
Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
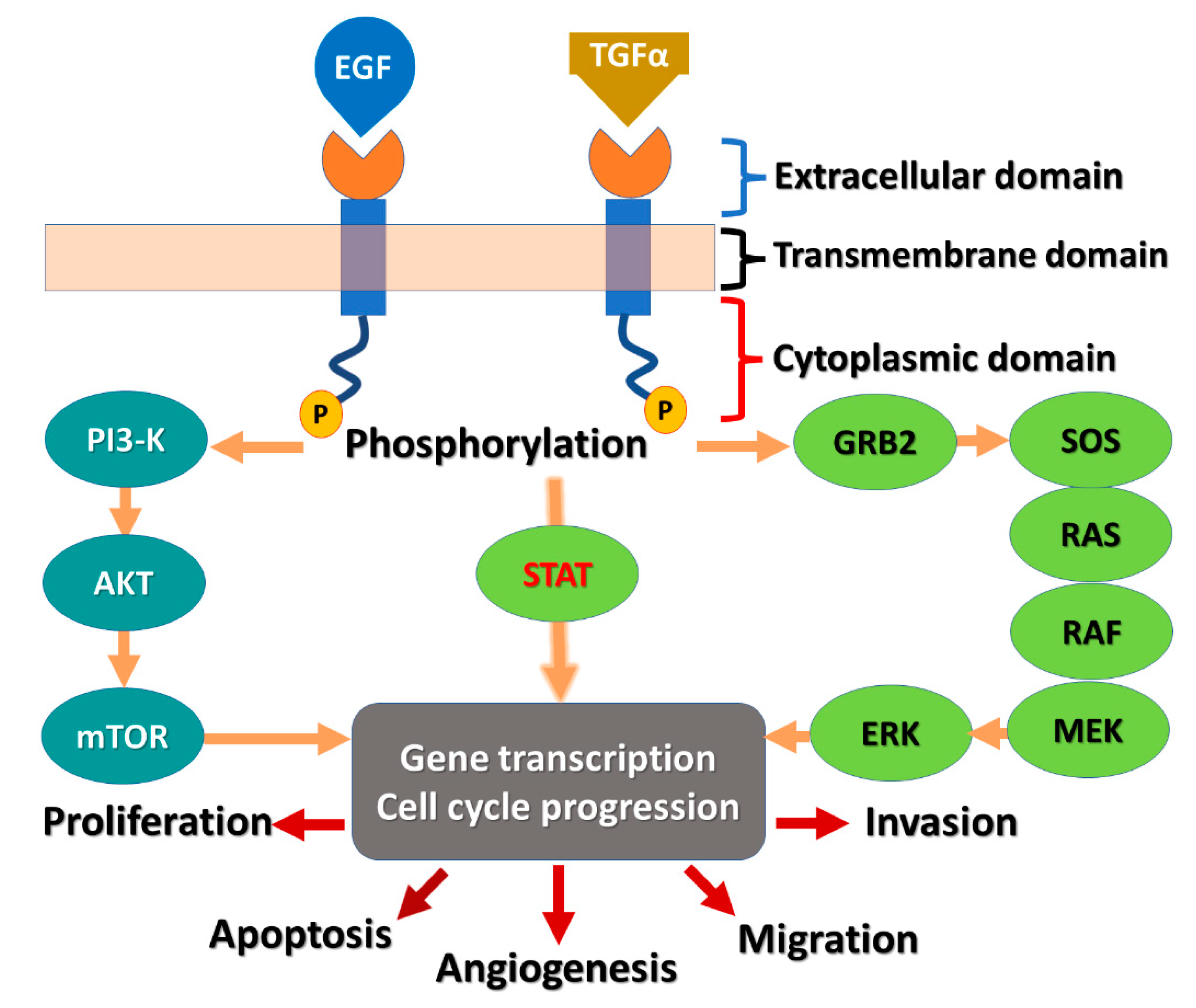
:1. Introduction
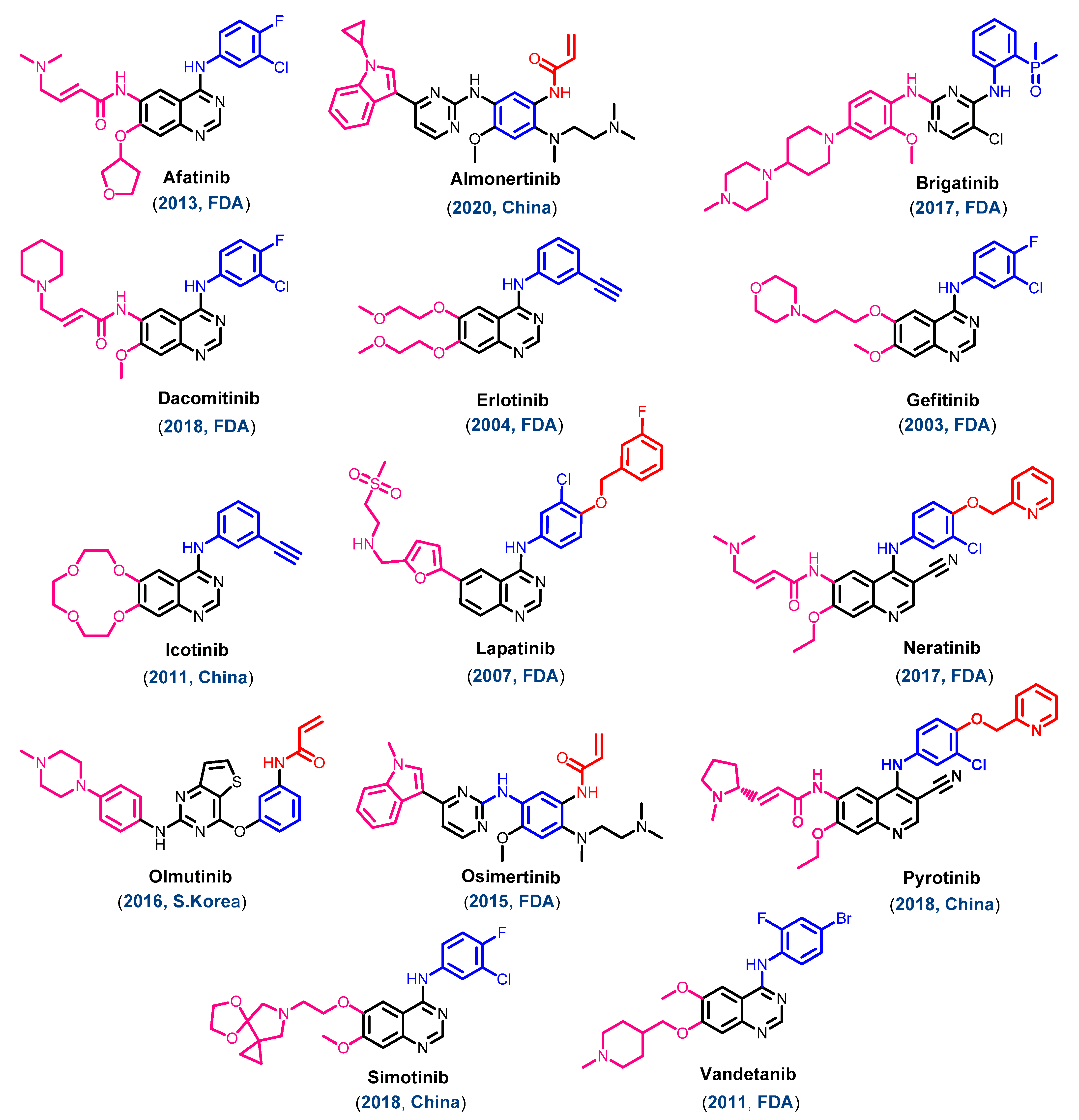
1.1. Classification of the Approved EGFR-TKIs
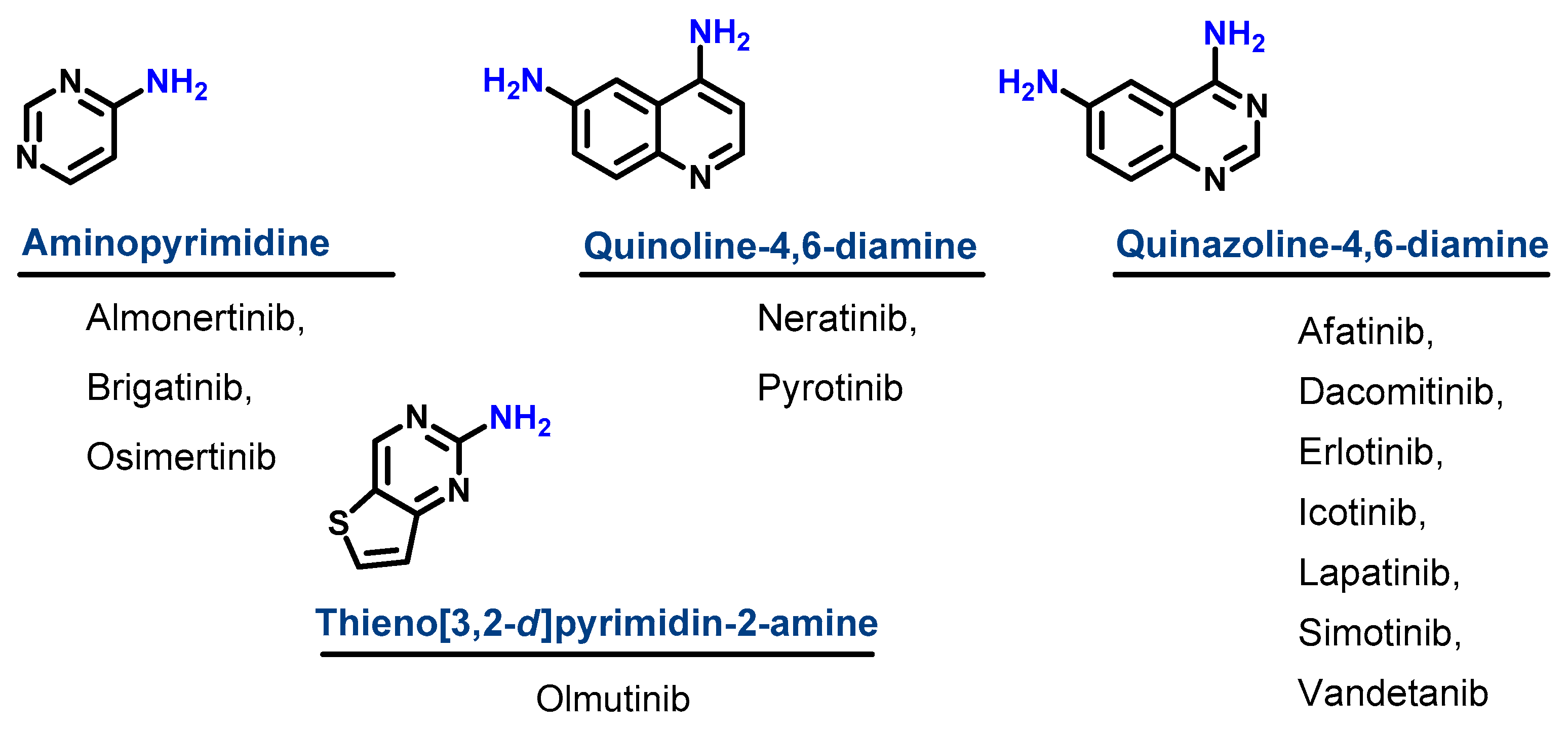
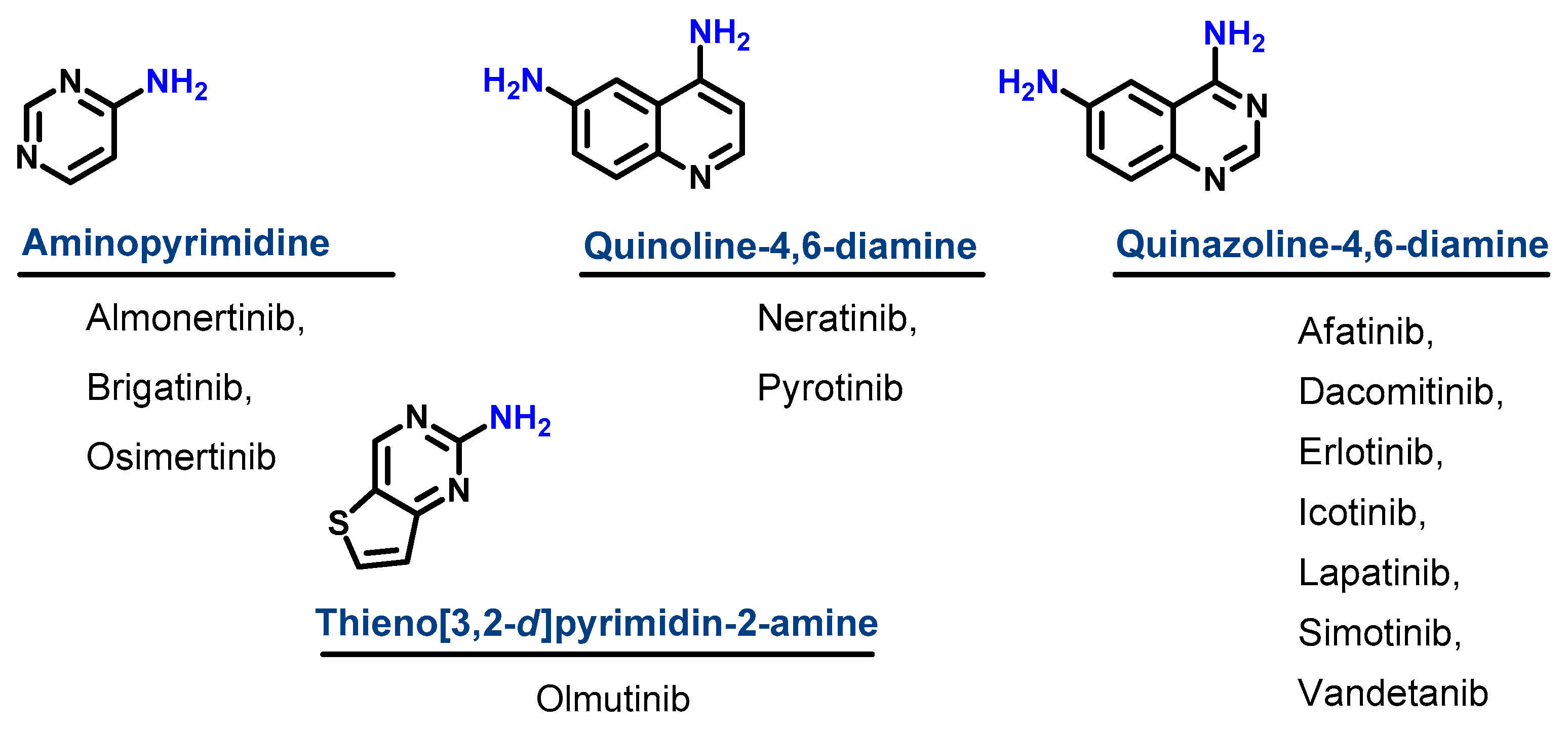
1.1.1. Chemical Classification
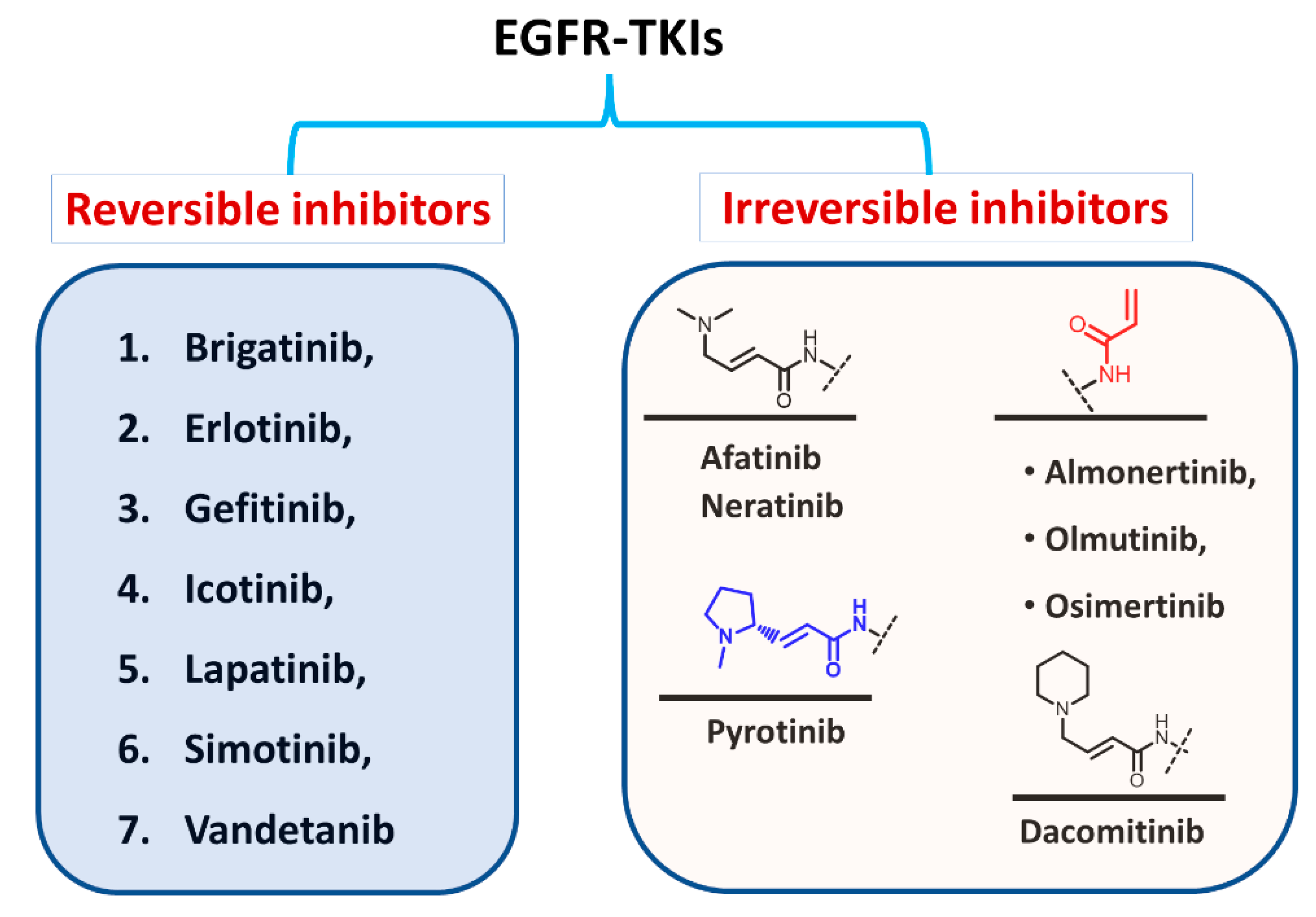
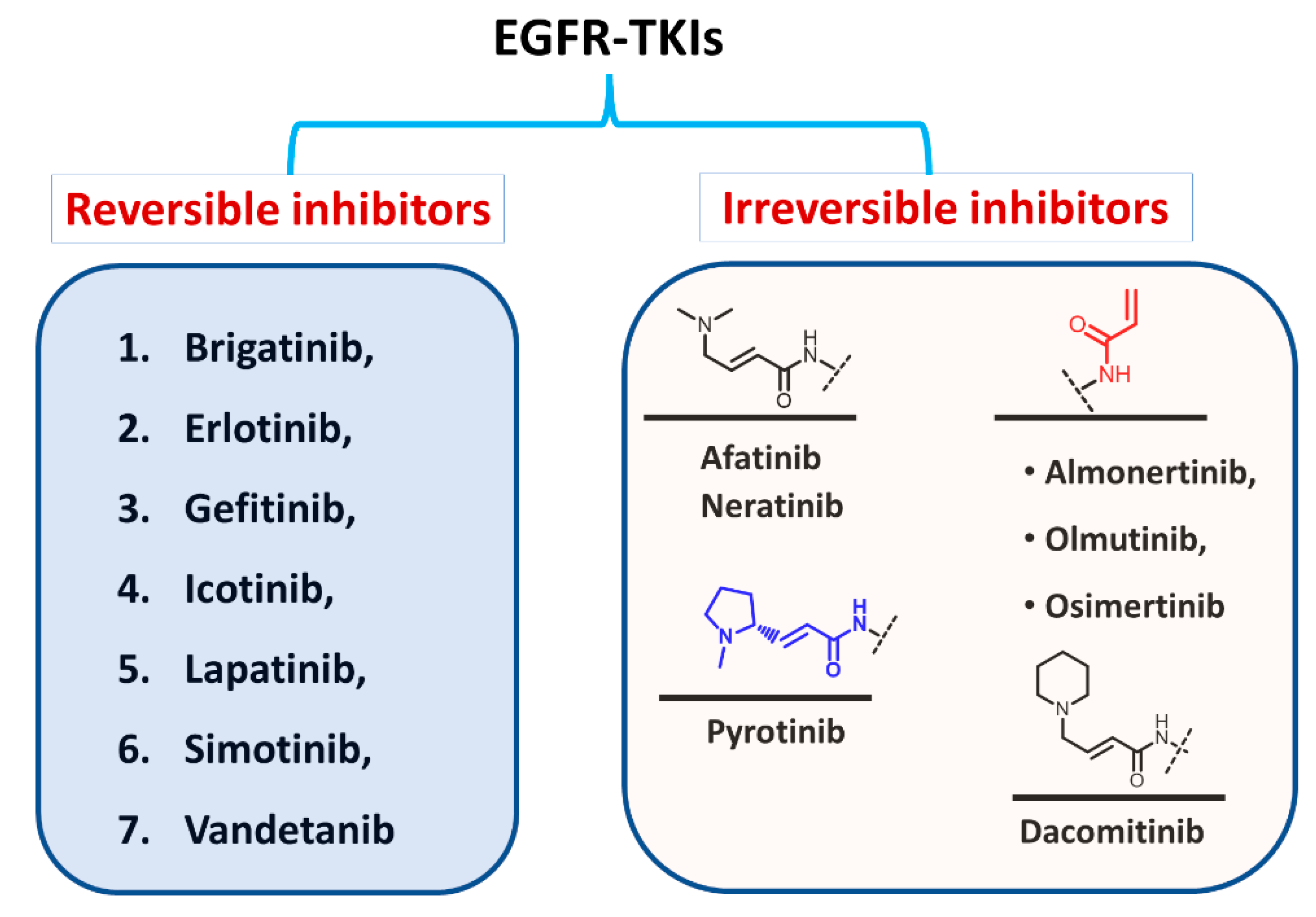
1.1.2. Classification Based on the Types of Interaction with EGFR
1.1.3. Classification Based on the Clinical Use
1.1.4. Classification Based on the Target Kinases
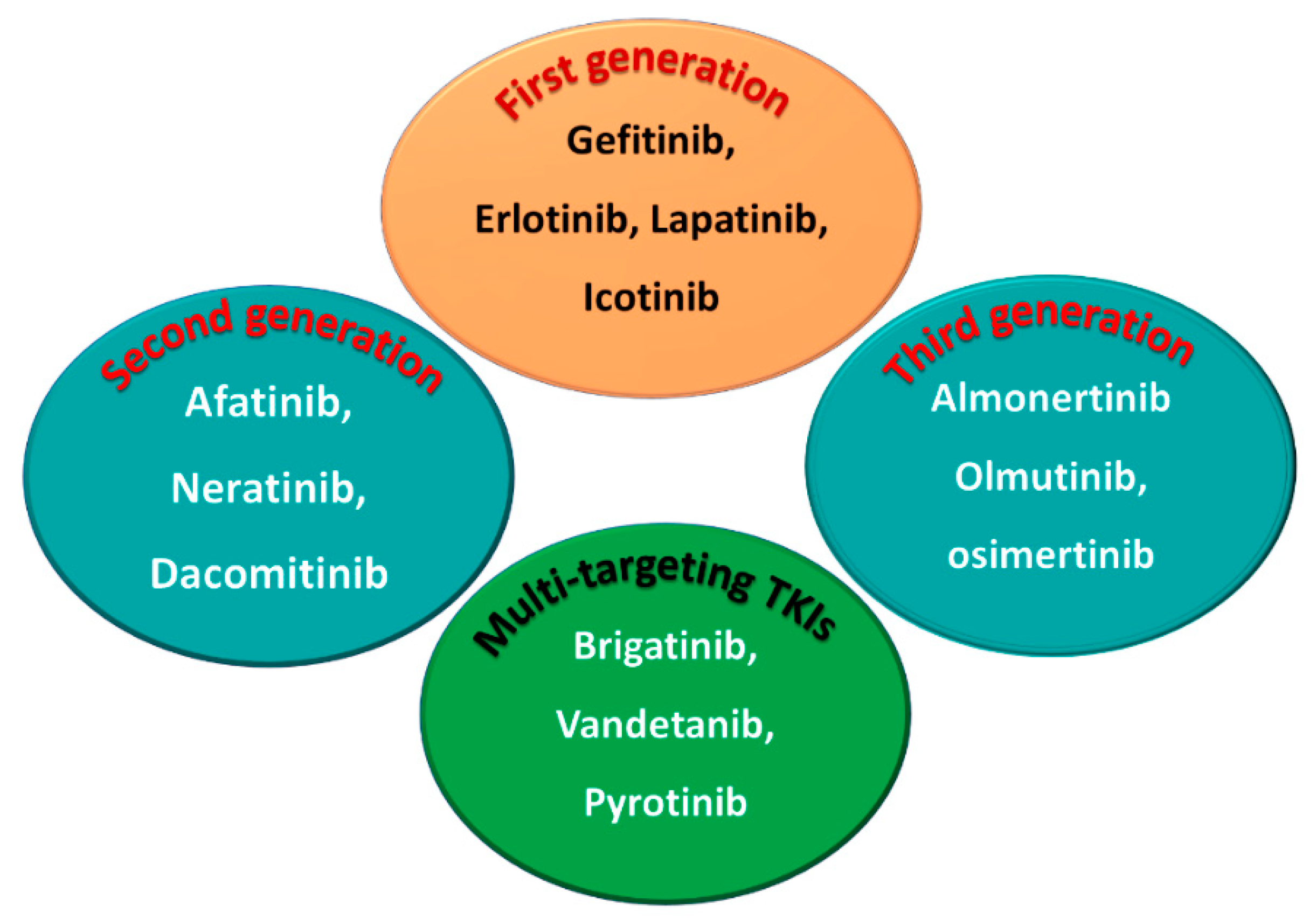
1.1.5. Classification into the First, Second, and Third Generation
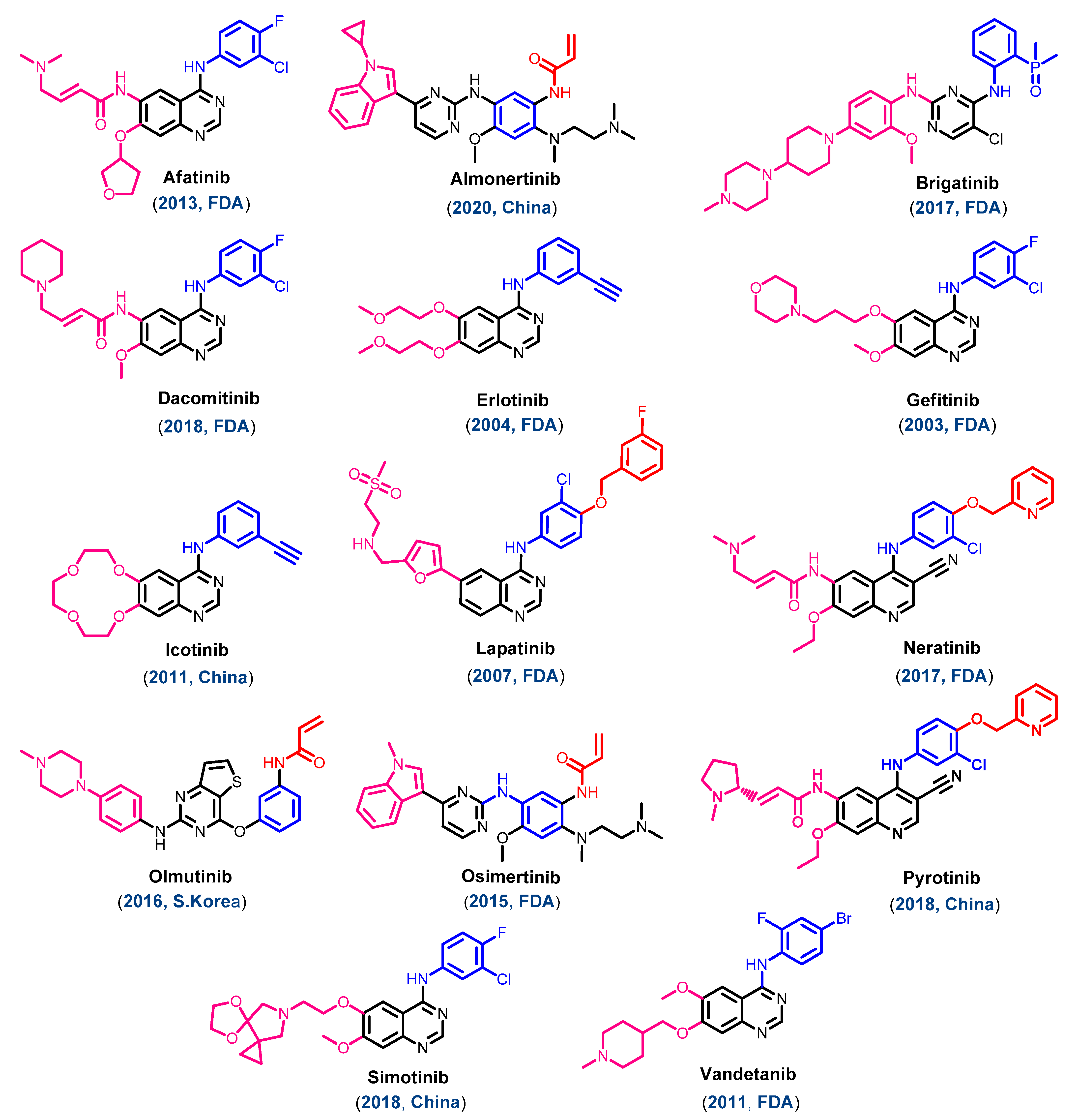
