
Interaction Energy Analysis of Monovalent Inorganic Anions in Bulk Water Versus Air/Water Interface



Abstract
1. Introduction
2. Computational Methods
2.1. Classical MD Simulations
2.2. Symmetry-Adapted Perturbation Theory
2.3. Polarization and Charge Transfer
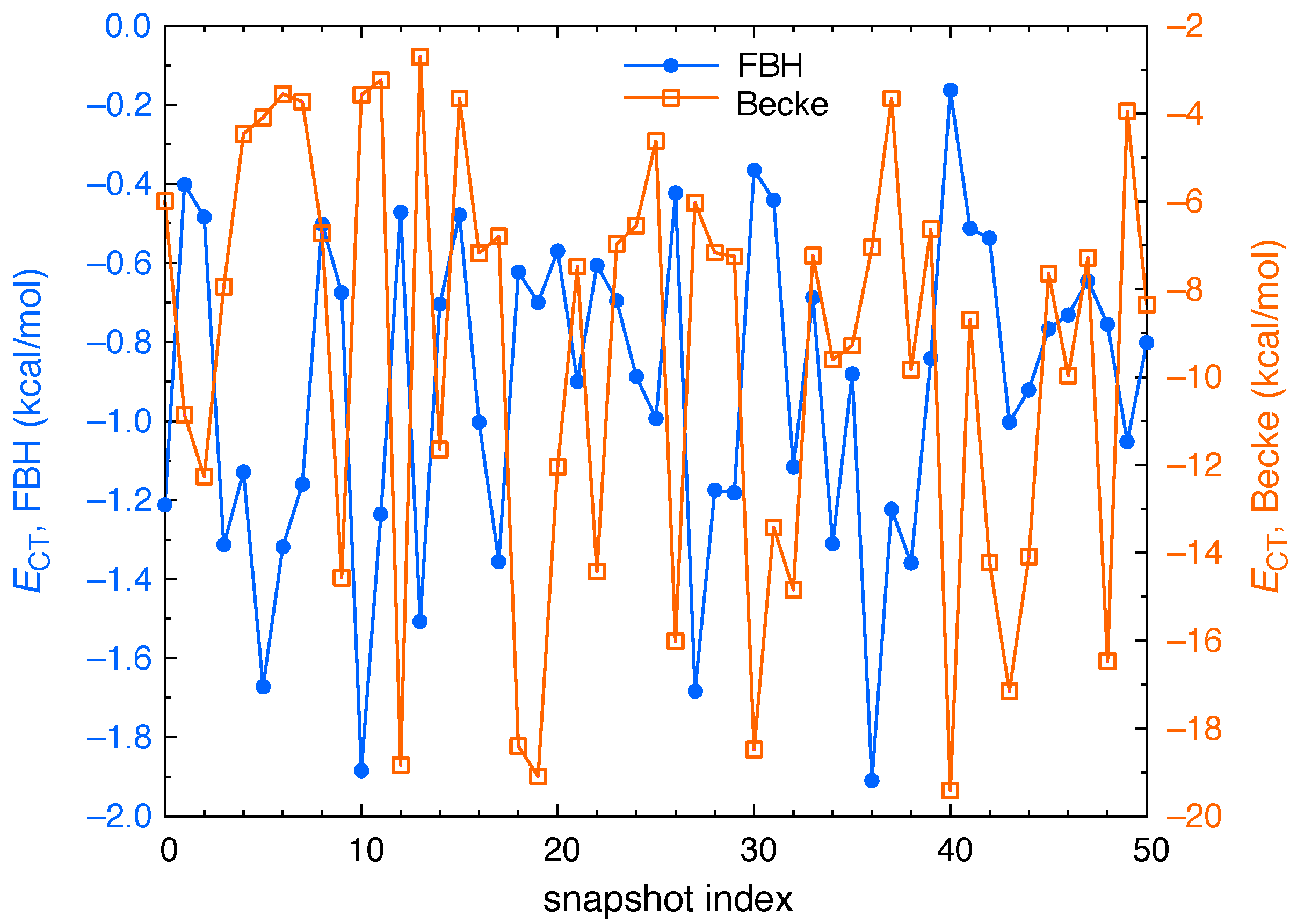
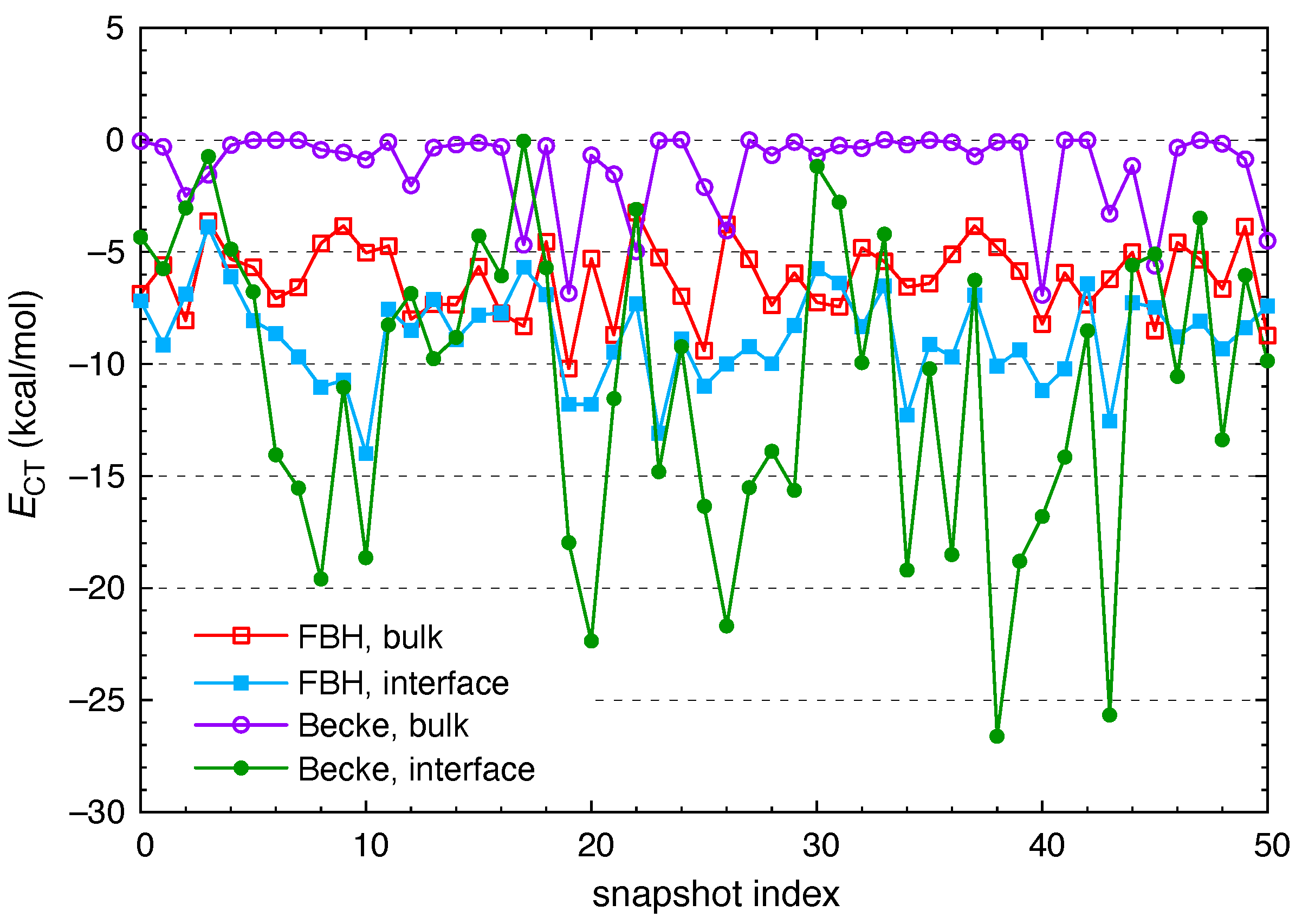
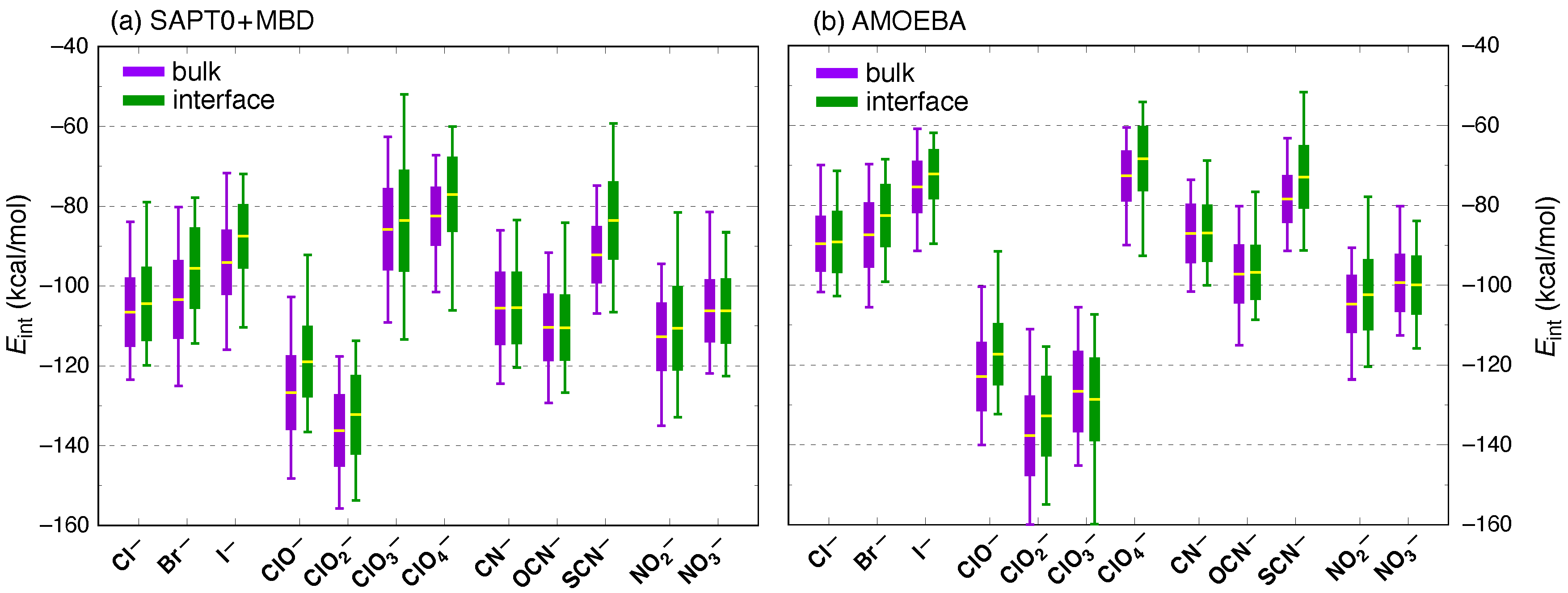
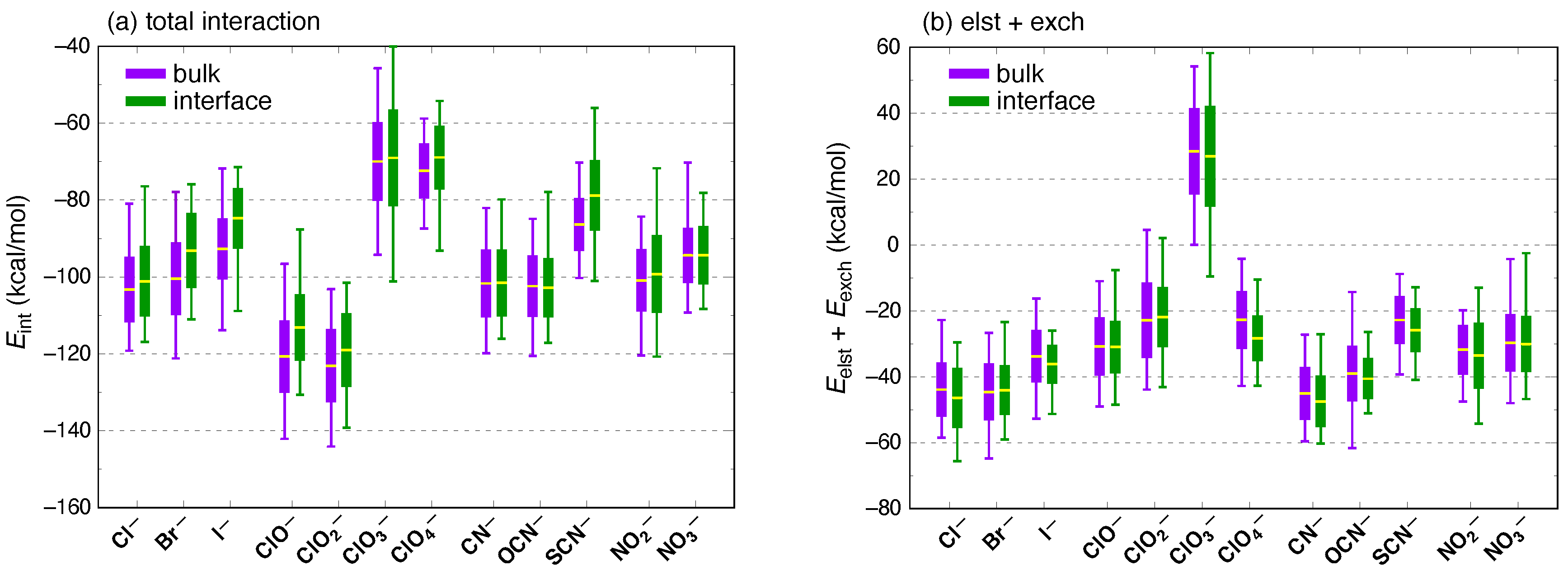
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Verreault, D.; Hua, W.; Allen, H.C. From conventional to phase-sensitive vibrational sum frequency generation spectroscopy: Probing water organization at aqueous interfaces. J. Phys. Chem. Lett. 2012, 3, 3012–3028. [Google Scholar] [CrossRef]
- Tang, F.; Ohto, T.; Sun, S.; Rouxel, J.R.; Imoto, S.; Backus, E.H.G.; Mukamel, S.; Bonn, M.; Nagata, Y. Molecular structure and modeling of water–air and ice–air interfaces monitored by sum-frequency generation. Chem. Rev. 2020, 120, 3633–3667. [Google Scholar] [CrossRef]
- Raymond, E.A.; Richmond, G.L. Probing the molecular structure and bonding of the surface of aqueous salt solutions. J. Phys. Chem. B 2004, 108, 5051–5059. [Google Scholar] [CrossRef]
- Liu, D.; Ma, G.; Levering, L.M.; Allen, H.C. Vibrational spectroscopy of aqueous sodium halide solutions and air–liquid interfaces: Observation of increased interfacial depth. J. Phys. Chem. B 2004, 108, 2252–2260. [Google Scholar] [CrossRef]
- Mucha, M.; Frigato, T.; Levering, L.M.; Allen, H.C.; Tobias, D.J.; Dang, L.X.; Jungwirth, P. Unified molecular picture of the surfaces of aqueous acid, base, and salt solutions. J. Phys. Chem. B 2005, 109, 7617–7623. [Google Scholar] [CrossRef]
- Garrett, B. Ions at the air/water interface. Science 2004, 303, 1146–1147. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.K.; Herbert, J.M. Probing interfacial effects on ionization energies: The surprising banality of anion–water hydrogen bonding at the air/water interface. J. Am. Chem. Soc. 2021, 143, 10189–10202. [Google Scholar] [CrossRef]
- Collins, K.D. Charge density-dependent strength of hydration and biological structure. Biophys. J. 1997, 72, 65–76. [Google Scholar] [CrossRef]
- Jungwirth, P.; Tobias, D.J. Ions at the air/water interface. J. Phys. Chem. B 2002, 106, 6361–6373. [Google Scholar] [CrossRef]
- Dang, L.X. Computational study of ion binding to the liquid interface of water. J. Phys. Chem. B 2002, 106, 10388–10394. [Google Scholar] [CrossRef]
- Herce, D.H.; Perera, L.; Darden, T.A.; Sagui, C. Surface solvation for an ion in a water cluster. J. Chem. Phys. 2005, 122, 024513. [Google Scholar] [CrossRef] [PubMed]
- Archontis, G.; Leontidis, E.; Andreou, G. Attraction of iodide ions by the free water surface, revealed by simulations with a polarizable force field based on Drude oscillators. J. Phys. Chem. B 2005, 109, 17957–17966. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jungwirth, P.; Tobias, D.J. Specific ion effects at the air/water interface. Chem. Rev. 2006, 106, 1259–1281. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.M.; Dang, L.X. Recent advances in molecular simulations of ion solvation at liquid interfaces. Chem. Rev. 2006, 106, 1305–1322. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, T.; Morita, A. Molecular dynamics study of gas–liquid aqueous sodium halide interfaces. I. Flexible and polarizable molecular modeling and interfacial properties. J. Phys. Chem. C 2007, 111, 721–737. [Google Scholar] [CrossRef]
