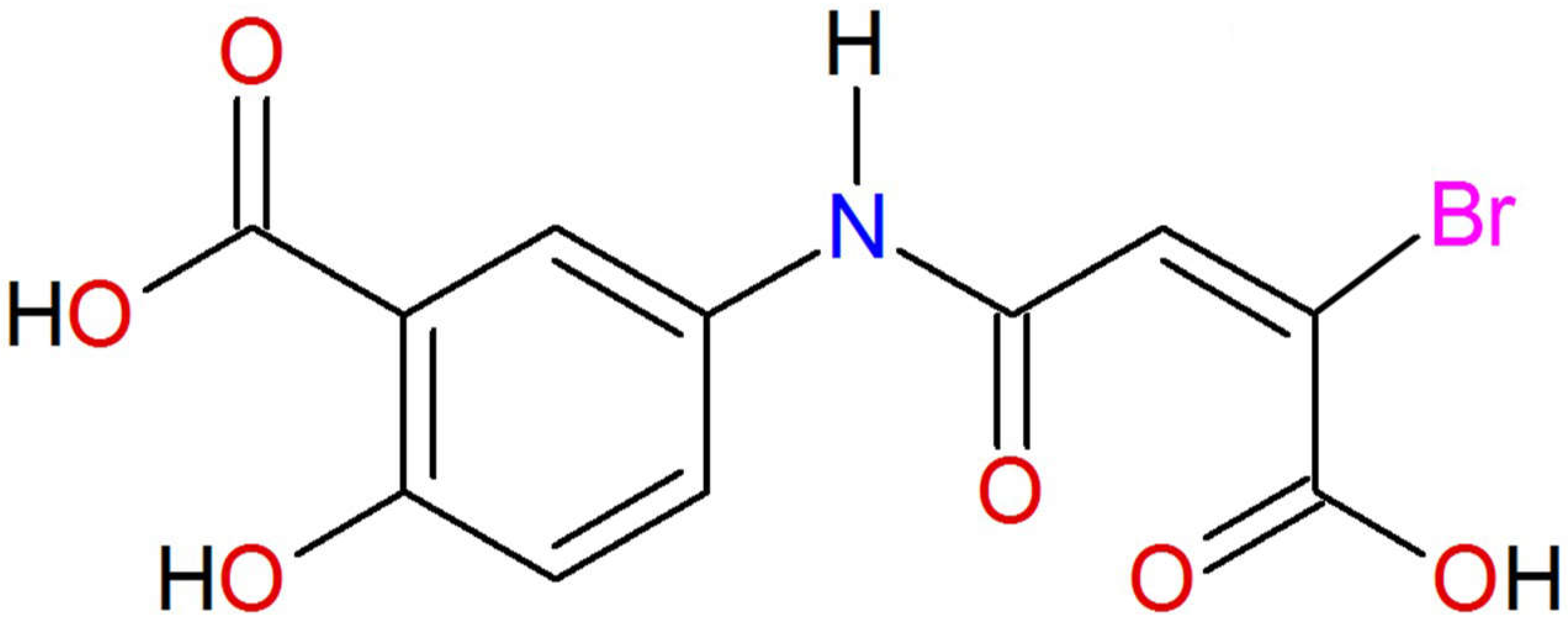
:
1. Introduction
2. Results
2.1. Acute Toxicity of C1
2.2. Validation of the Method for the Determination of C1
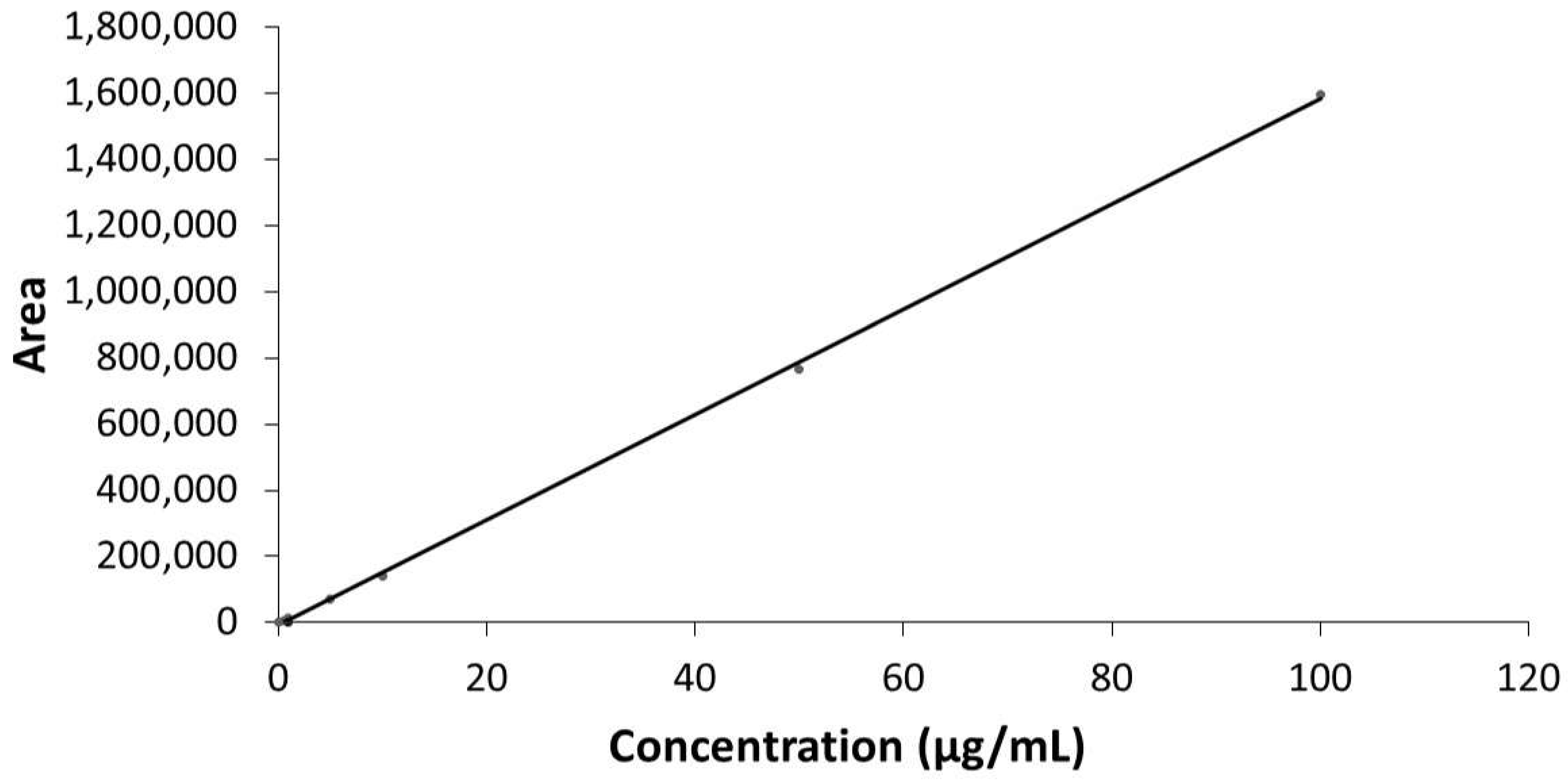
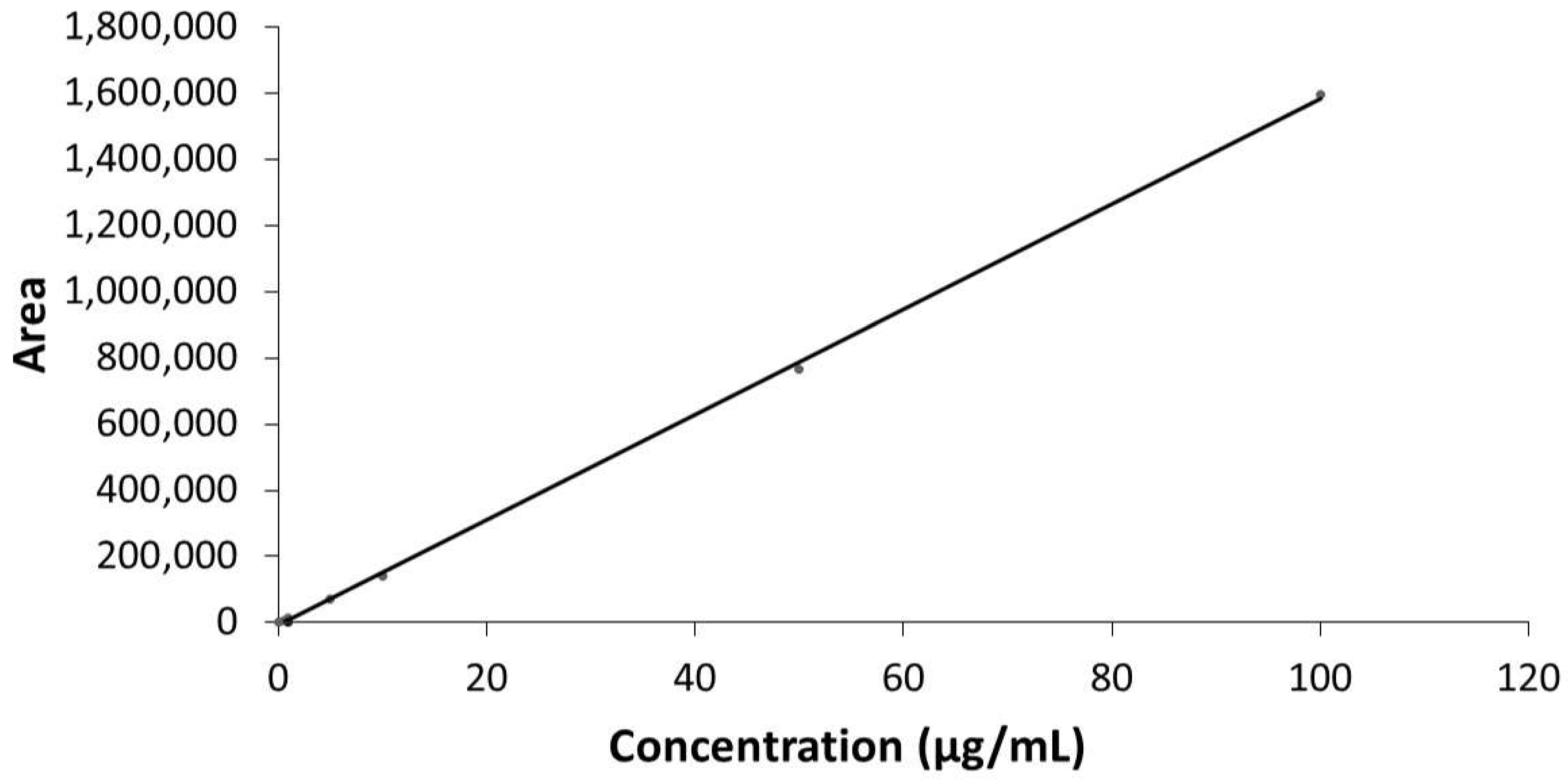
2.2.1. Linearity
2.2.2. Intra- and Inter-Day Accuracy and Precision
2.2.3. Recovery
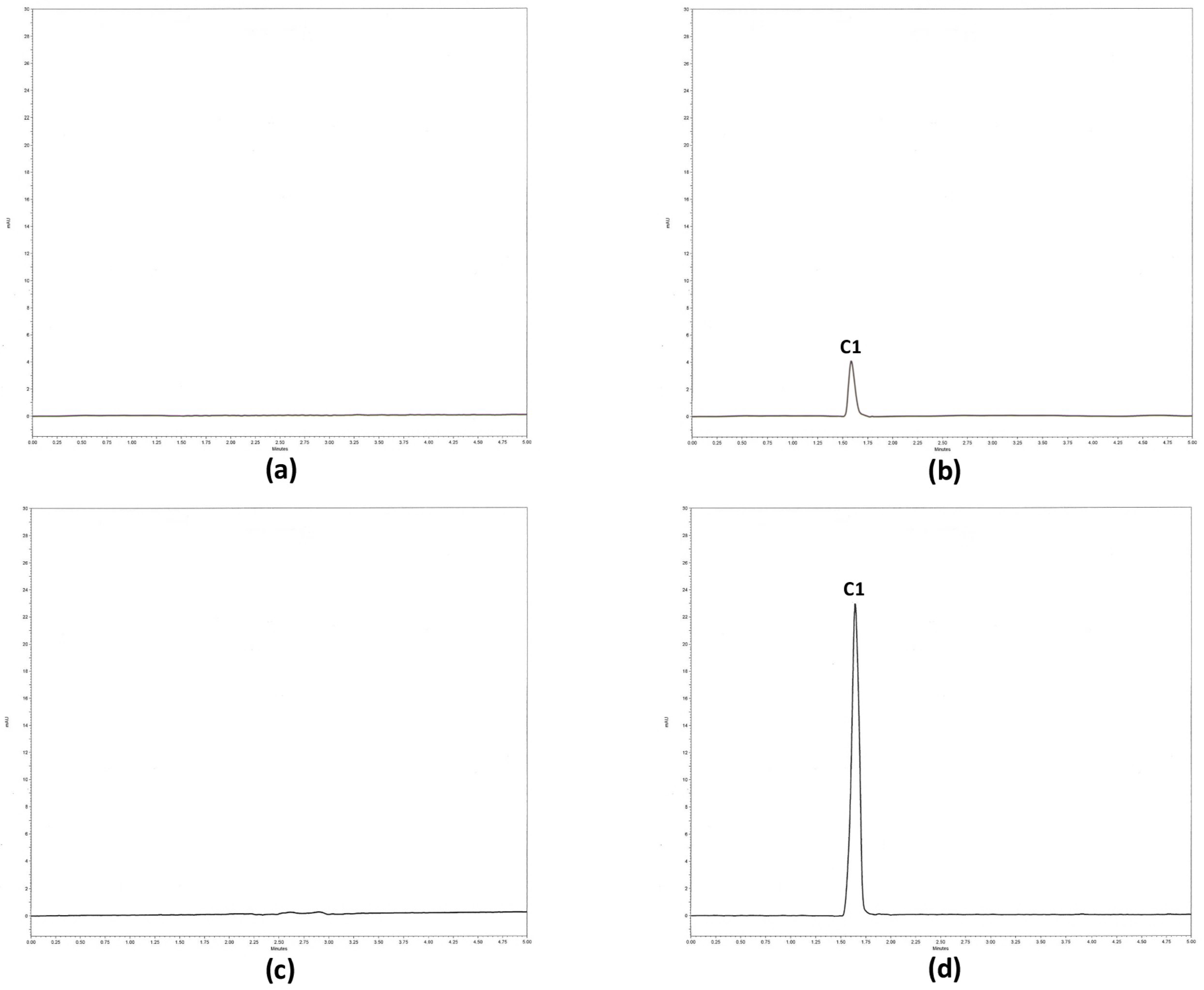
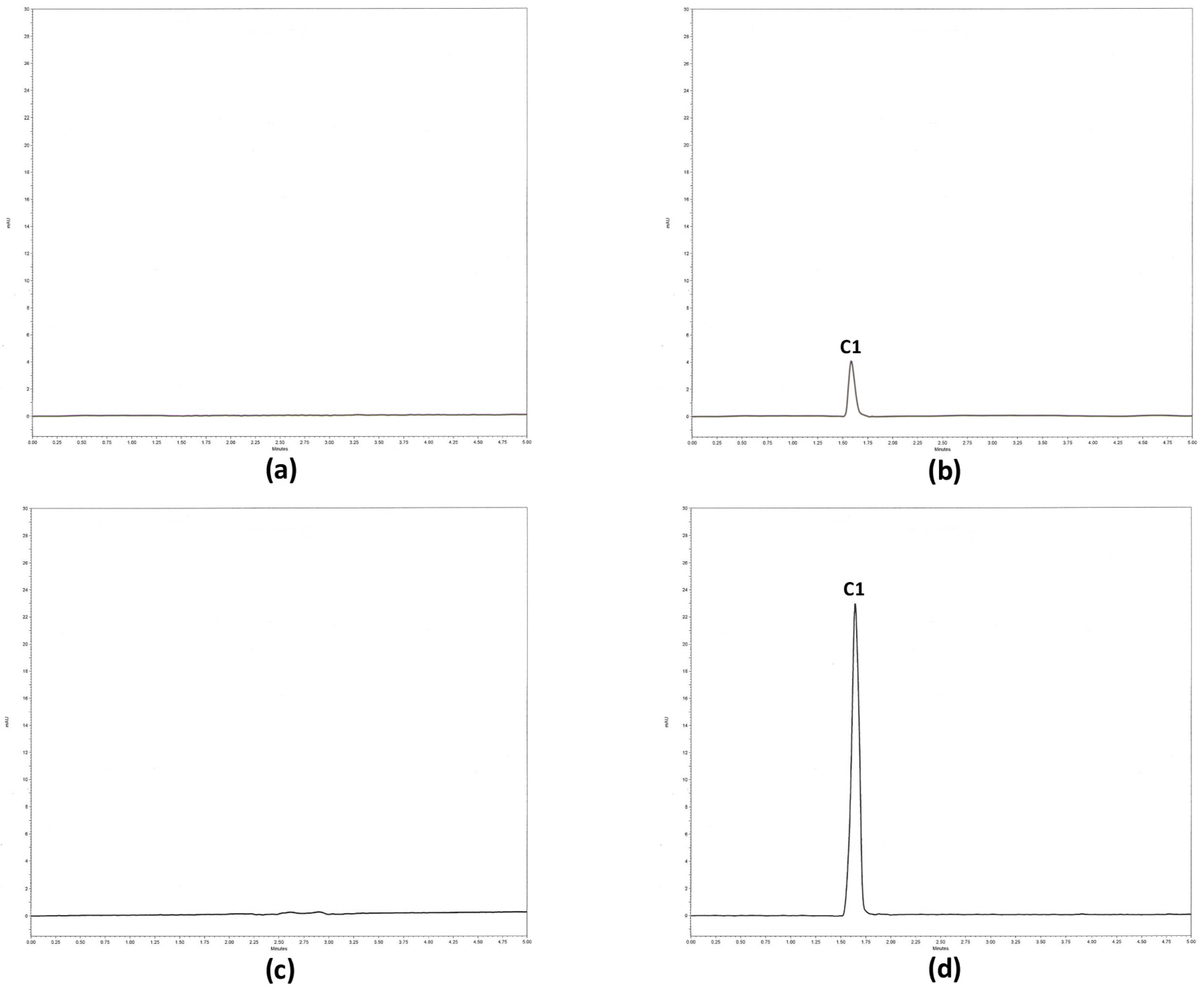
2.2.4. Selectivity
2.2.5. Stability
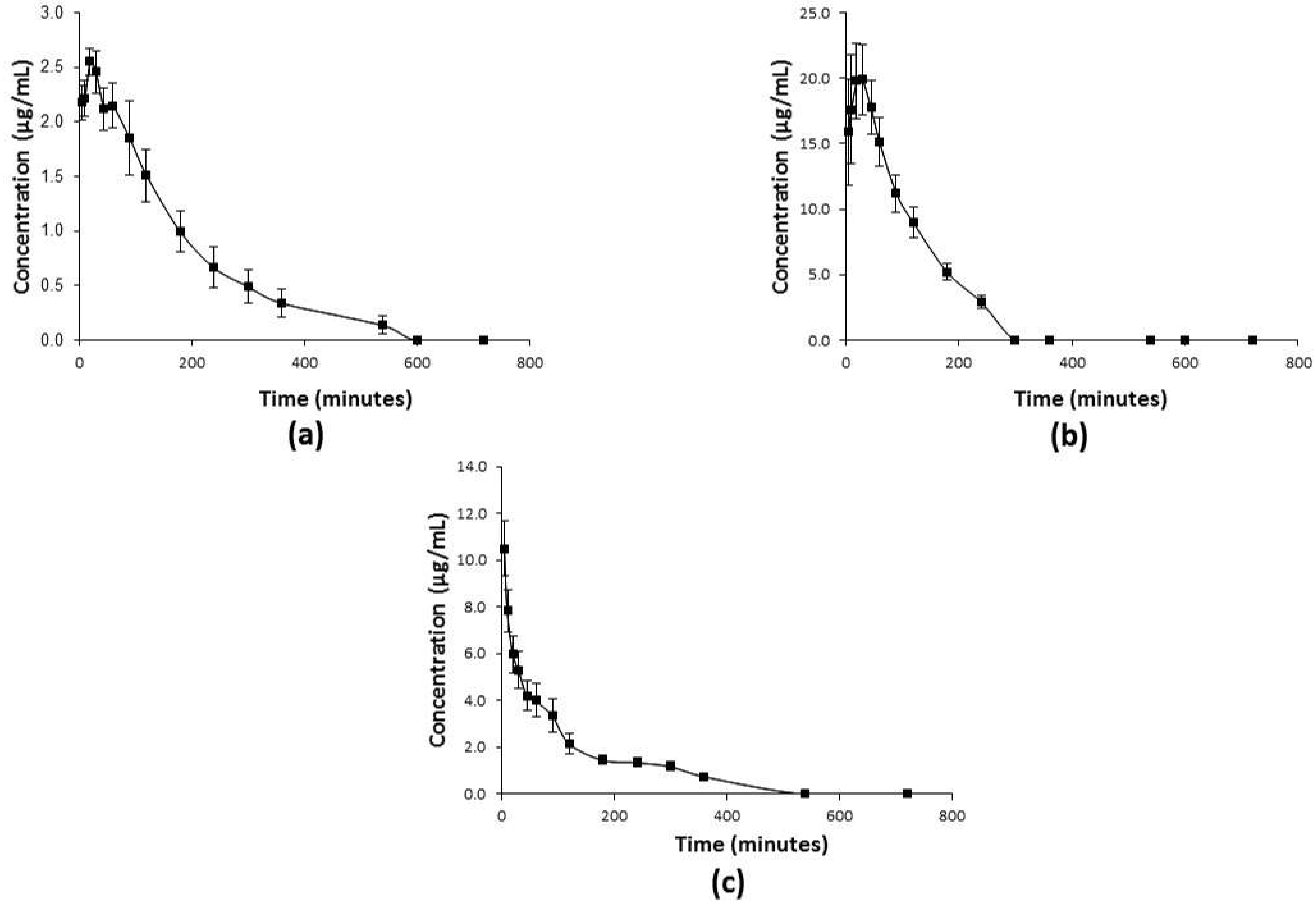
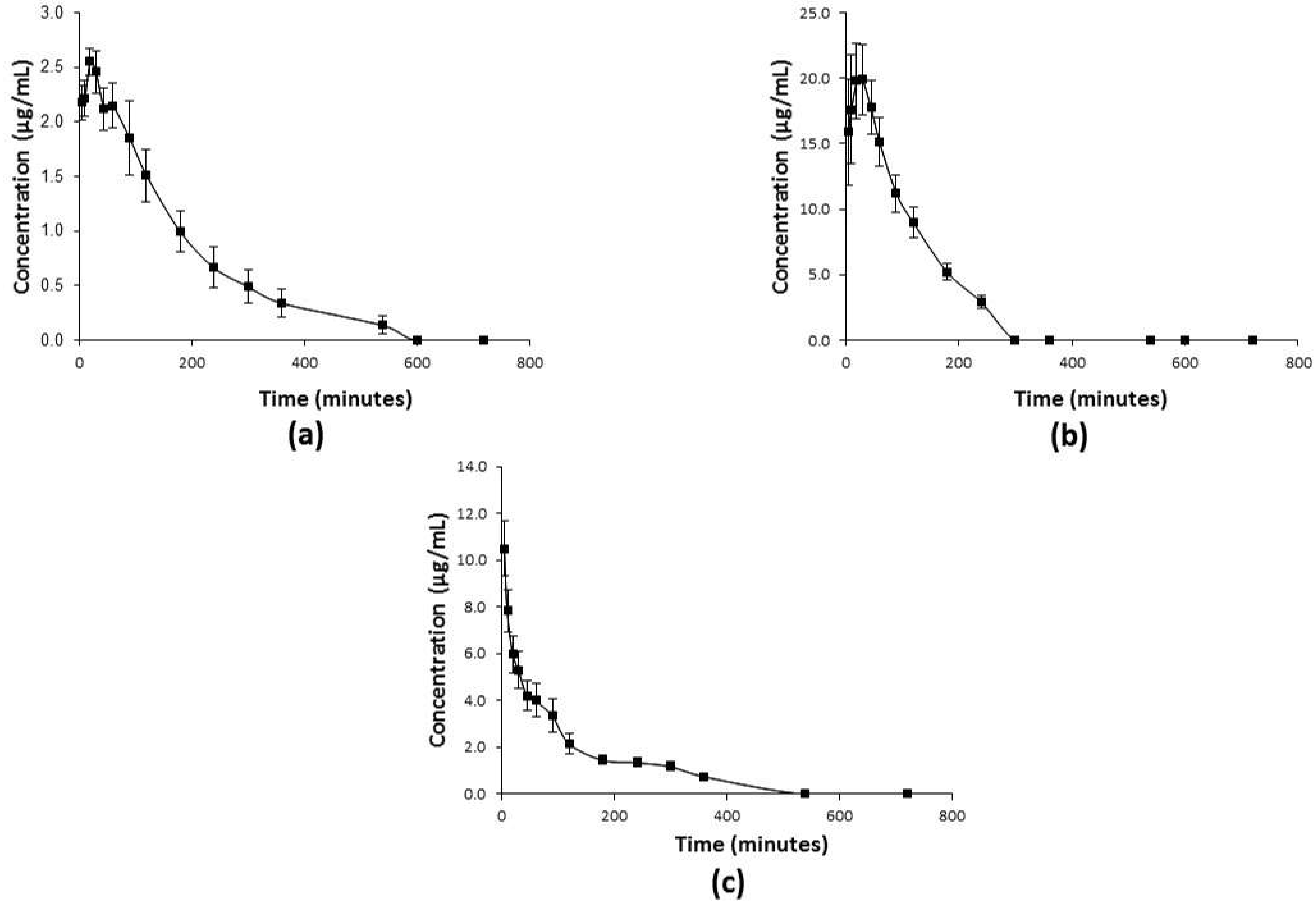
2.3. Pharmacokinetic Profiles of C1
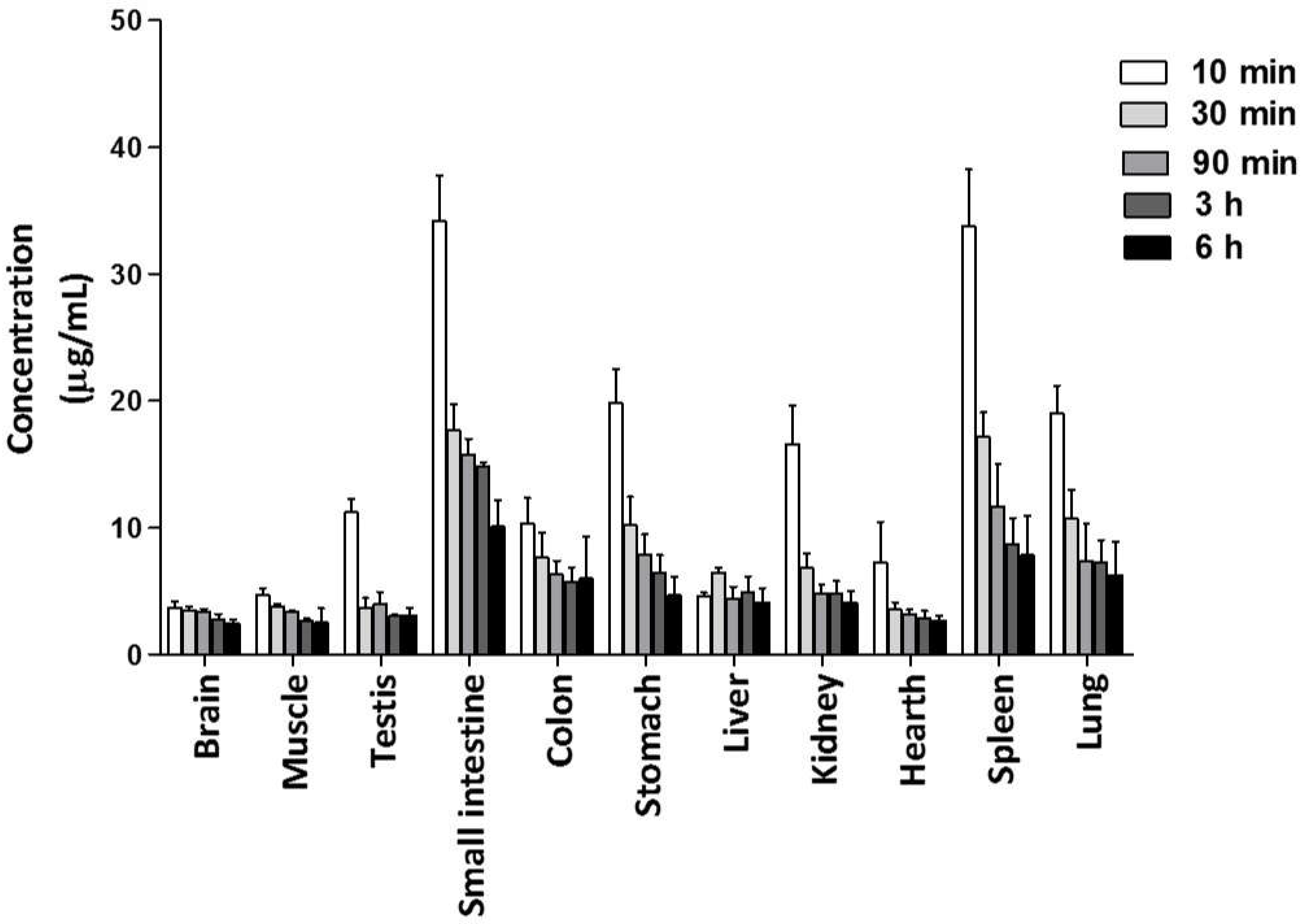
2.3.1. Distribution of C1 in Tissues
2.3.2. Plasma Protein Binding of C1
2.3.3. Blood/Plasma Partitioning to Find the Blood/Plasma Ratio of C1
3. Discussion
4. Material and Methods
4.1. Chemicals and Standards
4.2. The Formulation for Administering C1
4.3. Animals
4.4. Ethics Statement
4.5. Acute Toxicity of C1 Determined by the Up-and-Down Method
4.6. RP-HPLC Validation
4.6.1. Rat Plasma Sampling
4.6.2. Instrumentation and Analytical Conditions
4.6.3. Preparation of Calibration Standards
4.6.4. Quality Control Samples
4.6.5. Validation Parameters
4.7. Pharmacokinetic Studies of C1
4.7.1. Distribution of C1 in Tissues
4.7.2. Blood/Plasma Partitioning and Blood/Plasma Ratio of C1
4.7.3. Plasma Protein Binding Assay of C1
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ghosh, R.; Alajbegovic, A.; Gomes, A.V. NSAIDs and Cardiovascular Diseases: Role of Reactive Oxygen Species. Oxid. Med. Cell. Longev. 2015, 2015, 536962. [Google Scholar] [CrossRef] [Green Version]