1.2. EGFR Inhibitors Approved for Cancer Treatments
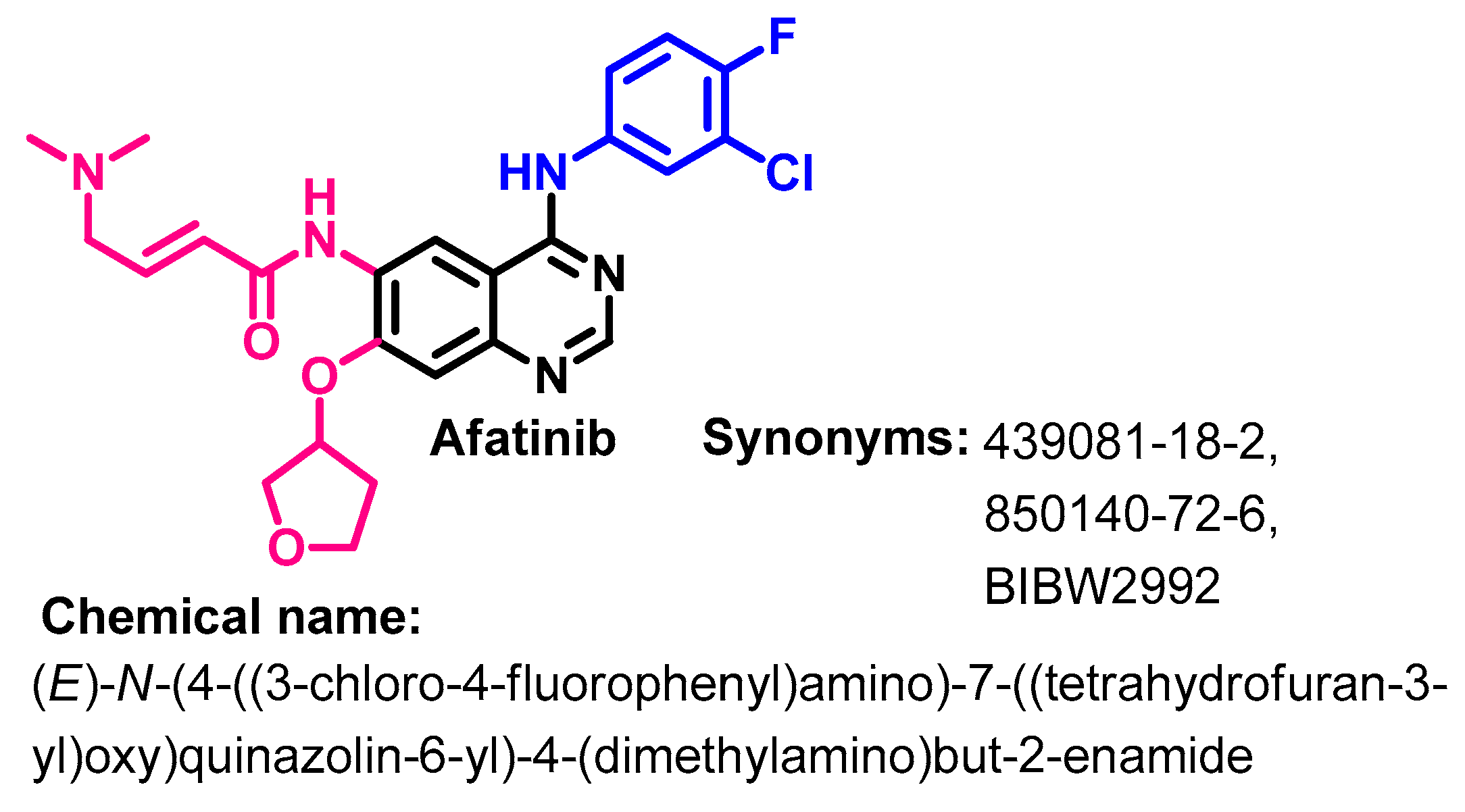
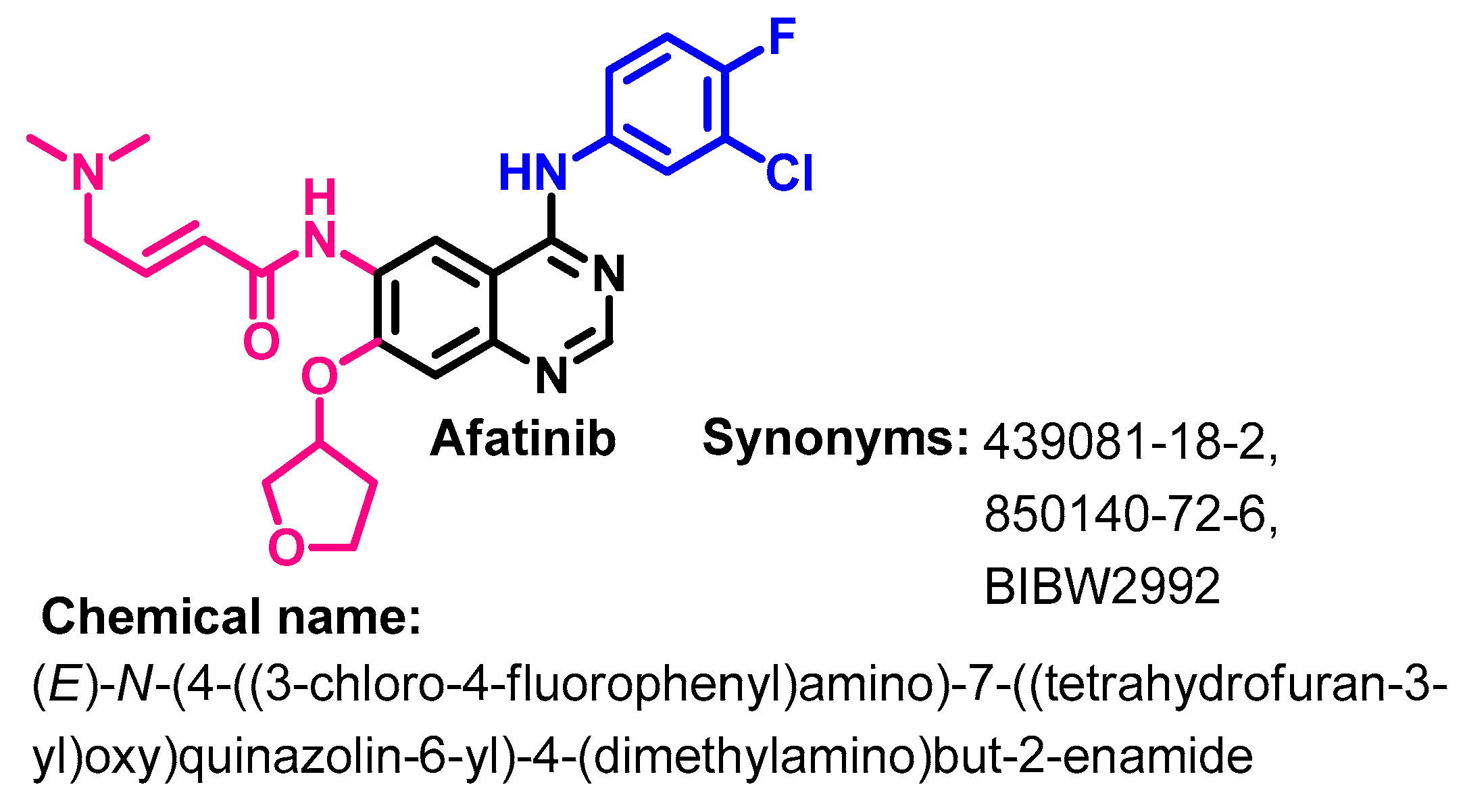
1.2.1. Afatinib
Approval History
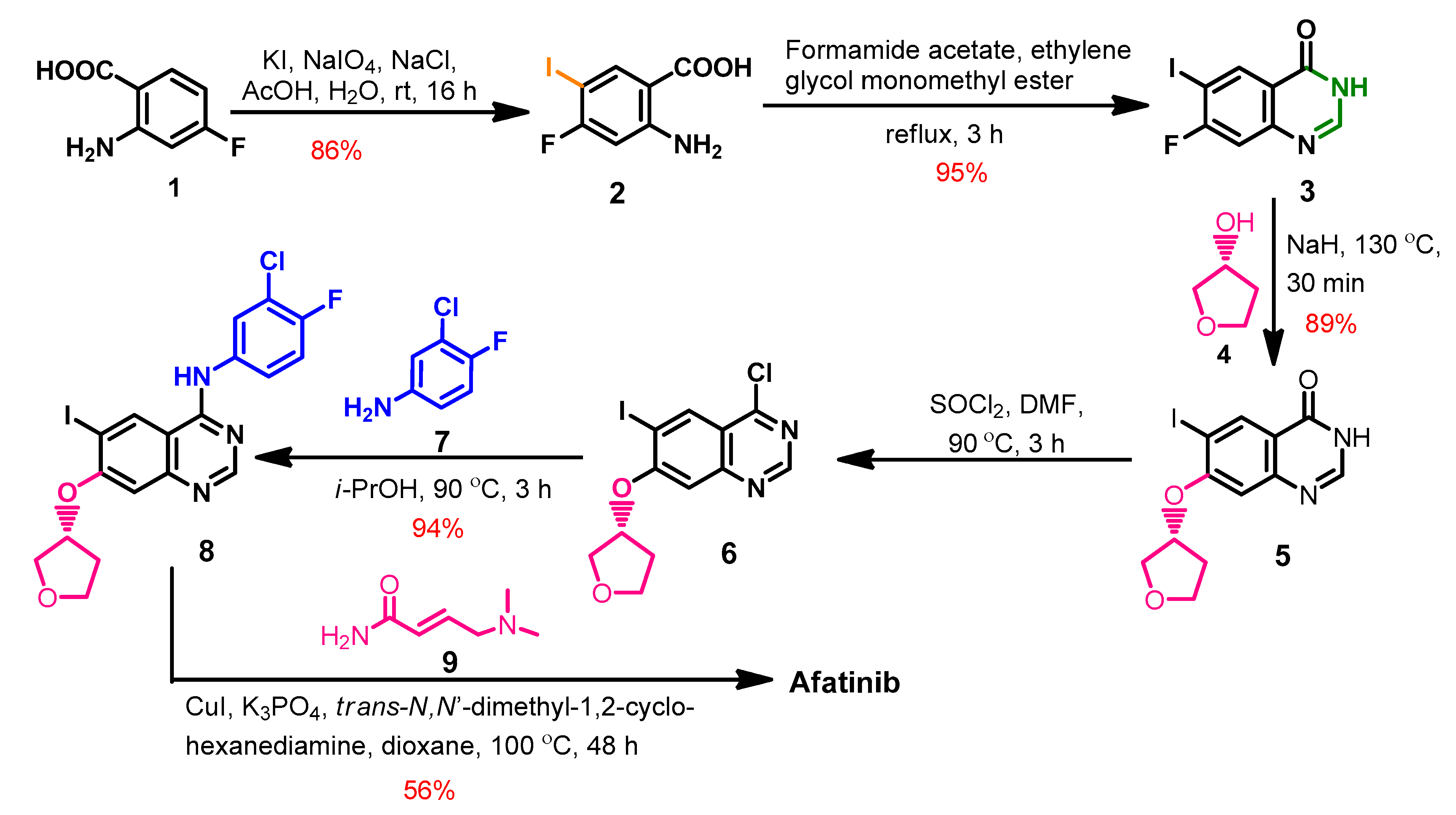
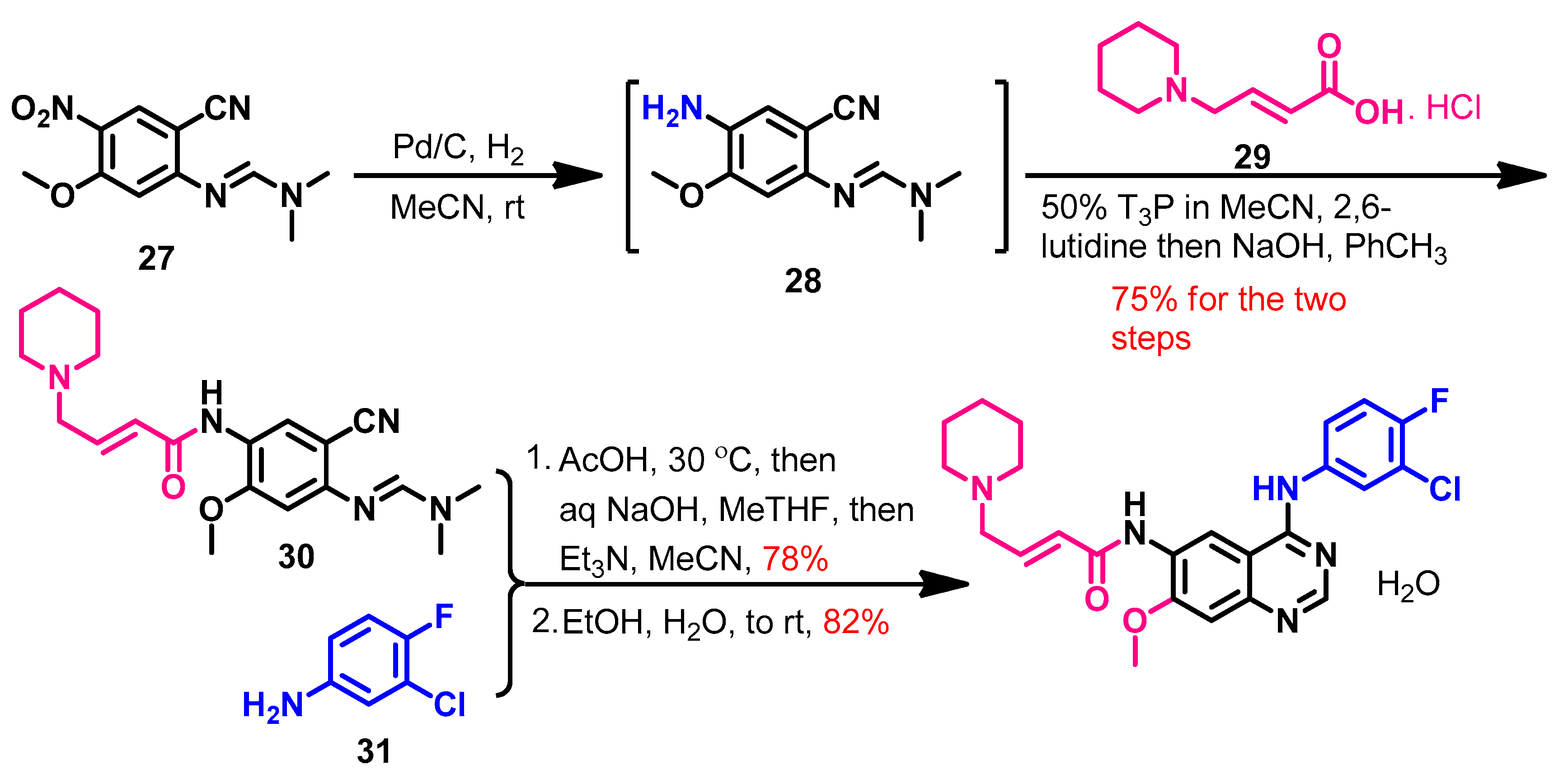
Synthesis
Target Kinases
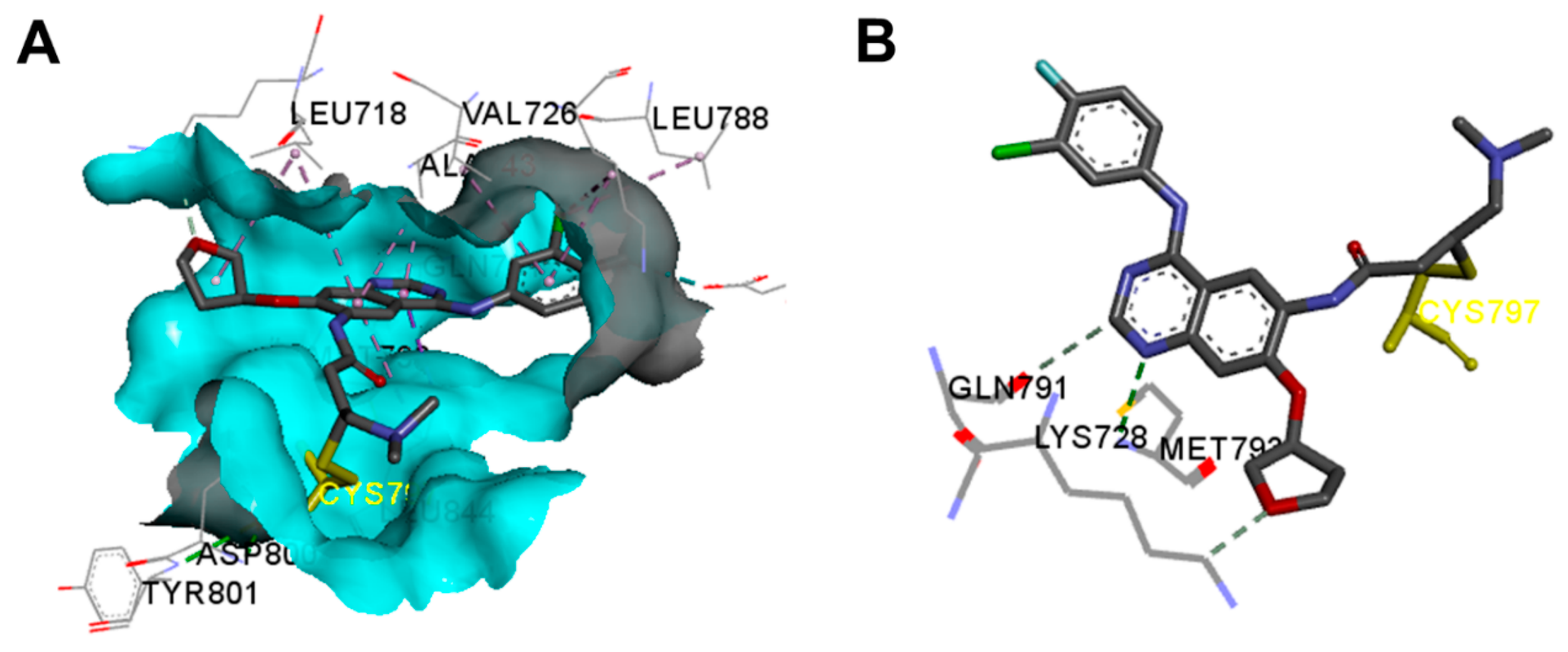
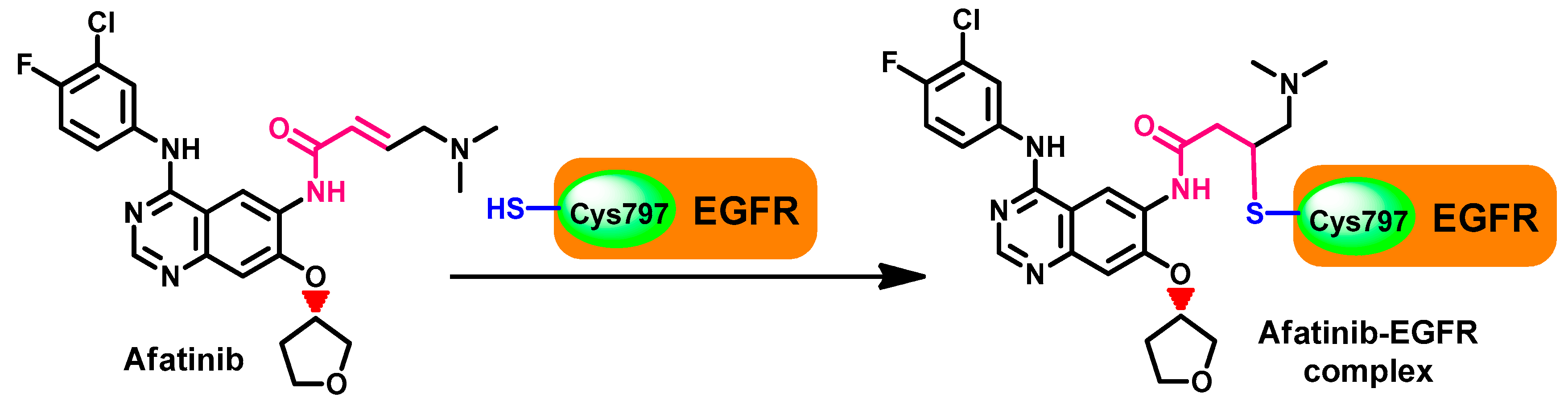
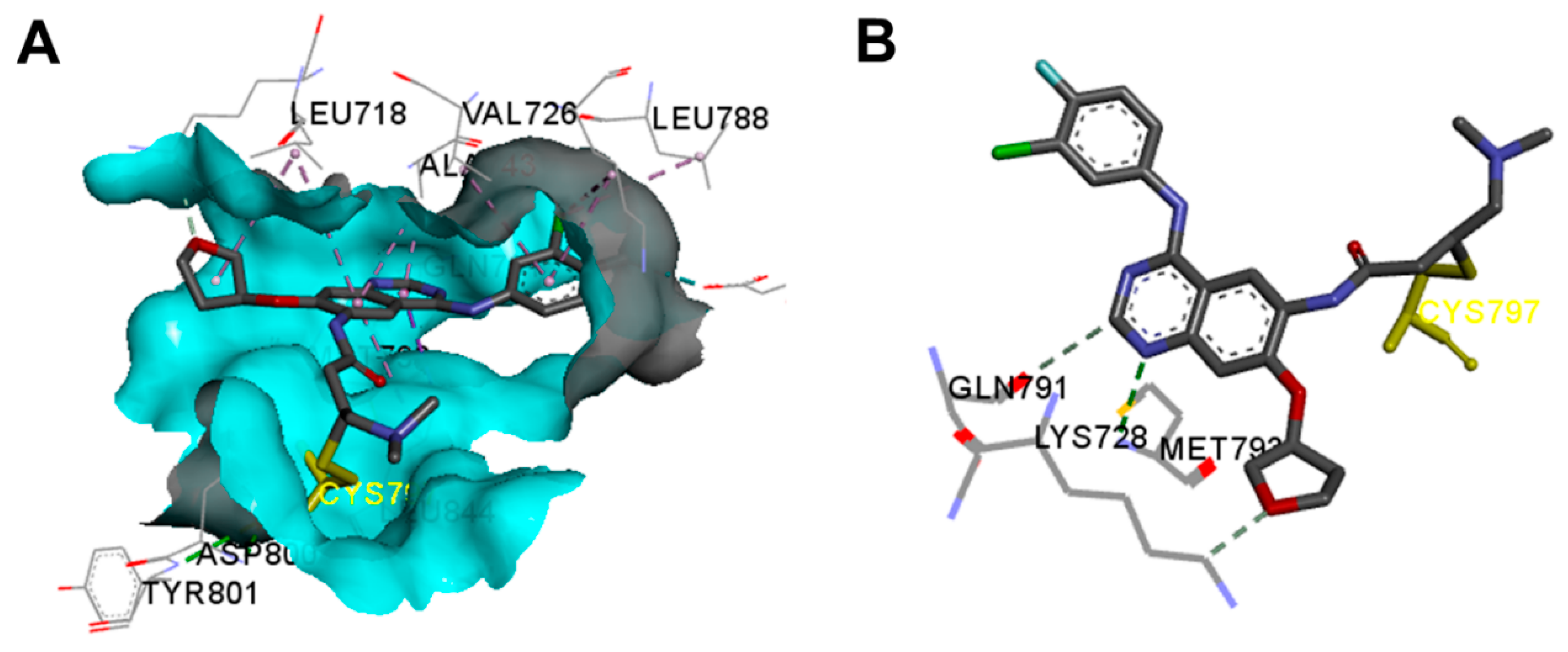
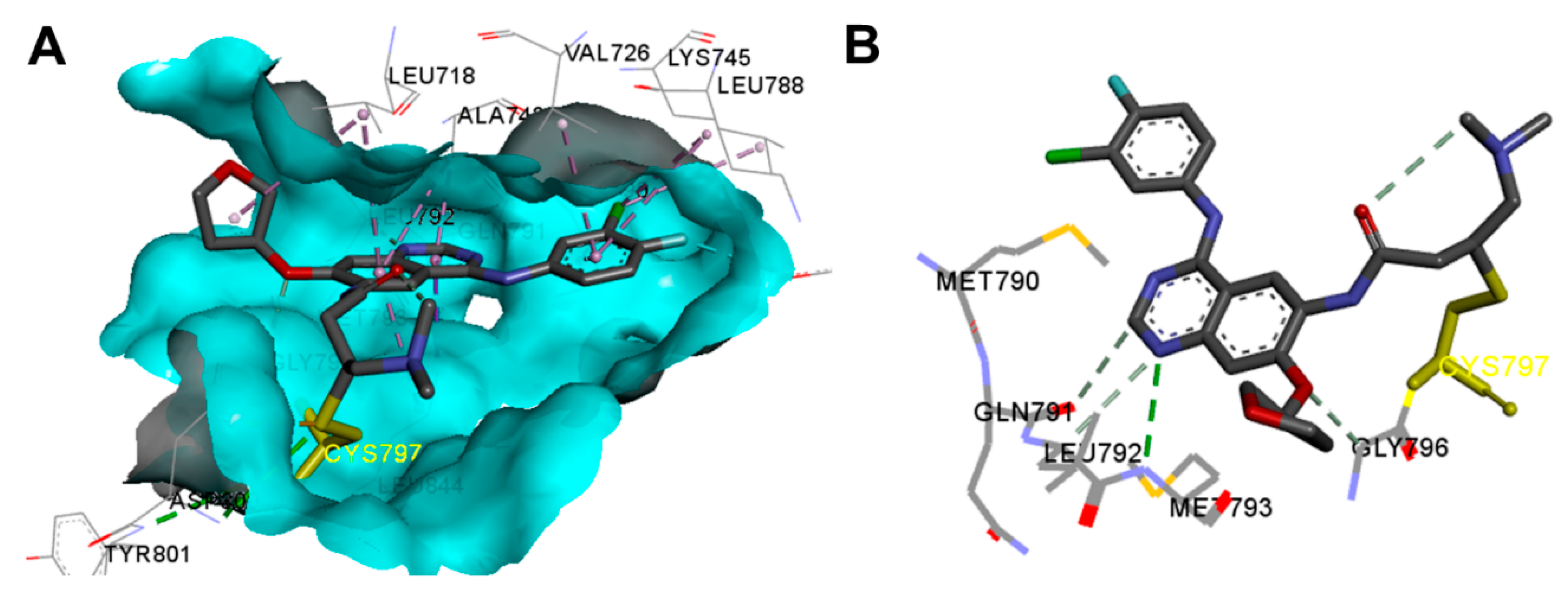
Crystal Structures and Binding Interactions
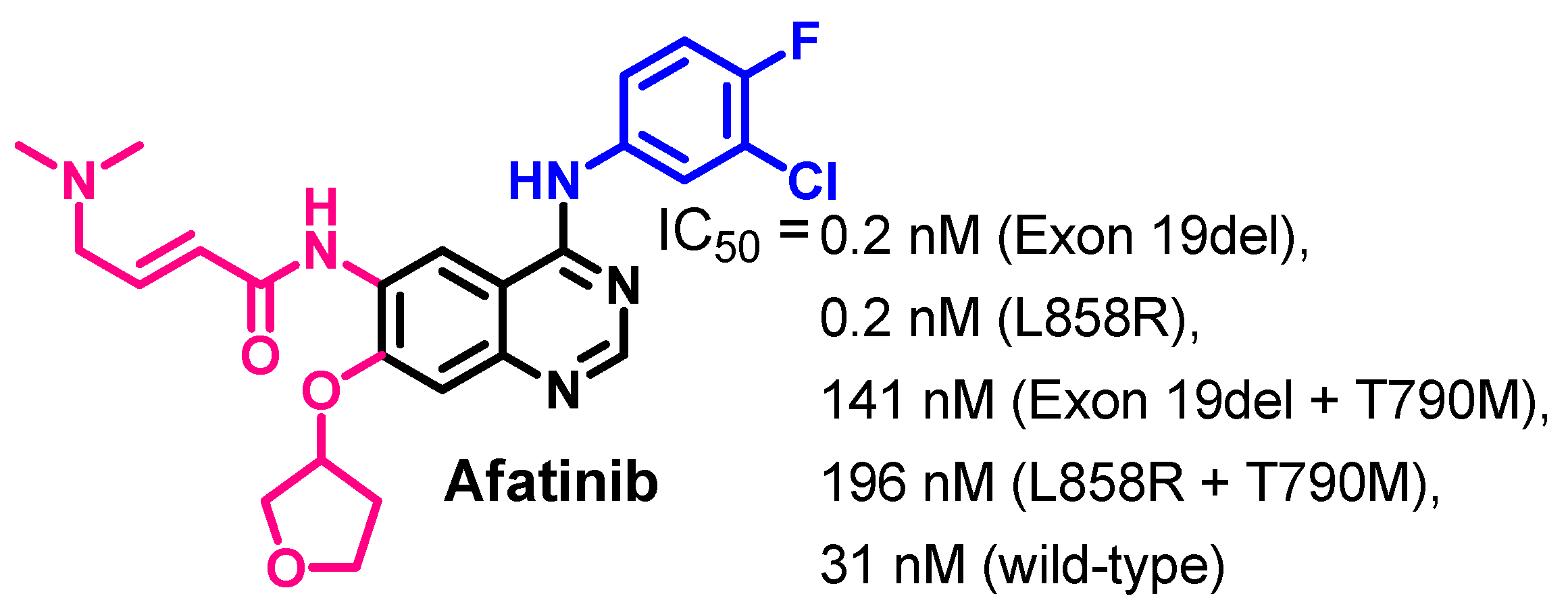
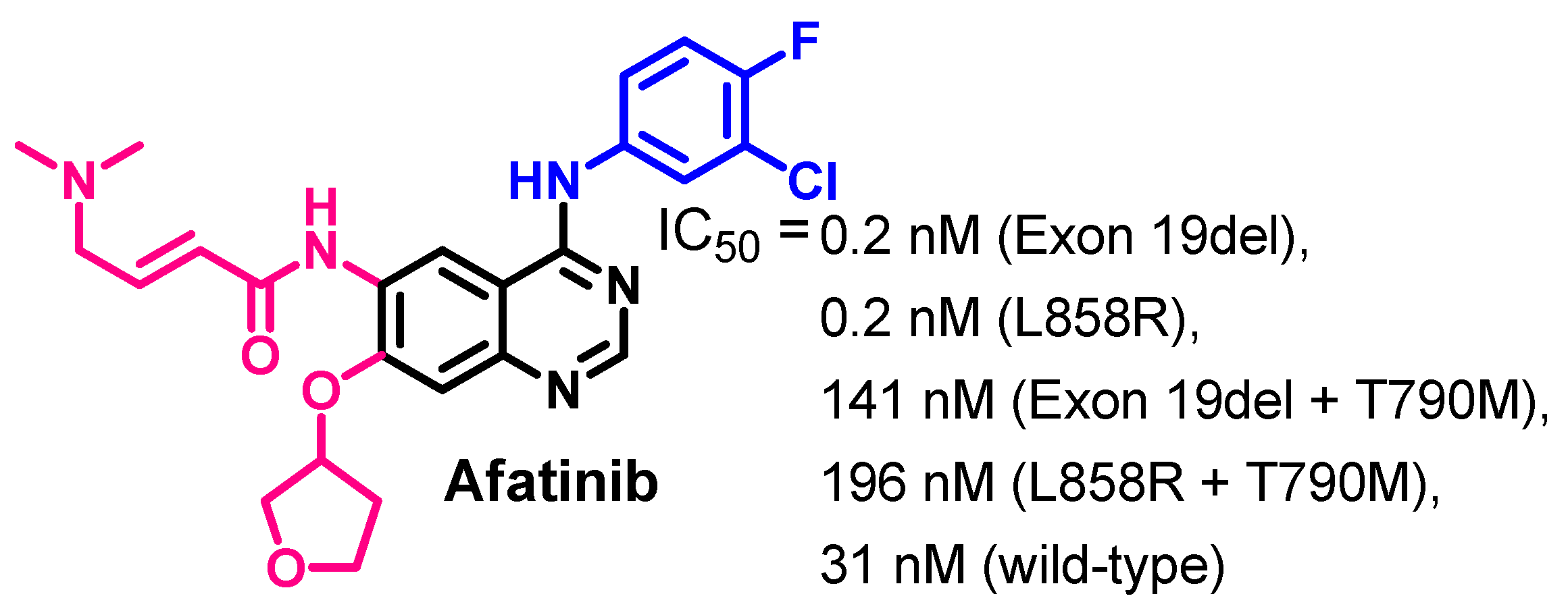
Biological Activity
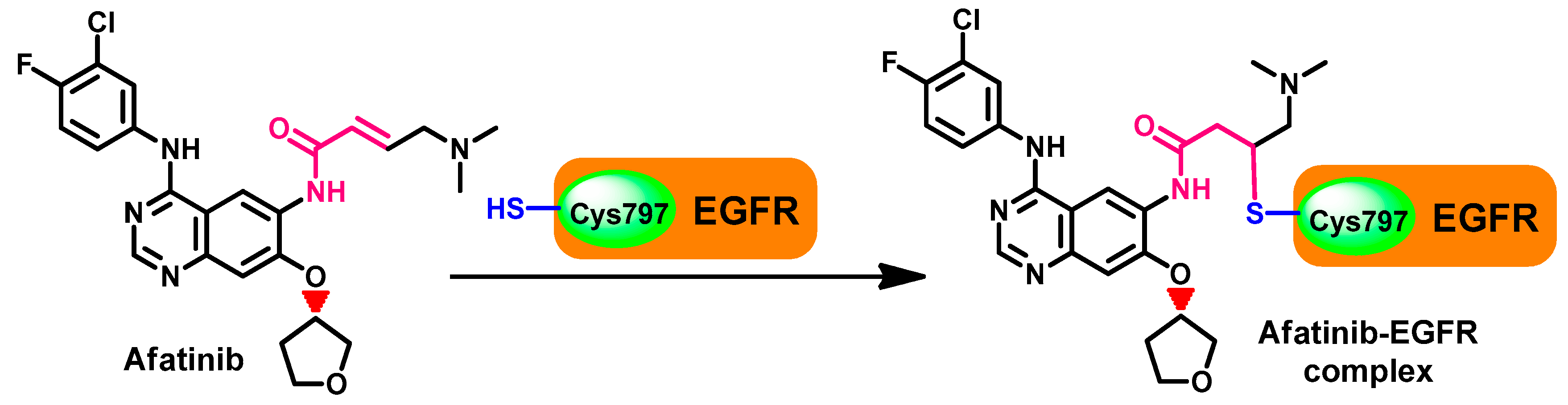
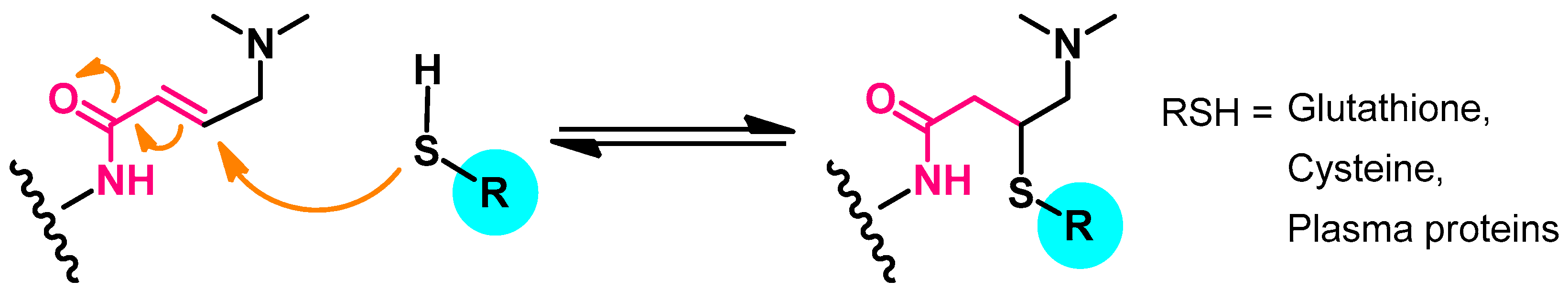
Metabolism