- Dweik, J.; Srour, M.; Karaky, K.; Kobeissi, M.; Joumaa, W.; Abou-Saleh, K. Molecular simulation of ion transport at the water/vapor interface. Open J. Phys. Chem. 2012, 2, 147–155. [Google Scholar] [CrossRef]
- Sun, L.; Li, X.; Tu, Y.; Ågren, H. Origin of ion selectivity at the air/water interface. Phys. Chem. Chem. Phys. 2015, 17, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, P.; Curtis, J.E.; Tobias, D.J. Polarizability and aqueous solvation of the sulfate dianion. Chem. Phys. Lett. 2003, 367, 704–710. [Google Scholar] [CrossRef]
- Jubb, A.M.; Hua, W.; Allen, H.C. Environmental chemistry at vapor/water interfaces: Insights from vibrational sum frequency generation spectroscopy. Annu. Rev. Phys. Chem. 2012, 63, 107–130. [Google Scholar] [CrossRef]
- Jungwirth, P.; Tobias, D.J. Molecular structure of salt solutions: A new view of the interface with implications for heterogeneous atmospheric chemistry. J. Phys. Chem. B 2001, 105, 10468–10472. [Google Scholar] [CrossRef]
- Caleman, C.; Hub, J.S.; van Maaren, P.J.; van der Spoel, D. Atomistic simulation of ion solvation in water explains surface preference of halides. Proc. Natl. Acad. Sci. USA 2011, 108, 6838–6842. [Google Scholar] [CrossRef]
- Petersen, P.B.; Saykally, R.J.; Mucha, M.; Jungwirth, P. Enhanced concentration of polarizable anions at the liquid water surface: SHG spectroscopy and MD simulations of sodium thiocyanide. J. Phys. Chem. B 2015, 109, 10915–10921, Erratum: ibid. 2015, 109, 13402. [Google Scholar] [CrossRef]
- Horinek, D.; Herz, A.; Vrbka, L.; Sedlmeier, F.; Mamatkulov, S.I.; Netz, R.R. Specific ion adsorption at the air/water interface: The role of hydrophobic solvation. Chem. Phys. Lett. 2009, 479, 173–183. [Google Scholar] [CrossRef]
- Netz, R.R.; Horinek, D. Progress in modeling of ion effects at the vapor/water interface. Annu. Rev. Phys. Chem. 2012, 63, 401–418. [Google Scholar] [CrossRef]
- Baer, M.D.; Mundy, C.J. Toward an understanding of the specific ion effect using density functional theory. J. Phys. Chem. Lett. 2011, 2, 1088–1093. [Google Scholar] [CrossRef]
- Baer, M.D.; Stern, A.C.; Levin, Y.; Tobias, D.J.; Mundy, C.J. Electrochemical surface potential due to classical point charge models drives anion adsorption to the air-water interface. J. Phys. Chem. Lett. 2012, 3, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Hu, Y.; Patel, S.; Wan, H. Spherical monovalent ions at aqueous liquid–vapor interfaces: Interfacial stability and induced interface fluctuations. J. Phys. Chem. B 2013, 117, 11732–11742. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ou, S.C.; Cui, D.; Patel, S. Molecular modeling of ions at interfaces: Exploring similarities to hydrophobic solvation through the lens of induced aqueous interfacial fluctuations. Phys. Chem. Chem. Phys. 2016, 18, 30357–30365. [Google Scholar] [CrossRef]
- Ishiyama, T.; Imamura, T.; Morita, A. Theoretical studies of structures and vibrational sum frequency generation spectra at aqueous interfaces. Chem. Rev. 2014, 114, 8447–8470. [Google Scholar] [CrossRef] [PubMed]