- Bello, A.E.; Holt, R.J. Cardiovascular risk with non-steroidal anti-inflammatory drugs: Clinical implications. Drug Saf. 2014, 37, 897–902. [Google Scholar] [CrossRef] [Green Version]
- Trevor, J.L.; Deshane, J.S. Refractory asthma: Mechanisms, targets, and therapy. Allergy 2014, 69, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Yu, R. Immune-Signaling Molecules and Obesity-Induced Inflammation. J. Nutr. Sci. Vitaminol. 2015, 61, S131–S132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribaldone, D.G.; Fagoonee, S.; Astegiano, M.; De Angelis, C.; Smedile, A.; Caviglia, G.P.; Petrini, E.; Greco, A.; Pellicano, R. Coxib’s Safety in Patients with Inflammatory Bowel Diseases: A Meta-analysis. Pain Physician 2015, 18, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Hiľovská, L.; Jendželovský, R.; Fedoročko, P. Potency of non-steroidal anti-inflammatory drugs in chemotherapy. Mol. Clin. Oncol. 2015, 3, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Al-Shidhani, A.; Al-Rawahi, N.; Al-Rawahi, A.; Murthi, P.S. Non-steroidal Anti-inflammatory Drugs (NSAIDs) Use in Primary Health Care Centers in A’Seeb, Muscat: A Clinical Audit. Oman Med. J. 2015, 30, 366–371. [Google Scholar] [PubMed]
- Thun, M.J.; Blackard, B. Pharmacologic effects of NSAIDs and implications for the risks and benefits of long-term prophylactic use of aspirin to prevent cancer. Recent Results Cancer Res. 2009, 181, 215–221. [Google Scholar]