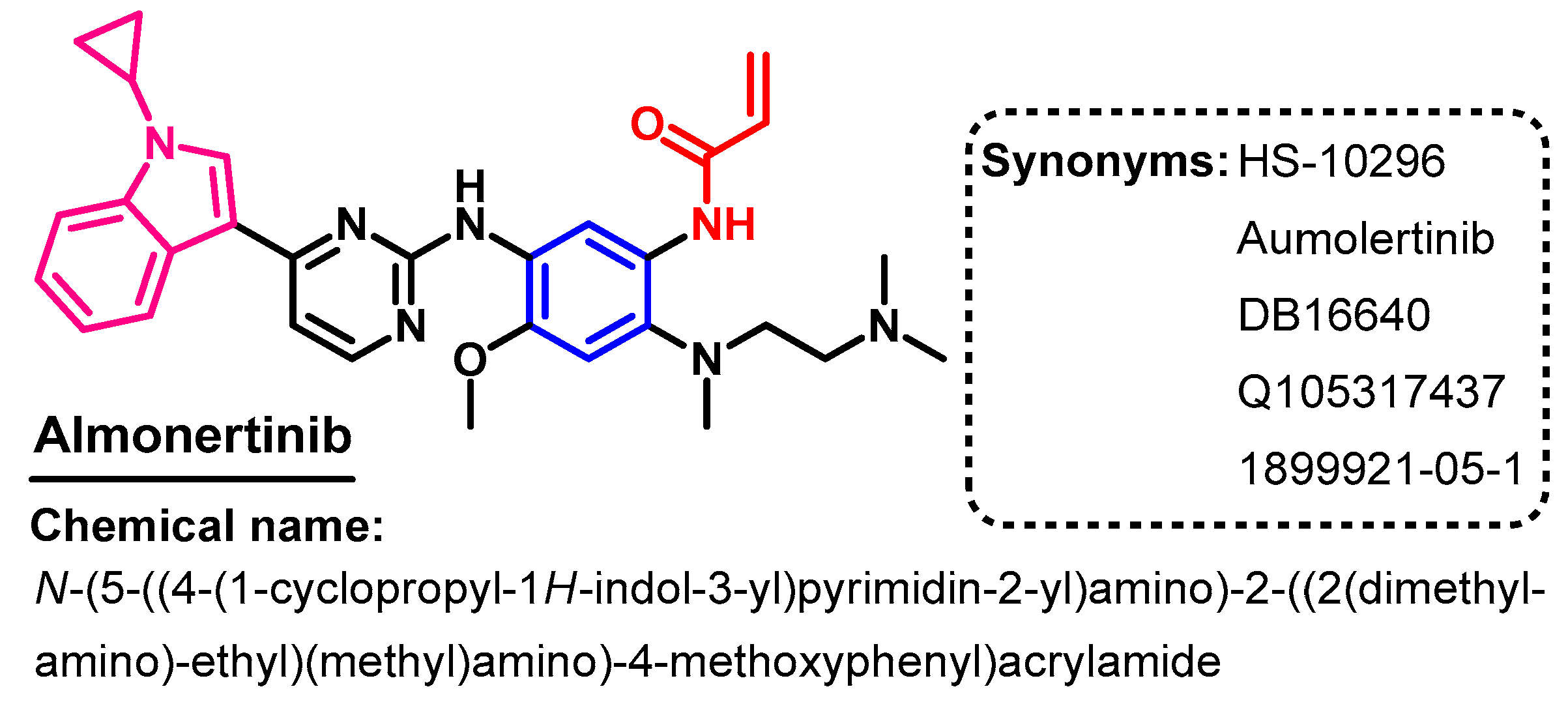
1.2.2. Almonertinib
Approval History
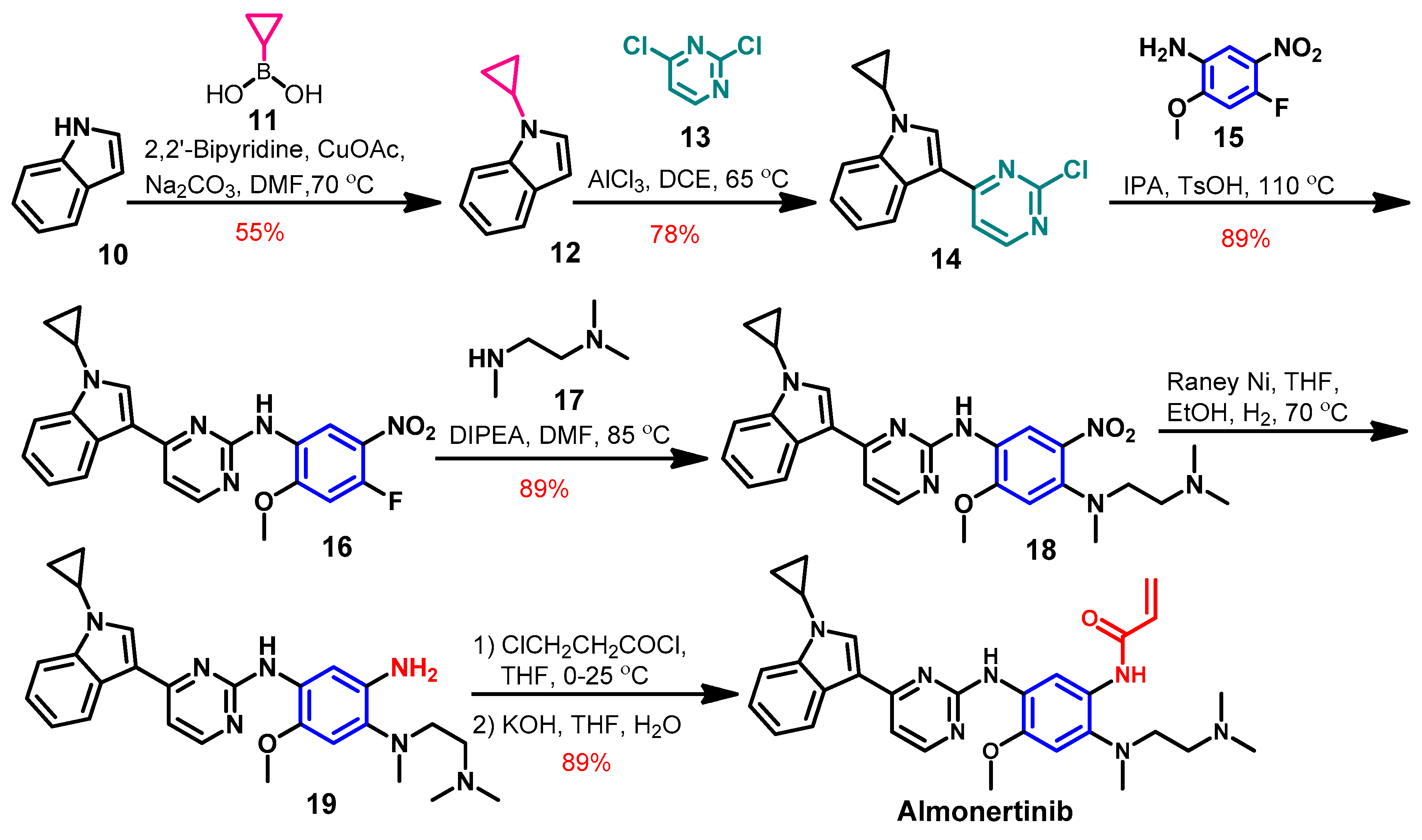
Synthesis
Target Kinases
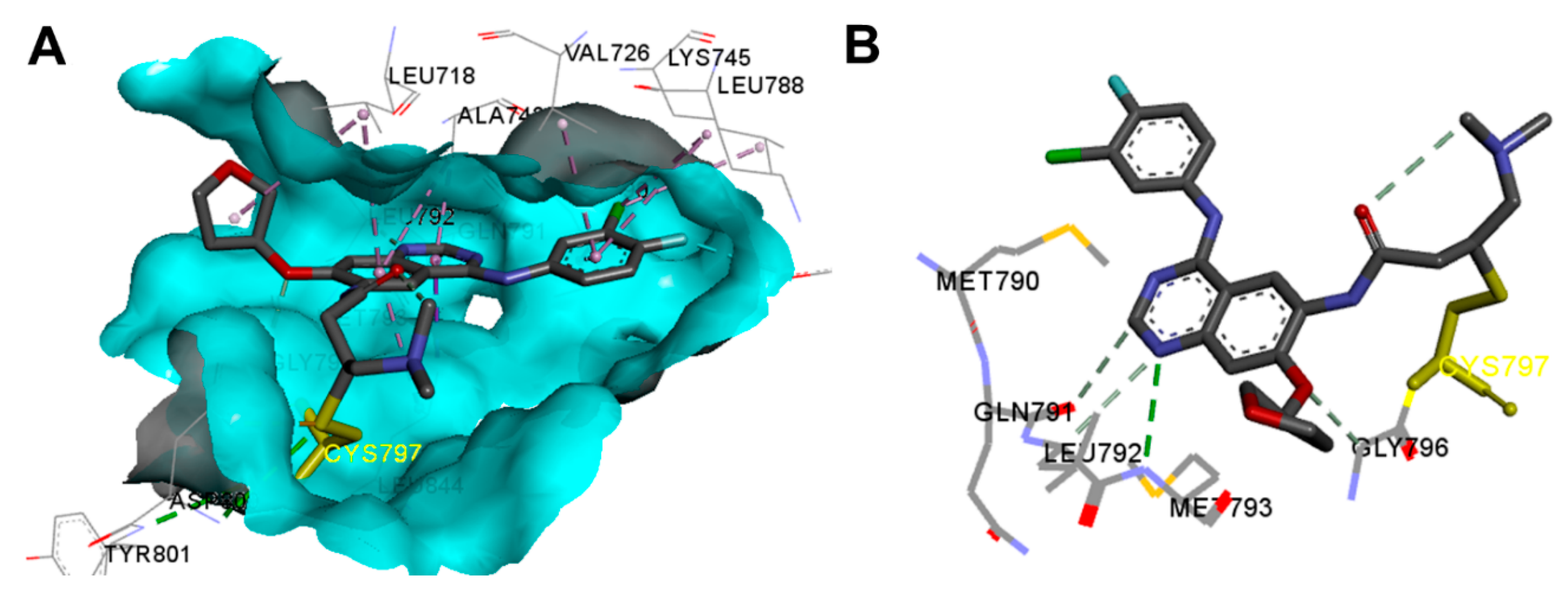
Crystal Structures and Binding Interactions
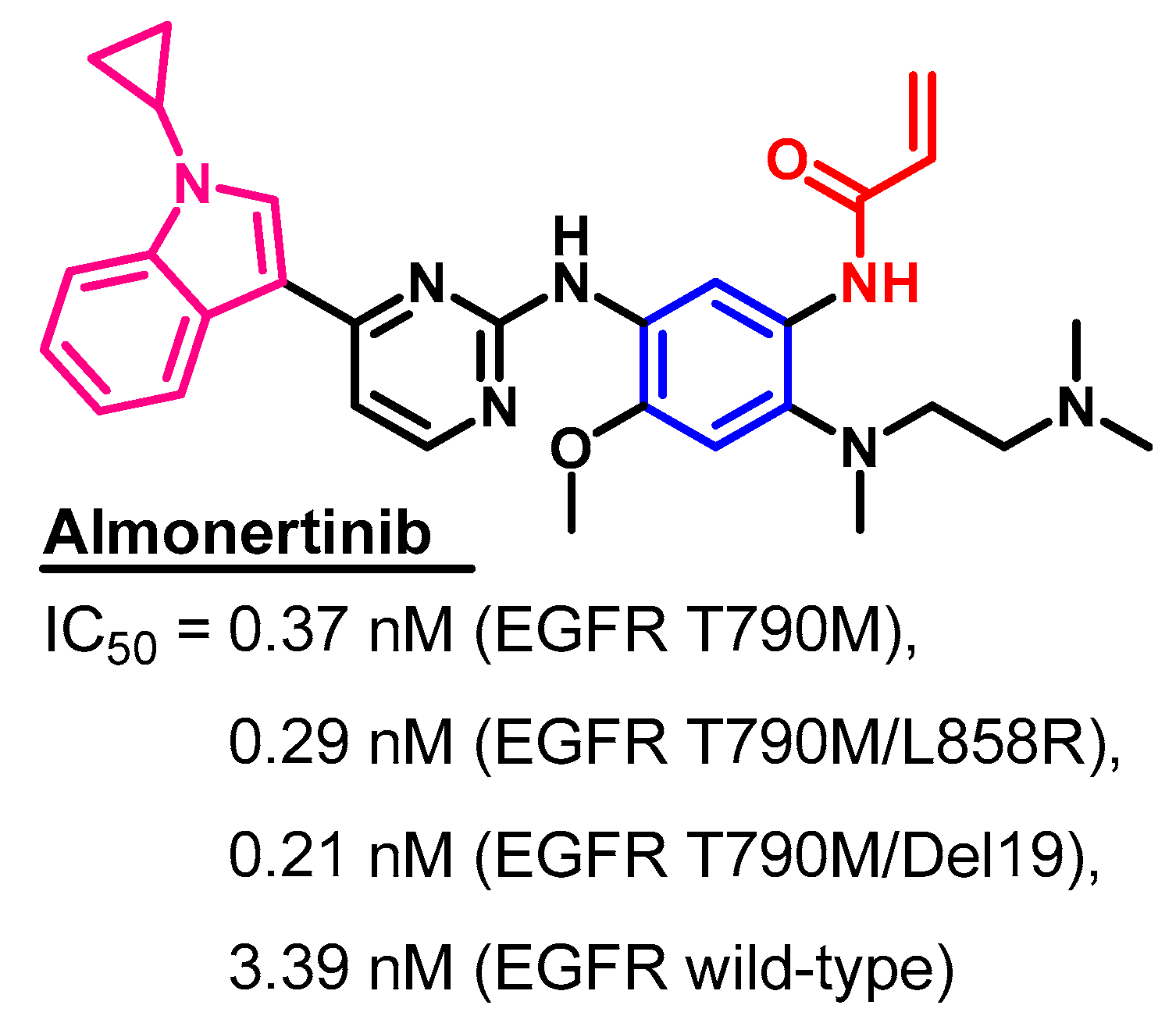
Biological Activity
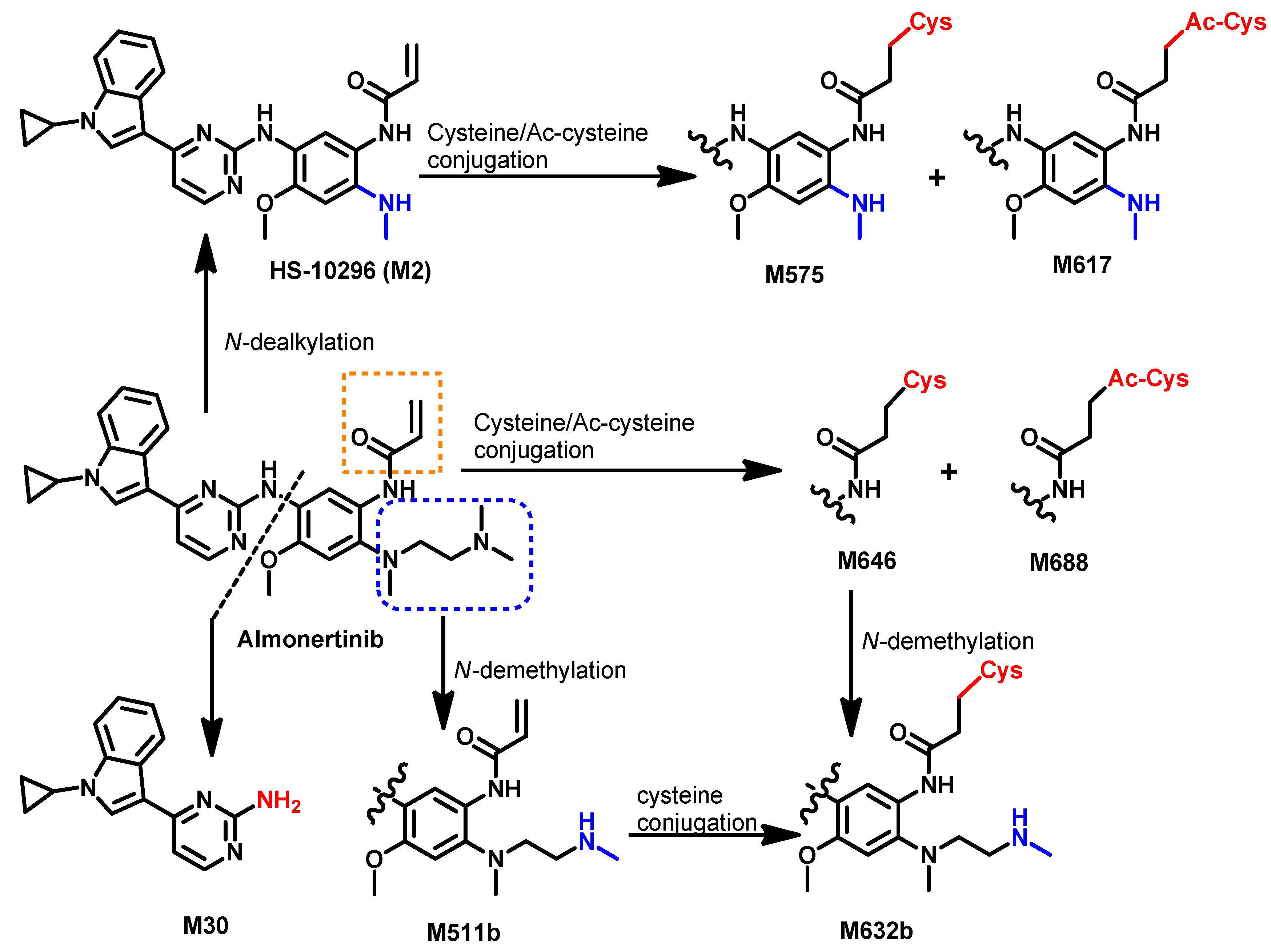
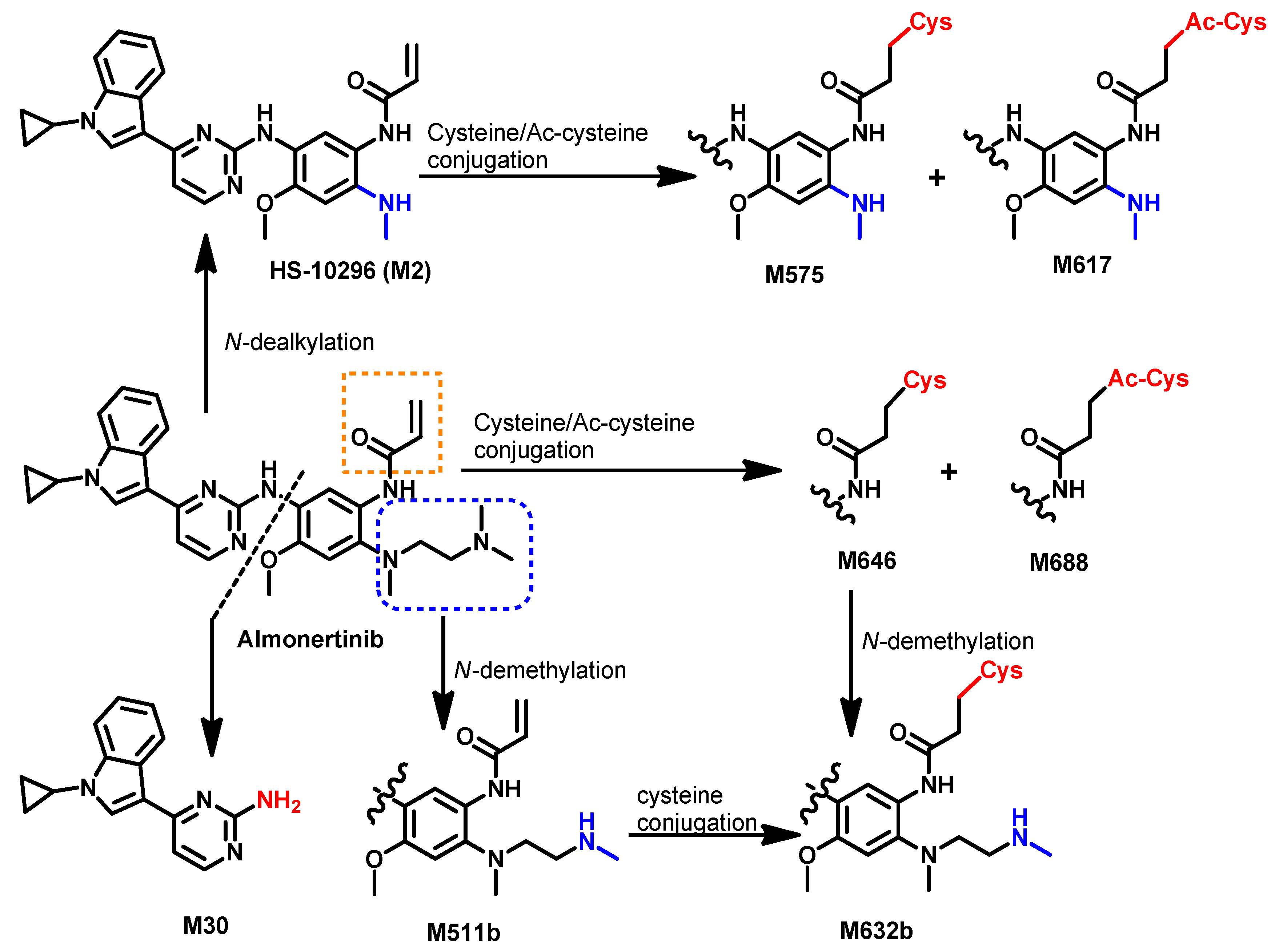
Metabolism
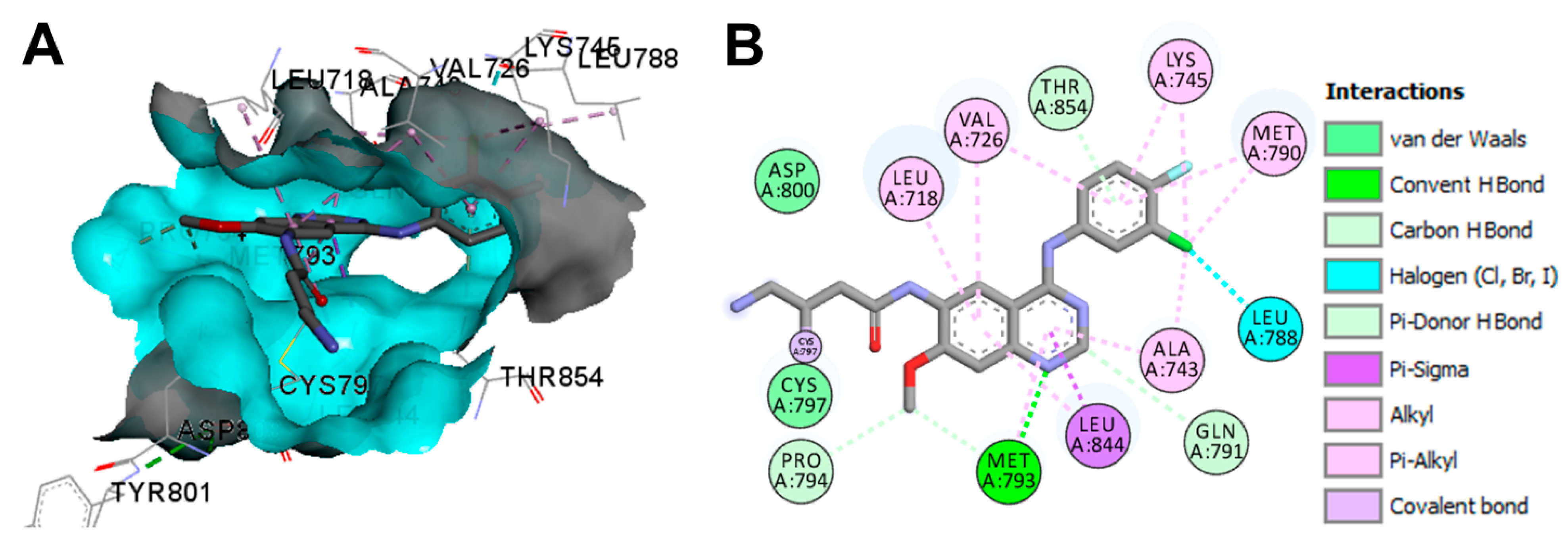
1.2.3. Brigatinib
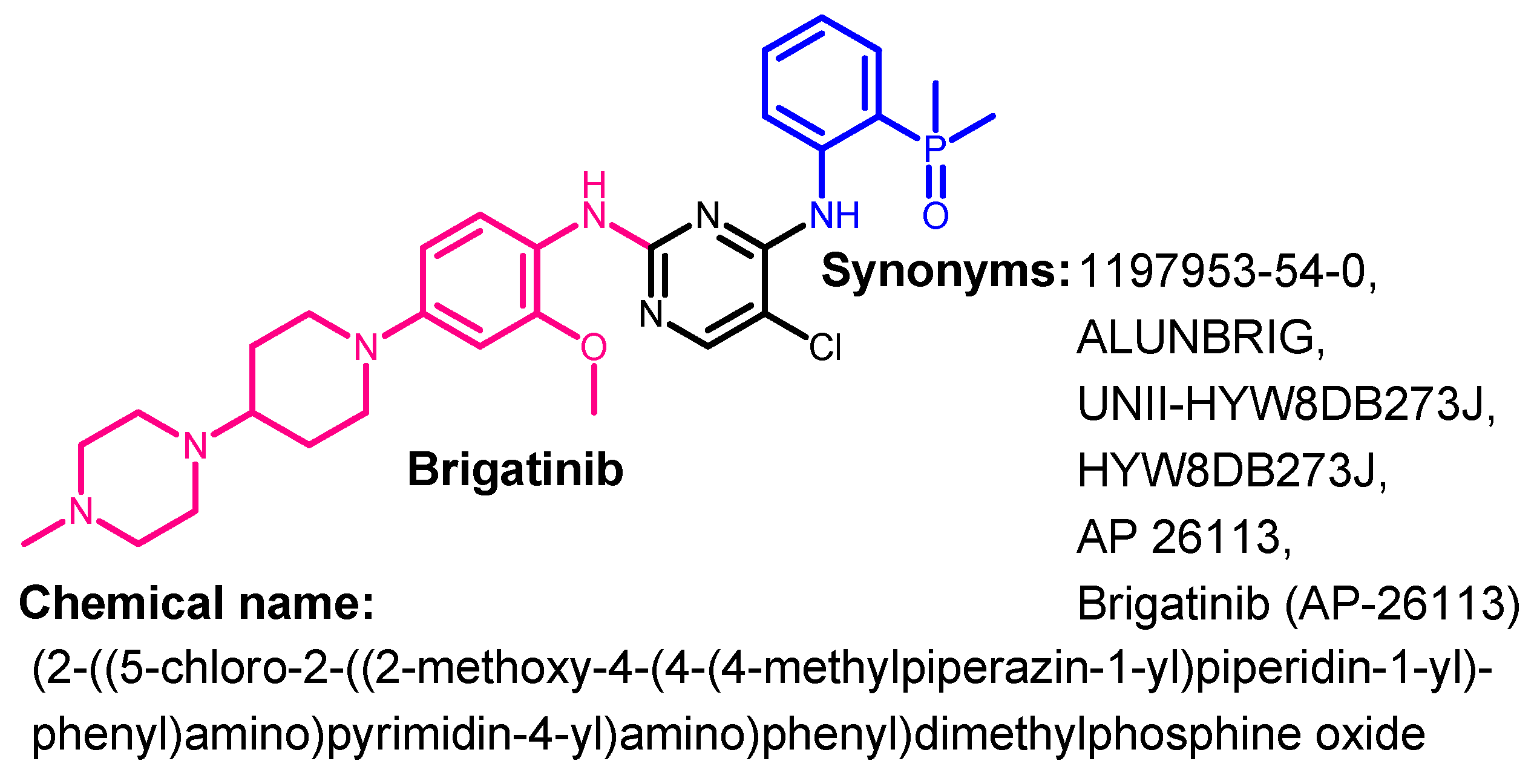
Approval History
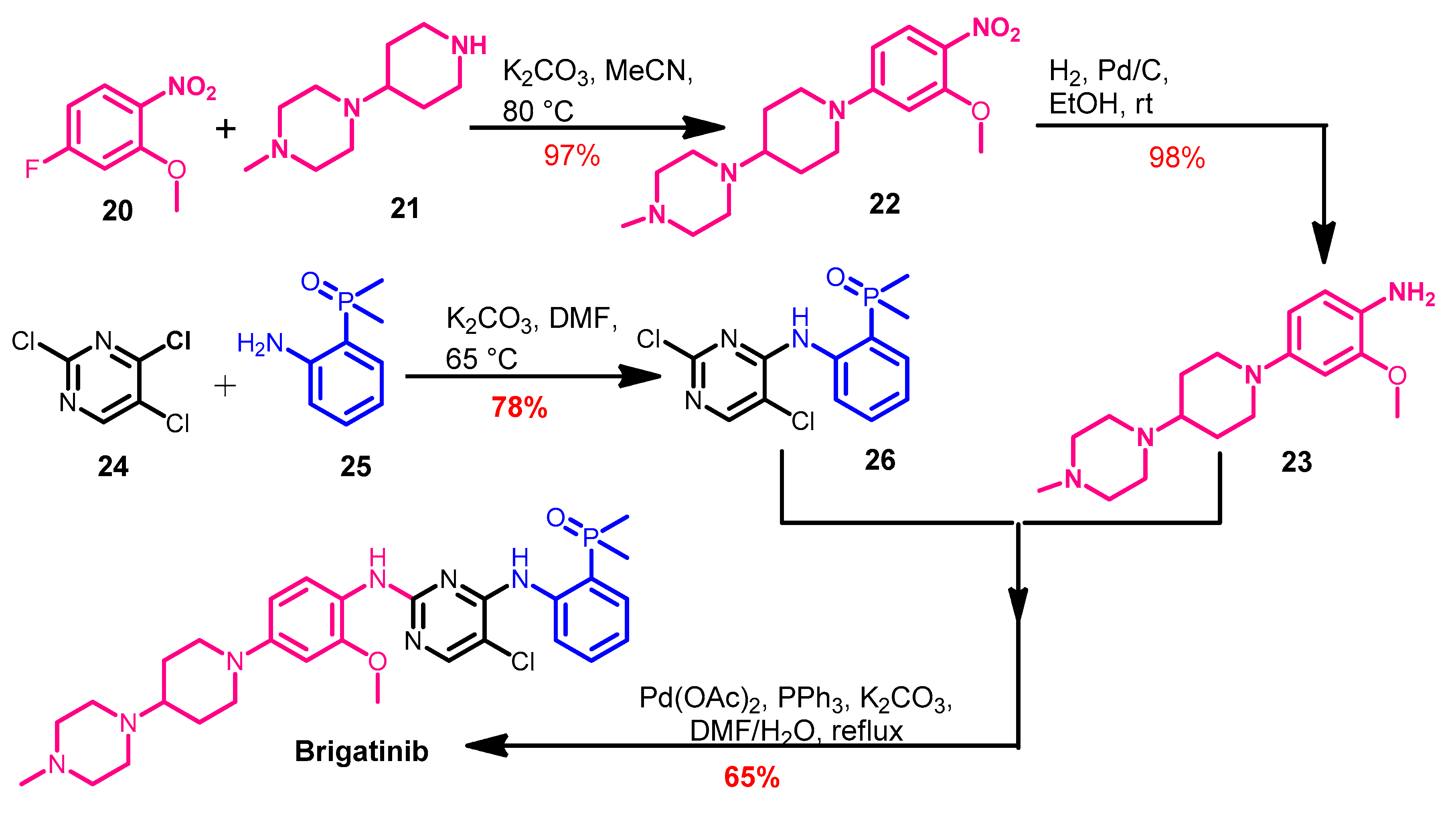
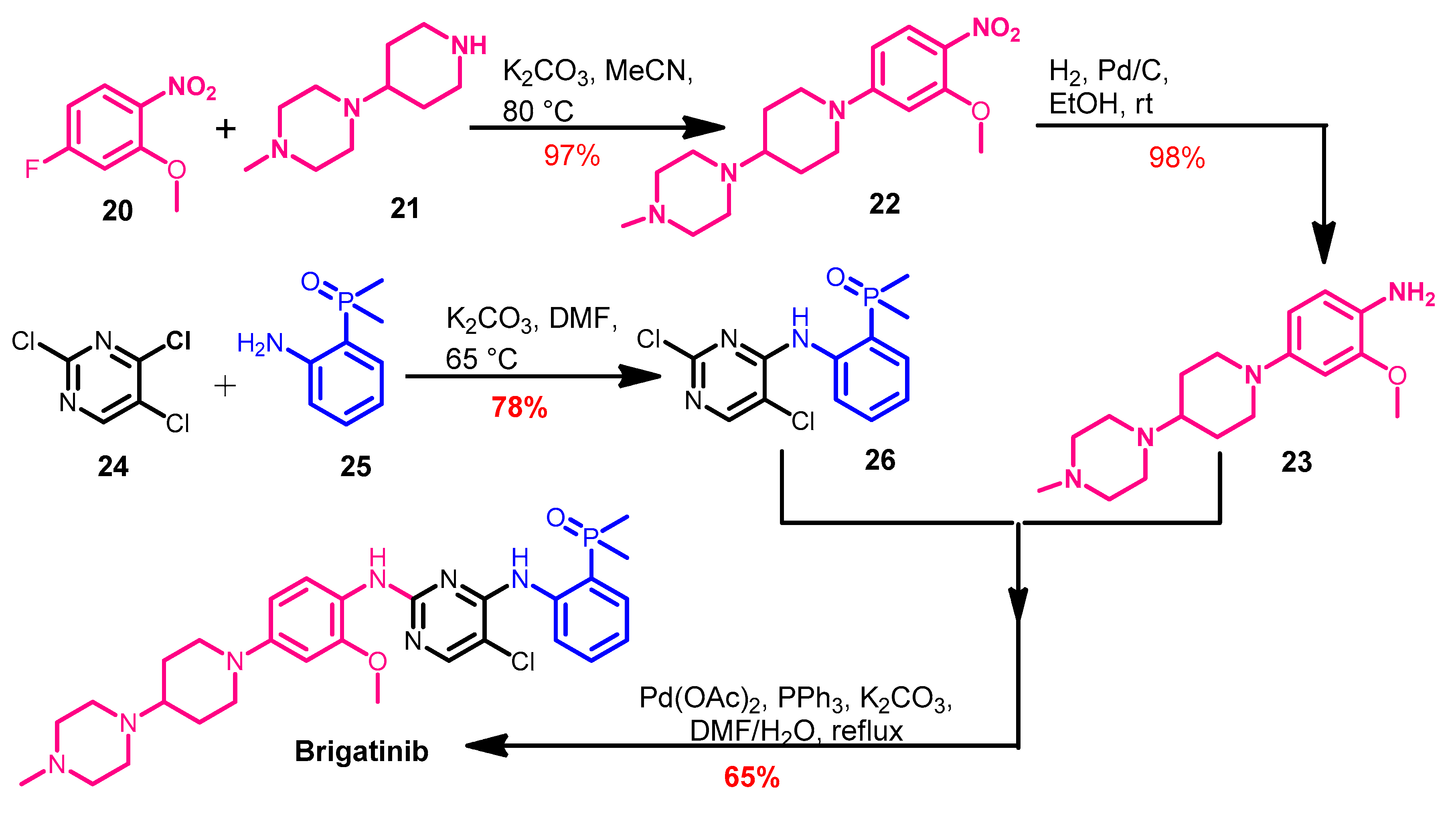
Synthesis
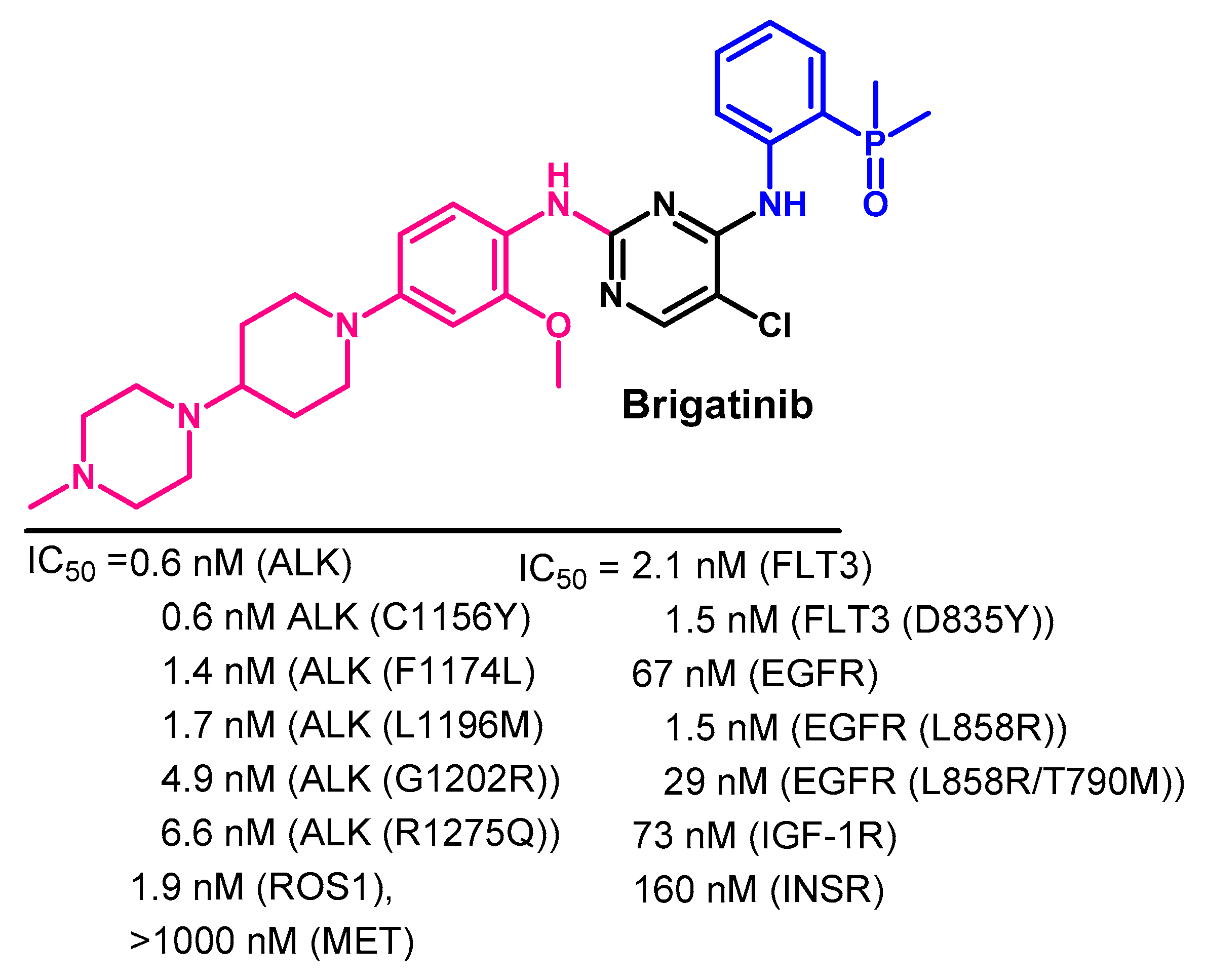
Target Kinases
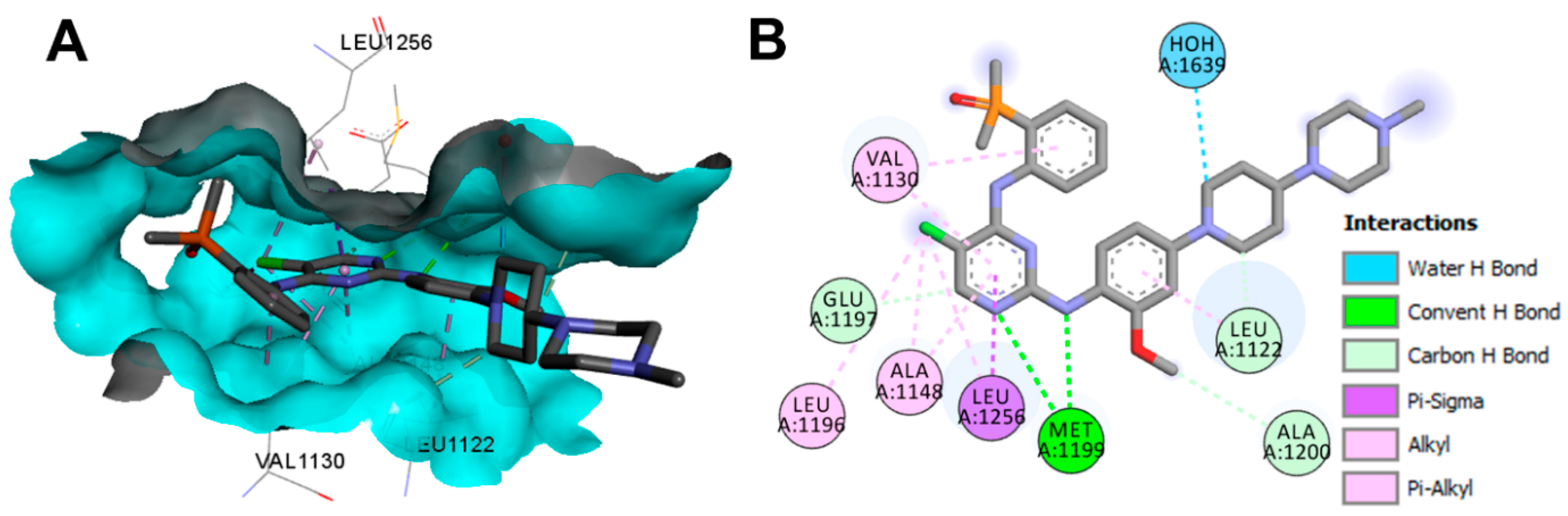