- Levin, Y.; dos Santos, A.P.; Diehl, A. Ions at the air-water interface: An end to a hundred-year-old mystery? Phys. Rev. Lett. 2009, 103, 257802. [Google Scholar] [CrossRef]
- Levin, Y.; dos Santos, A.P. Ions at hydrophobic interfaces. J. Phys. Condens. Matt. 2014, 26, 203101. [Google Scholar] [CrossRef] [PubMed]
- Wick, C.D.; Xantheas, S.S. Computational investigation of the first solvation shell structure of interfacial and bulk aqueous chloride and iodide ions. J. Phys. Chem. B 2009, 113, 4141–4146. [Google Scholar] [CrossRef] [PubMed]
- Baer, M.D.; Mundy, C.J. An ab initio approach to understanding the specific ion effect. Faraday Discuss. 2013, 160, 89–101. [Google Scholar] [CrossRef]
- Viswanath, P.; Motschmann, H. Oriented thiocyanate anions at the air–electrolyte interface and its implications on interfacial water—A vibrational sum frequency spectroscopy study. J. Phys. Chem. C 2007, 111, 4484–4486. [Google Scholar] [CrossRef]
- Viswanath, P.; Motschmann, M. Effect of interfacial presence of oriented thiocyanate on water structure. J. Phys. Chem. C 2008, 112, 2099–2103. [Google Scholar] [CrossRef]
- Cooper, R.J.; O’Brien, J.T.; Chang, T.M.; Williams, E.R. Structural and electrostatic effects at the surfaces of size- and charge-selected aqueous nanodrops. Chem. Sci. 2017, 8, 5201–5213. [Google Scholar] [CrossRef]
- Bakker, H.J. Structural dynamics of aqueous salt solutions. Chem. Rev. 2008, 108, 1456–1473. [Google Scholar] [CrossRef]
- Omta, A.W.; Kropman, M.F.; Woutersen, S.; Bakker, H.J. Negligible effect of ions on the hydrogen-bond structure in liquid water. Science 2003, 301, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Fayer, M.D. Hydrogen bond dynamics in aqueous NaBr solutions. Proc. Natl. Acad. Sci. USA 2007, 104, 16731–16738. [Google Scholar] [CrossRef]
- Moilanen, D.E.; Wong, D.; Rosenfeld, D.E.; Fenn, E.E.; Fayer, M.D. Ion–water hydrogen-bond switching observed with 2D IR vibrational echo chemical exchange spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 375–380. [Google Scholar] [CrossRef]
- Botti, A.; Bruni, F.; Imberti, S.; Ricci, M.A.; Soper, A.K. Ions in water: The microscopic structure of concentrated NaOH solutions. J. Chem. Phys. 2004, 120, 10154–10162. [Google Scholar] [CrossRef]
- Botti, A.; Bruni, F.; Imberti, S.; Ricci, M.A.; Soper, A.K. Ions in water: The microscopic structure of a concentrated HCl solution. J. Chem. Phys. 2004, 121, 7840–7848. [Google Scholar] [CrossRef] [PubMed]
- Imberti, S.; Botti, A.; Bruni, F.; Cappa, G.; Ricci, M.A.; Soper, A.K. Ions in water: The microscopic structure of concentrated hydroxide solutions. J. Chem. Phys. 2005, 122, 194509. [Google Scholar] [CrossRef]
- Mancinelli, R.; Botti, A.; Bruni, F.; Ricci, M.A.; Soper, A.K. Perturbation of water structure due to monovalent ions in solution. Phys. Chem. Chem. Phys. 2007, 9, 2959–2967. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A. Effects of ion atmosphere on hydrogen-bond dynamics in aqueous electrolyte solutions. Phys. Rev. Lett. 2000, 85, 768–771. [Google Scholar] [CrossRef]