- Bushra, R.; Aslam, N. An overview of clinical pharmacology of Ibuprofen. Oman Med. J. 2010, 25, 155–1661. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.S.; Daugherty, A.; Herrington, D.; Greenland, P.; Roberts, H.; Taubert, K.A. The use of nonsteroidal anti-inflammatory drugs (NSAIDs): A science advisory from the American Heart Association. Circulation 2005, 111, 1713–1716. [Google Scholar]
- Moore, N.; Pollack, C.; Butkerait, P. Adverse drug reactions and drug-drug interactions with over-the-counter NSAIDs. Ther. Clin. Risk Manag. 2015, 11, 1061–1075. [Google Scholar]
- Brune, K.; Patrignani, P. New insights into the use of currently available non-steroidal anti-inflammatory drugs. J. Pain Res. 2015, 8, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; Cryer, B. Gastrointestinal injury associated with NSAID use: A case study and review of risk factors and preventative strategies. Drug Healthc. Patient Saf. 2015, 7, 31–41. [Google Scholar]
- Scarpignato, C.; Lanas, A.; Blandizzi, C.; Lems, W.F.; Hermann, M.; Hunt, R.H. International NSAID Consensus Group. Safe prescribing of non-steroidal anti-inflammatory drugs in patients with osteoarthritis--an expert consensus addressing benefits as well as gastrointestinal and cardiovascular risks. BMC Med. 2015, 13, 55. [Google Scholar]
- Turajane, T.; Wongbunnak, R.; Patcharatrakul, T.; Ratansumawong, K.; Poigampetch, Y.; Songpatanasilp, T. Gastrointestinal and cardiovascular risk of non-selective NSAIDs and COX-2 inhibitors in elderly patients with knee osteoarthritis. J. Med. Assoc. Thai. 2009, 92, S19–S26. [Google Scholar] [PubMed]