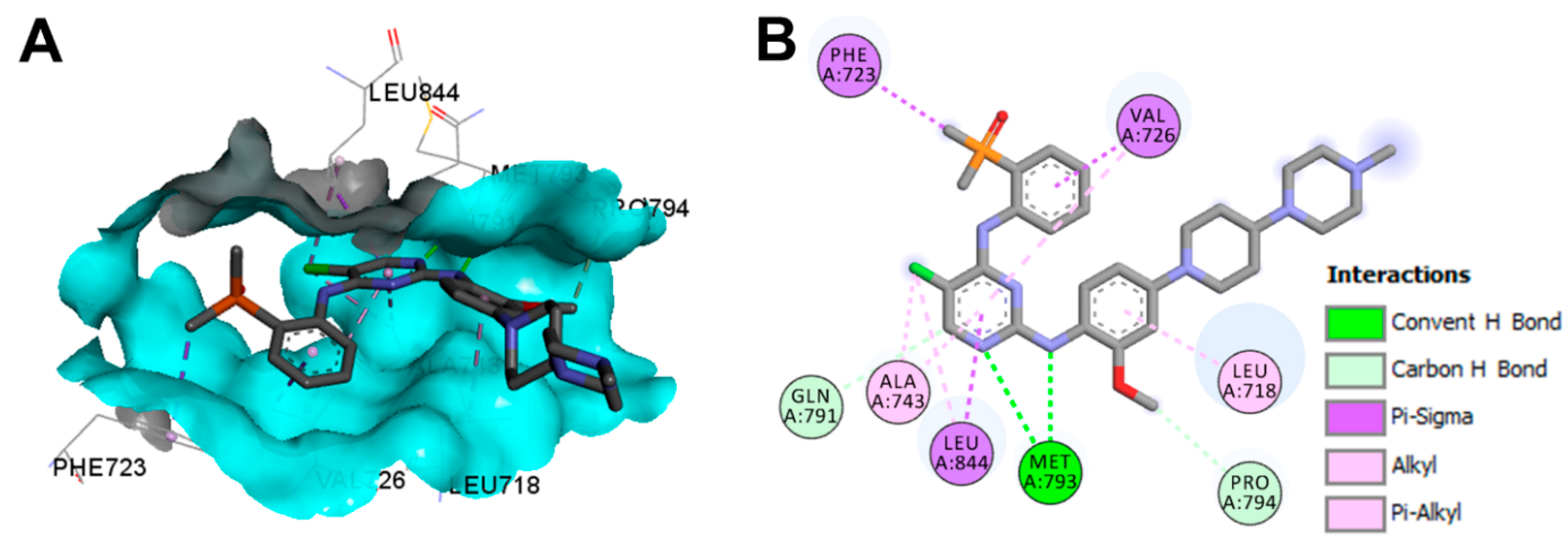
Crystal Structures and Binding Interactions
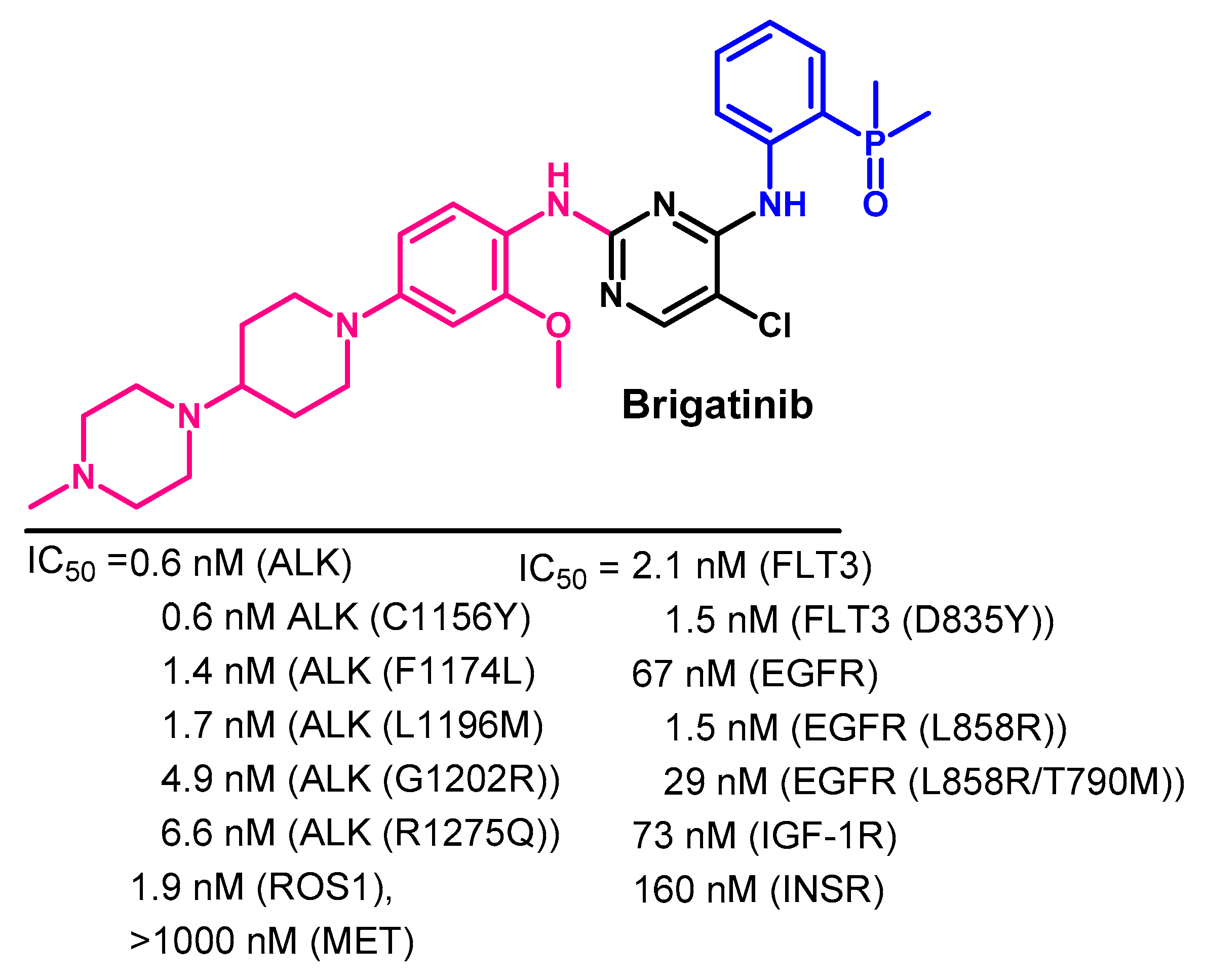
Biological Activity
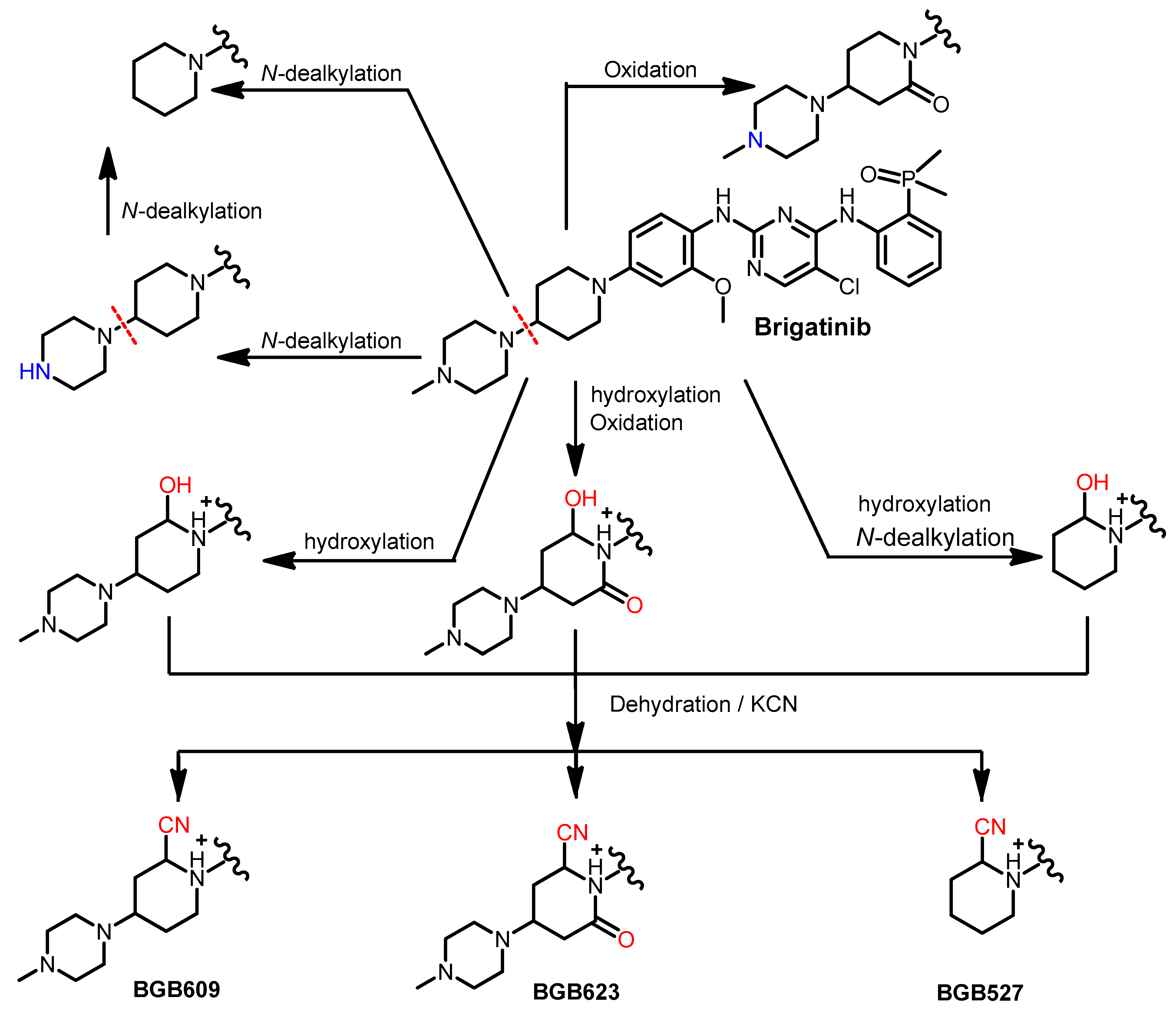
Metabolism
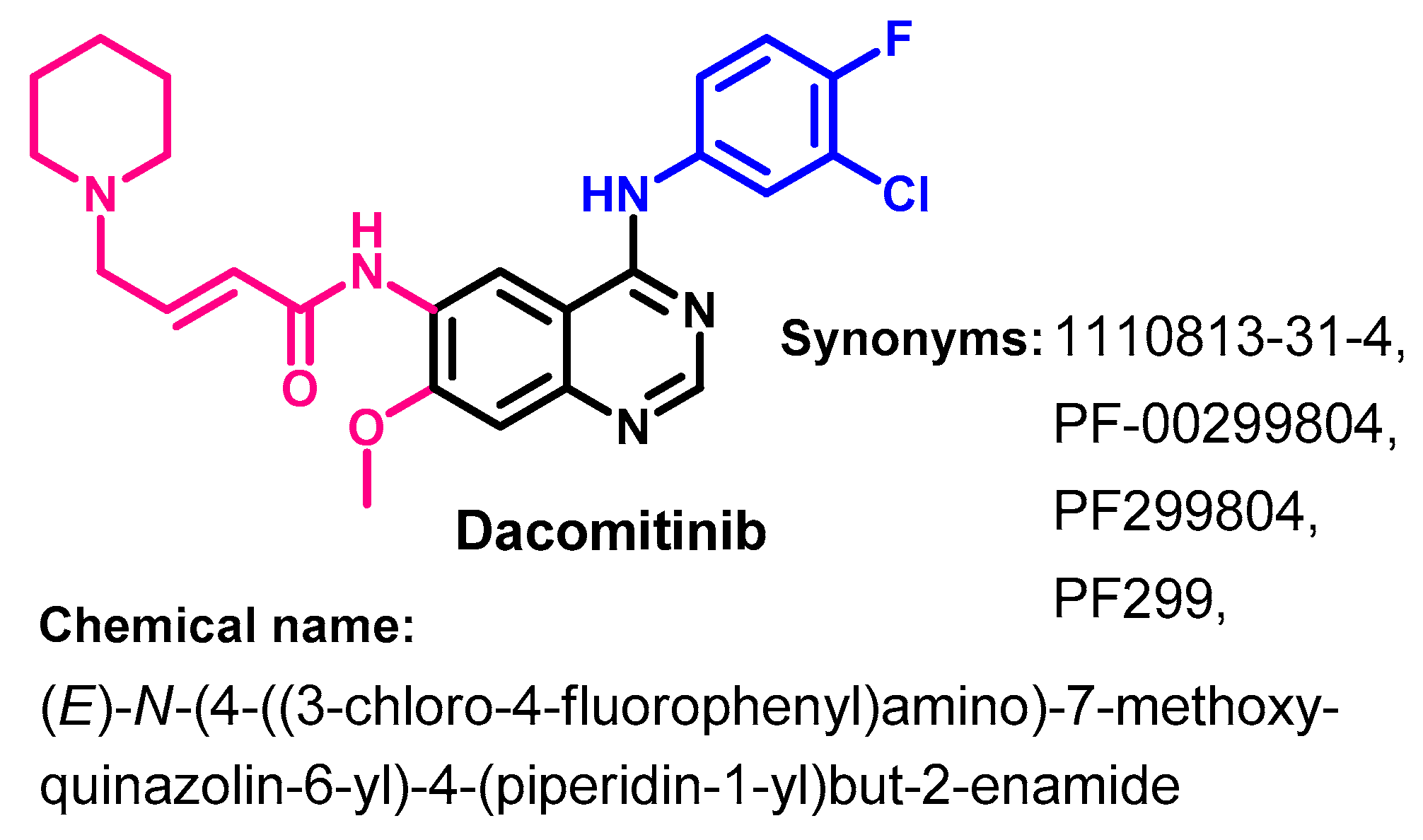
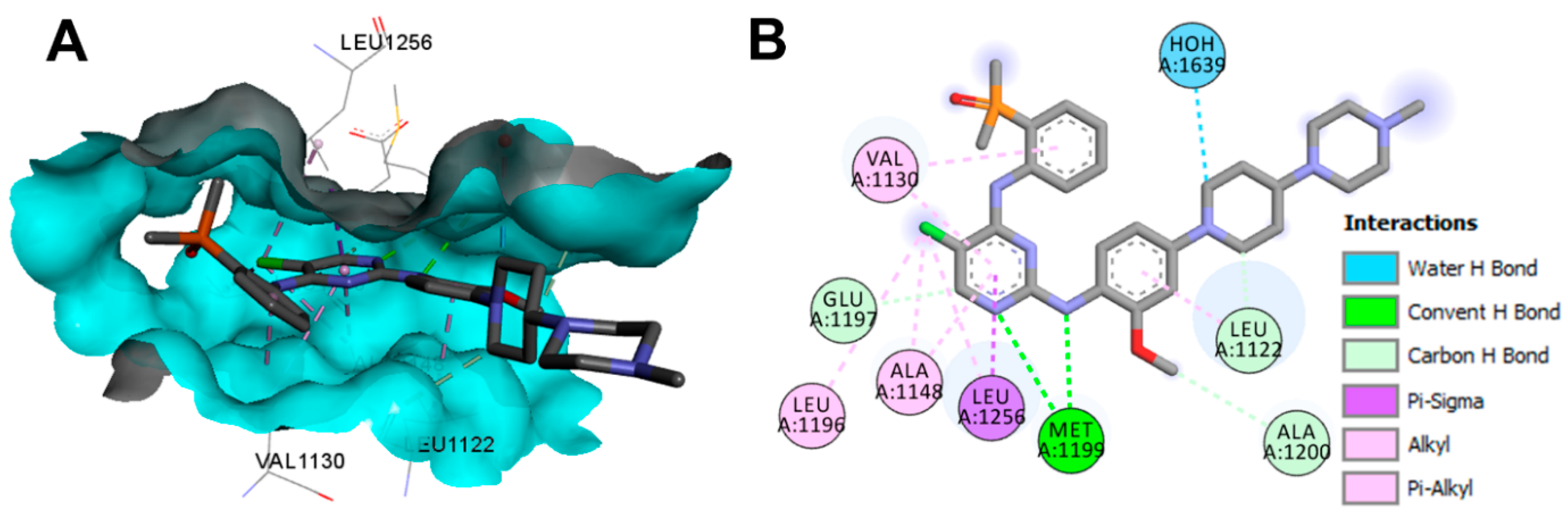
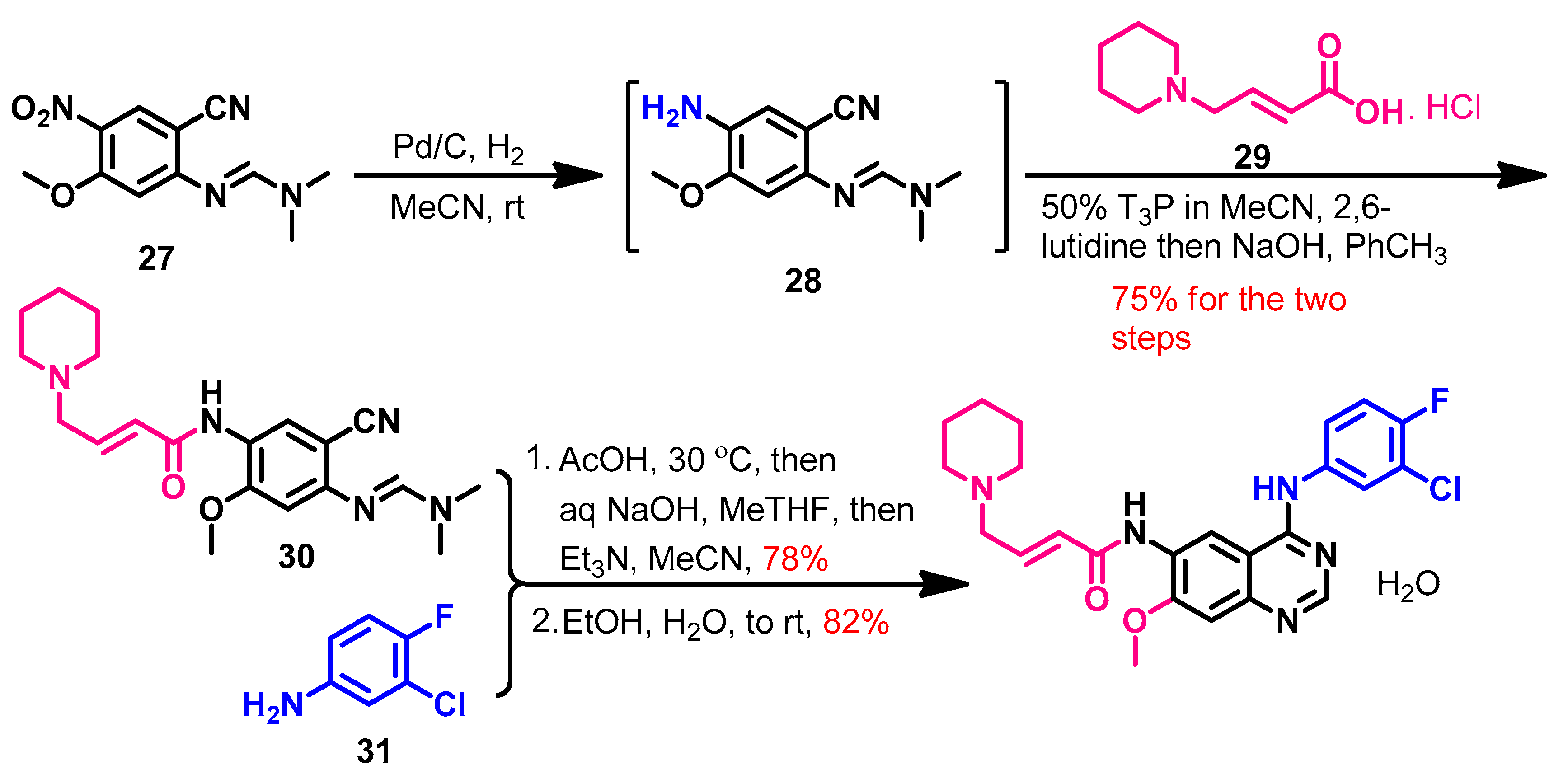
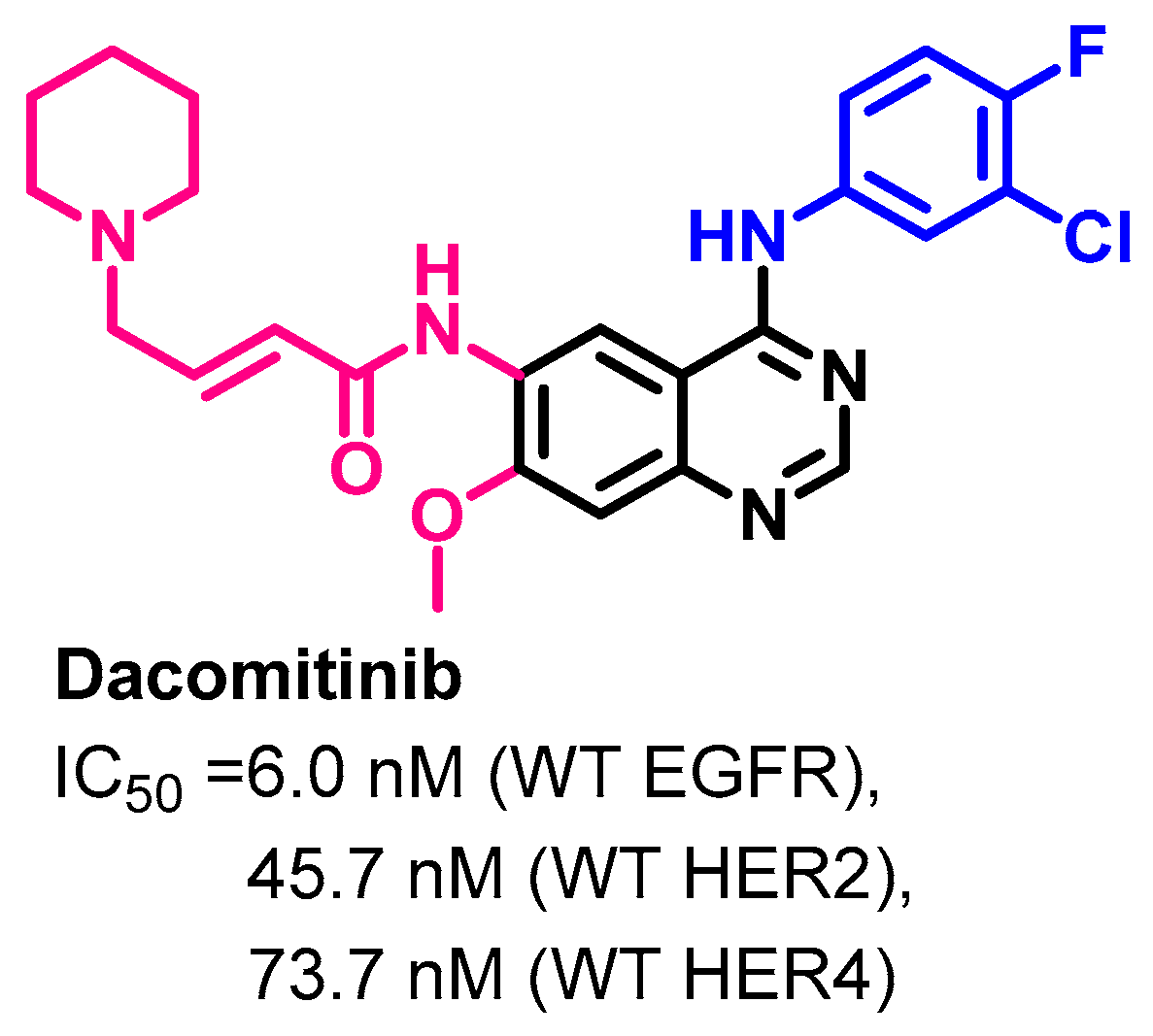
1.2.4. Dacomitinib
Approval History
Synthesis
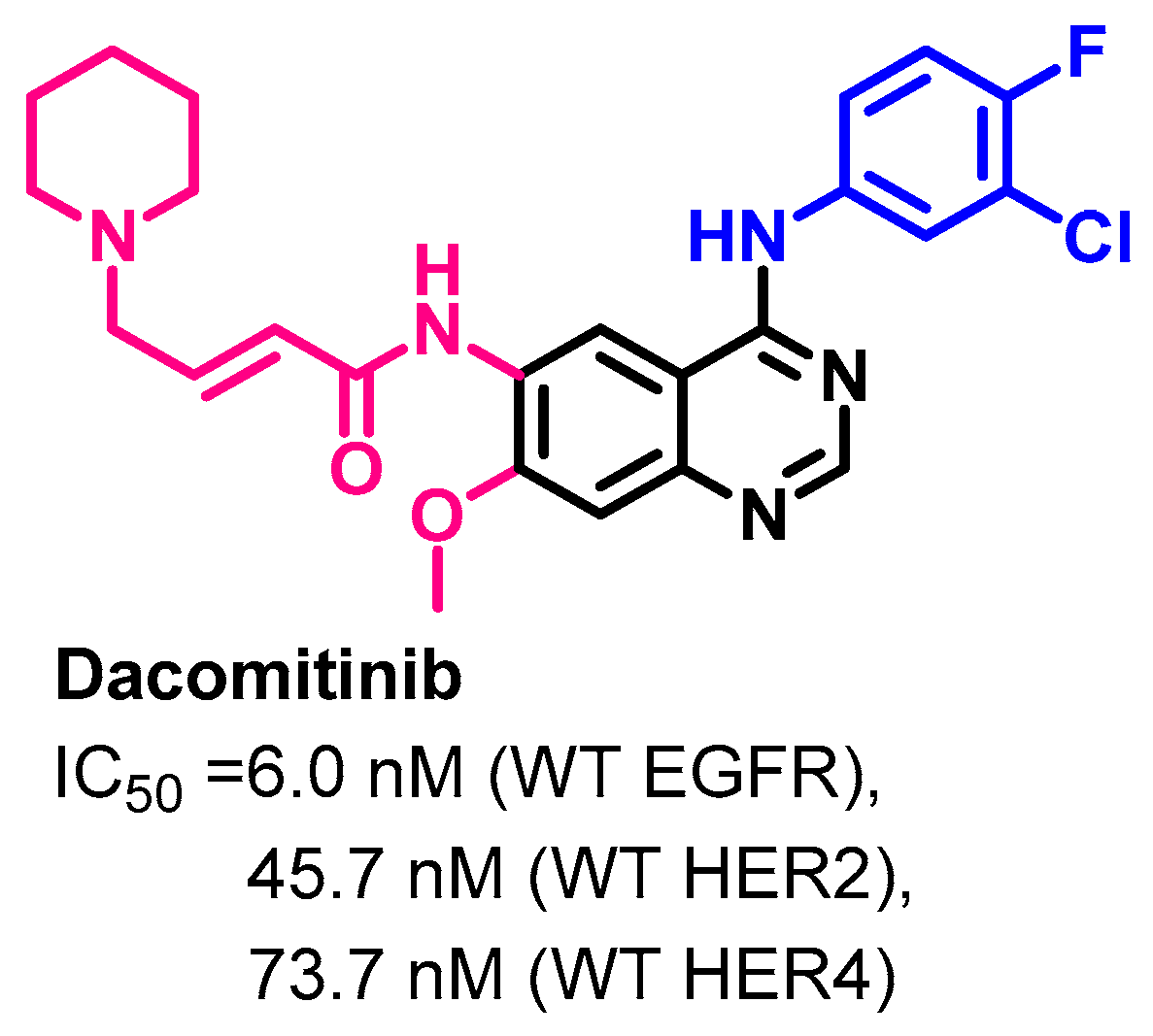
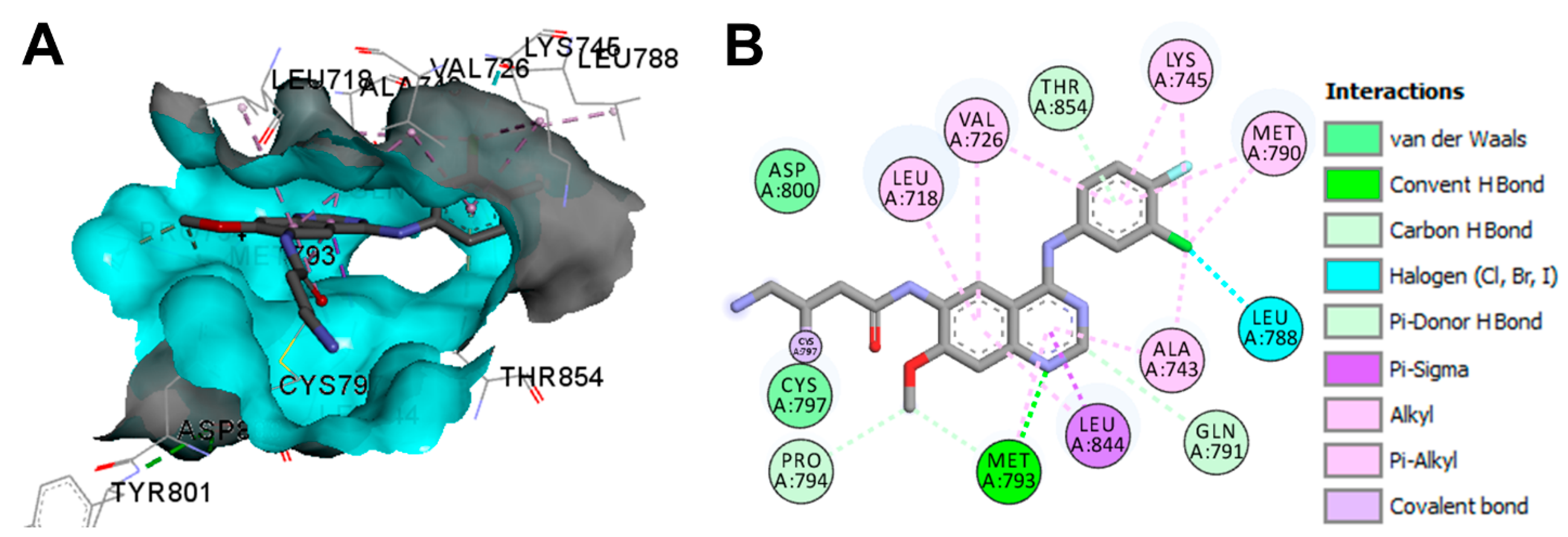
Target Kinases
Crystal Structures and Binding Interactions
Biological Activity
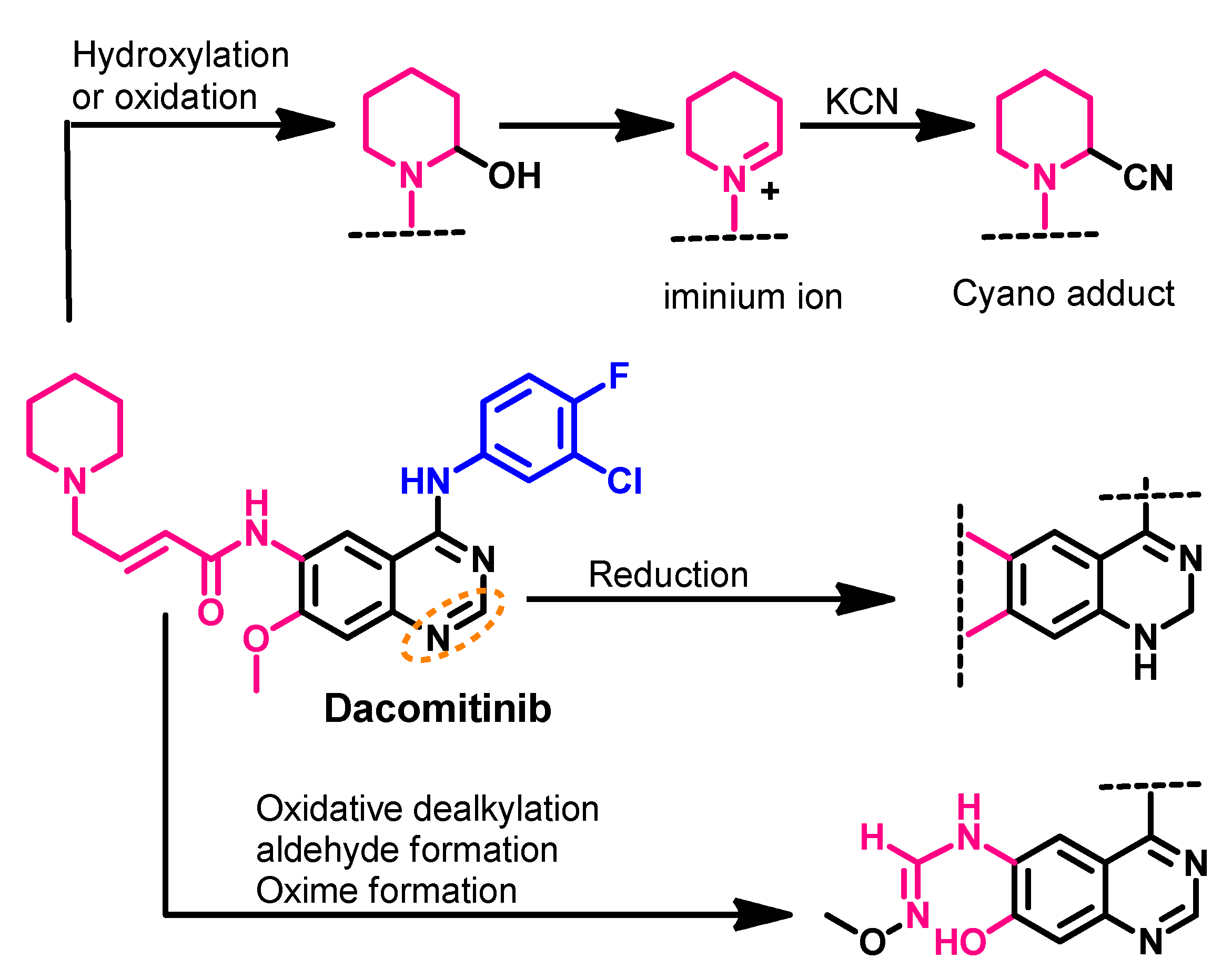
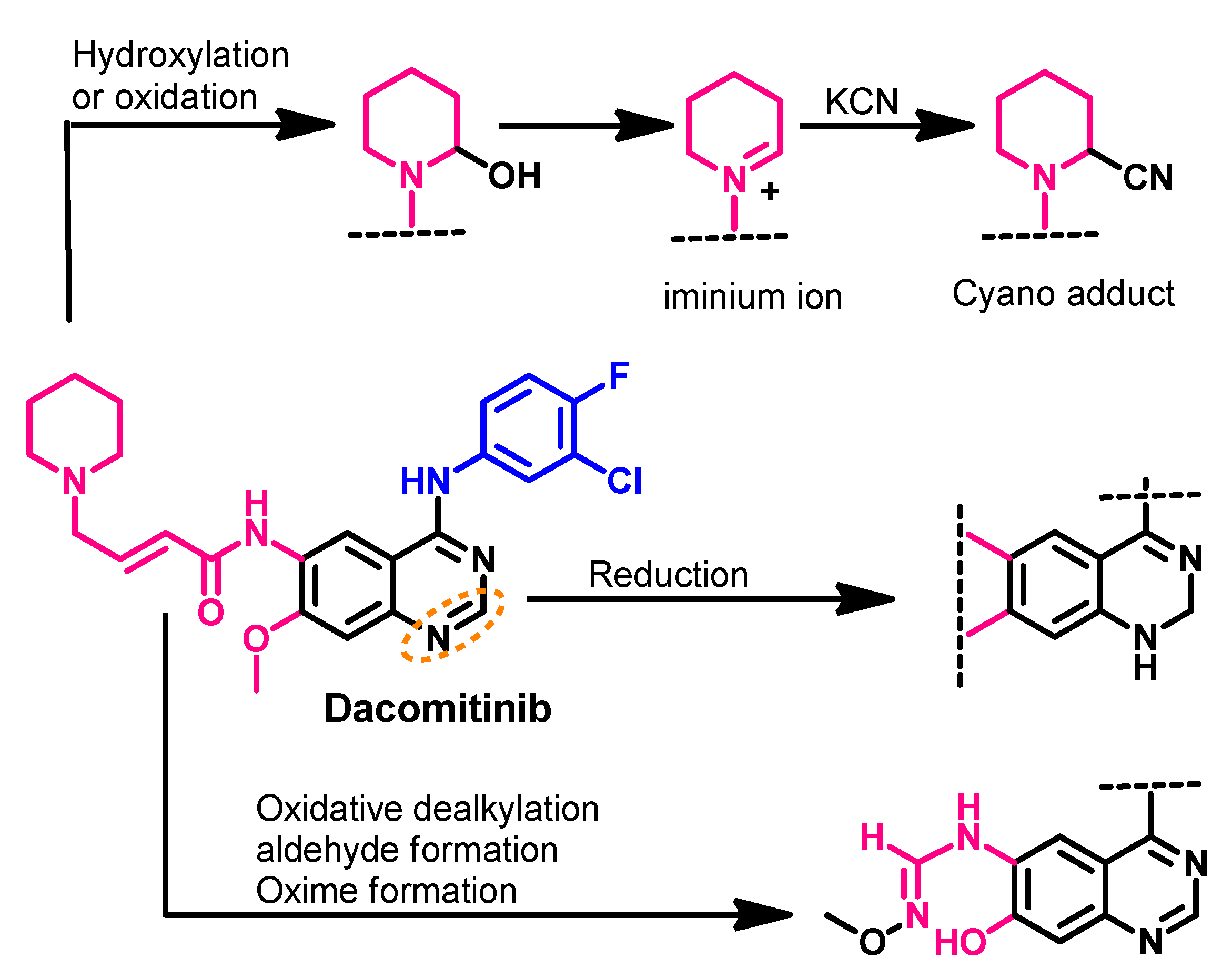
Metabolism
1.2.5. Erlotinib
Approval History