- Chialvo, A.A.; Simonson, J.M. The effect of salt concentration on the structure of water in CaCl2 aqueous solutions. J. Mol. Liq. 2004, 112, 99–105. [Google Scholar] [CrossRef]
- Tielrooij, K.J.; Garcia-Araez, N.; Bonn, M.; Bakker, H.J. Cooperativity in ion hydration. Science 2010, 328, 1006–1009. [Google Scholar] [CrossRef]
- Migliorati, V.; Mancini, G.; Chillemi, G.; Zitolo, A.; D’Angelo, P. Effect of the Zn2+ and Hg2+ ions on the structure of liquid water. J. Phys. Chem. A 2011, 115, 4798–4803. [Google Scholar] [CrossRef] [PubMed]
- Seidel, R.; Winter, B.; Bradforth, S.E. Valence electronic structure of aqueous solutions: Insights from photoelectron spectroscopy. Annu. Rev. Phys. Chem. 2016, 67, 283–305. [Google Scholar] [CrossRef] [PubMed]
- Lao, K.U.; Herbert, J.M. Accurate and efficient quantum chemistry calculations of noncovalent interactions in many-body systems: The XSAPT family of methods. J. Phys. Chem. A 2015, 119, 235–253. [Google Scholar] [CrossRef]
- Carter-Fenk, K.; Lao, K.U.; Herbert, J.M. Predicting and understanding non-covalent interactions using novel forms of symmetry-adapted perturbation theory. Acc. Chem. Res. 2021, 54, 3679–3690. [Google Scholar] [CrossRef] [PubMed]
- Francisco, E.; Pendás, A.M. Energy partition analyses: Symmetry-adapted perturbation theory and other techniques. In Non-Covalent Interactions in Quantum Chemistry and Physics; de la Roza, A.O., DiLabio, G.A., Eds.; Elsevier: Amsterdam, The Netherlandas, 2017; Chapter 2; pp. 27–64. [Google Scholar] [CrossRef]
- Patkowski, K. Recent developments in symmetry-adapted perturbation theory. WIREs Comput. Mol. Sci. 2020, 10, e1452. [Google Scholar] [CrossRef]
- Ren, P.; Ponder, J.W. Polarizable atomic multipole water model for molecular mechanics simulation. J. Phys. Chem. B 2003, 107, 5933–5947. [Google Scholar] [CrossRef]
- Grossfield, A.; Ren, P.; Ponder, J.W. Ion solvation thermodynamics from simulation with a polarizable force field. J. Am. Chem. Soc. 2003, 125, 15671–15682. [Google Scholar] [CrossRef]
- Ren, P.; Wu, C.; Ponder, J.W. Polarizable atomic multipole-based molecular mechanics for organic molecules. J. Chem. Theory Comput. 2011, 7, 3143–3161. [Google Scholar] [CrossRef]
- Rackers, J.A.; Wang, Z.; Lu, C.; Laury, M.L.; Lagardère, L.; Schnieders, M.J.; Piquemal, J.P.; Ren, P.; Ponder, J.W. Tinker 8: Software tools for molecular design. J. Chem. Theory Comput. 2018, 14, 5273–5289. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]
- Gray, M.; Herbert, J.M. Simplified tuning of long-range corrected density functionals for symmetry-adapted perturbation theory. J. Chem. Phys. 2021, 155, 034103. [Google Scholar] [CrossRef]
- Lao, K.U.; Herbert, J.M. Atomic orbital implementation of extended symmetry-adapted perturbation theory (XSAPT) and benchmark calculations for large supramolecular complexes. J. Chem. Theory Comput. 2018, 14, 2955–2978. [Google Scholar] [CrossRef] [PubMed]
- Ambrosetti, A.; Reilly, A.M.; DiStasio, R.A., Jr.; Tkatchenko, A. Long-range correlation energy calculated from coupled atomic response functions. J. Chem. Phys. 2014, 140, 18A508. [Google Scholar] [CrossRef] [PubMed]
- Carter-Fenk, K.; Lao, K.U.; Liu, K.Y.; Herbert, J.M. Accurate and efficient ab initio calculations for supramolecular complexes: Symmetry-adapted perturbation theory with many-body dispersion. J. Phys. Chem. Lett. 2019, 10, 2706–2714. [Google Scholar] [CrossRef]