- Asghar, L.F.A.; Chure, C.B.; Chandran, S. Colon specific delivery of indomethacin: Effect of incorporating pH sensitive polymers in xanthan gum matrix bases. AAPS PharmSciTech 2009, 10, 418–429. [Google Scholar]
- Nagai, N.; Yoshioka, C.; Ito, Y. Topical therapies for rheumatoid arthritis by gel ointments containing indomethacin nanoparticles in adjuvant-induced arthritis rat. J. Oleo Sci. 2015, 64, 337–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, M.; Blobaum, A.L.; Marnett, L.J.; Hey-Hawkins, E. Synthesis and evaluation of carbaborane derivatives of indomethacin as cyclooxygenase inhibitors. Bioorg. Med. Chem. 2011, 19, 3242–3248. [Google Scholar] [CrossRef] [Green Version]
- Sood, B.G.; Lulic-Botica, M.; Holzhausen, K.A.; Pruder, S.; Kellogg, H.; Salari, V.; Thomas, R. The risk of necrotizing enterocolitis after indomethacin tocolysis. Pediatrics 2011, 128, e54–e62. [Google Scholar]
- Kim, J.-H.; Kim, B.-W.; Kwon, H.-J.; Nam, S.-W. Curative effect of selenium against indomethacin-induced gastric ulcers in rats. J. Microbiol. Biotechnol. 2011, 21, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, S.; Nakamura, T.; Yasui, H.; Nishikawa, C.; Miki, I.; Inoue, J.; Horibe, S.; Hamaguchi, T.; Tanahashi, T.; Mizuno, S. Intestinal and hepatic expression of cytochrome P450s and mdr1a in rats with indomethacin-induced small intestinal ulcers. Int. J. Med. Sci. 2014, 11, 1208–1217. [Google Scholar]