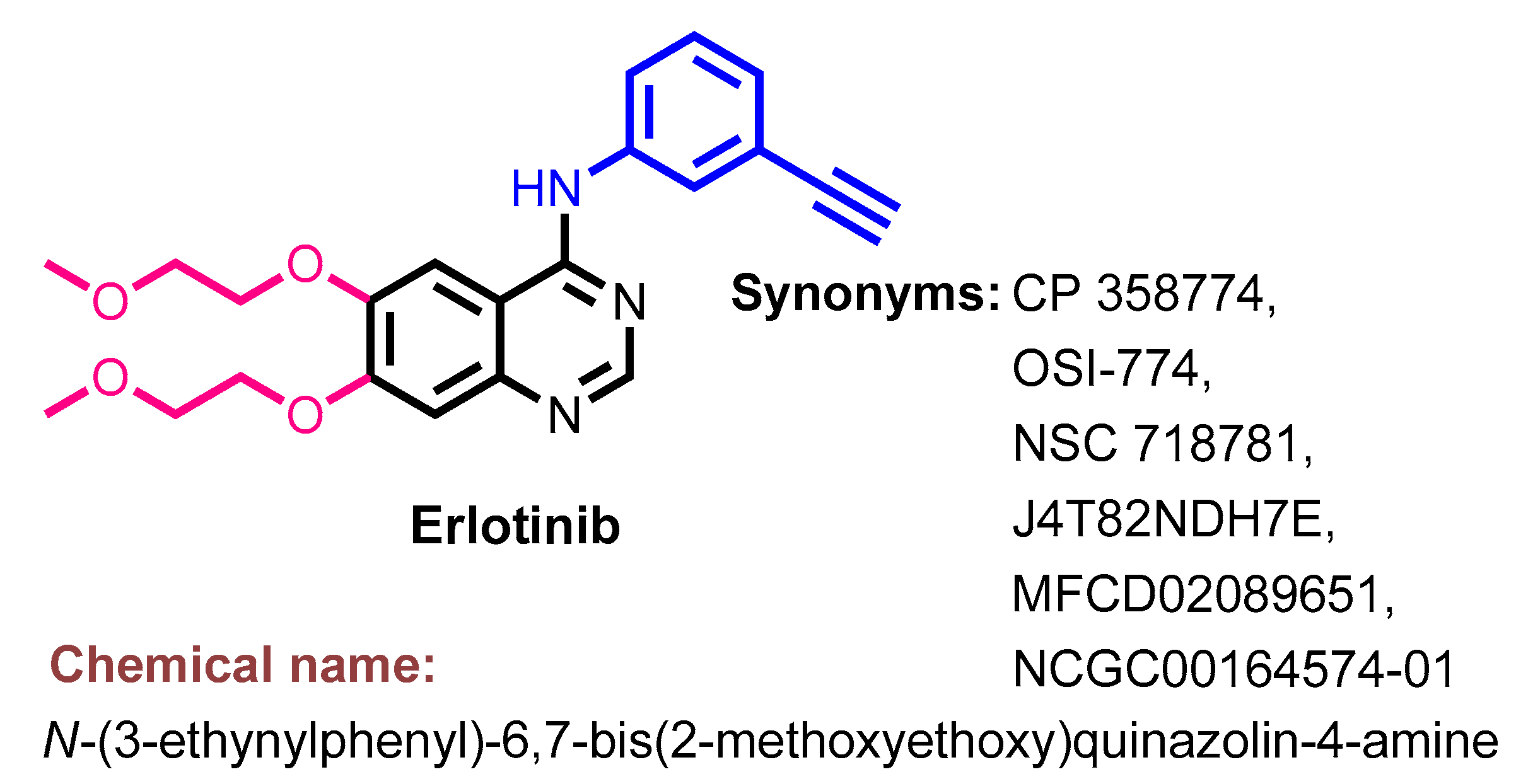
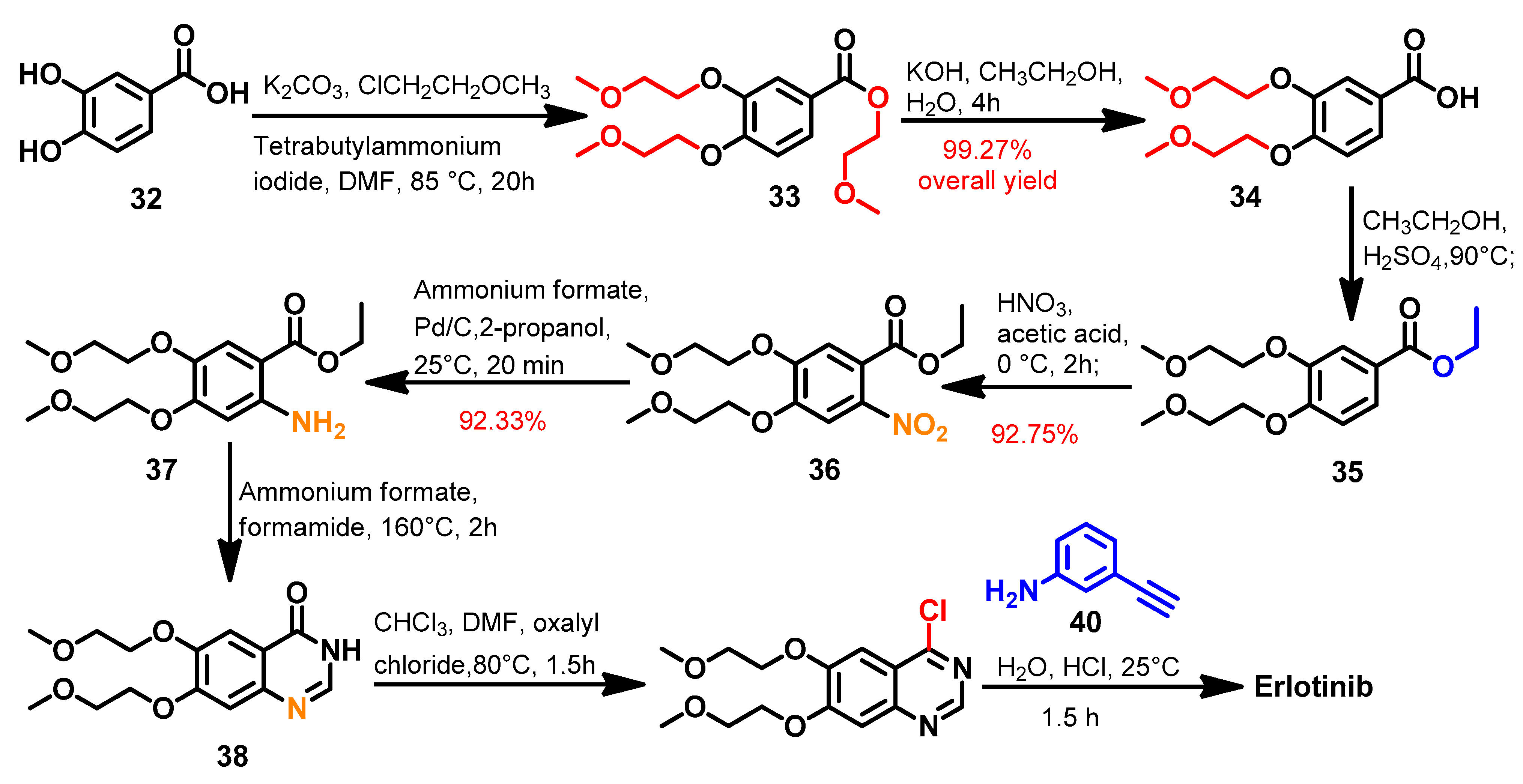
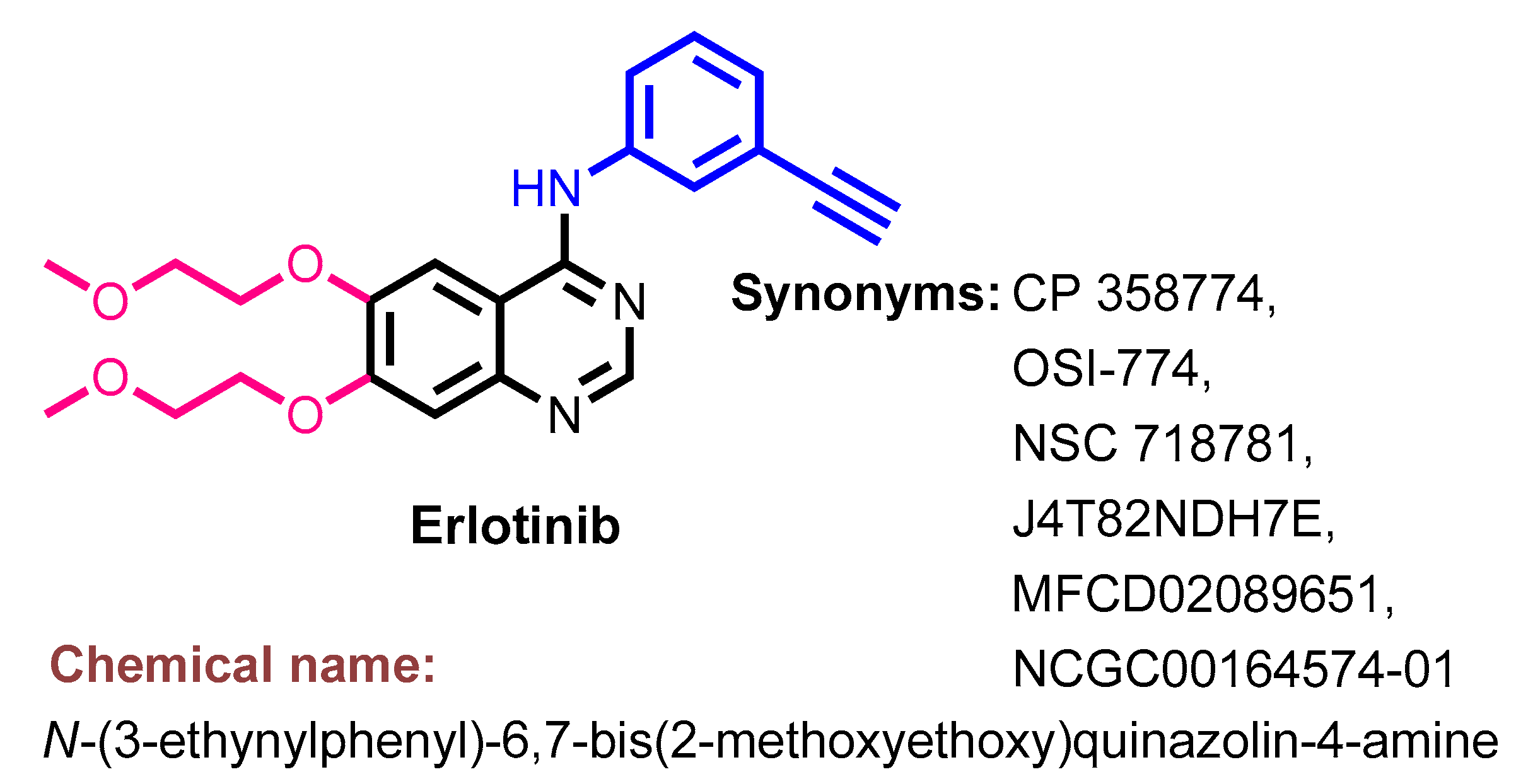
Synthesis
Target Kinases
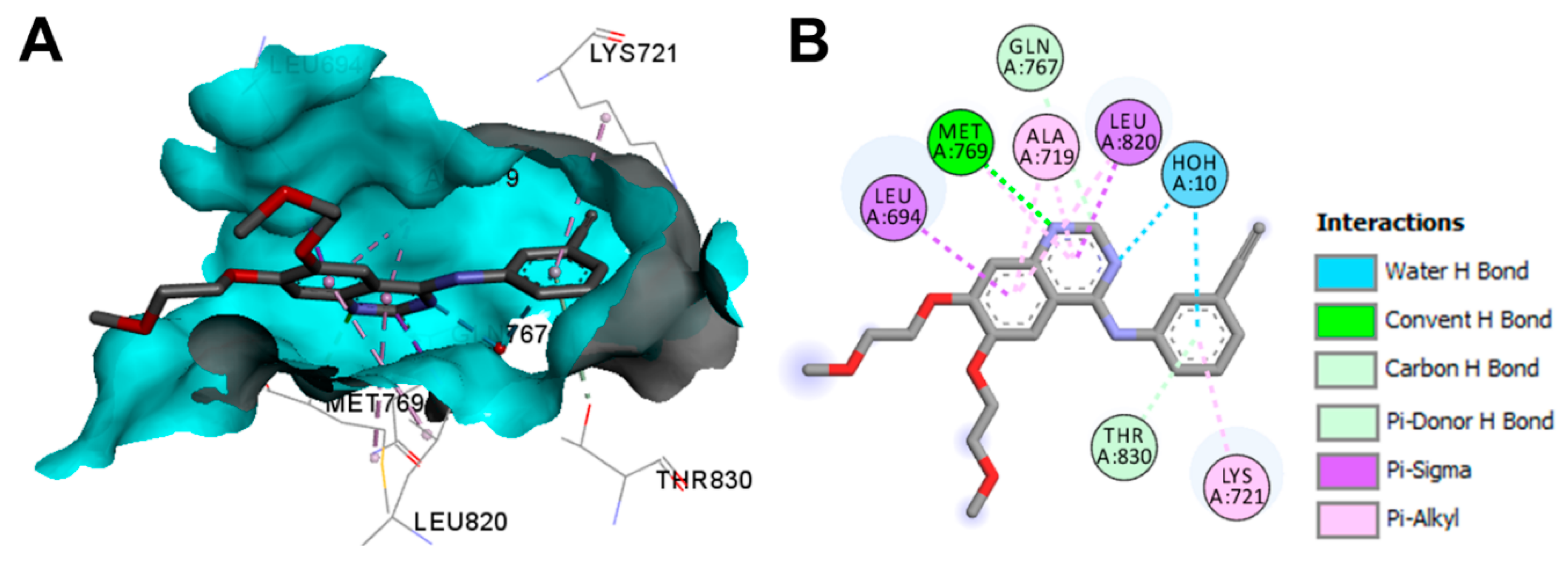
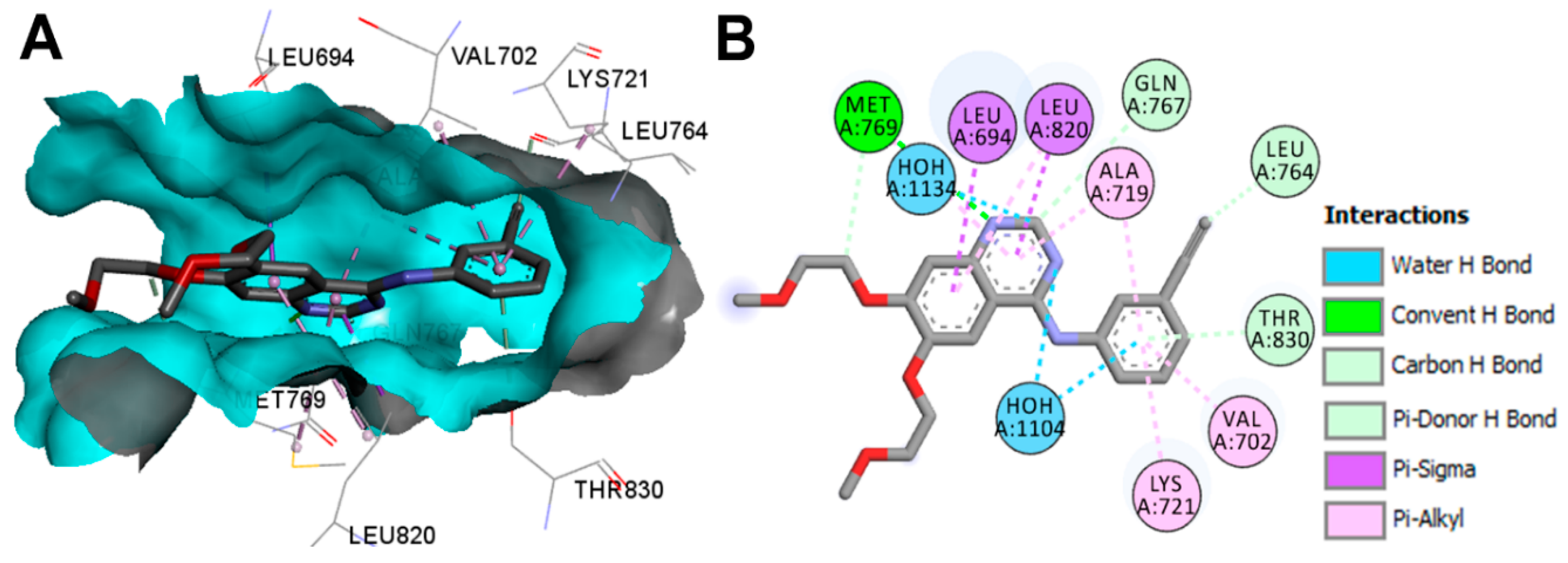
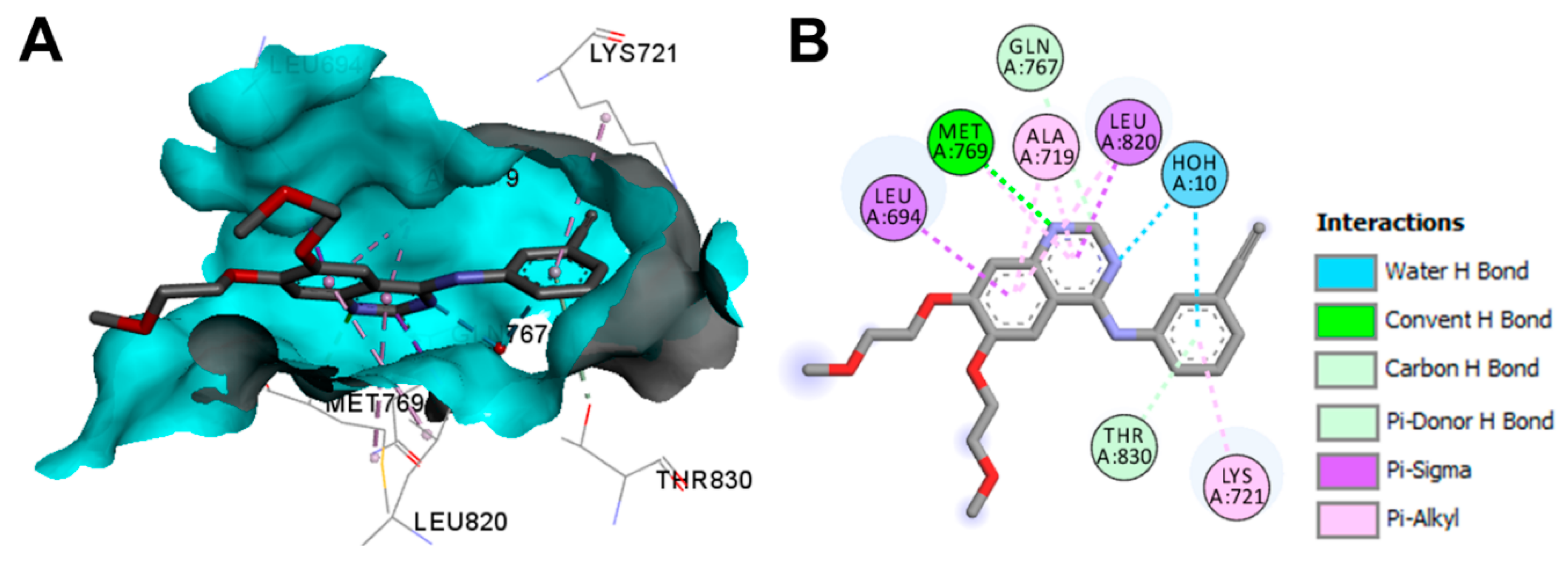
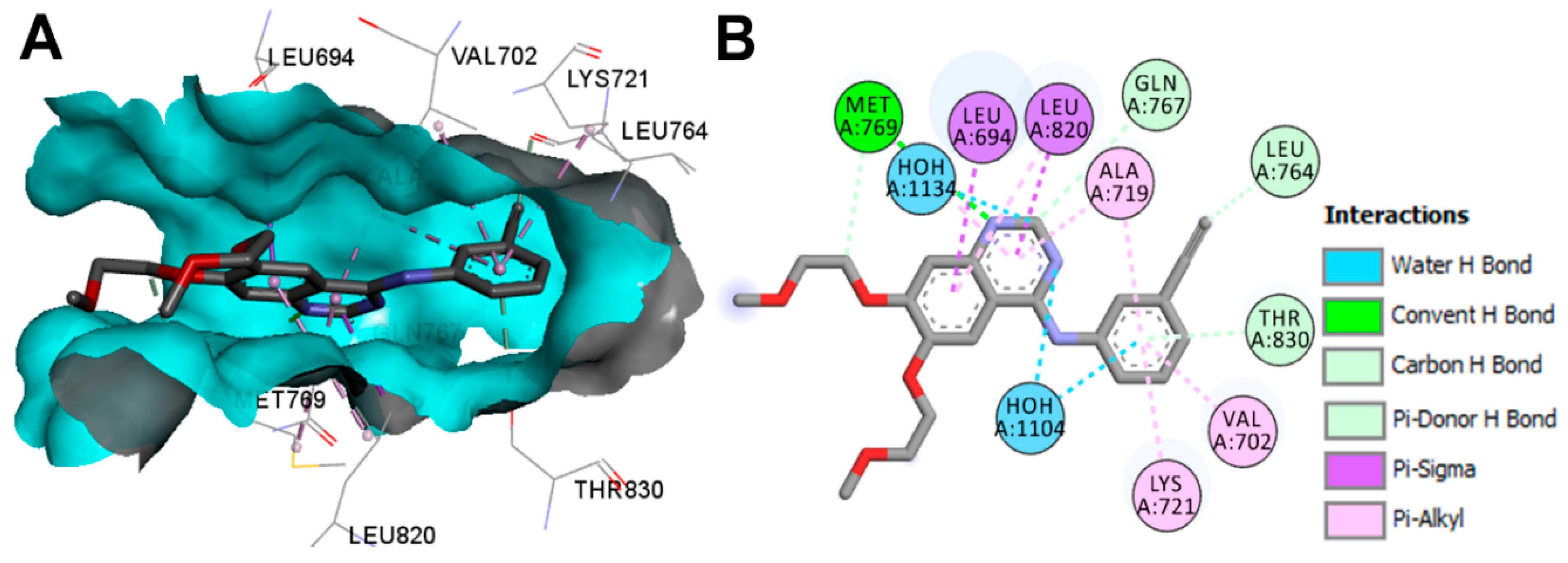
Crystal Structures and Binding Interactions
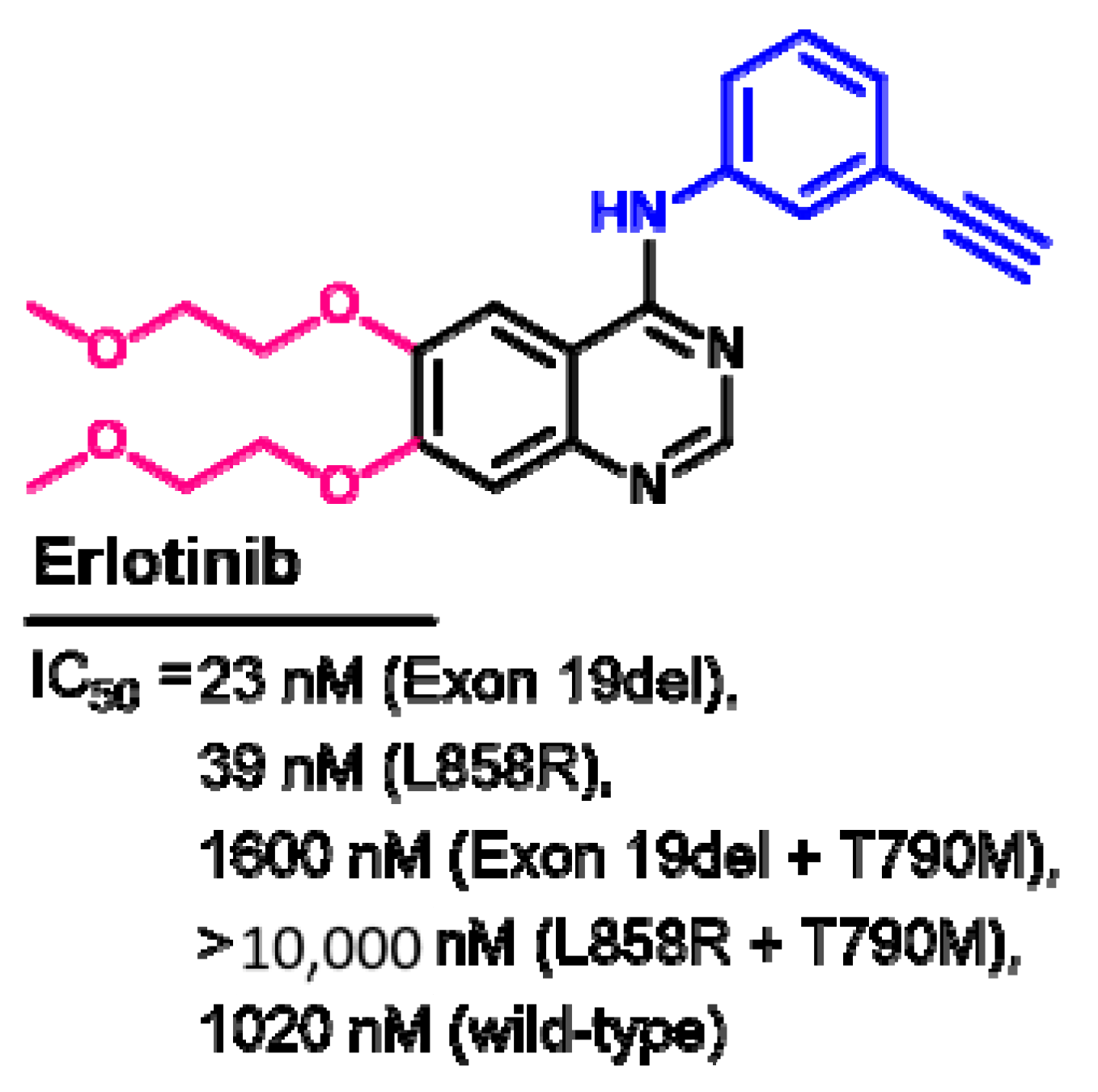
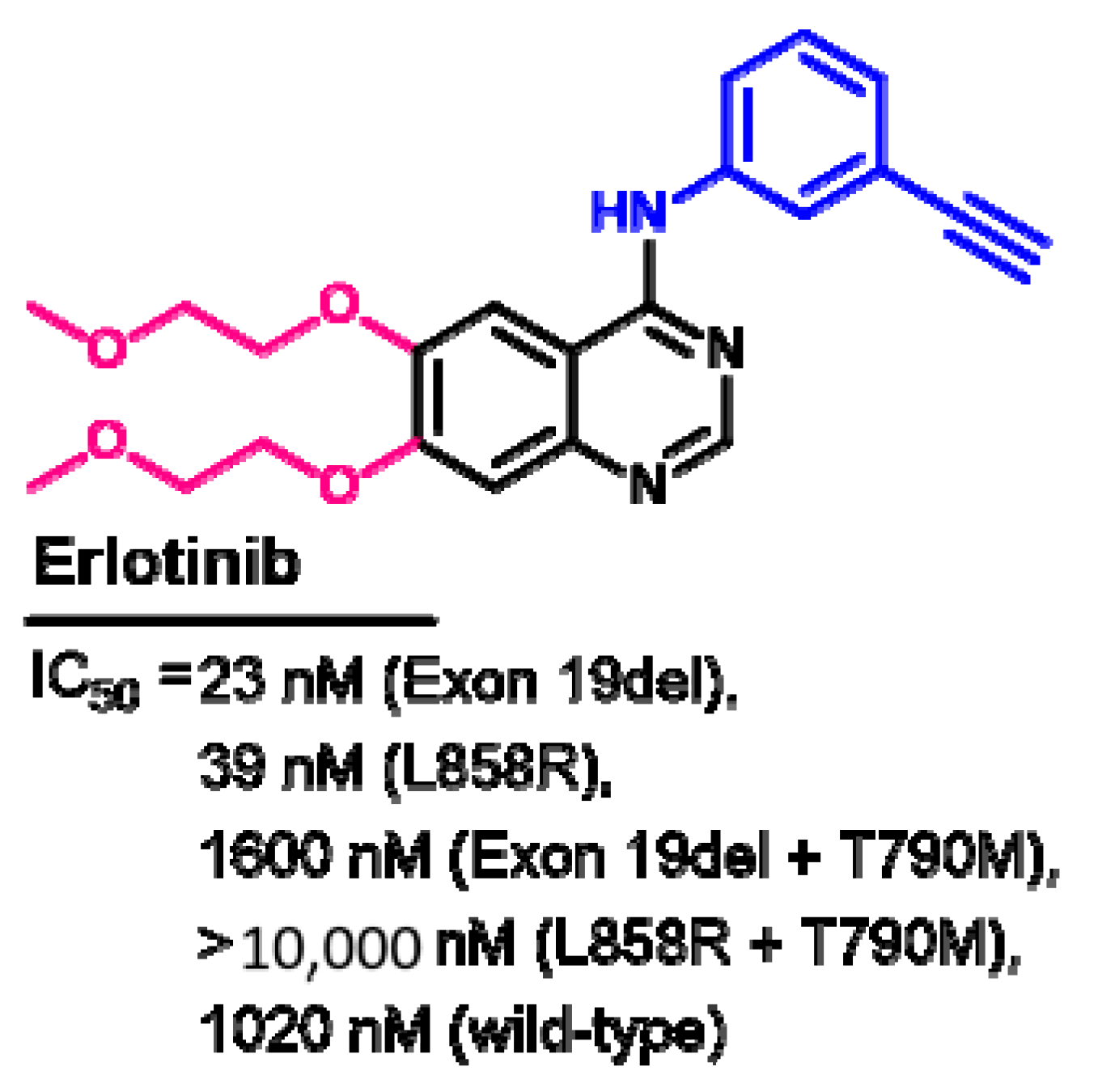
Biological Activity
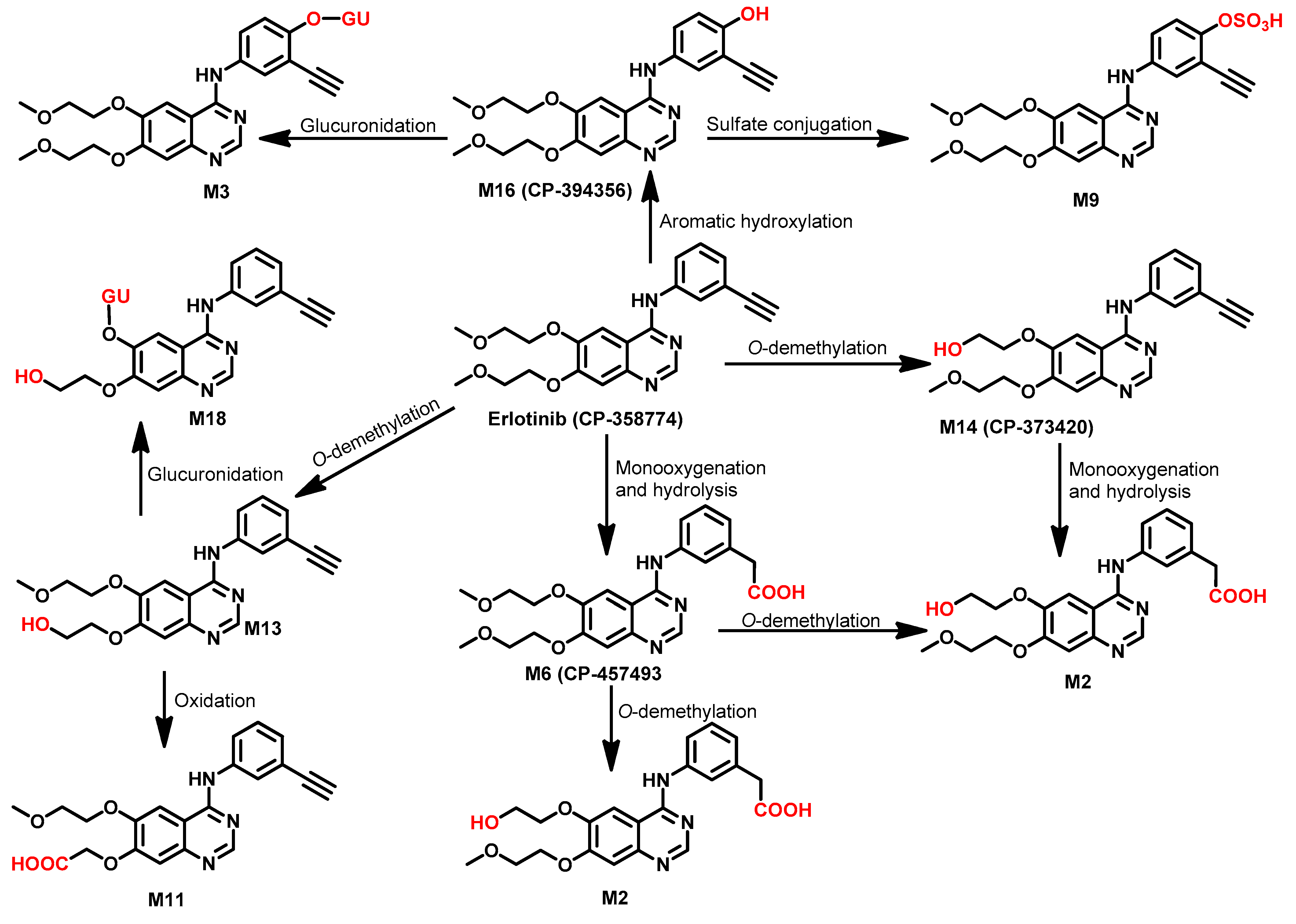
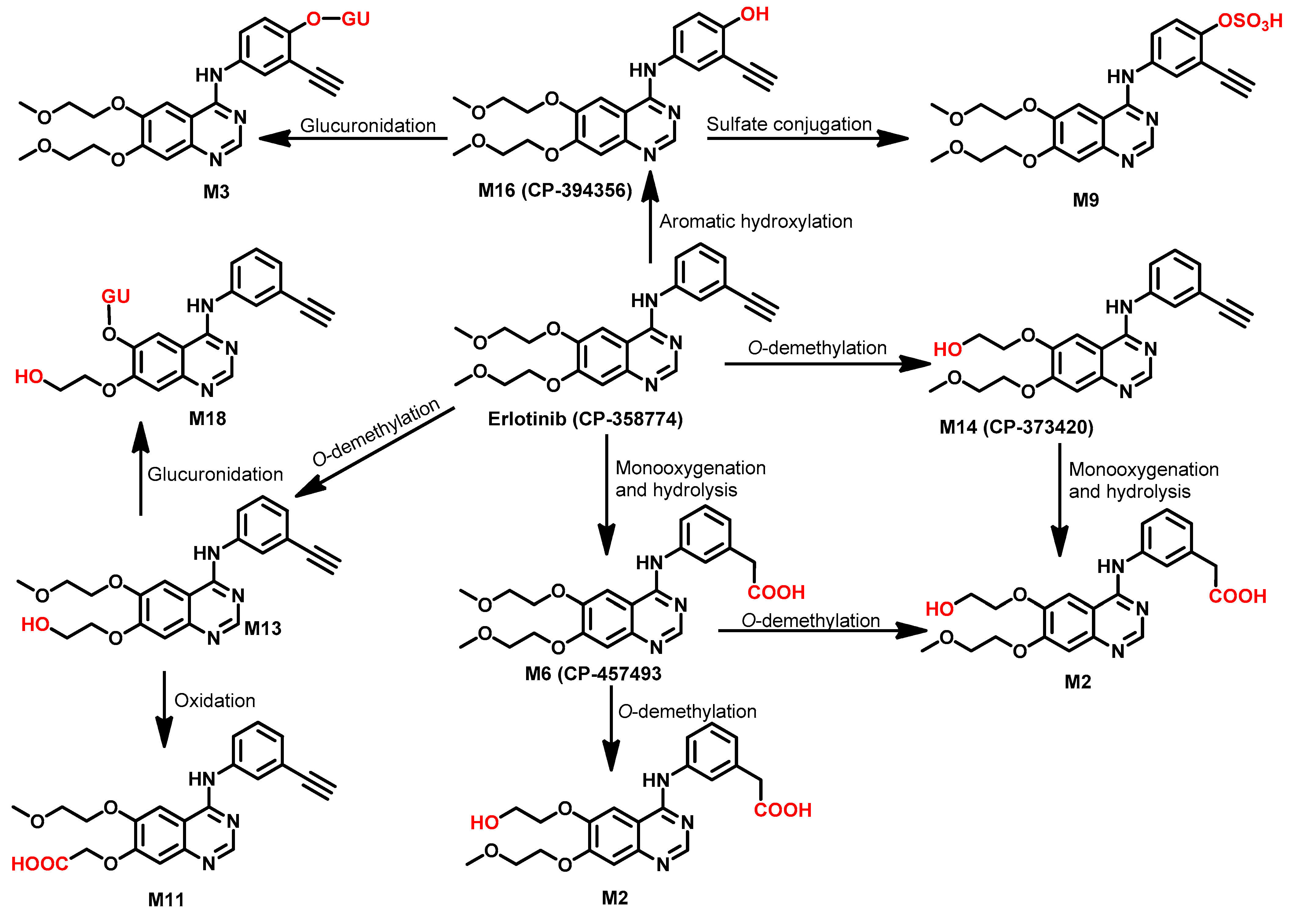
Metabolism
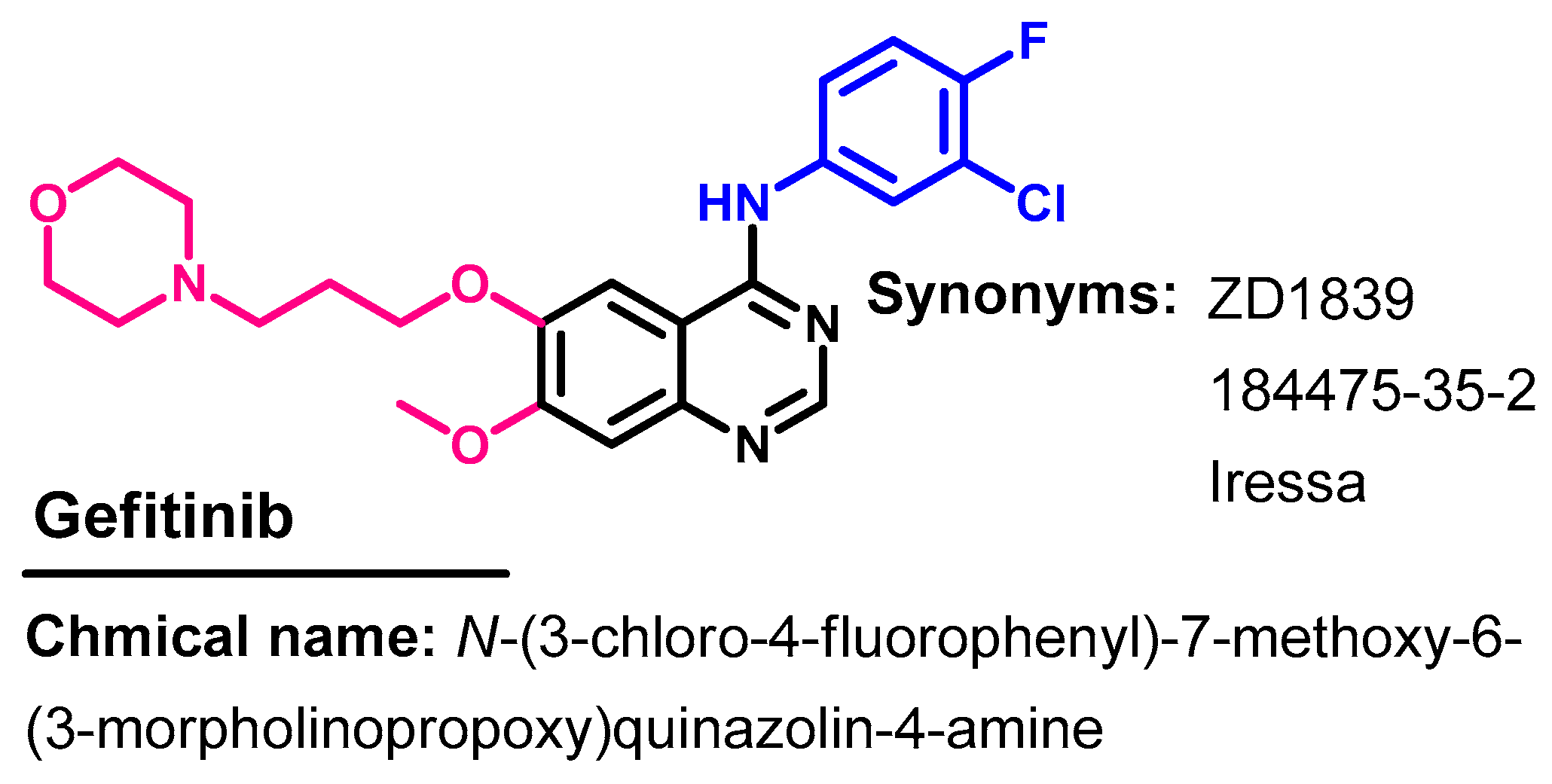
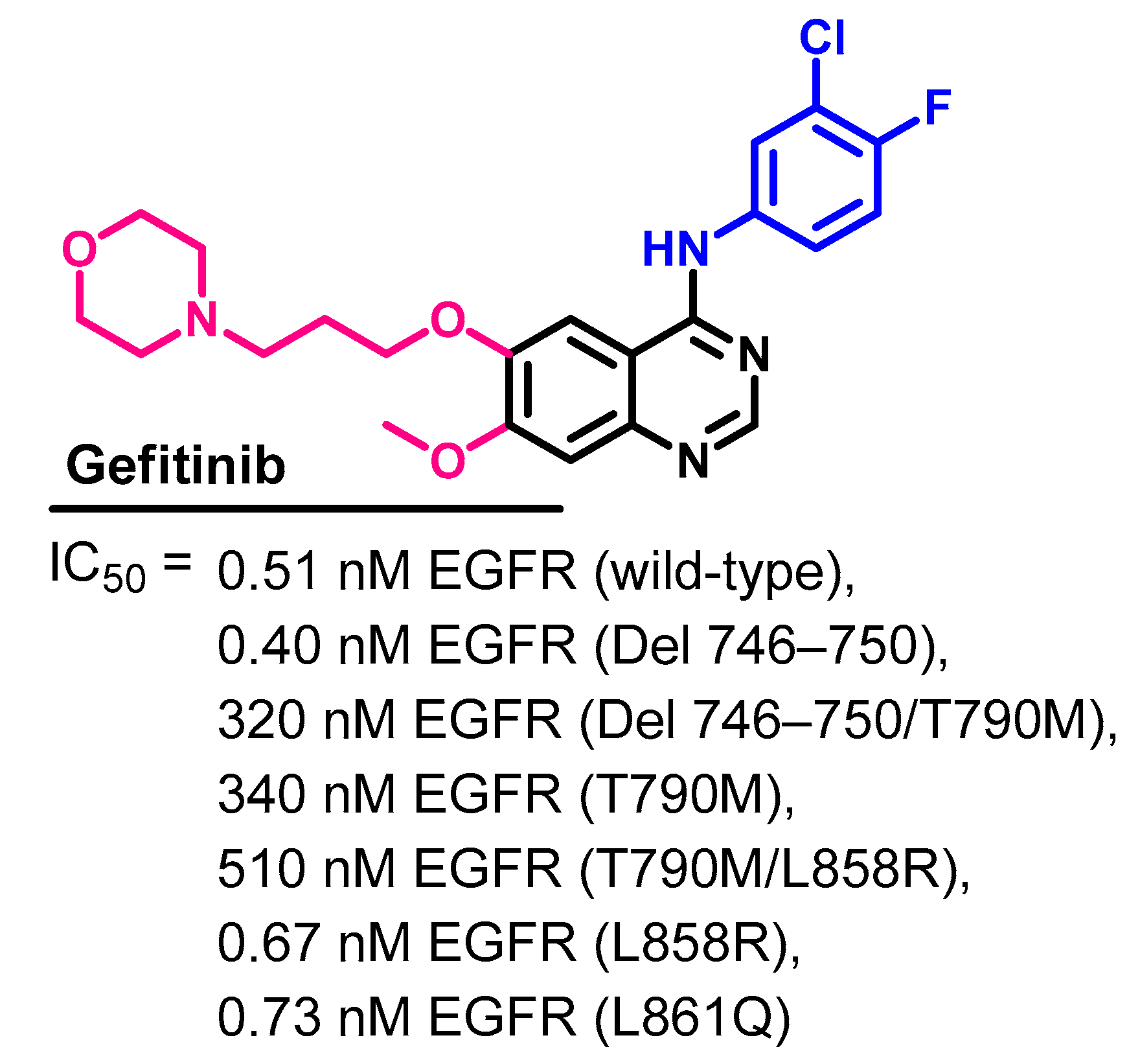
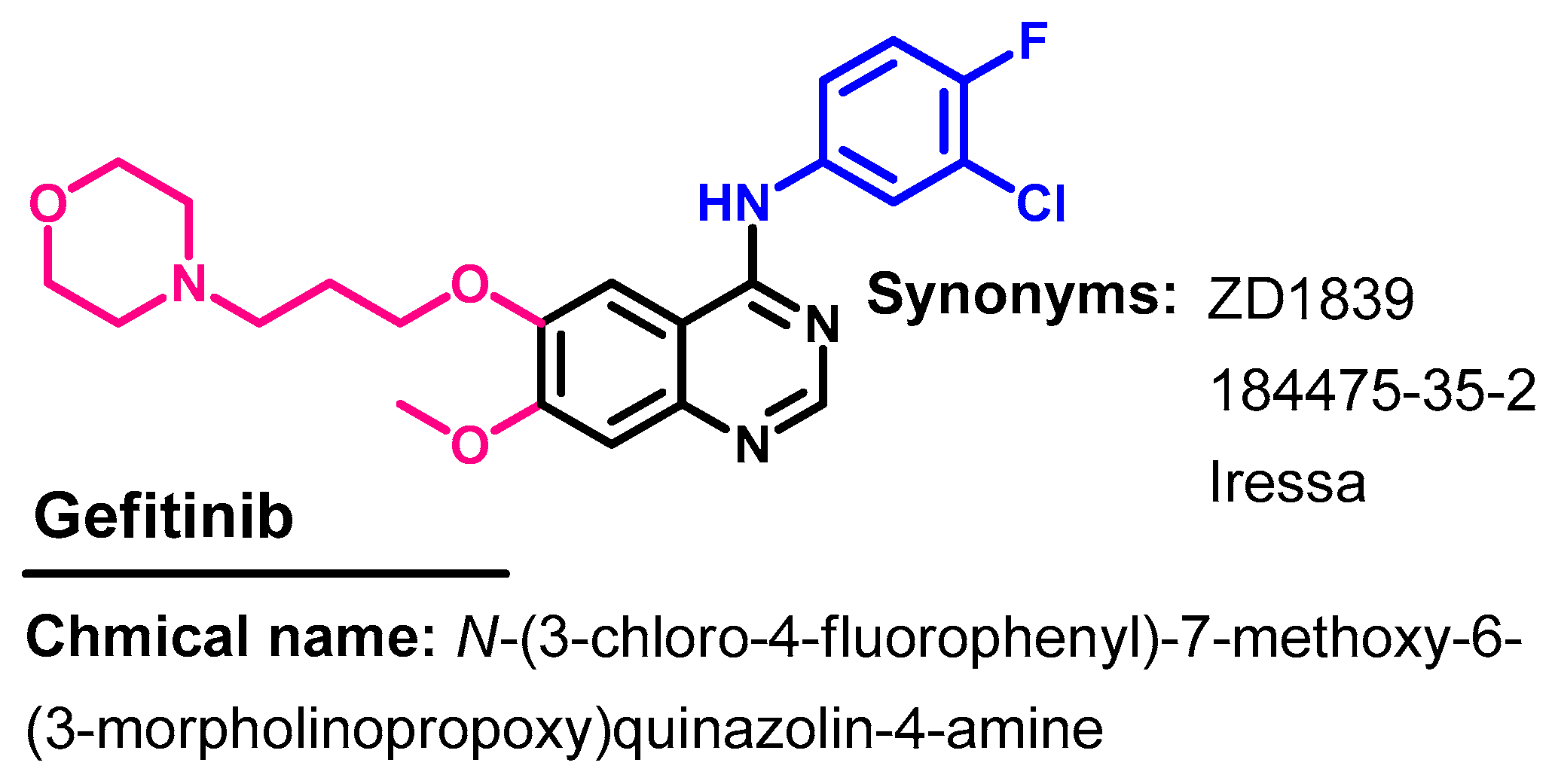
1.2.6. Gefitinib
Approval History