- Gray, M.; Herbert, J.M. Comprehensive basis-set testing of extended symmetry-adapted perturbation theory. 2021. 2021. [Google Scholar] [CrossRef]
- Epifanovsky, E.; Gilbert, A.T.B.; Feng, X.; Lee, J.; Mao, Y.; Mardirossian, N.; Pokhilko, P.; White, A.F.; Coons, M.P.; Dempwolff, A.L.; et al. Software for the frontiers of quantum chemistry: An overview of developments in the Q-Chem 5 package. J. Chem. Phys. 2021, 155, 084801. [Google Scholar] [CrossRef] [PubMed]
- Herbert, J.M. Neat, simple, and wrong: Debunking electrostatic fallacies regarding noncovalent interactions. J. Phys. Chem. A 2021, 125, 7125–7137. [Google Scholar] [CrossRef]
- Rohrdanz, M.A.; Martins, K.M.; Herbert, J.M. A long-range-corrected density functional that performs well for both ground-state properties and time-dependent density functional theory excitation energies, including charge-transfer excited states. J. Chem. Phys. 2009, 130, 054112. [Google Scholar] [CrossRef] [PubMed]
- Lao, K.U.; Herbert, J.M. Symmetry-adapted perturbation theory with Kohn-Sham orbitals using non-empirically tuned, long-range-corrected density functionals. J. Chem. Phys. 2014, 140, 044108. [Google Scholar] [CrossRef] [PubMed]
- Hapka, M.; Rajchel, L.; Modrzejewski, M.; Chałasiǹski, G.; Szczȩśniak, M.M. Tuned range-separated hybrid functionals in the symmetry-adapted perturbation theory. J. Chem. Phys. 2014, 141, 134120. [Google Scholar] [CrossRef] [PubMed]
- Baer, R.; Livshits, E.; Salzner, U. Tuned range-separated hybrids in density functional theory. Annu. Rev. Phys. Chem. 2010, 61, 85–109. [Google Scholar] [CrossRef] [PubMed]
- Alam, B.; Morrison, A.F.; Herbert, J.M. Charge separation and charge transfer in the low-lying excited states of pentacene. J. Phys. Chem. C 2020, 124, 24653–24666. [Google Scholar] [CrossRef]
- Uhlig, F.; Herbert, J.M.; Coons, M.P.; Jungwirth, P. Optical spectroscopy of the bulk and interfacial hydrated electron from ab initio calculations. J. Phys. Chem. A 2014, 118, 7507–7515. [Google Scholar] [CrossRef]
- Jacobson, L.D.; Herbert, J.M. An efficient, fragment-based electronic structure method for molecular systems: Self-consistent polarization with perturbative two-body exchange and dispersion. J. Chem. Phys. 2011, 134, 094118. [Google Scholar] [CrossRef]
- Herbert, J.M.; Jacobson, L.D.; Lao, K.U.; Rohrdanz, M.A. Rapid computation of intermolecular interactions in molecular and ionic clusters: Self-consistent polarization plus symmetry-adapted perturbation theory. Phys. Chem. Chem. Phys. 2012, 14, 7679–7699. [Google Scholar] [CrossRef]
- Jacobson, L.D.; Richard, R.M.; Lao, K.U.; Herbert, J.M. Efficient monomer-based quantum chemistry methods for molecular and ionic clusters. Annu. Rep. Comput. Chem. 2013, 9, 25–58. [Google Scholar] [CrossRef]
- Liu, K.Y.; Carter-Fenk, K.; Herbert, J.M. Self-consistent charge embedding at very low cost, with application to symmetry-adapted perturbation theory. J. Chem. Phys. 2019, 151, 031102. [Google Scholar] [CrossRef] [PubMed]
- Moszyński, R.; Cybulski, S.M.; Chałasiński, G. Many-body theory of intermolecular induction interactions. J. Chem. Phys. 1994, 100, 4998–5010. [Google Scholar] [CrossRef]