- Dursun, H.; Albayrak, F.; Bilici, M.; Koc, F.; Alp, H.H.; Candar, T.; Kukula, O. Gastroprotective and antioxidant effects of opipramol on indomethacin-induced ulcers in rats. Yakugaku Zasshi 2009, 129, 861–869. [Google Scholar] [PubMed] [Green Version]
- Suemasu, S.; Tanaka, K.-I.; Namba, T.; Ishihara, T.; Katsu, T.; Fujimoto, M.; Adachi, H.; Sobue, G.; Takeuchi, K.; Nakai, A.; et al. A role for HSP70 in protecting against indomethacin-induced gastric lesions. J. Biol. Chem. 2009, 284, 19705–19715. [Google Scholar] [PubMed] [Green Version]
- Antonisamy, P.; Arasu, M.V.; Dhanasekaran, M.; Choi, K.C.; Aravinthan, A.; Kim, N.S.; Kang, C.-W.; Kim, J.-H. Protective effects of trigonelline against indomethacin-induced gastric ulcer in rats and potential underlying mechanisms. Food Funct. 2016, 7, 398–408. [Google Scholar] [CrossRef]
- Sales-Campos, H.; Basso, P.J.; Alves, V.B.F.; Fonseca, M.T.C.; Bonfá, G.; Nardini, V.; Cardoso, C.R.B. Classical and recent advances in the treatment of inflammatory bowel diseases. Braz. J. Med. Biol. Res. 2015, 48, 96–107. [Google Scholar] [CrossRef]
- Iskandar, H.N.; Dhere, T.; Farraye, F.A. Ulcerative Colitis: Update on Medical Management. Curr. Gastroenterol. Rep. 2015, 17, 44. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.I.; Escobar, L.; García, X.A.; Van Domselaar, M.; Achecar, L.M.; Luján, D.R.; García, M.J. Mesalazine-induced eosinophilic pneumonia in a patient with ulcerative colitis disease: A case report and literature review. Int. J. Colorectal Dis. 2016, 31, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Alskaf, E.; Aljoudeh, A.; Edenborough, F. Mesalazine-induced lung fibrosis. BMJ Case Rep. 2013, 2013, bcr2013008724. [Google Scholar] [CrossRef]