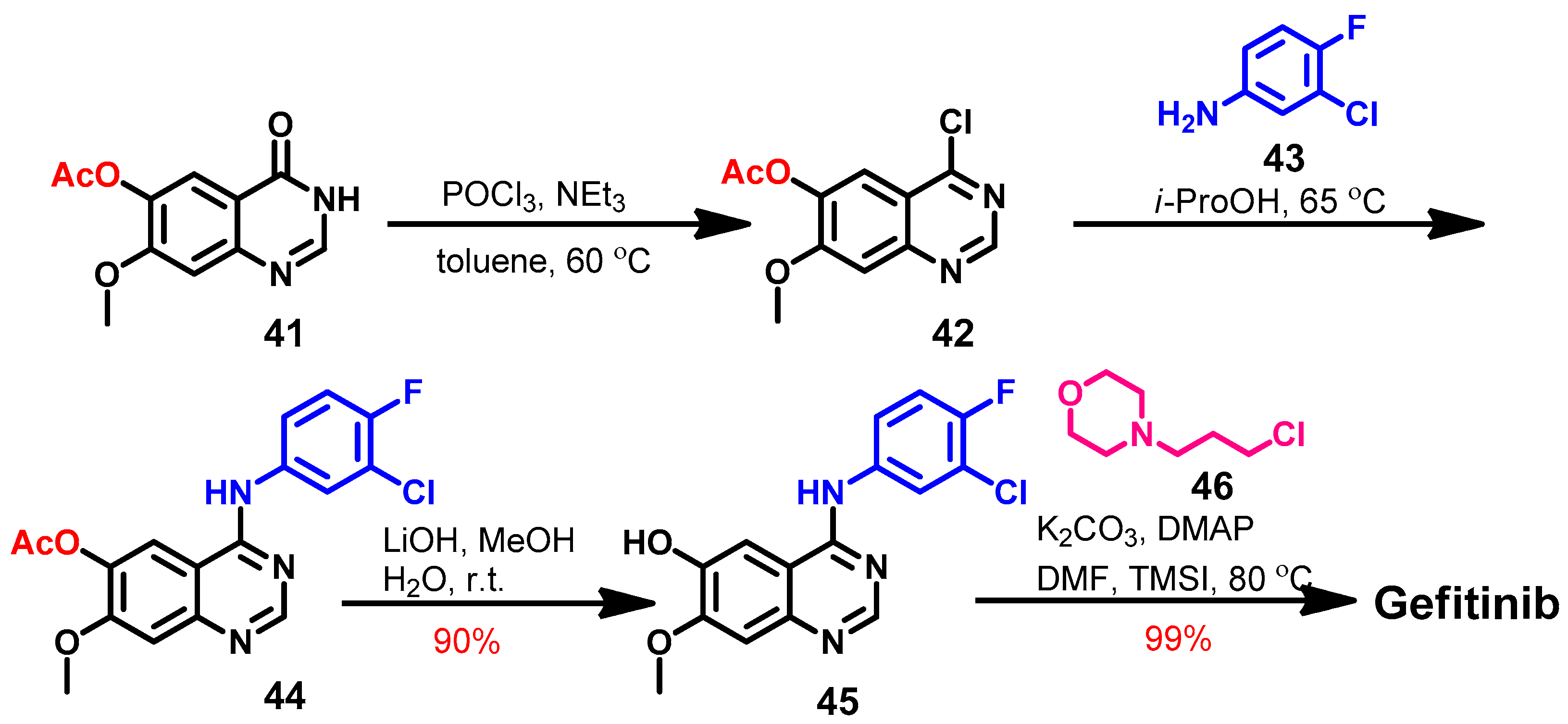
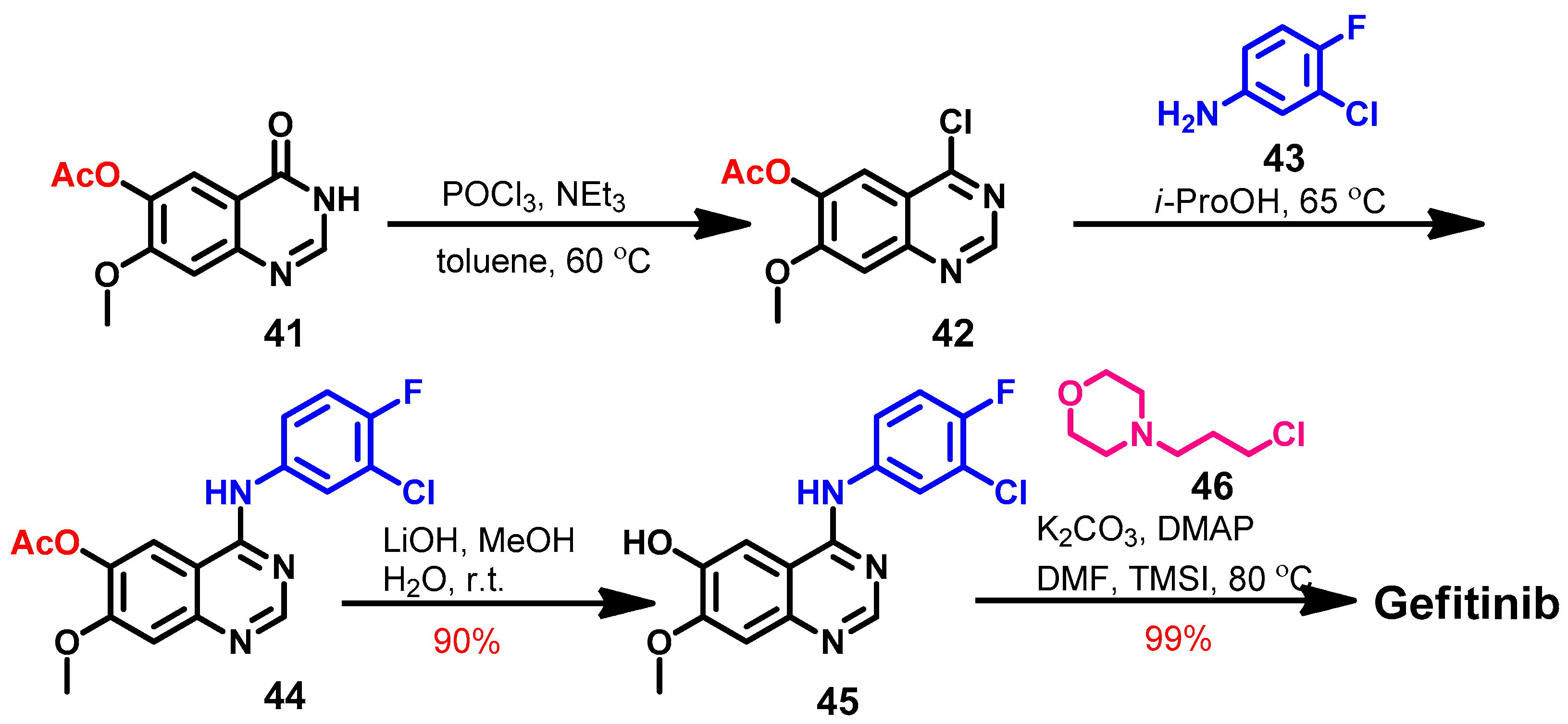
Synthesis
Target Kinases
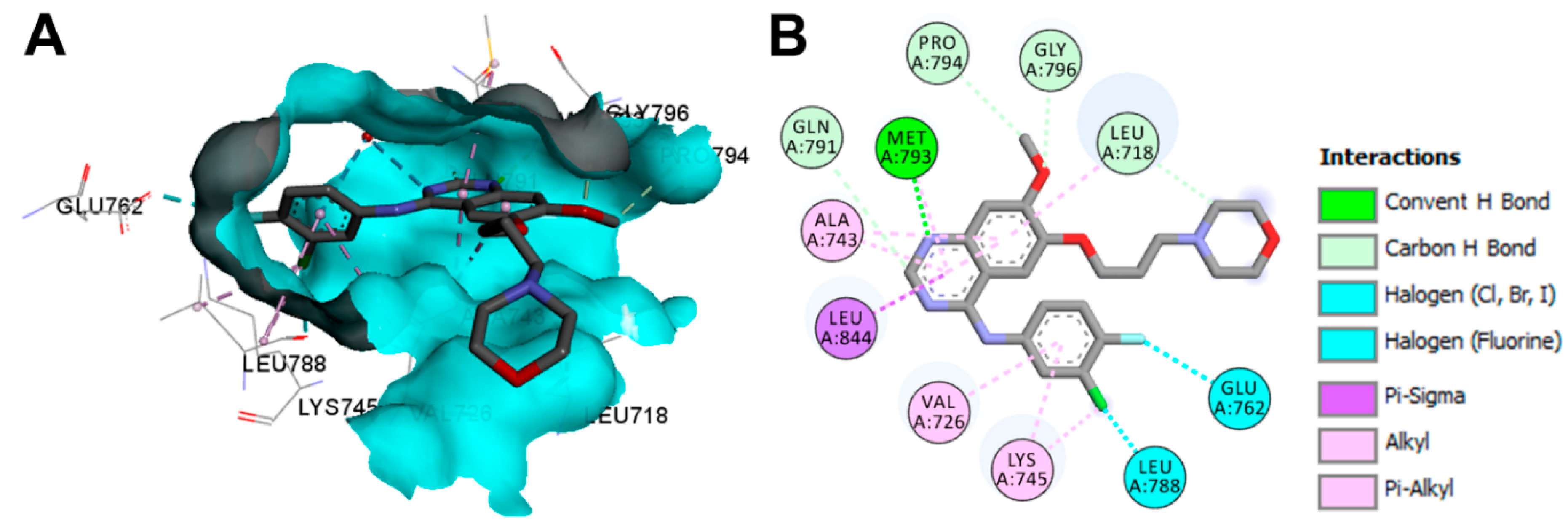
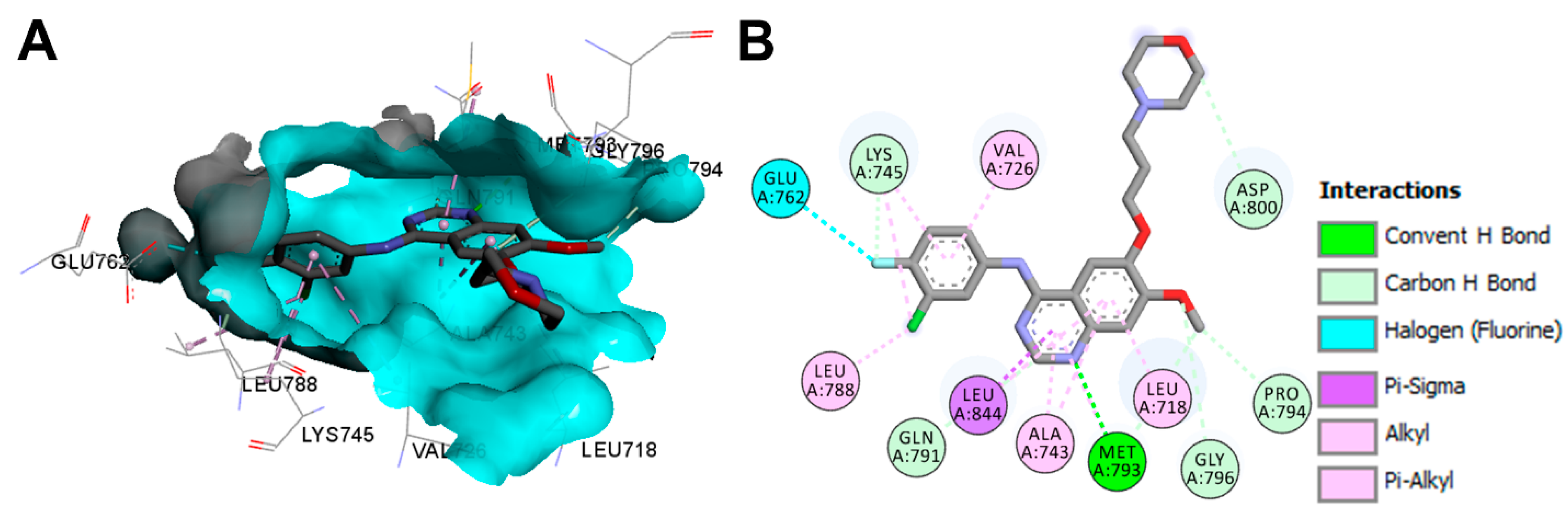
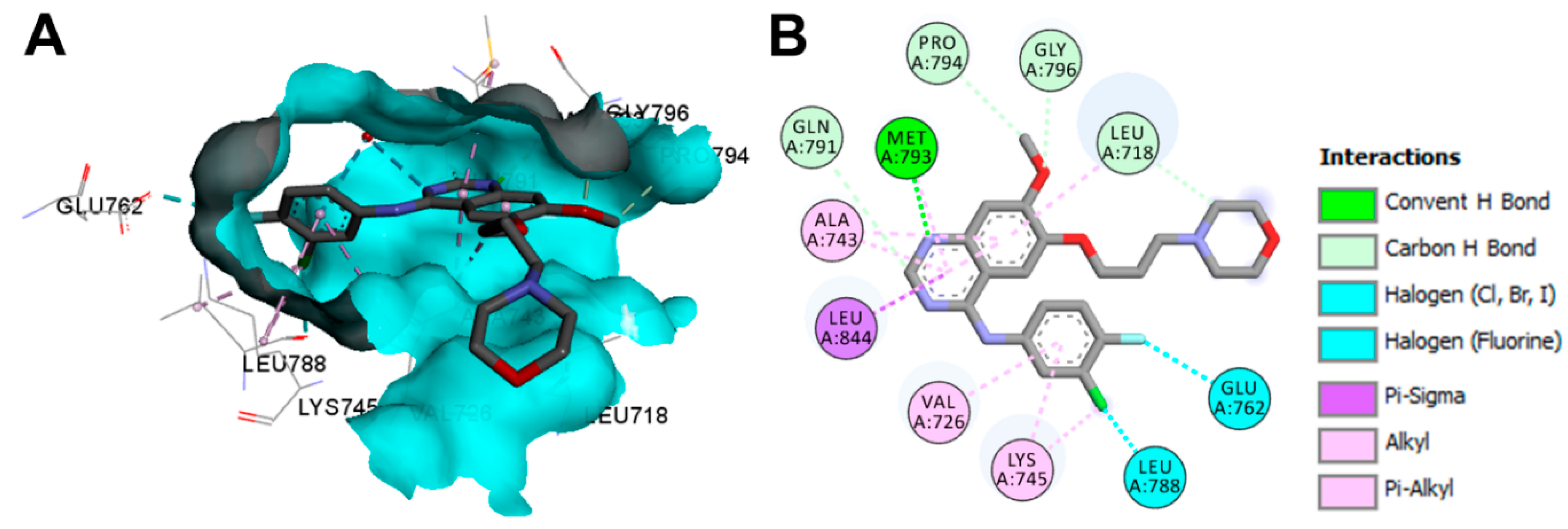
Crystal Structures and Binding Interactions
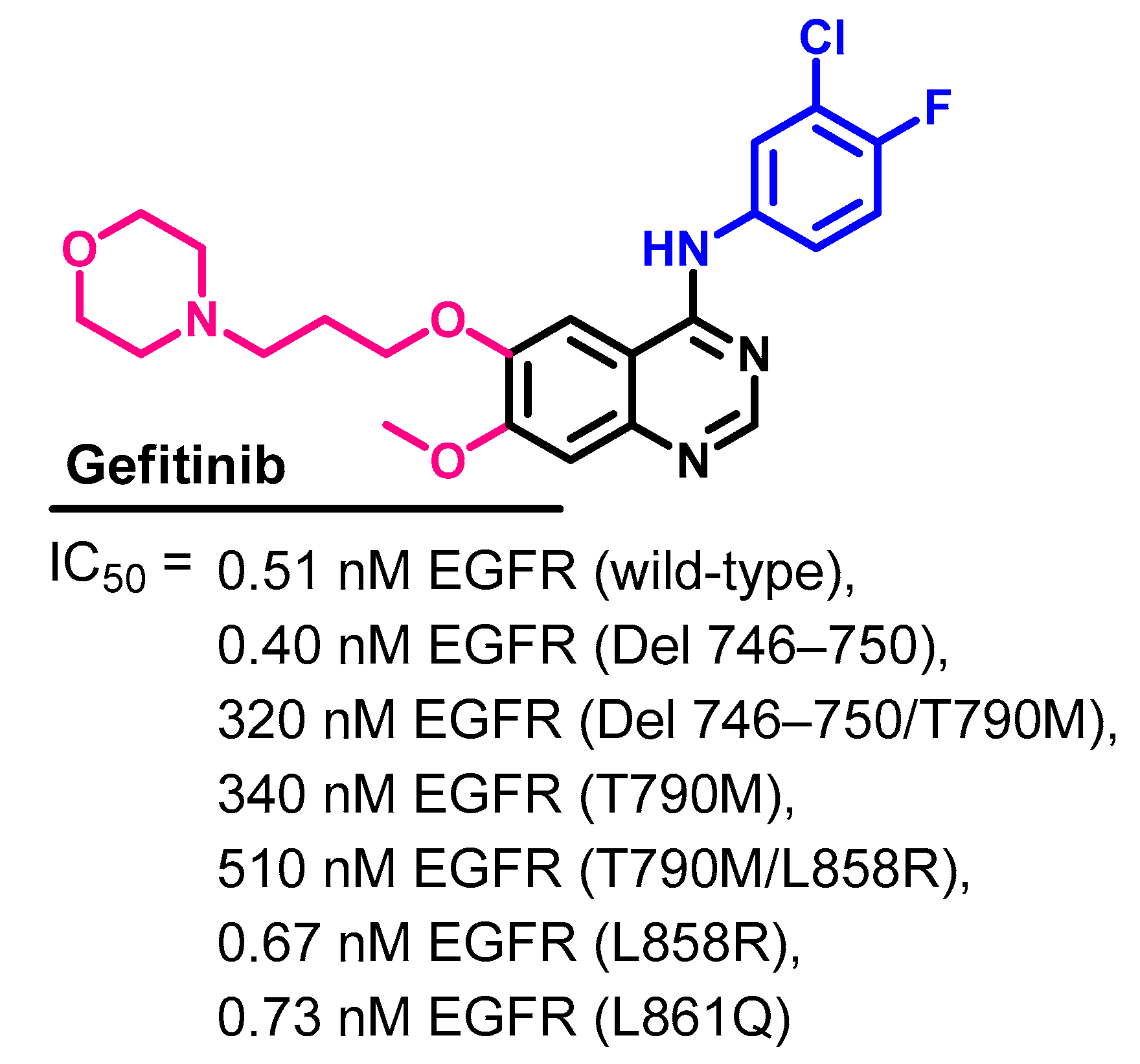
Biological Activity
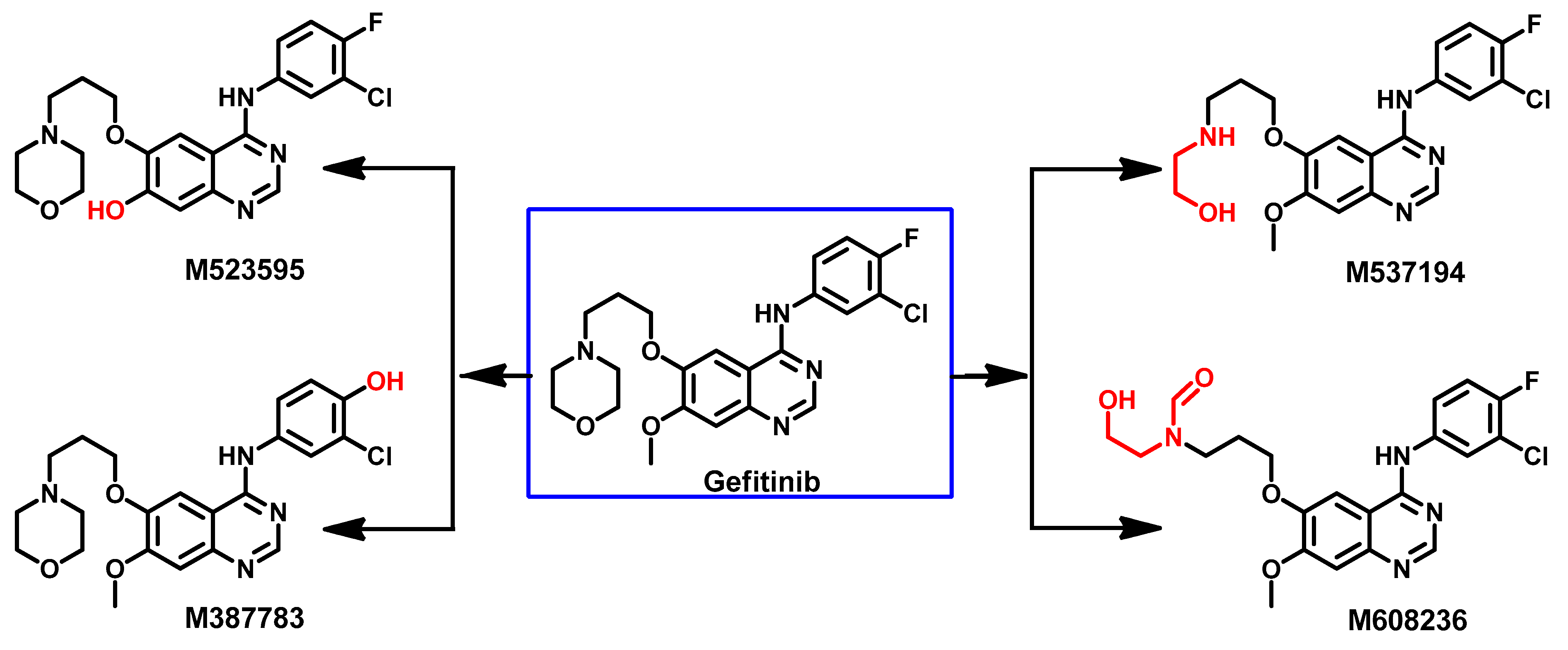
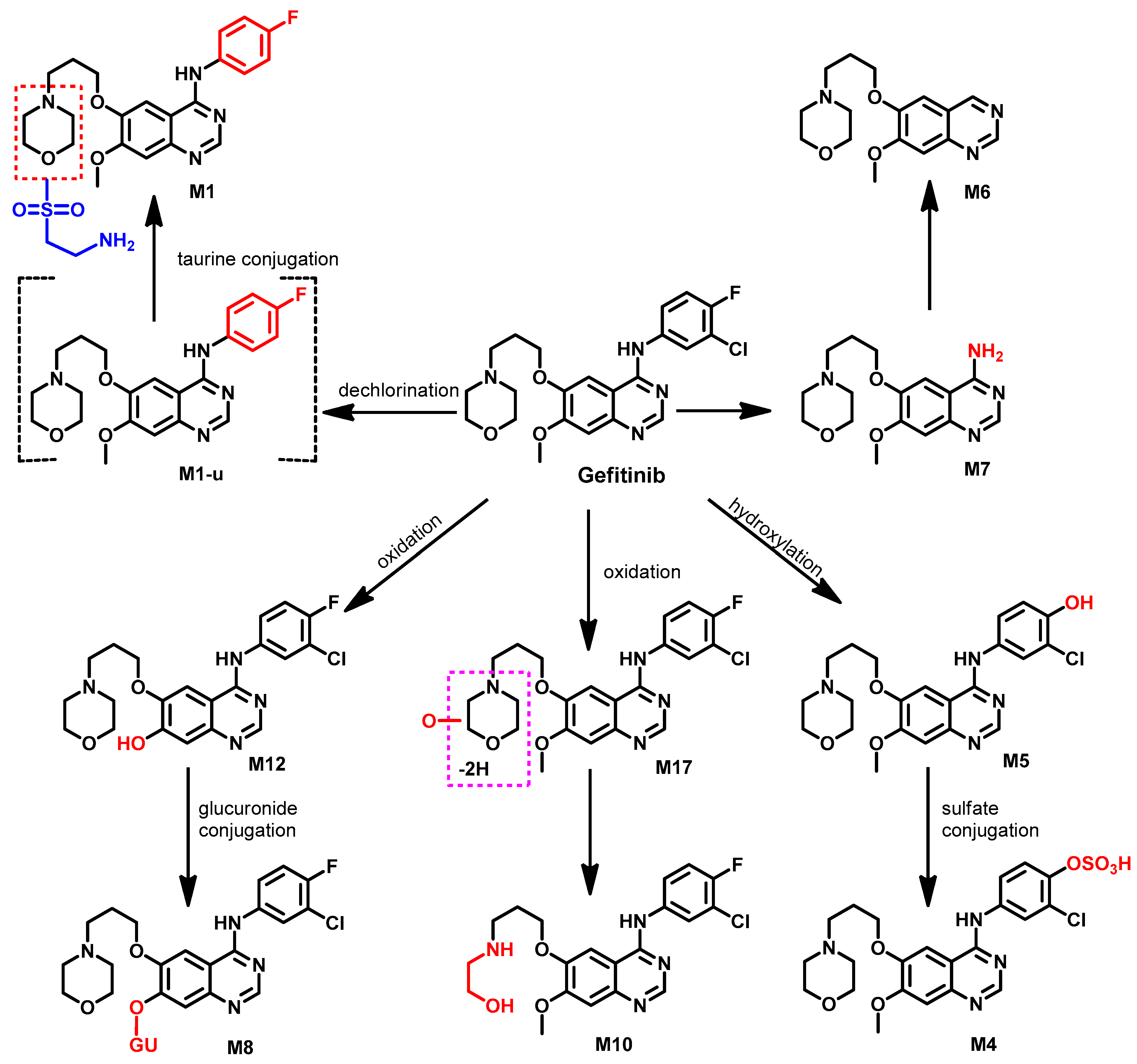
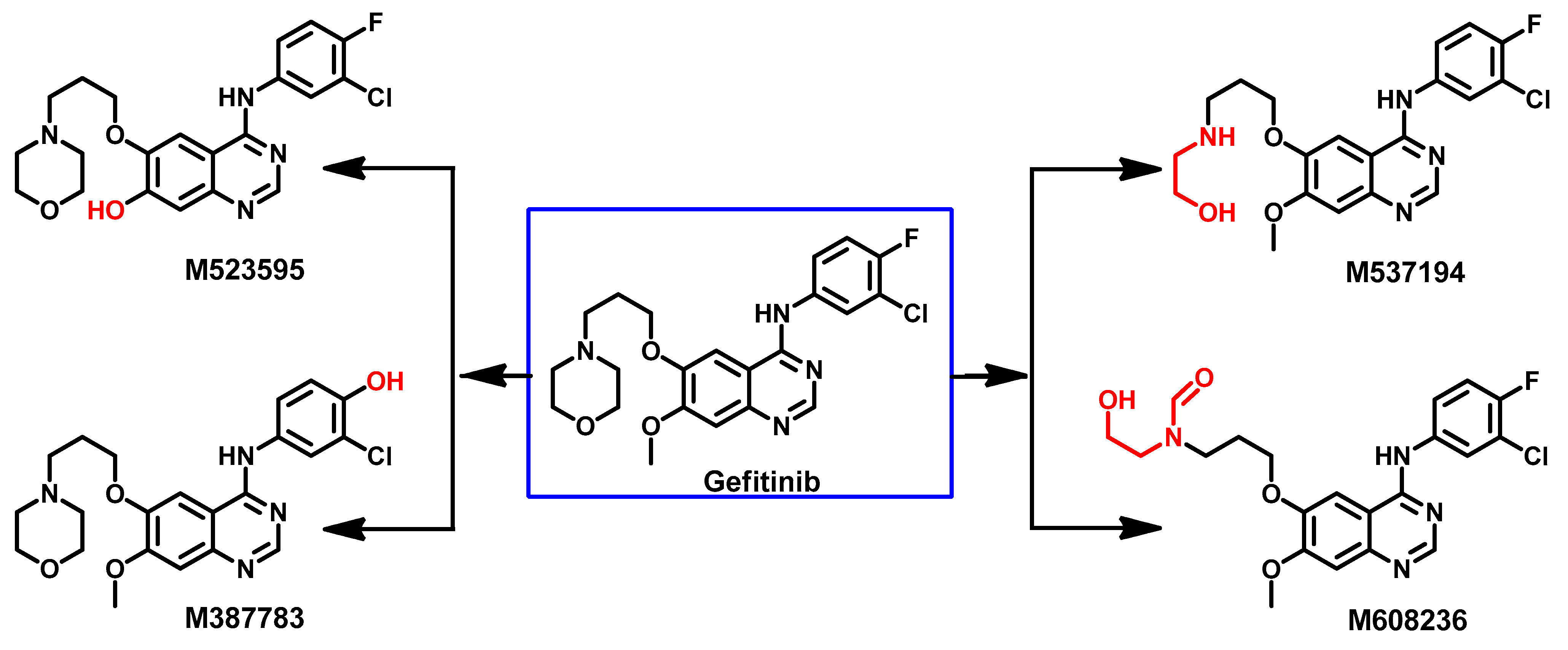
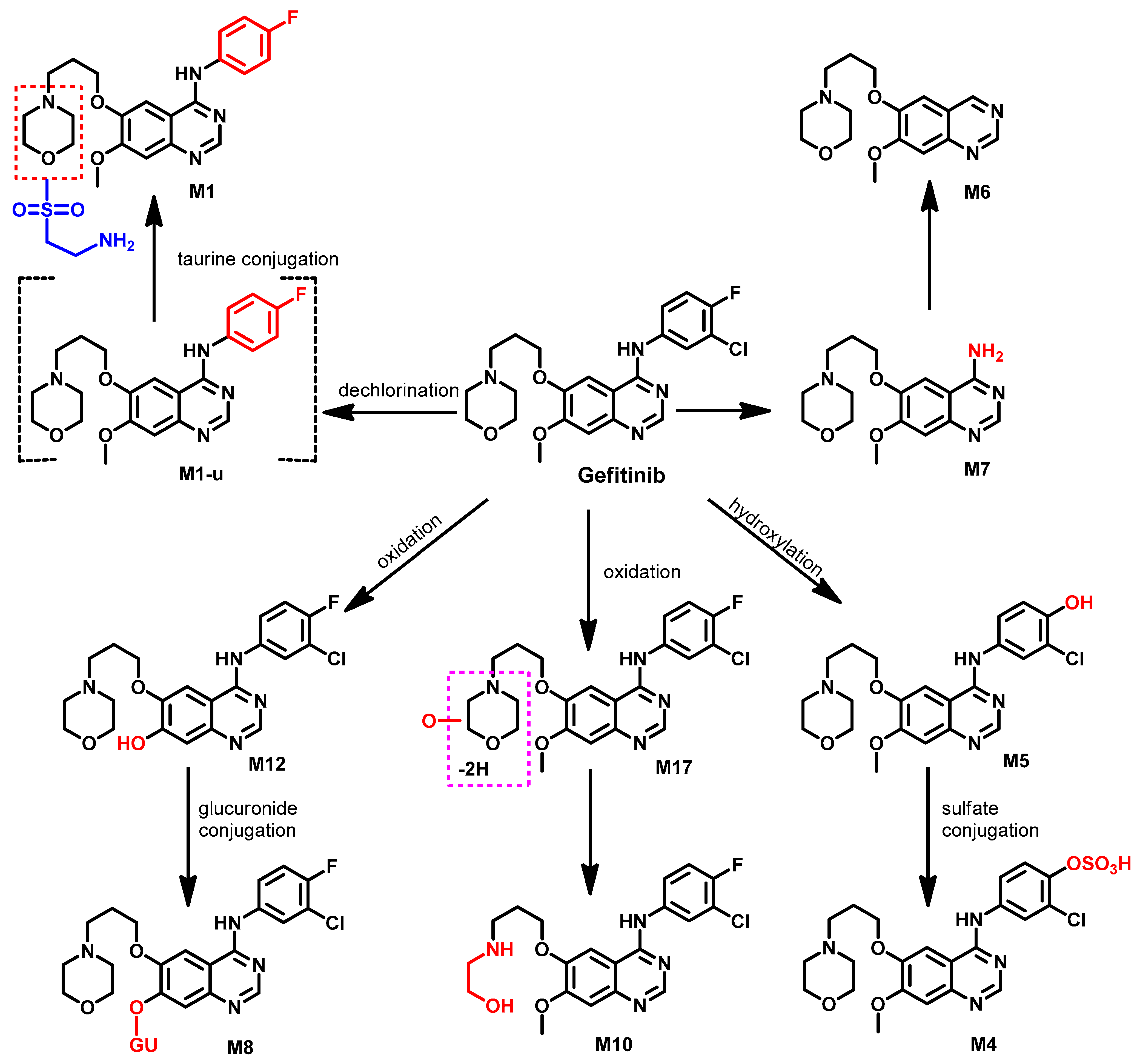
Metabolism
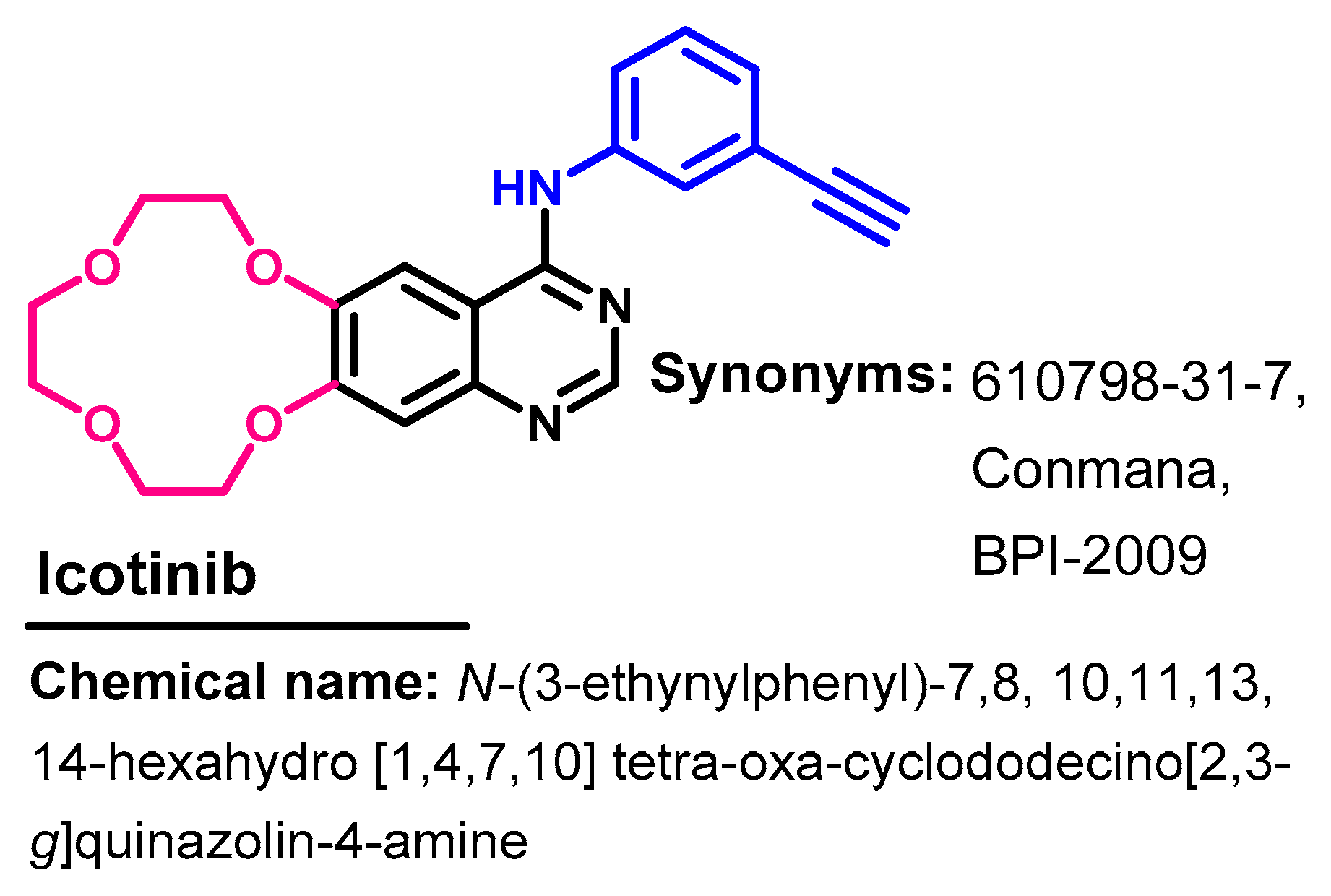
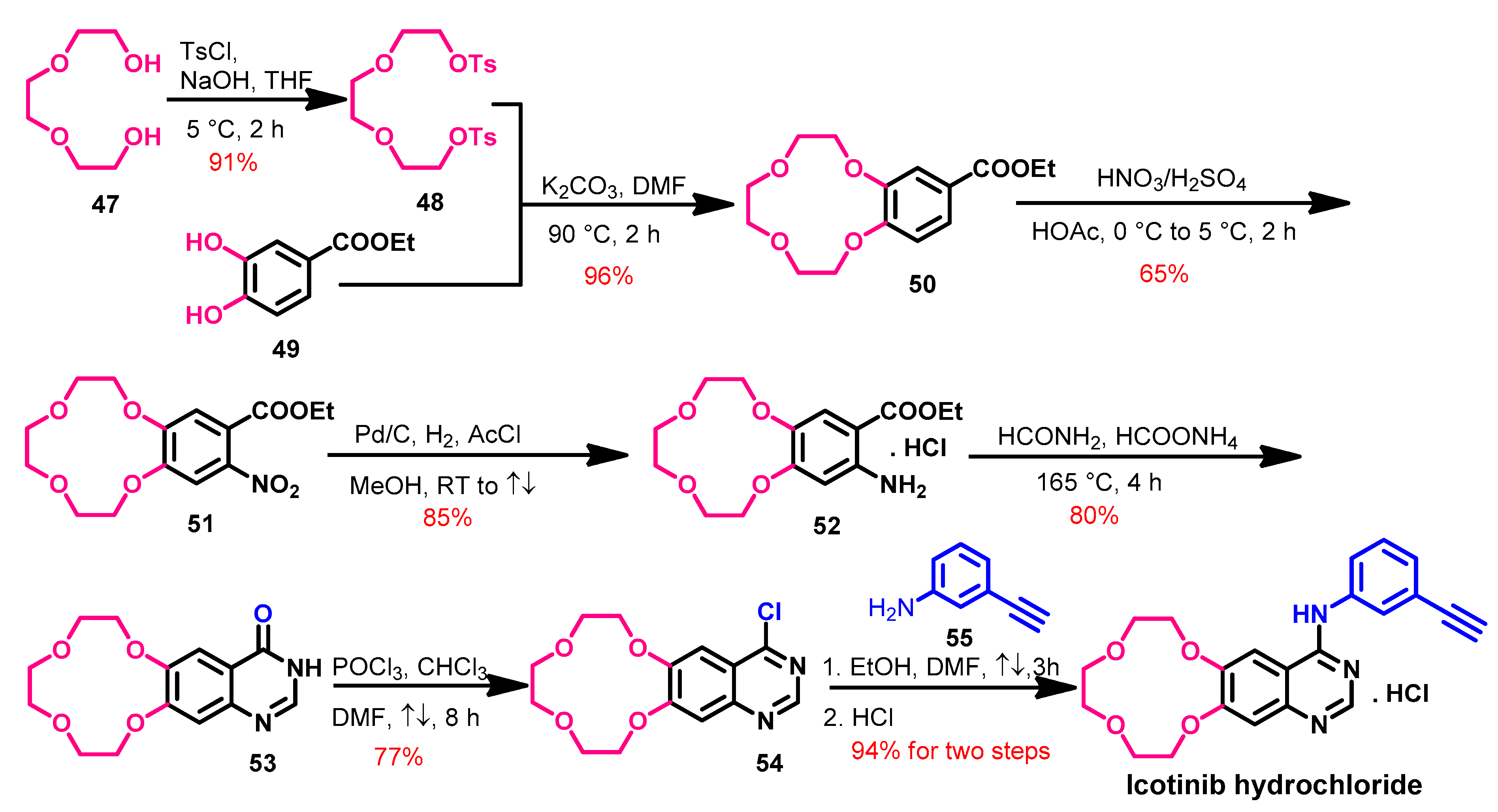
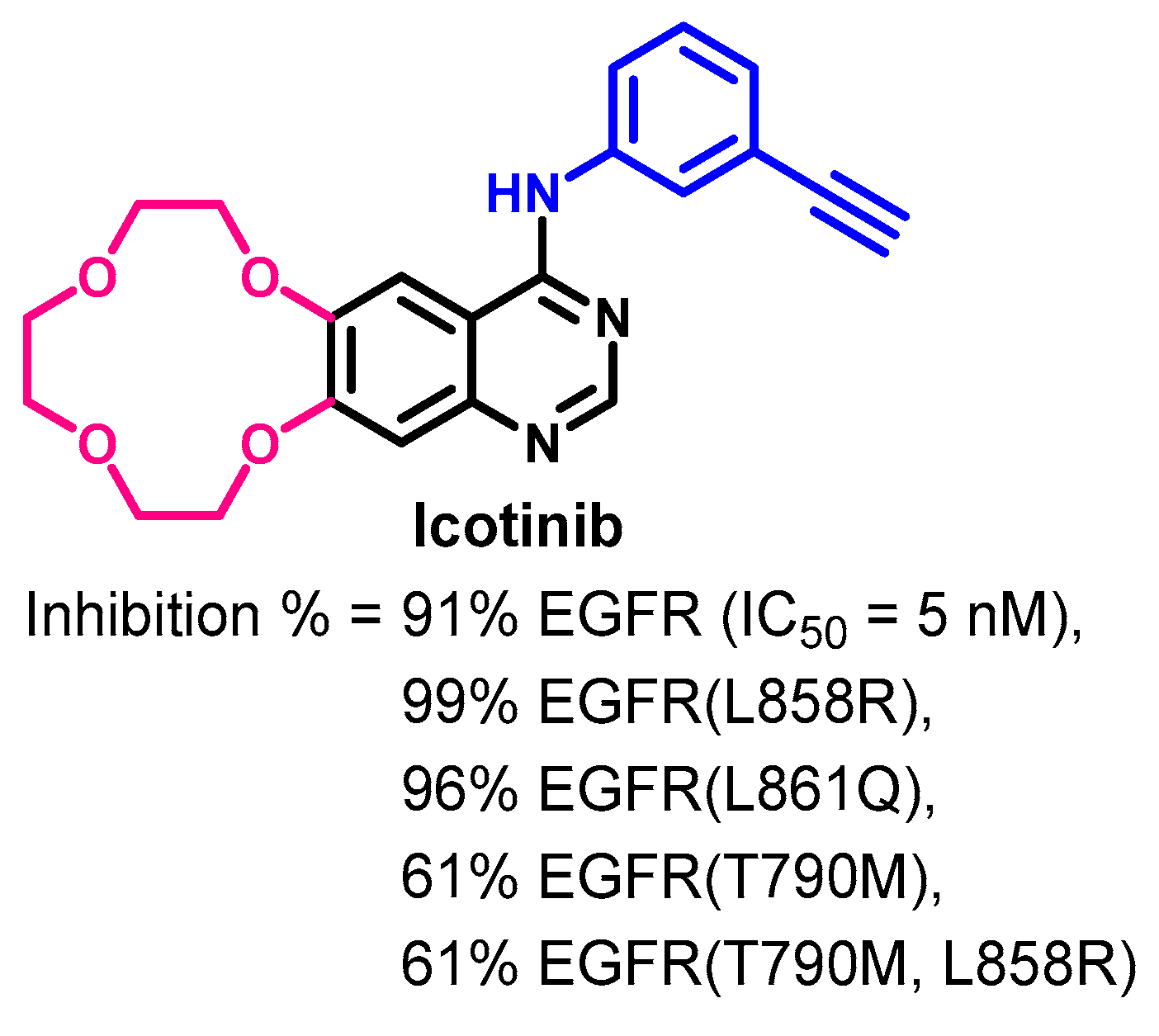
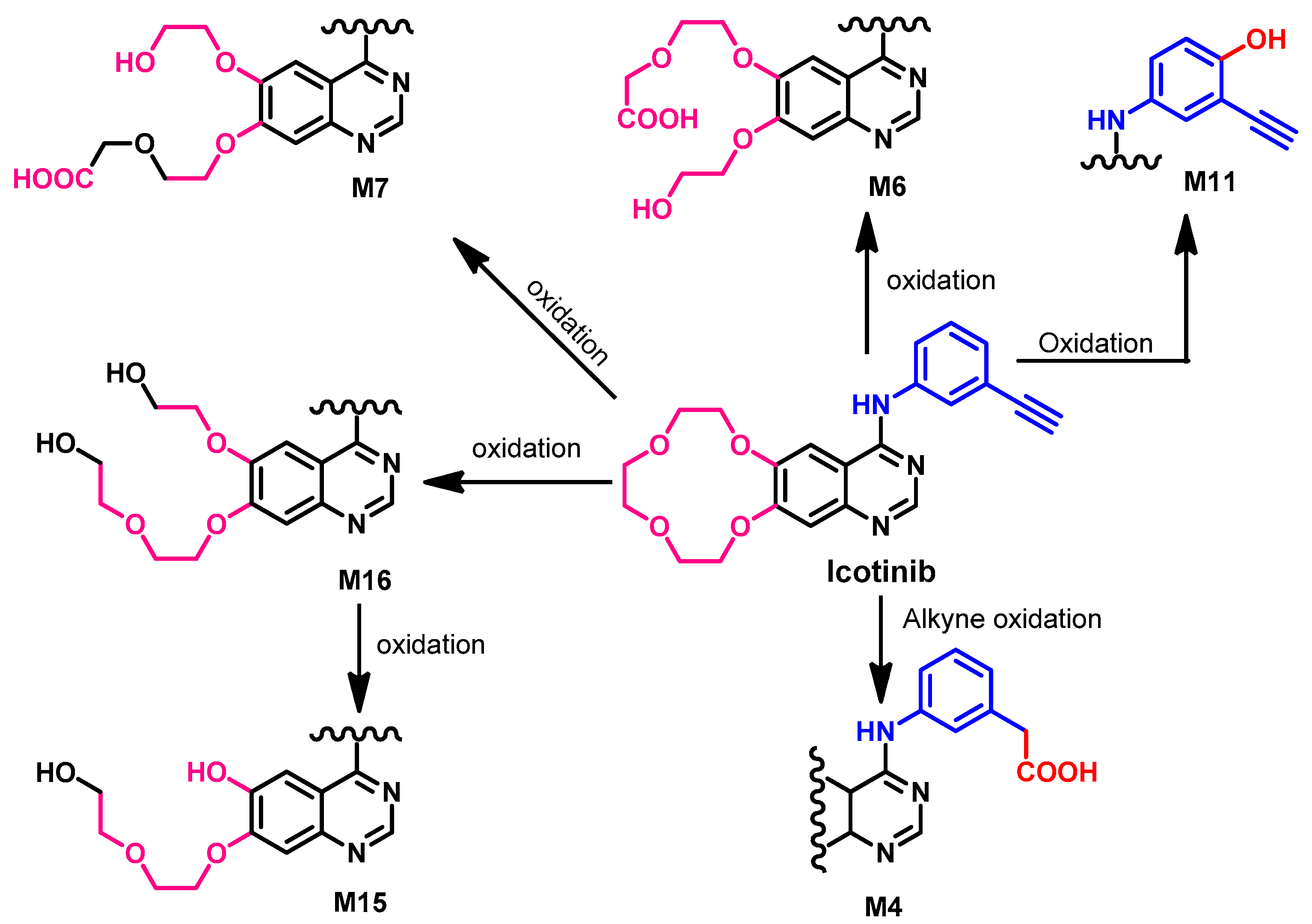
1.2.7. Icotinib