- Lao, K.U.; Herbert, J.M. Energy decomposition analysis with a stable charge-transfer term for interpreting intermolecular interactions. J. Chem. Theory Comput. 2016, 12, 2569–2582. [Google Scholar] [CrossRef]
- Kaduk, B.; Kowalczyk, T.; Van Voorhis, T. Constrained density functional theory. Chem. Rev. 2012, 112, 321–370. [Google Scholar] [CrossRef] [PubMed]
- Řezáč, J.; de la Lande, A. Robust, basis-set independent method for the evaluation of charge-transfer energy in noncovalent complexes. J. Chem. Theory Comput. 2015, 11, 528–537. [Google Scholar] [CrossRef]
- Řezác, J.; de la Lande, A. On the role of charge transfer in halogen bonding. Phys. Chem. Chem. Phys. 2017, 19, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Herbert, J.M.; Carter-Fenk, K. Electrostatics, charge transfer, and the nature of the halide–water hydrogen bond. J. Phys. Chem. A 2021, 125, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Medvedev, N.N. The algorithm for three-dimensional Voronoi polyhedra. J. Comput. Phys. 1986, 67, 223–229. [Google Scholar] [CrossRef]
- Slater, J.C. Atomic radii in crystals. J. Chem. Phys. 1964, 41, 3199–3204. [Google Scholar] [CrossRef]
- Dasgupta, S.; Herbert, J.M. Standard grids for high-precision integration of modern density functionals: SG-2 and SG-3. J. Comput. Chem. 2017, 38, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Chen, C.L.; Van Voorhis, T. Configuration interaction based on constrained density functional theory: A multireference method. J. Chem. Phys. 2007, 127, 164119. [Google Scholar] [CrossRef] [PubMed]
- Richard, R.M.; Lao, K.U.; Herbert, J.M. Aiming for benchmark accuracy with the many-body expansion. Acc. Chem. Res. 2014, 47, 2828–2836. [Google Scholar] [CrossRef]
- Lao, K.U.; Liu, K.Y.; Richard, R.M.; Herbert, J.M. Understanding the many-body expansion for large systems. II. Accuracy considerations. J. Chem. Phys. 2016, 144, 164105. [Google Scholar] [CrossRef]
- Liu, K.Y.; Herbert, J.M. Understanding the many-body expansion for large systems. III. Critical role of four-body terms, counterpoise corrections, and cutoffs. J. Chem. Phys. 2017, 147, 161729. [Google Scholar] [CrossRef]
- Herbert, J.M. Fantasy versus reality in fragment-based quantum chemistry. J. Chem. Phys. 2019, 151, 170901. [Google Scholar] [CrossRef]
- Heindel, J.P.; Xantheas, S.S. The many-body expansion for aqueous systems revisited: I. Water–water interactions. J. Chem. Theory Comput. 2020, 16, 6843–6855. [Google Scholar] [CrossRef]
- Heindel, J.P.; Xantheas, S.S. The many-body expansion for aqueous systems revisited: II. Alkali metal and halide ion–water interactions. J. Chem. Theory Comput. 2021, 17, 2200–2216. [Google Scholar] [CrossRef]
- Tissandier, M.D.; Cowen, K.A.; Feng, W.Y.; Gundlach, E.; Cohen, M.H.; Earhart, A.D.; Coe, J.V.; Tuttle, T.R., Jr. The proton’s absolute aqueous enthalpy and Gibbs free energy of solvation from cluster-ion solvation data. J. Phys. Chem. A 1998, 102, 7787–7794, Erratum: ibid. 1998, 102, 9308. [Google Scholar] [CrossRef]