- Klotz, U. Clinical pharmacokinetics of sulphasalazine, its metabolites and other prodrugs of 5-aminosalicylic acid. Clin. Pharmacokinet. 1985, 10, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Hawkins, C.L.; Pattison, D.I.; Rees, M.D. Mammalian heme peroxidases: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 1199–1234. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, B.S.; de Winther, M.P.J.; Heeringa, P. Myeloperoxidase: Molecular mechanisms of action and their relevance to human health and disease. Antioxid. Redox Signal. 2009, 11, 2899–2937. [Google Scholar] [CrossRef]
- Cabrera Pérez, L.C.; Gutiérrez Sánchez, M.; Mendieta Wejebe, J.E.; Hernández Rodríguez, M.; Fragoso Vázquez, M.J.; Salazar, J.R.; Correa Basurto, J.; Padilla Martínez, I.I.; Rosales Hernández, M.C. Novel 5-aminosalicylic derivatives as anti-inflammatories and myeloperoxidase inhibitors evaluated in silico, in vitro and ex vivo. Arabian J. Chem. 2019, 12, 5278–5291. [Google Scholar] [CrossRef]
- Correa Basurto, J.; Rosales Hernández, M.C.; Mendieta Wejebe, J.E.; Trujillo Ferrara, J.G.; Padilla Martínez, I.I.; Ramírez Durán, L.A.; Alemán González Duhart, D. Síntesis de un derivado del ácido 5-aminosalicílico con actividad antioxidante. Patente del Instituto Politécnico Nacional. Título de Patente No. 376240 y Solicitud No. MX/a/2013/007745. Instituto Mexicano de la Propiedad Industrial 2020. Available online: http://www.gob.mx/impi (accessed on 8 September 2021).