Approval History
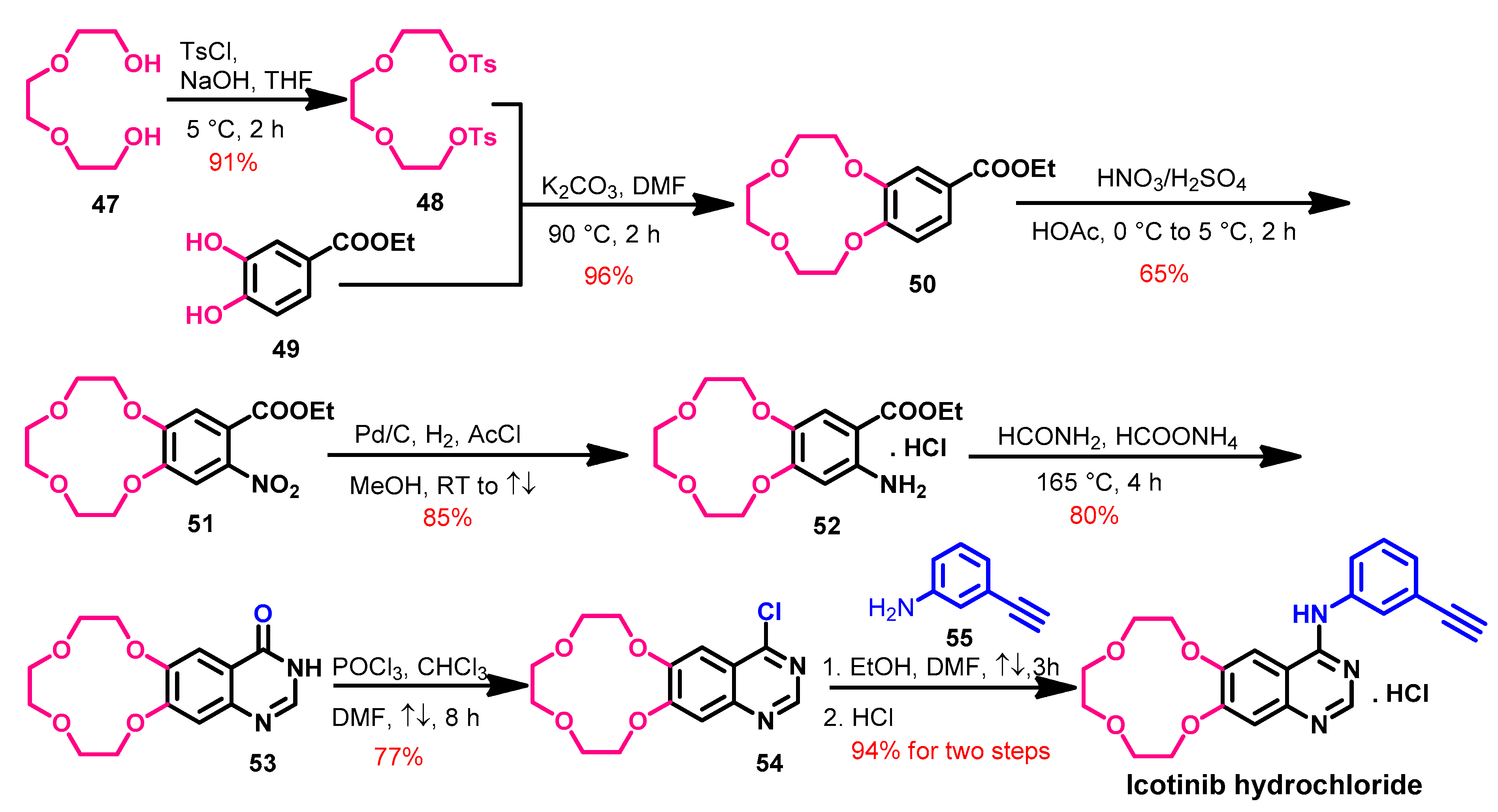
Synthesis
Target Kinases
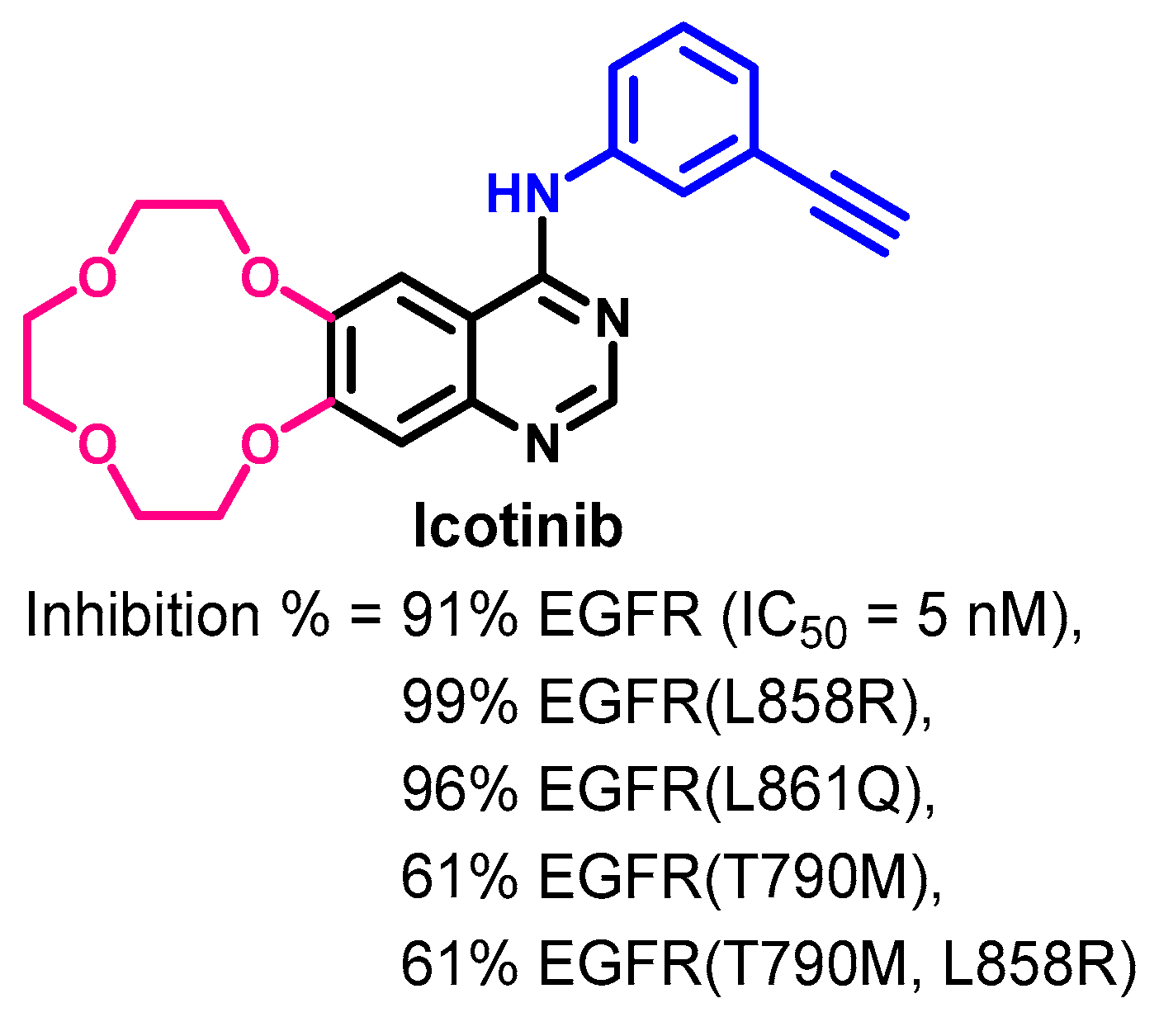
Crystal Structures and Binding Interactions
Biological Activity
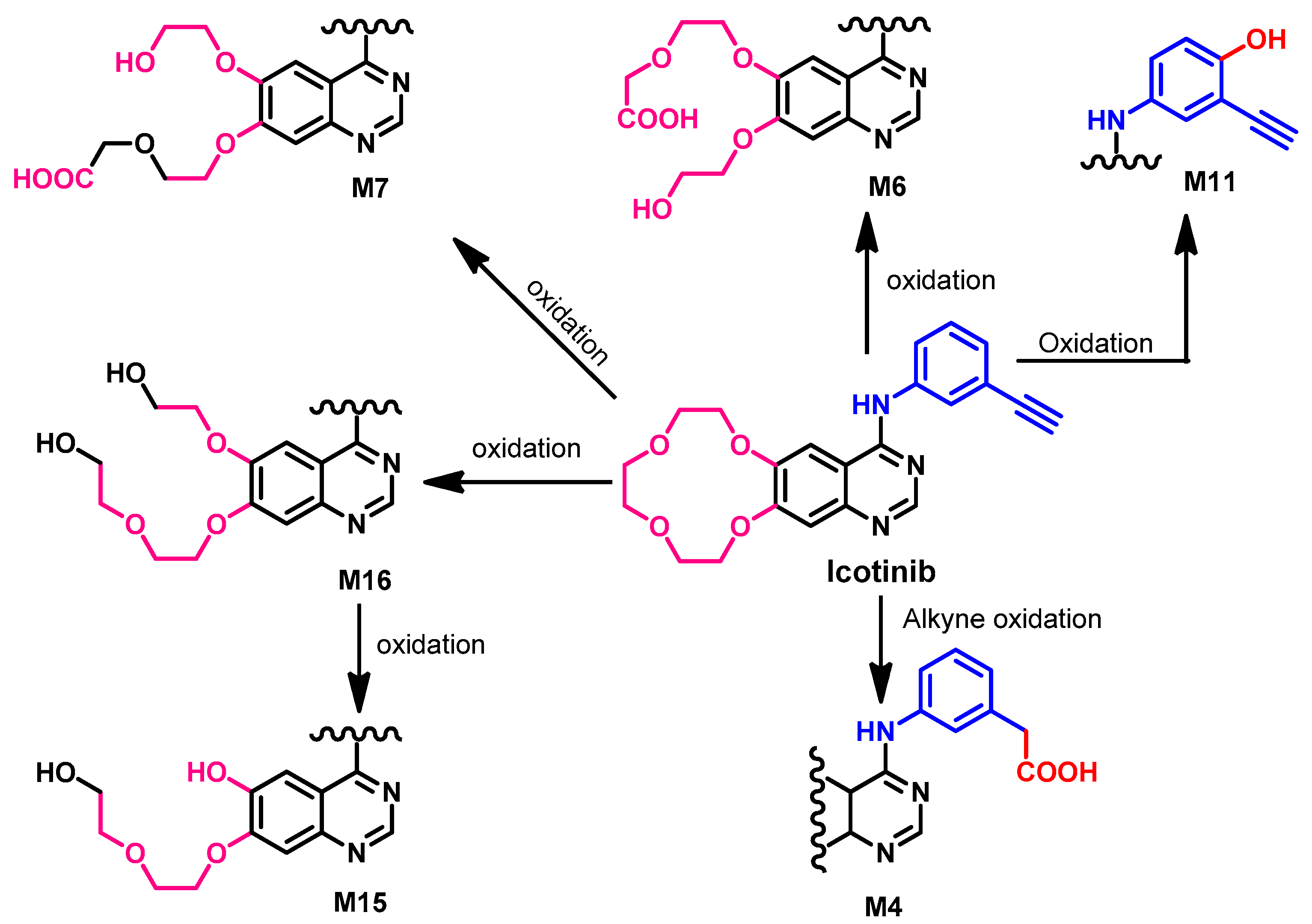
Metabolism
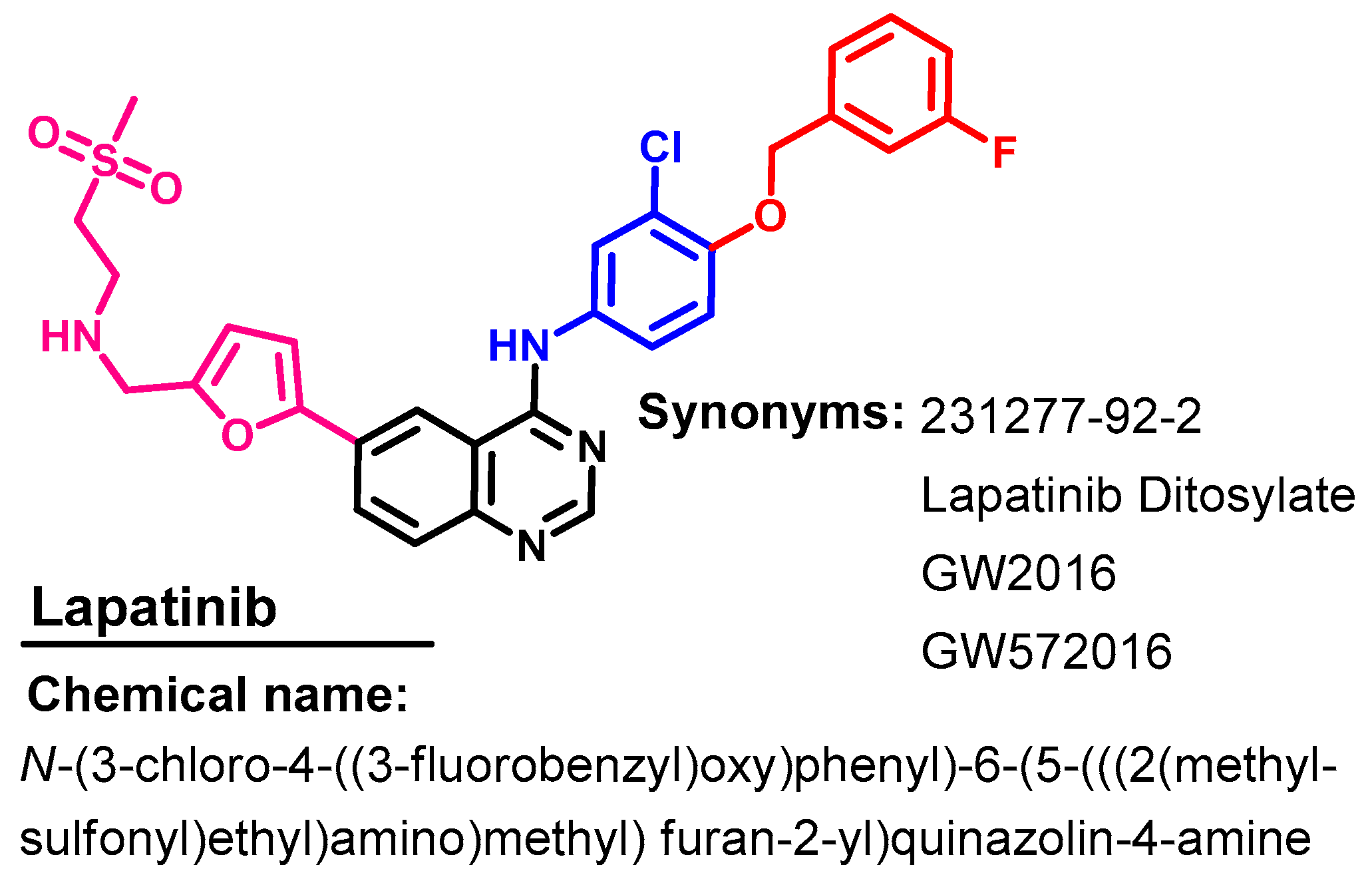
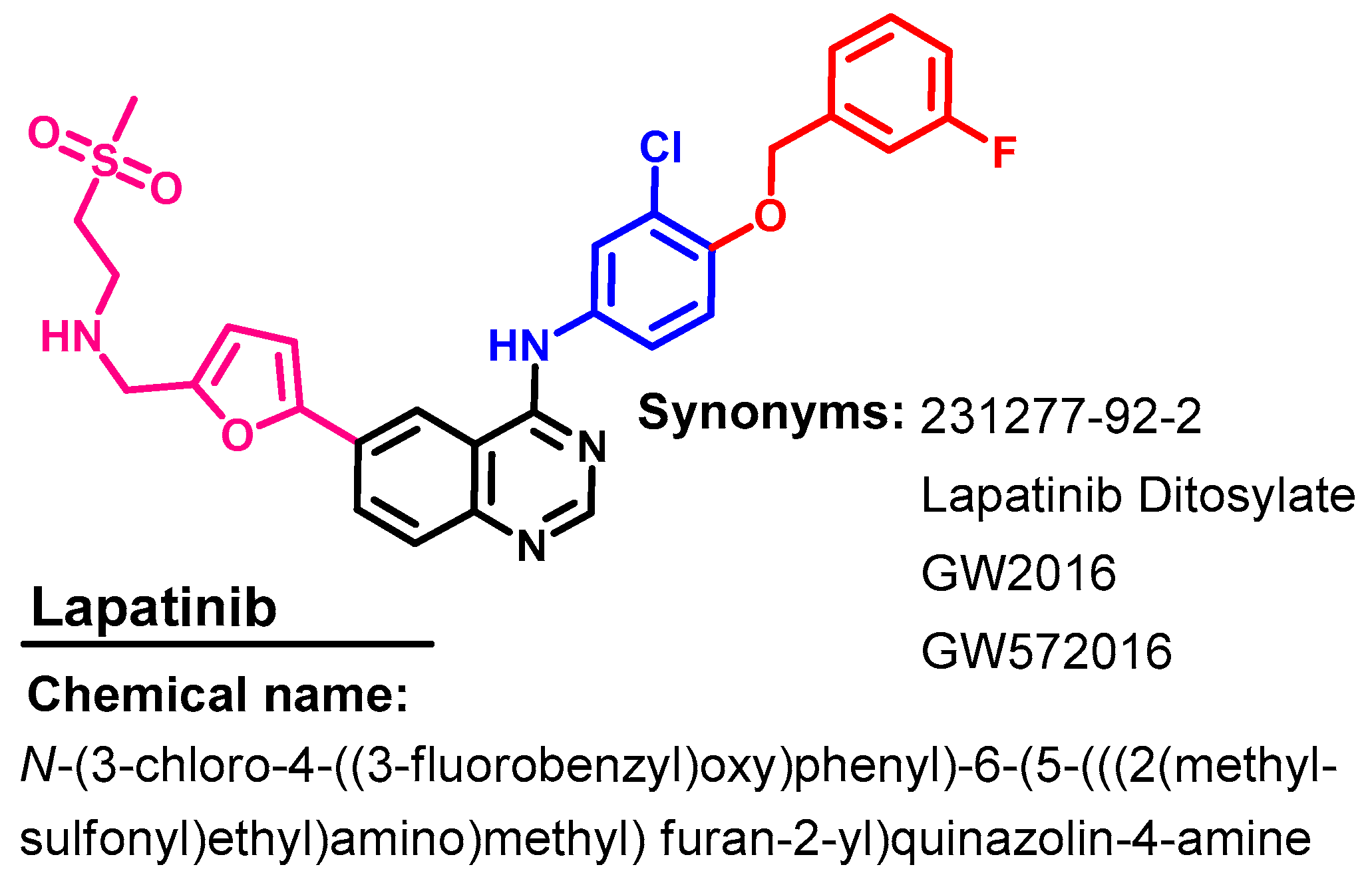
1.2.8. Lapatinib
Approval History
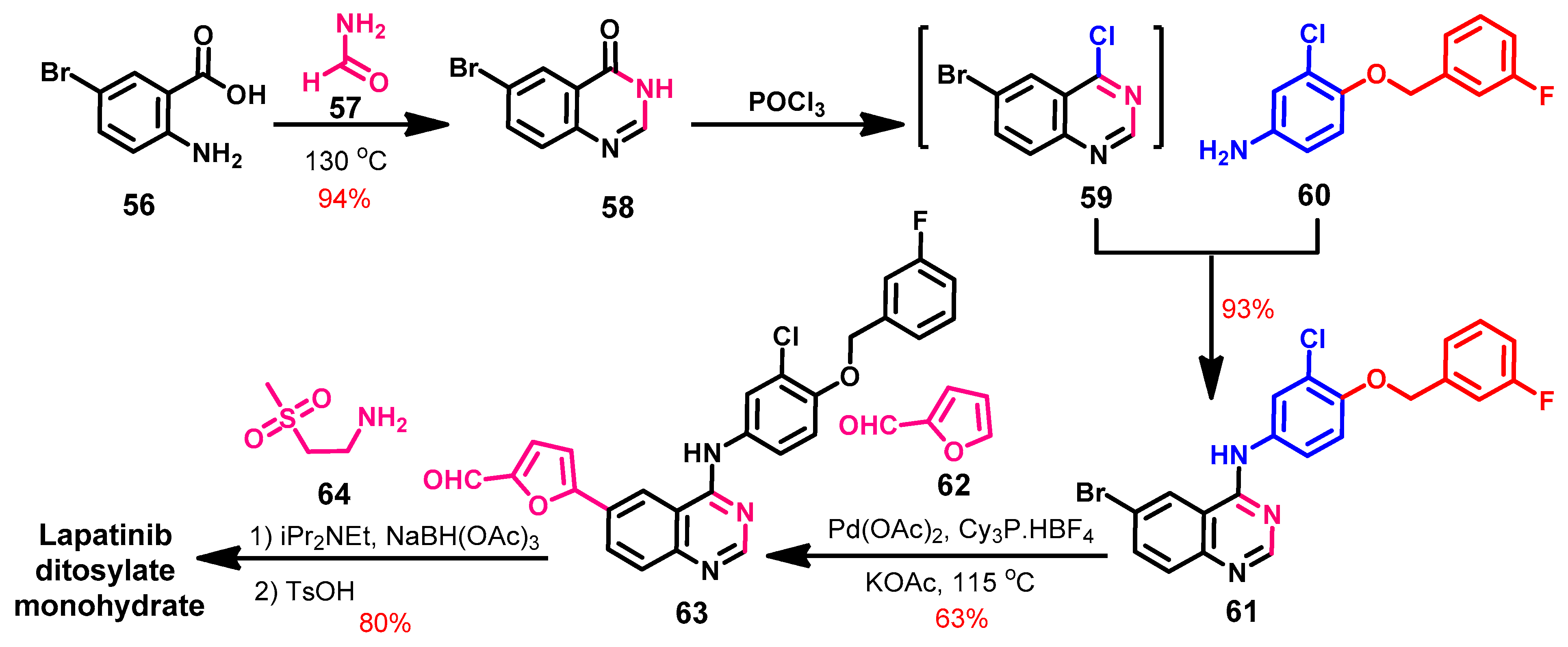
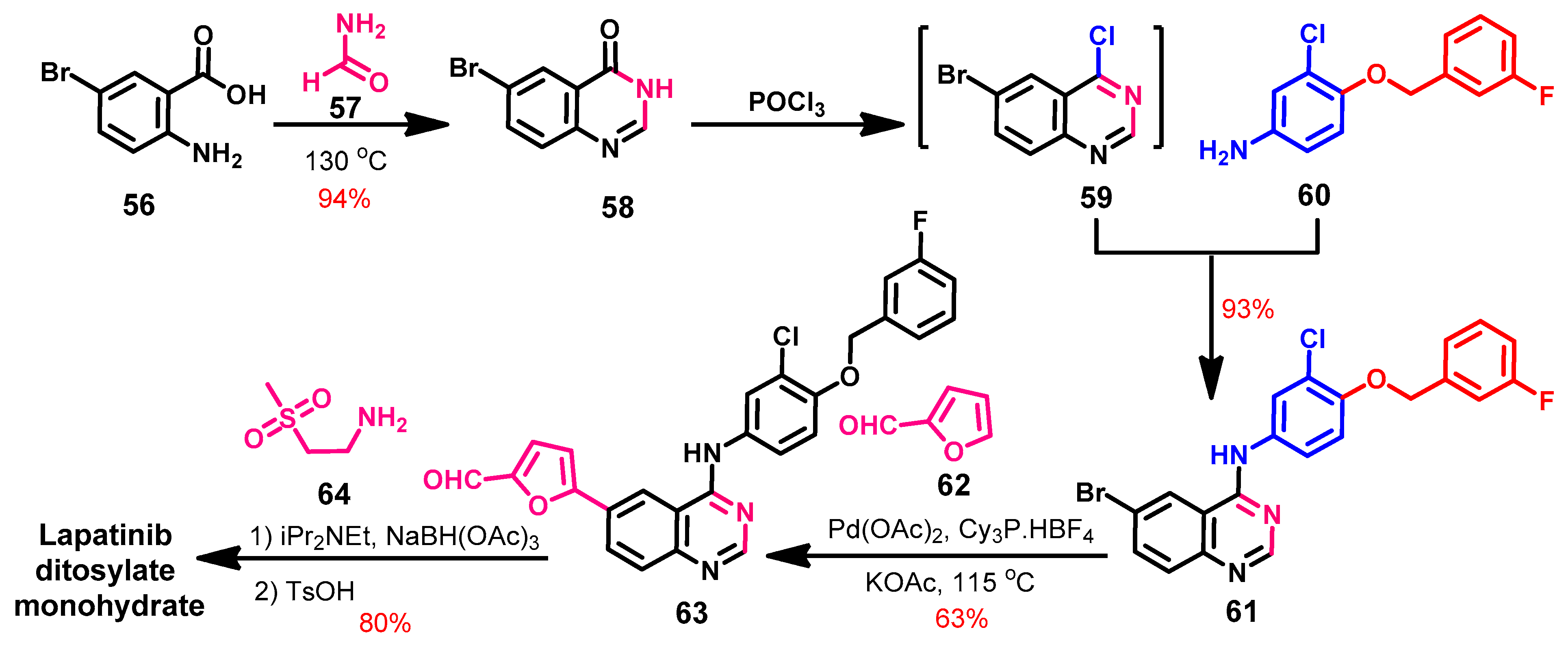
Synthesis
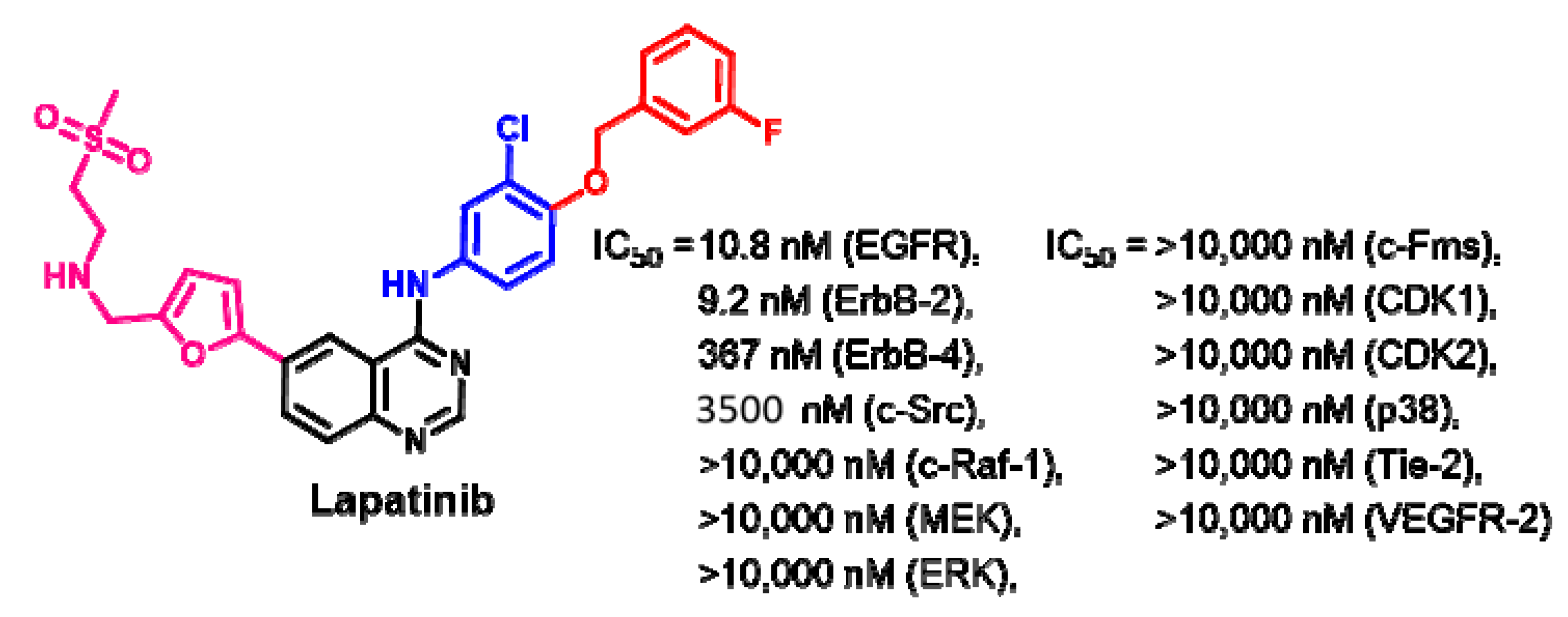
Target Kinases
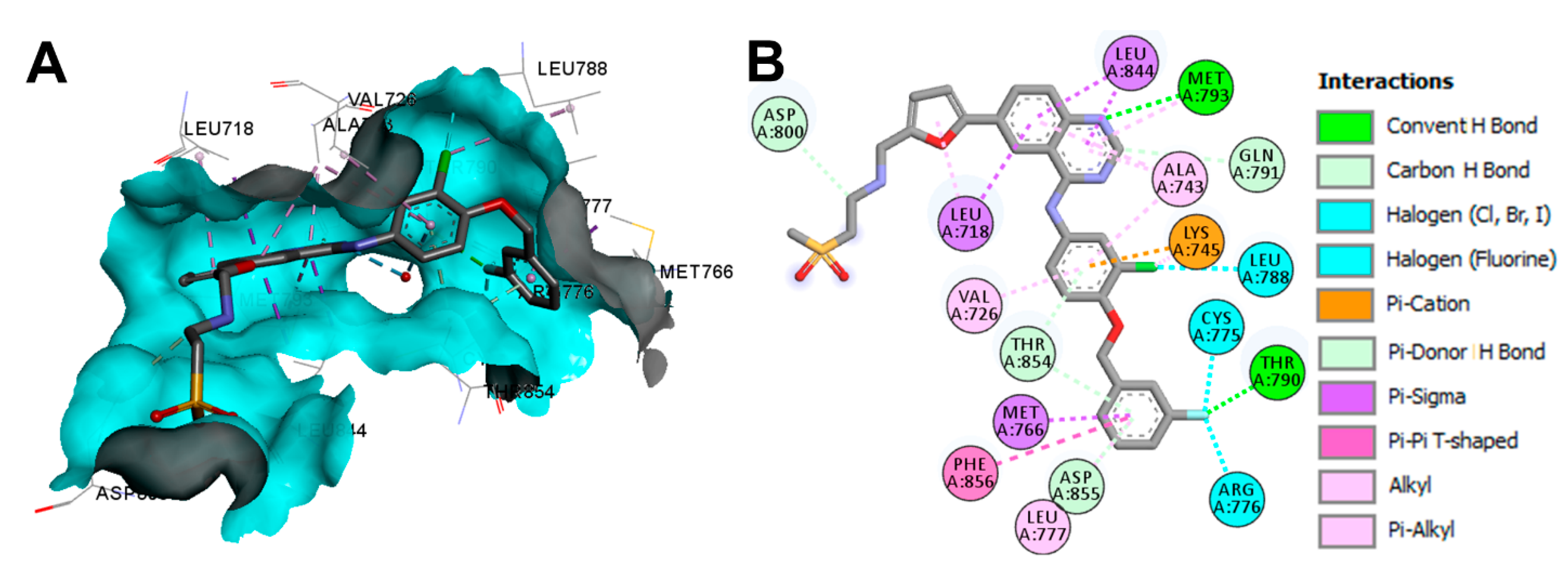
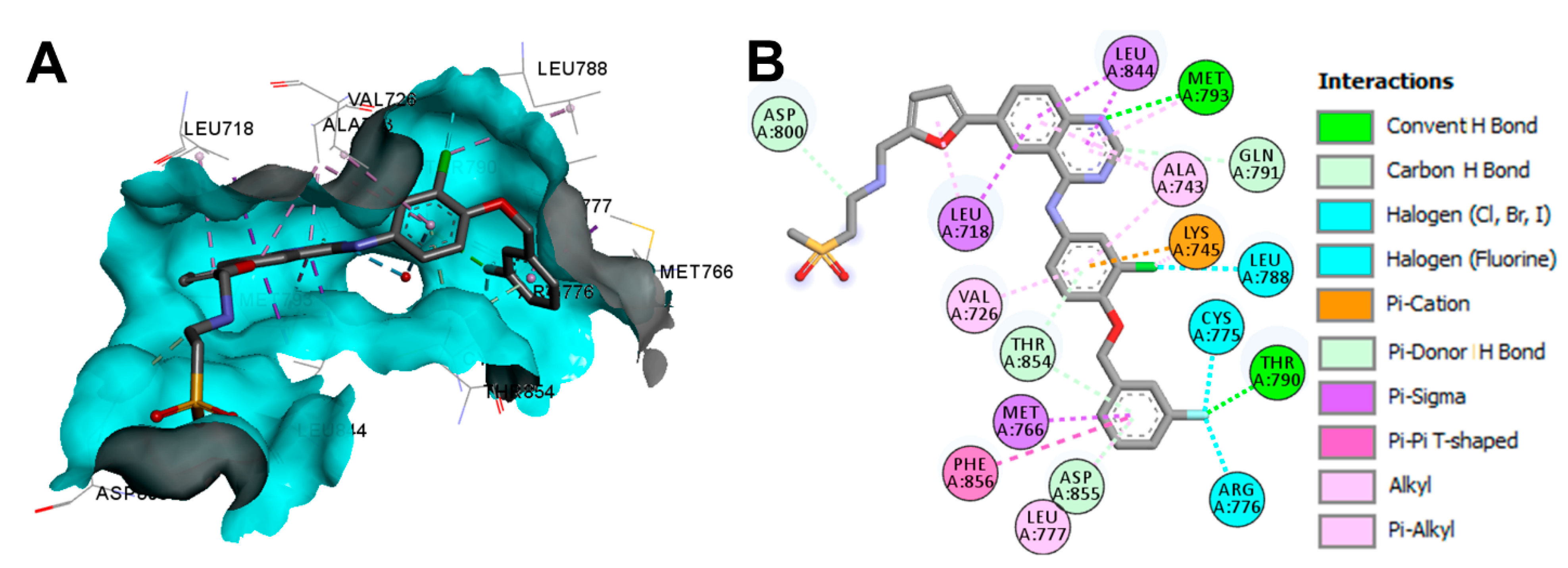
Crystal Structures and Binding Interactions
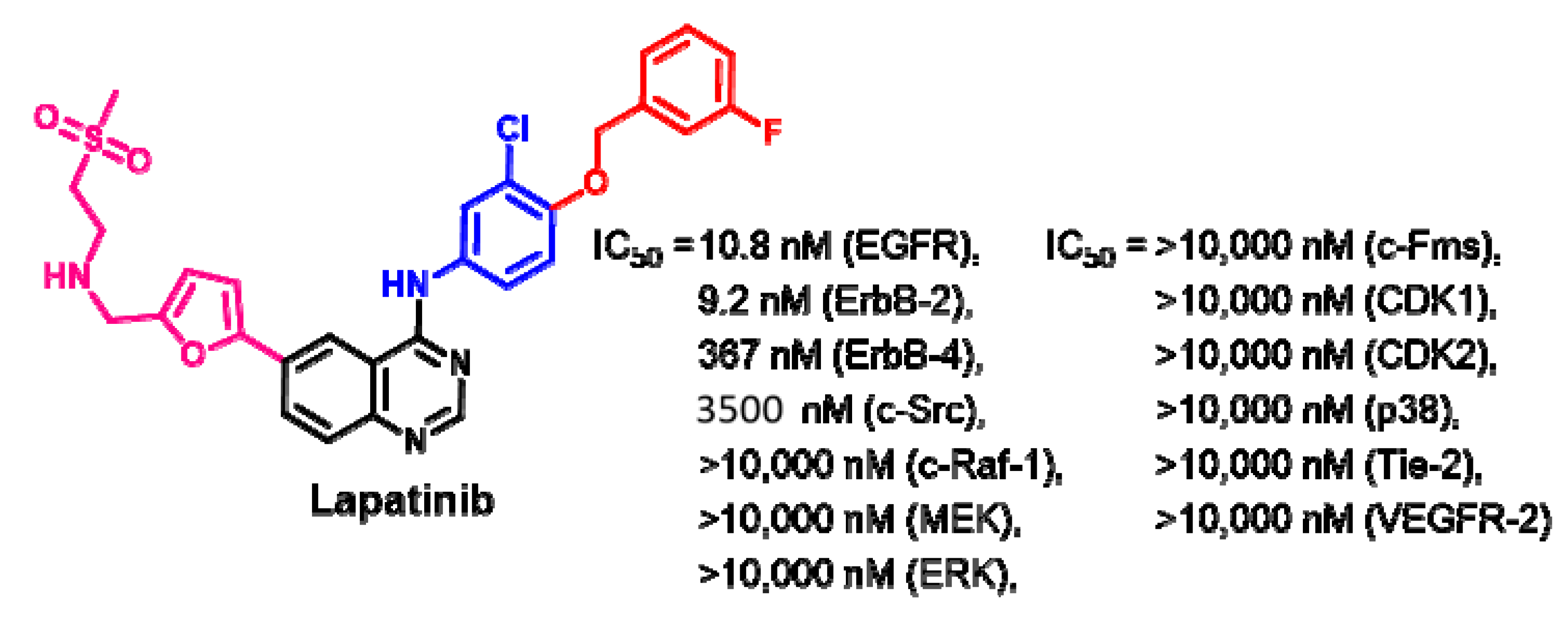
Biological Activity