- Prasetyo, N.; Hünenberger, P.H.; Hofer, T.S. Single-ion thermodynamics from first principles: Calculation of the absolute hydration free energy and single-electrode potential of aqueous Li+ using ab initio quantum mechanical/molecular mechanical dynamics simulations. J. Chem. Theory Comput. 2018, 14, 6443–6459. [Google Scholar] [CrossRef]
- Malloum, A.; Fifen, J.J.; Conradie, J. Determination of the absolute solvation free energy and enthalpy of the proton in solutions. J. Mol. Liq. 2021, 322, 114919. [Google Scholar] [CrossRef]
- Herbert, J.M. Dielectric continuum methods for quantum chemistry. WIREs Comput. Mol. Sci. 2021, 11, e1519. [Google Scholar] [CrossRef]
- Rackers, J.A.; Wang, Q.; Liu, C.; Piquemal, J.P.; Ren, P.; Ponder, J.W. An optimized charge penetration model for use with the AMOEBA force field. Phys. Chem. Chem. Phys. 2017, 19, 276–291. [Google Scholar] [CrossRef]
- Deng, S.; Wang, Q.; Ren, P. Estimating and modeling charge transfer from the SAPT induction energy. J. Comput. Chem. 2017, 38, 2222–2231. [Google Scholar] [CrossRef]
- Jing, Z.; Liu, C.; Ren, P. Advanced electrostatic model for monovalent ions based on ab initio energy decompoosition. J. Chem. Inf. Model. 2021, 61, 2806–2817. [Google Scholar] [CrossRef]
- Elgengehi, S.M.; El-Taher, S.; Ibrahim, M.A.A.; El-Kelany, K.E. Unexpected favourable noncovalent interaction between chlorine oxyanions (ClOx−; x=1–4) and benzene: Benchmarking DFT and SAPT methods with respect to CCSD(T). Comput. Theor. Chem. 2021, 1199, 113214. [Google Scholar] [CrossRef]
- Eklund, L.; Hofer, T.S.; Persson, I. Structure and water exchange dynamics of hydrated oxo halo ions in aqueous solution using QMCF MD simulations, large angle X-ray scattering and EXAFS. Dalton Trans. 2015, 44, 1816–1828. [Google Scholar] [CrossRef] [PubMed]
- Eklund, L.; Hofer, T.S.; Pribil, A.B.; Rode, B.M.; Persson, I. On the structure and dynamics of the hydrated sulfite ion in aqueous solution—An ab initio QMCF MD simulation and large angle x-ray scattering study. Dalton Trans. 2012, 41, 5209–5216. [Google Scholar] [CrossRef]
- Lao, K.U.; Herbert, J.M. A simple correction for nonadditive dispersion within extended symmetry-adapted perturbation theory (XSAPT). J. Chem. Theory Comput. 2018, 14, 5128–5142. [Google Scholar] [CrossRef]
- Dobson, J.F. Beyond pairwise additivity in London dispersion interactions. Int. J. Quantum Chem. 2014, 114, 1157–1161. [Google Scholar] [CrossRef]
- Tong, Y.; Zhang, I.Y.; Campen, R.K. Experimentally quantifying anion polarizability at the air/water interface. Nat. Commun. 2018, 9, 1313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cremer, P.S. Interactions between macromolecules and ions: The Hofmeister series. Curr. Opin. Struc. Biol. 2006, 10, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, P.; Cremer, P.S. Beyond Hofmeister. Nat. Chem. 2014, 6, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Rembert, K.B.; Paterová, J.; Heyda, J.; Hilty, C.; Jungwirth, P.; Cremer, P.S. Molecular mechanisms of ion-specific effects on proteins. J. Am. Chem. Soc. 2012, 134, 10039–10046. [Google Scholar] [CrossRef] [PubMed]
- Mancinelli, R.; Botti, A.; Bruni, F.; Ricci, M.A.; Soper, A.K. Hydration of sodium, potassium, and chloride ions in solution and the concept of structure maker/breaker. J. Phys. Chem. B 2007, 111, 13570–13577. [Google Scholar] [CrossRef] [PubMed]