- Lin, J.H.; Lu, A.Y. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmaco. Rev. 1997, 49, 403–449. [Google Scholar]
- International Conference on Harmonization. Harmonized Tripartite Guideline: Validation of Analytical Procedures: Text and Methodology, Q2 (R1). 2005, pp. 1–13. Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 7 September 2021).
- Talbi, A.; Zhao, D.; Liu, Q.; Li, J.; Fan, A.; Yang, W.; Han, X.; Chen, X. Pharmacokinetics, tissue distribution, excretion and plasma protein binding studies of wogonin in rats. Molecules 2014, 19, 5538–5549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skopp, G.; Pötsch, L.; Mauden, M.; Richter, B. Partition coefficient, blood to plasma ratio, protein binding and short-term stability of 11-nor-Delta(9)-carboxy tetrahydrocannabinol glucuronide. Forensic Sci. Int. 2002, 126, 17–23. [Google Scholar]
- DiMasi, J.A. Risks in new drug development: Approval success rates for investigational drugs. Clin. Pharmacol. Ther. 2001, 69, 297–307. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 4075, Mesalamine. PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Mesalamine (accessed on 7 September 2021).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 3715, Indomethacin. PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Indomethacin (accessed on 7 September 2021).
- Sehgal, P.; Colombel, J.F.; Aboubakr, A.; Narula, N. Systematic review: Safety of mesalazine in ulcerative colitis. Aliment. Pharmacol. Ther. 2018, 47, 1597–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargent, E.V.; Naumann, B.D.; Dolan, D.G.; Faria, E.C.; Schulman, L. The Importance of Human Data in the Establishment of Occupational Exposure Limits. Hum. Ecol. Risk Assess. 2002, 8, 805–822. [Google Scholar] [CrossRef]
- Tozaki, H.; Fujita, T.; Odoriba, T.; Terabe, A.; Okabe, S.; Muranishi, S.; Yamamoto, A. Validation of a pharmacokinetic model of colon-specific drug delivery and the therapeutic effects of chitosan capsules containing 5-aminosalicylic acid on 2,4,6-trinitrobenzenesulphonic acid-induced colitis in rats. J. Pharm. Pharmacol. 1999, 51, 1107–1112. [Google Scholar] [CrossRef]
- Fan, J.; de Lannoy, I.A.M. Pharmacokinetics. Biochem. Pharmacol. 2014, 87, 93–120. [Google Scholar]
- Kendrick, J. Basic Pharmacokinetics. Can. J. Hosp. Pharm. 2010, 63, 55. [Google Scholar] [CrossRef]
- Cataldi, M.; Vigliotti, C.; Mosca, T.; Cammarota, M.; Capone, D. Emerging Role of the Spleen in the Pharmacokinetics of Monoclonal Antibodies, Nanoparticles and Exosomes. Int. J. Mol. Sci. 2017, 18, 1249. [Google Scholar] [CrossRef] [Green Version]
- Steiniger, B.; Bette, M.; Schwarzbach, H. The open microcirculation in human spleens: A three-dimensional approach. J. Histochem. Cytochem. 2011, 59, 639–648. [Google Scholar]
- Li, X.; Lu, C.; Yang, Y.; Yu, C.; Rao, Y. Site-specific targeted drug delivery systems for the treatment of inflammatory bowel disease. Biomed. Pharmacother. 2020, 129, 110486. [Google Scholar] [CrossRef]
- Sinha, V.R.; Kumria, R. Colonic drug delivery: Prodrug approach. Pharm. Res. 2001, 18, 557–564. [Google Scholar]
- Davies, J.E. The pharmacological basis of therapeutics. Occup. Environ. Med. 2007, 64, e2. [Google Scholar] [CrossRef]
- Hallare, J.; Gerriets, V. Half Life. Available online: https://pubmed.ncbi.nlm.nih.gov/32119385/ (accessed on 9 September 2021).
- Oliveira, L.; Cohen, R.D. Maintaining remission in ulcerative colitis--role of once daily extended-release mesalamine. Drug Des. Devel. Ther. 2011, 5, 111–116. [Google Scholar]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef]
- Ferdowsian, H.R.; Beck, N. Ethical and scientific considerations regarding animal testing and research. PLoS ONE 2011, 6, e24059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, D.B. Humane Endpoints in Animal Experimentation for Biomedical Research: Ethical, Legal and Practical Aspects. 1998, pp. 5–12. Available online: https://tierschutz.charite.de/fileadmin/user_upload/microsites/sonstige/tierschutz/Humane_Endpoints-MORTON.pdf (accessed on 9 September 2021).
- Organization for Economic Cooperation and Development. OECD, Test No. 425: Acute Oral Toxicity: Up-and-Down Procedure; OECD: Paris, France, 2008; pp. 1–27. [Google Scholar]
- Food and Drug Administration. Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services. Fed. Regist. 2001, 66, 28526–28527. [Google Scholar]
- Singh, S.S. Preclinical pharmacokinetics: An approach towards safer and efficacious drugs. Curr. Drug Metab. 2006, 7, 165–182. [Google Scholar]
- Allinson, J.L. Clinical biomarker validation. Bioanalysis 2018, 10, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Sjöquist, B.; Ahnfelt, N.; Andersson, S.; D’argy, R.; Fjellner, G.; Hatsuoka, M.; Ljungstedt-Påhlman, I. Pharmacokinetics of Salazosulfapyridine (Sulfasalazine, SASP). V. Pharmacokinetics of SASP after a single intravenous or oral administration in the dog. Drug Metab. Pharmacokinet. 1991, 6, 491–507. [Google Scholar]
- Romero-Castro, A.; Gutiérrez-Sánchez, M.; Correa-Basurto, J.; Rosales Hernández, M.C.; Padilla, Martínez, I.I.; Mendieta-Wejebe, J.E. Pharmacokinetics in Wistar Rats of 5-[(4-Carboxybutanoyl)Amino]-2-Hydroxybenzoic Acid: A Novel Synthetic Derivative of 5-Aminosalicylic Acid (5-ASA) with Possible Anti-Inflammatory Activity. PLoS ONE 2016, 11, e0159889. [Google Scholar] [CrossRef] [PubMed]
- Scheife, R.T. Protein binding: What does it mean? DICP 1989, 23, S27–S31. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nominal Concentration (µg/mL) | Measured Concentration (µg/mL) Expressed as the Mean ± SD | Accuracy (%) | Precision (C.V., %) |
|---|---|---|---|
| Intra-day | |||
| 0.1 | 0.09 ± 0.01 | 89.3 | 14.9 |
| 10 | 9.0 ± 0.4 | 89.5 | 4.4 |
| 100 | 99.5 ± 6.6 | 99.5 | 6.6 |
| Inter-day | |||
| 0.1 | 0.09 ± 0.01 | 89.1 | 11.1 |
| 10 | 8.9 ± 0.5 | 81.5 | 5.9 |
| 100 | 99.3 ± 3.7 | 94.9 | 3.7 |
| Nominal Concentration (µg/mL) | Recovery (%), Expressed as the Mean ± SD | C.V. (%) |
|---|---|---|
| 0.1 | 89.3 ± 0.0 | 14.9 |
| 10 | 90.5 ± 0.4 | 4.4 |
| 100 | 99.5 ± 6.6 | 6.6 |
| Condition 1, Room Temperature (25 °C) | ||||||
| Concentration (µg/mL) | Difference at 12 h (%) * | C.V. (%) | Difference at 24 h (%) * | C.V. (%) | Difference at 48 h (%) * | C.V. (%) |
| 0.1 | 10.7 | 14.9 | 3.2 | 14.5 | 3.8 | 8.9 |
| 0.5 | 3.2 | 11.9 | 7.8 | 9.3 | 4.1 | 6.2 |
| 10 | 9.5 | 4.4 | 6.5 | 8.8 | 3.7 | 4.0 |
| 100 | 0.5 | 6.6 | 6.5 | 7.2 | 5.4 | 1.5 |
| Condition 2, Refrigeration (4 °C) | ||||||
| Concentration (µg/mL) | Difference at 12 h (%) * | C.V. (%) | Difference at 24 h (%) * | C.V. (%) | Difference at 48 h (%) * | C.V. (%) |
| 0.1 | 5.1 | 13.4 | 14.0 | 11.3 | 4.3 | 6.8 |
| 0.5 | 4.0 | 6.7 | 3.6 | 7.2 | 5.3 | 8.9 |
| 10 | 10.1 | 7.9 | 13.1 | 5.3 | 9.3 | 6.4 |
| 100 | 1.2 | 5.3 | 0.2 | 6.3 | 5.2 | 2.3 |
| Condition 3, Three Freeze–Thaw Cycles (From −20 to +25 °C) | ||||||
| Concentration (µg/mL) | Difference at 12 h (%) * | C.V. (%) | Difference at 24 h (%) * | C.V. (%) | Difference at 48 h (%) * | C.V. (%) |
| 0.1 | 5.1 | 5.3 | 1.76 | 14.2 | 3.4 | 12.7 |
| 0.5 | 0.1 | 7.9 | 0.3 | 11.5 | 1.0 | 2.2 |
| 10 | 0.7 | 6.7 | 1.2 | 2.6 | 3.2 | 1.9 |
| 100 | 0.1 | 13.4 | 0.6 | 1.6 | 2.0 | 1.0 |
| Parameters * | Units | p.o. | i.p. | i.v. |
|---|---|---|---|---|
| Ka | h−1 | 0.9 ± 0.1 | 1.4 ± 0.2 | - |
| Cmax | µg/mL | 2.8 ± 0.1 | 22.0 ± 1.0 | - |
| tmax | min | 33 ± 3 | 33 ± 5 | - |
| Cp0 | µg/mL | - | - | 11.0 ± 1.9 |
| Ke | h−1 | 0.4 ± 0.1 | 0.5 ± 0.1 | 0.8 ± 0.3 |
| Vd | L/kg | - | - | 5.2 ± 0.9 |
| CL | mL/min | - | - | 24 ± 5 |
| t1/2a | h | 1.4 ± 0.3 | 1.3 ± 0.5 | - |
| t1/2e | h | 2.5 ± 0.6 | 1.4 ± 0.2 | 0.9 ± 0.2 |
| MRT | h | 1.7 ± 0.2 | 3.1 ± 0.9 | 1.7 ± 0.4 |
| AUCtot | µg min−1/mL | 618 ± 63 | 2780 ± 69 | 806 ± 72 |
| Concentration (µg/mL) | Percentage of Unbound Fraction ± SD (%) | Percentage of Bound Fraction ± SD (%) |
|---|---|---|
| 1 | 96.3 ± 1.4 | 3.7 ± 1.1 |
| 5 | 94.2 ± 2.4 | 5.8 ± 2.4 |
| 10 | 93.4 ± 1.5 | 6.6 ± 1.5 |
| 20 | 94.8 ± 2.3 | 5.2 ± 2.3 |
| Concentration (µg/mL) | BP Ratio ± SD |
|---|---|
| 5 | 0.54 ± 0.02 |
| 10 | 0.40 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutiérrez-Sánchez, M.; Romero-Castro, A.; Correa-Basurto, J.; Rosales-Hernández, M.C.; Padilla-Martínez, I.I.; Mendieta-Wejebe, J.E. Preclinical Pharmacokinetics and Acute Toxicity in Rats of 5-{[(2E)-3-Bromo-3-carboxyprop-2-enoyl]amino}-2-hydroxybenzoic Acid: A Novel 5-Aminosalicylic Acid Derivative with Potent Anti-Inflammatory Activity. Molecules 2021, 26, 6801. https://doi.org/10.3390/molecules26226801
Gutiérrez-Sánchez M, Romero-Castro A, Correa-Basurto J, Rosales-Hernández MC, Padilla-Martínez II, Mendieta-Wejebe JE. Preclinical Pharmacokinetics and Acute Toxicity in Rats of 5-{[(2E)-3-Bromo-3-carboxyprop-2-enoyl]amino}-2-hydroxybenzoic Acid: A Novel 5-Aminosalicylic Acid Derivative with Potent Anti-Inflammatory Activity. Molecules. 2021; 26(22):6801. https://doi.org/10.3390/molecules26226801
Chicago/Turabian StyleGutiérrez-Sánchez, Mara, Aurelio Romero-Castro, José Correa-Basurto, Martha Cecilia Rosales-Hernández, Itzia Irene Padilla-Martínez, and Jessica Elena Mendieta-Wejebe. 2021. "Preclinical Pharmacokinetics and Acute Toxicity in Rats of 5-{[(2E)-3-Bromo-3-carboxyprop-2-enoyl]amino}-2-hydroxybenzoic Acid: A Novel 5-Aminosalicylic Acid Derivative with Potent Anti-Inflammatory Activity" Molecules 26, no. 22: 6801. https://doi.org/10.3390/molecules26226801
APA StyleGutiérrez-Sánchez, M., Romero-Castro, A., Correa-Basurto, J., Rosales-Hernández, M. C., Padilla-Martínez, I. I., & Mendieta-Wejebe, J. E. (2021). Preclinical Pharmacokinetics and Acute Toxicity in Rats of 5-{[(2E)-3-Bromo-3-carboxyprop-2-enoyl]amino}-2-hydroxybenzoic Acid: A Novel 5-Aminosalicylic Acid Derivative with Potent Anti-Inflammatory Activity. Molecules, 26(22), 6801. https://doi.org/10.3390/molecules26226801