- Wick, C.D.; Lee, A.J.; Rick, S.W. How intermolecular charge transfer influences the air-water interface. J. Chem. Phys. 2012, 137, 154701. [Google Scholar] [CrossRef]
- Vácha, R.; Marsalek, O.; Willard, A.P.; Bonthuis, D.J.; Netz, R.R.; Jungwirth, P. Charge transfer between water molecules as the possible origin of the observed charging at the surface of pure water. J. Phys. Chem. Lett. 2012, 3, 107–111. [Google Scholar] [CrossRef]
- Samson, J.S.; Scheu, R.; Smolentsev, N.; Rick, S.W.; Roke, S. Sum frequency spectroscopy of the hydrophobic nanodroplet/water interface: Absence of hydroxyl ion and dangling OH bond signatures. Chem. Phys. Lett. 2014, 615, 124–131. [Google Scholar] [CrossRef]
- Poli, E.; Jong, K.H.; Hassanali, A. Charge transfer as a ubiquitous mechanism in determining the negative charge at hydrophobic interfaces. Nat. Commun. 2020, 11, 901. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ion | (kcal/mol) | (kcal/mol) | |||
|---|---|---|---|---|---|
| Bulk | Interface | Bulk | Interface | ||
| Cl− | −106.6 ± 8.4 | −104.4 ± 9.0 | −26.4 ± 2.4 | −24.6 ± 2.7 | |
| Br− | −103.4 ± 9.6 | −95.5 ± 9.9 | −24.6 ± 2.7 | −22.4 ± 3.3 | |
| I− | −94.1 ± 7.9 | −87.5 ± 7.8 | −22.3 ± 2.7 | −20.3 ± 2.8 | |
| ClO− | −126.7 ± 9.1 | −118.9 ± 8.7 | −47.0 ± 5.1 | −44.5 ± 4.3 | |
| −136.2 ± 8.8 | −132.2 ± 9.7 | −52.3 ± 4.7 | −50.2 ± 5.3 | ||
| −85.8 ± 10.0 | −83.6 ± 12.5 | −42.1 ± 4.1 | −41.7 ± 4.4 | ||
| −82.5 ± 7.1 | −77.0 ± 9.1 | −18.8 ± 1.8 | −16.2 ± 2.1 | ||
| −105.6 ± 8.9 | −105.4 ± 8.8 | −25.0 ± 2.6 | −23.8 ± 2.2 | ||
| −110.4 ± 8.2 | −110.4 ± 8.0 | −29.9 ± 3.0 | −29.3 ± 2.9 | ||
| −92.2 ± 6.9 | −83.5 ± 9.5 | −24.6 ± 2.2 | −22.4 ± 3.5 | ||
| −112.7 ± 8.3 | −110.5 ± 10.3 | −34.5 ± 3.2 | −33.0 ± 3.5 | ||
| −106.2 ± 7.6 | −106.2 ± 7.9 | −29.9 ± 3.2 | −29.5 ± 3.3 | ||
| Ion | Dipole | Energy Components (kcal/mol) a | ||||
|---|---|---|---|---|---|---|
| Moment (D) b | ||||||
| ClO− | 3.04 | −126.7 | −137.6 | 90.2 | −47.0 | −32.2 |
| 3.20 | −136.2 | −149.6 | 103.6 | −52.3 | −37.8 | |
| 2.46 | −85.8 | −120.6 | 125.1 | −42.1 | −48.2 | |
| 0.00 | −82.5 | −78.1 | 41.1 | −18.8 | −26.7 | |
| System | (kcal/mol) a | ||
|---|---|---|---|
| aiD3 | MBD b | ||
| ClO− | bulk | ||
| ClO− | interface | ||
| bulk | |||
| interface | |||
| bulk | |||
| interface | |||
| bulk | |||
| interface | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herbert, J.M.; Paul, S.K. Interaction Energy Analysis of Monovalent Inorganic Anions in Bulk Water Versus Air/Water Interface. Molecules 2021, 26, 6719. https://doi.org/10.3390/molecules26216719
Herbert JM, Paul SK. Interaction Energy Analysis of Monovalent Inorganic Anions in Bulk Water Versus Air/Water Interface. Molecules. 2021; 26(21):6719. https://doi.org/10.3390/molecules26216719
Chicago/Turabian StyleHerbert, John M., and Suranjan K. Paul. 2021. "Interaction Energy Analysis of Monovalent Inorganic Anions in Bulk Water Versus Air/Water Interface" Molecules 26, no. 21: 6719. https://doi.org/10.3390/molecules26216719
APA StyleHerbert, J. M., & Paul, S. K. (2021). Interaction Energy Analysis of Monovalent Inorganic Anions in Bulk Water Versus Air/Water Interface. Molecules, 26(21), 6719. https://doi.org/10.3390/molecules26216719