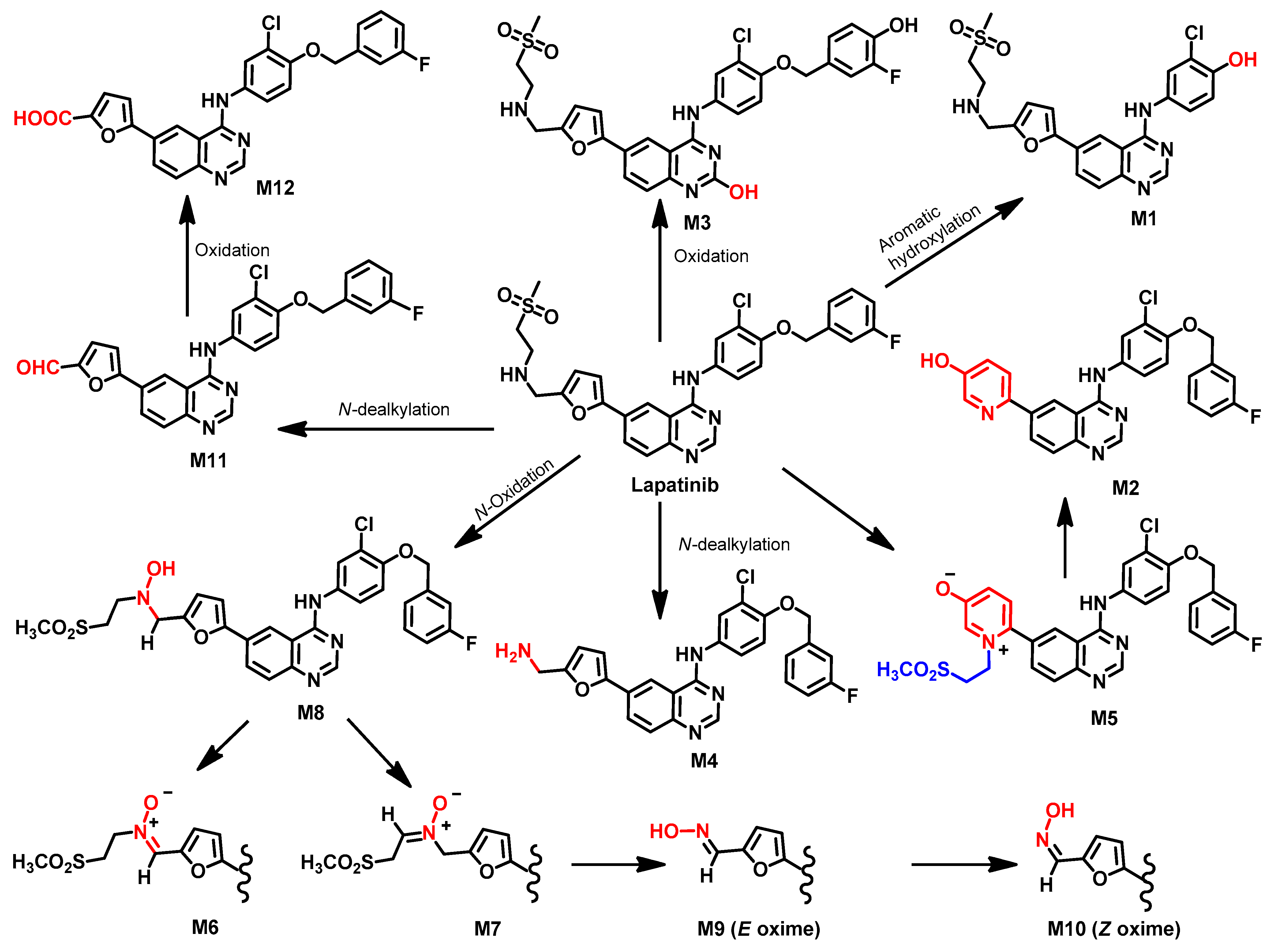
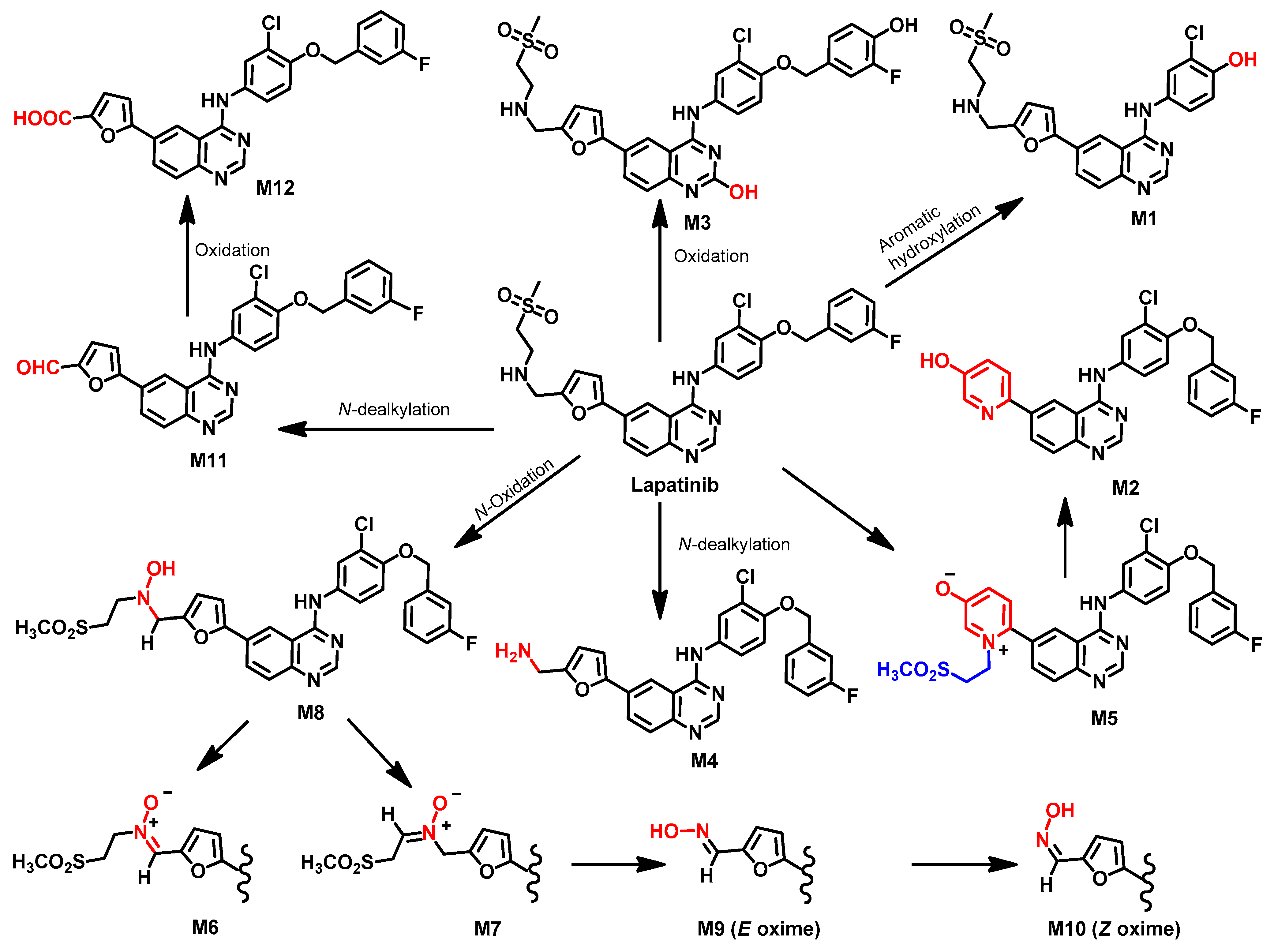
Metabolism
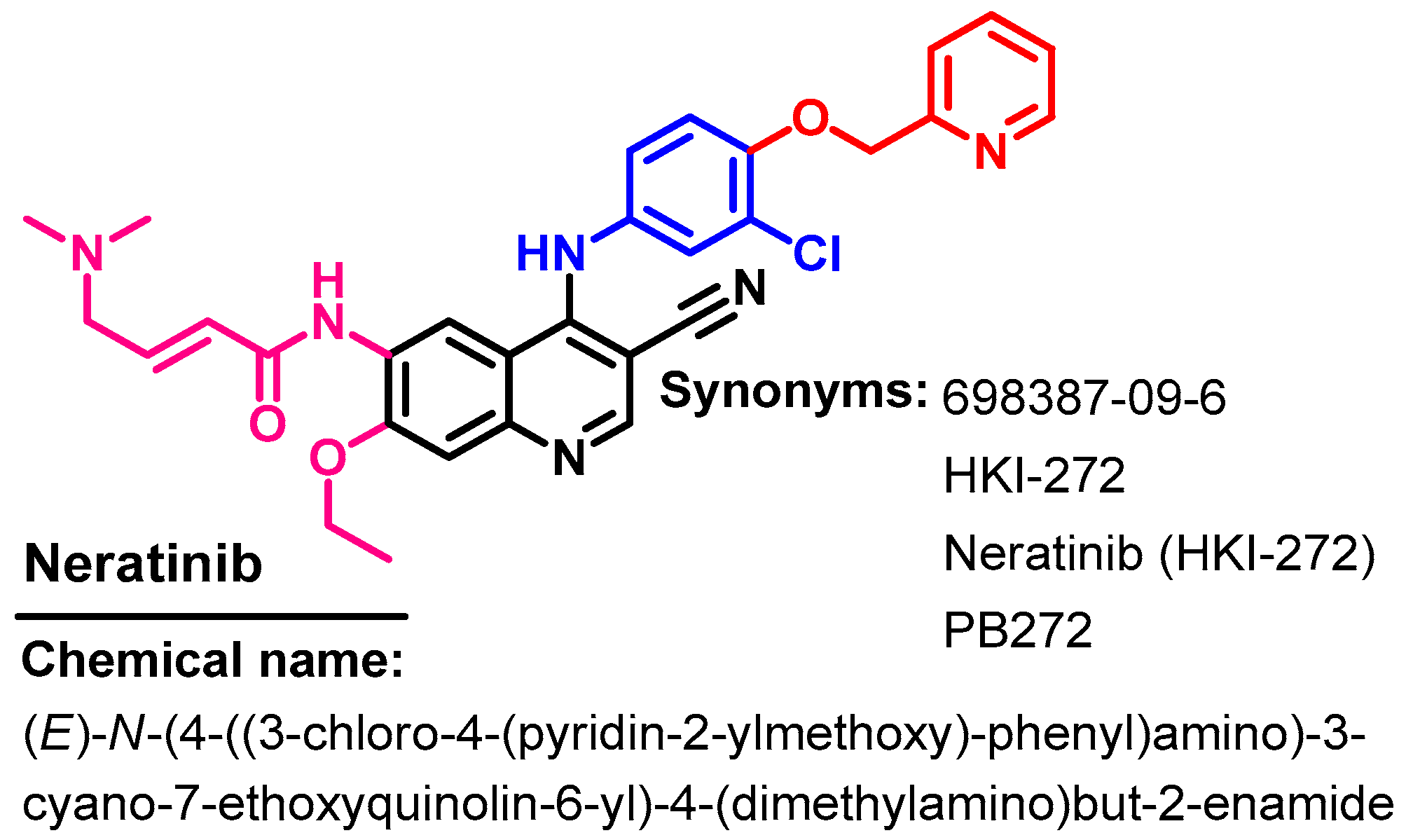
1.2.9. Neratinib
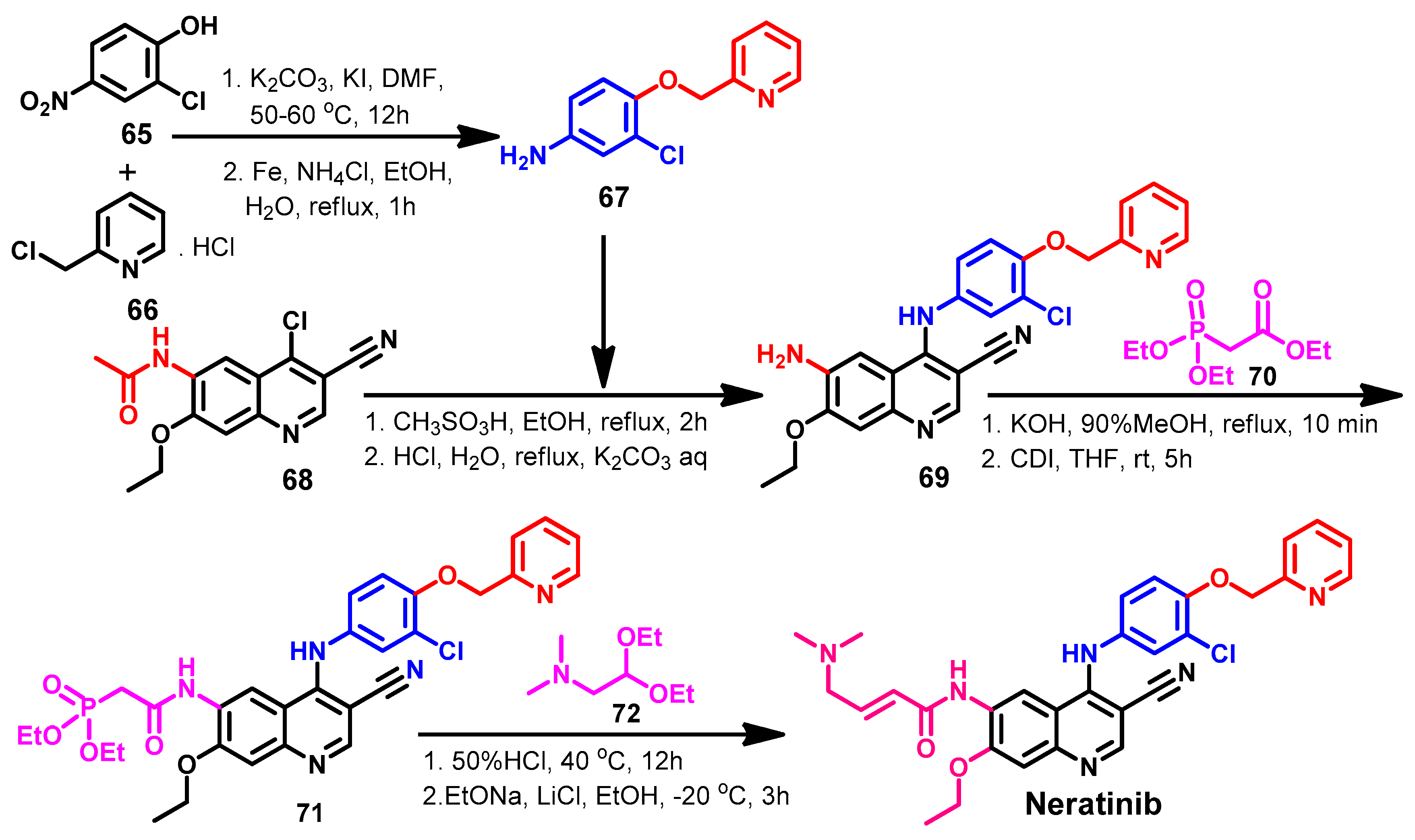
Approval History
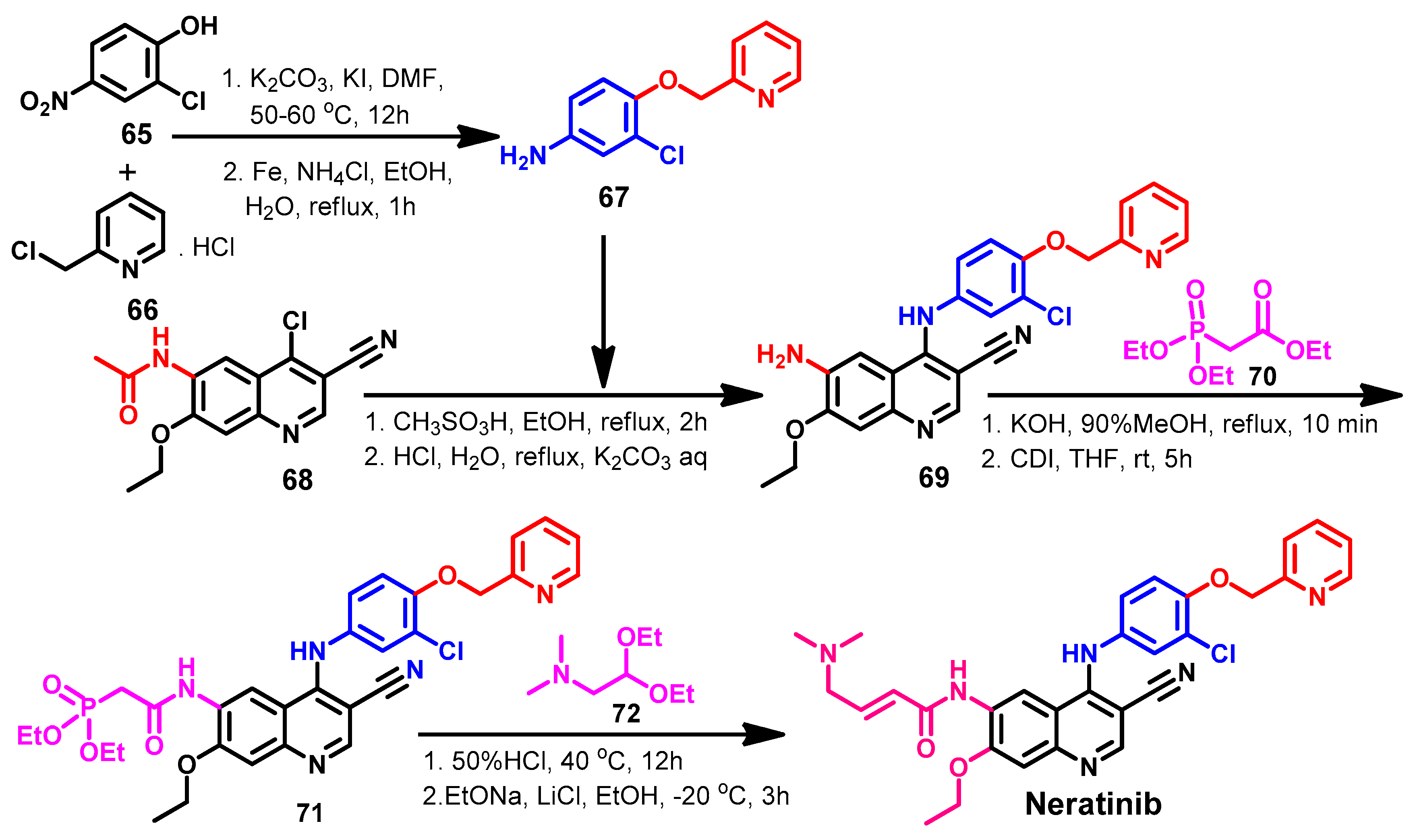
Synthesis
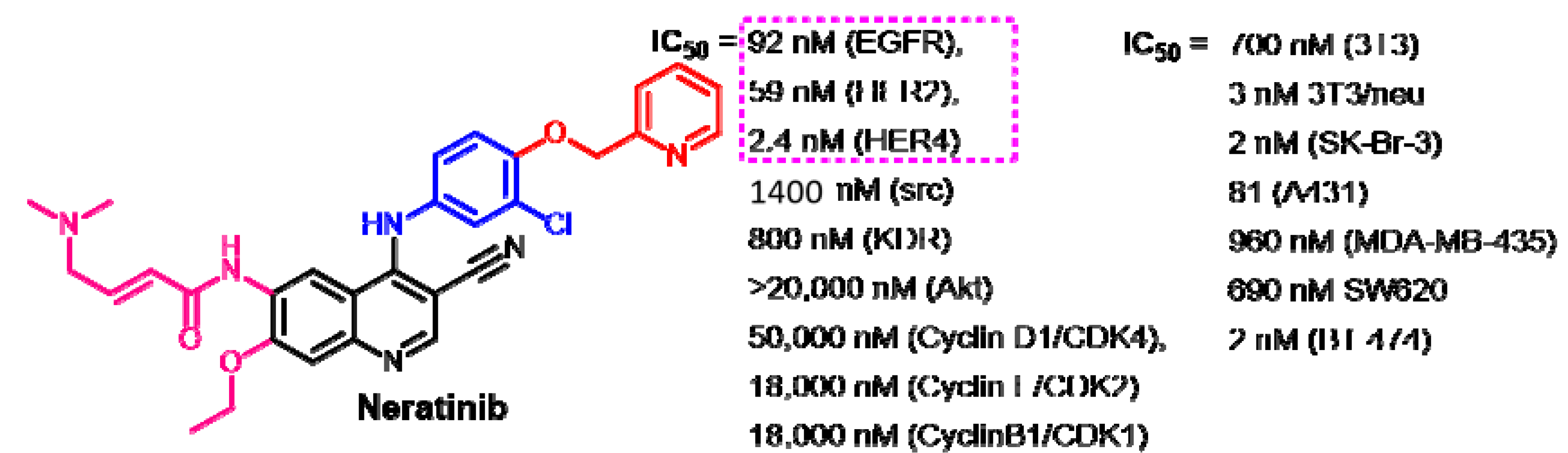
Target Kinases
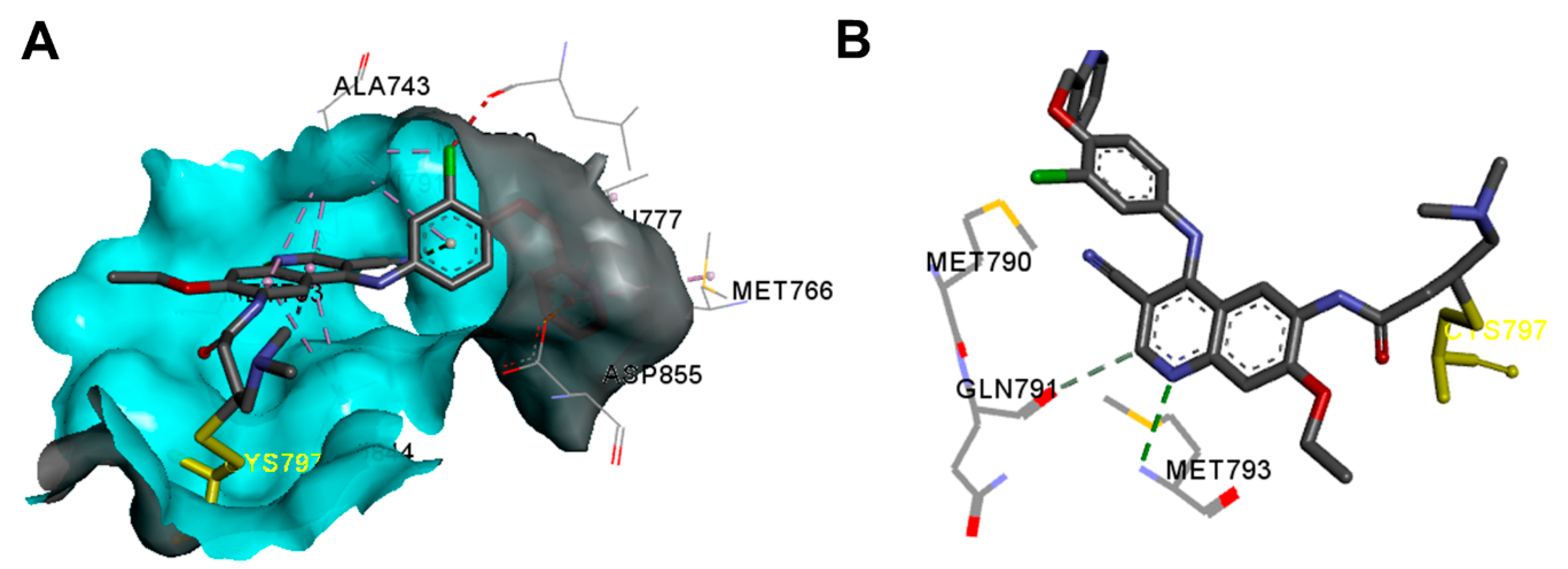
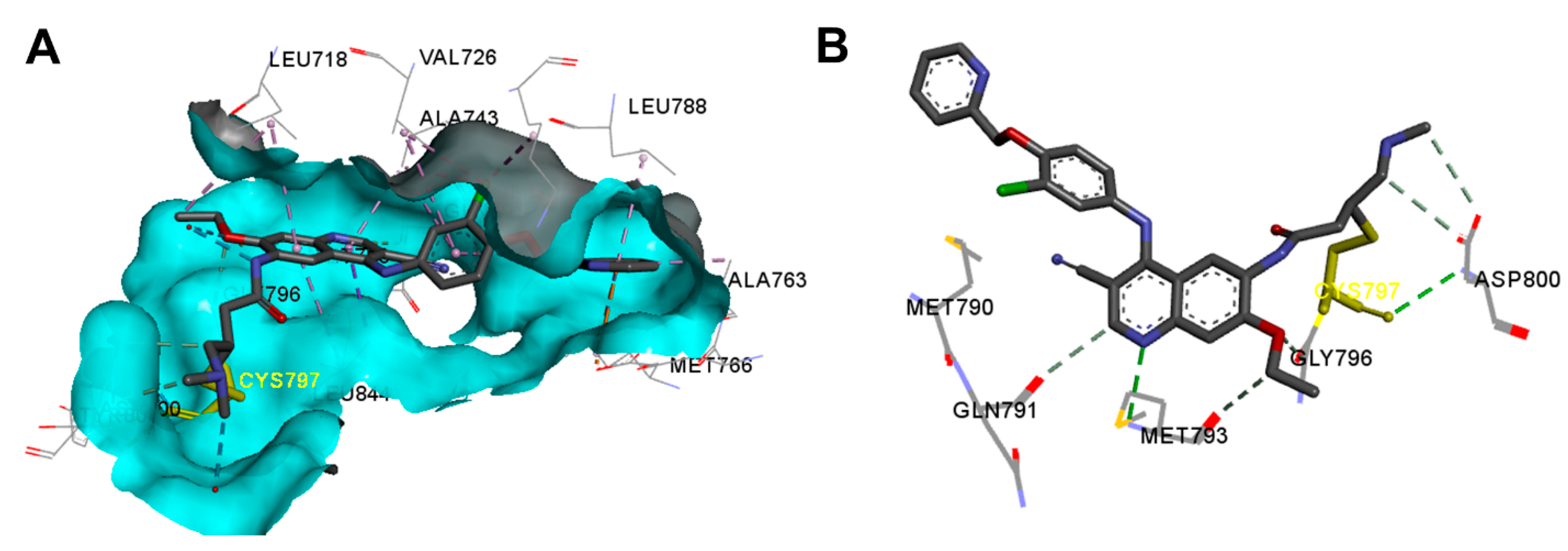
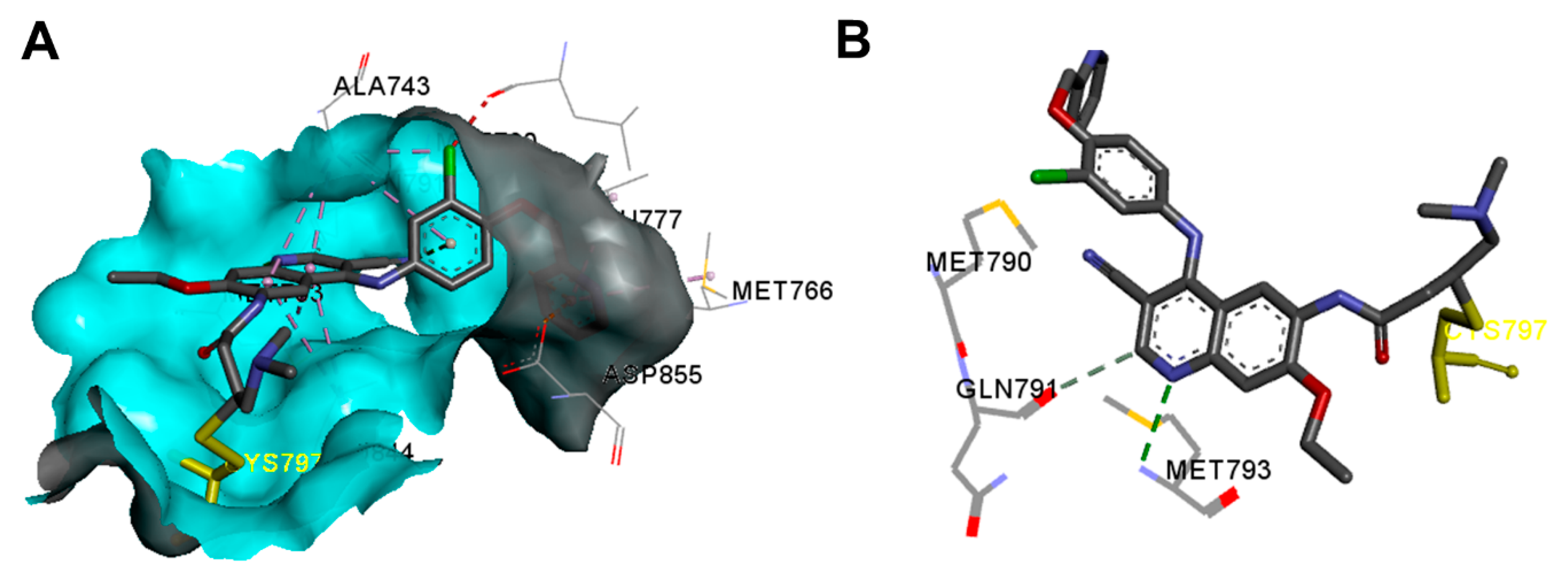
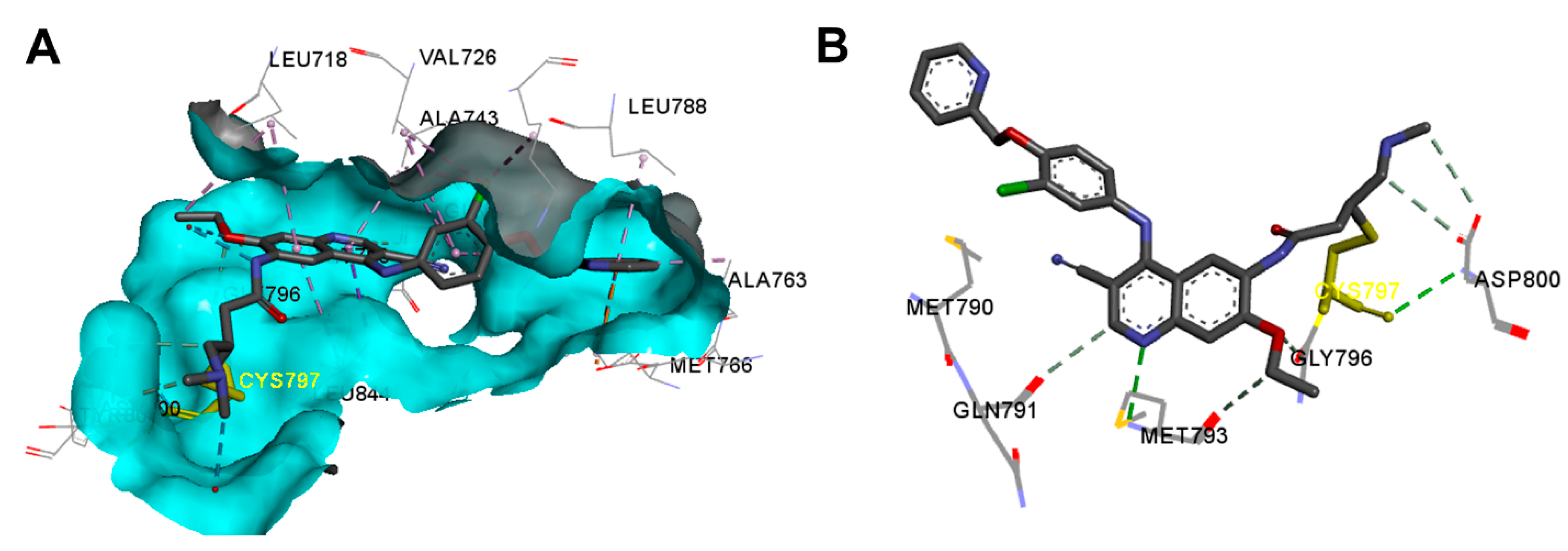
Crystal Structures and Binding Interactions
Biological Activity
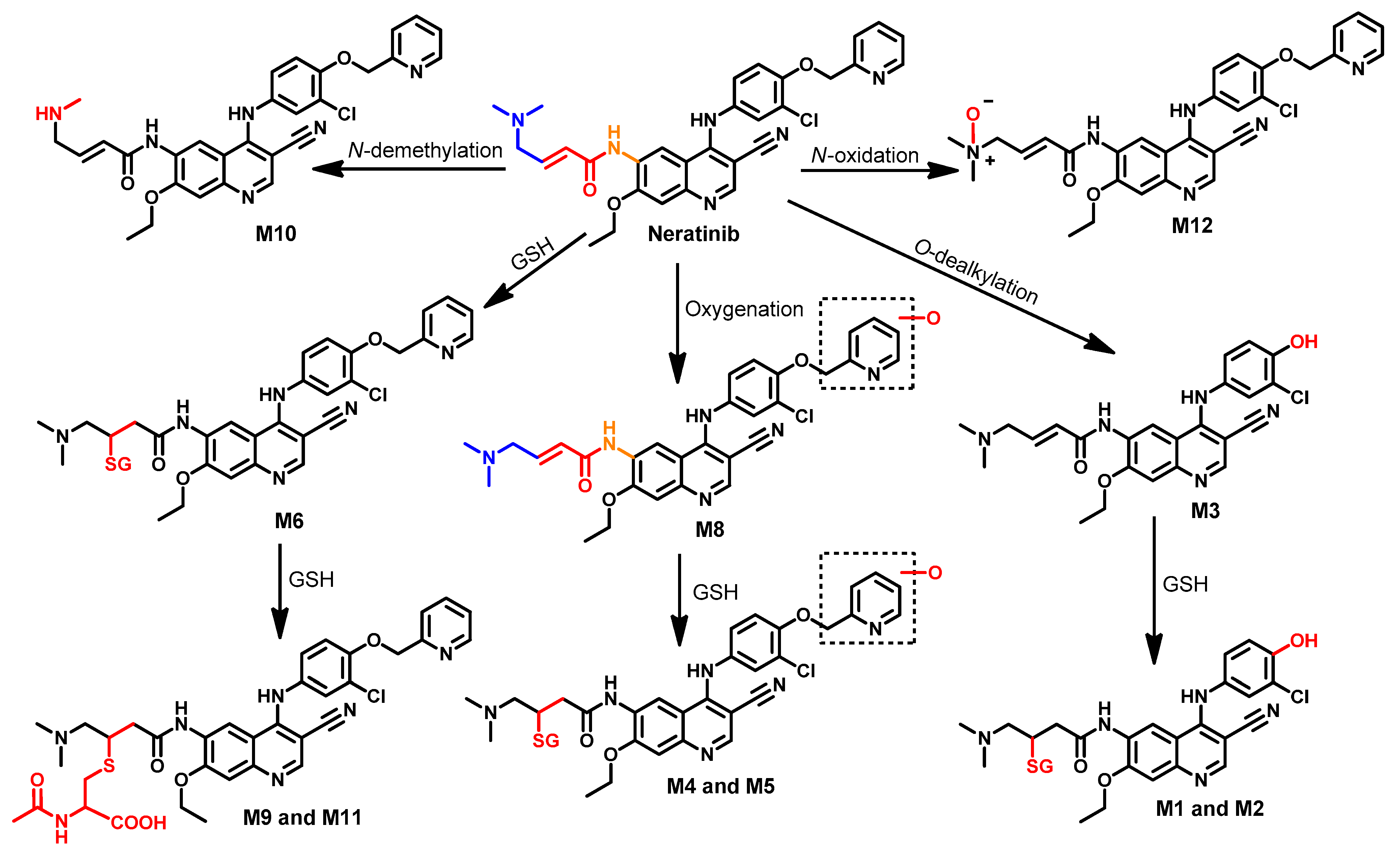
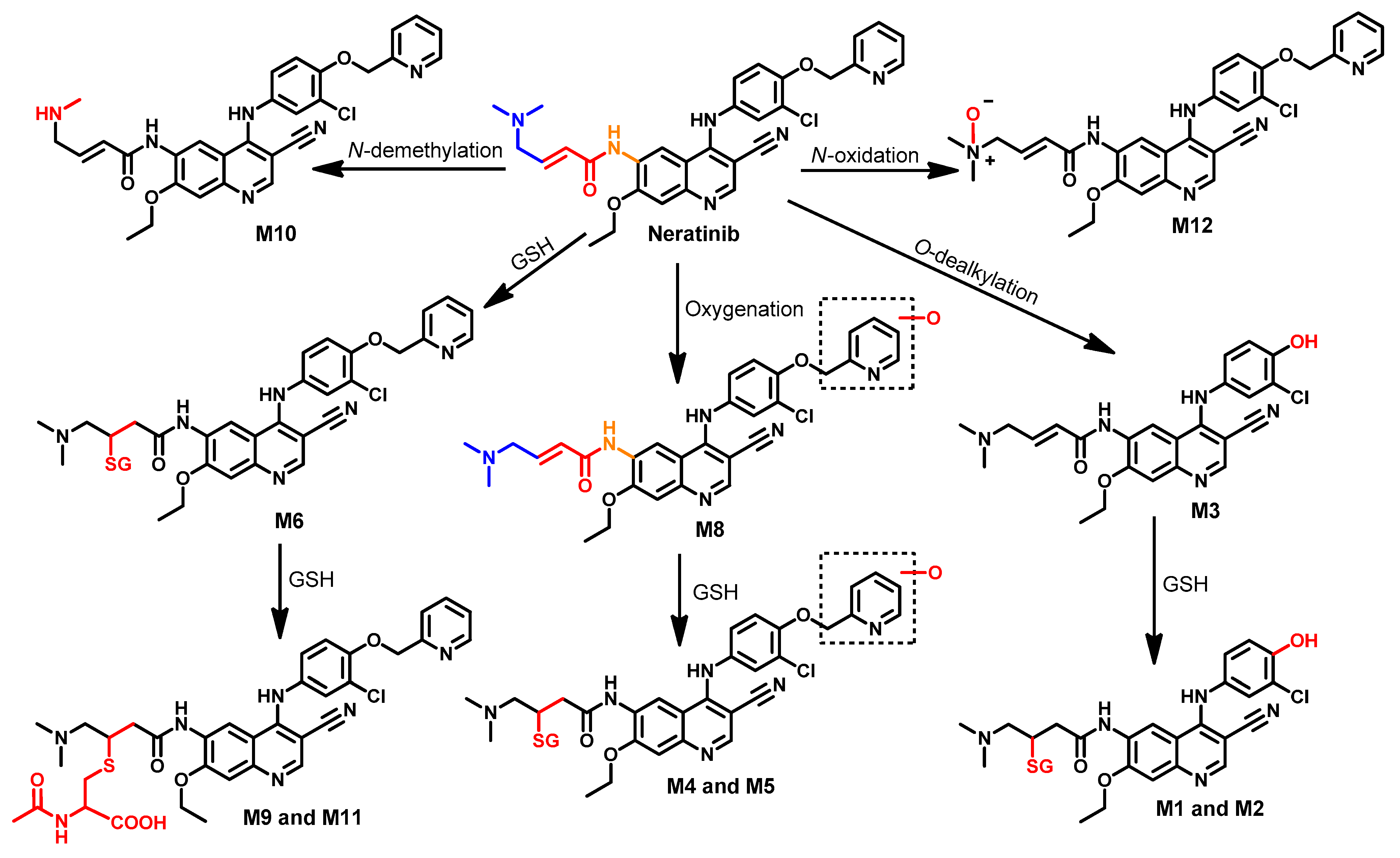
Metabolism
1.2.10. Olmutinib
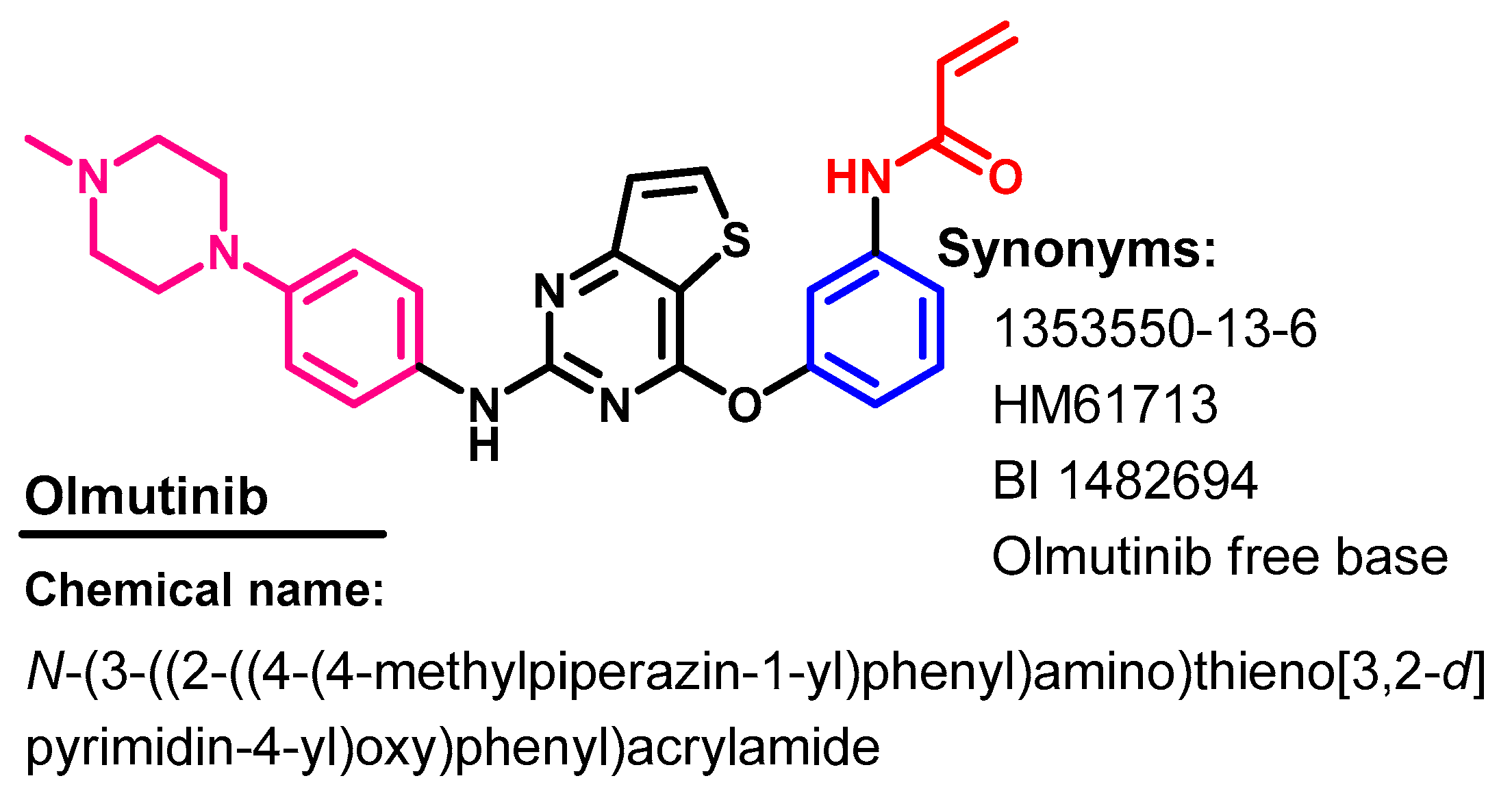
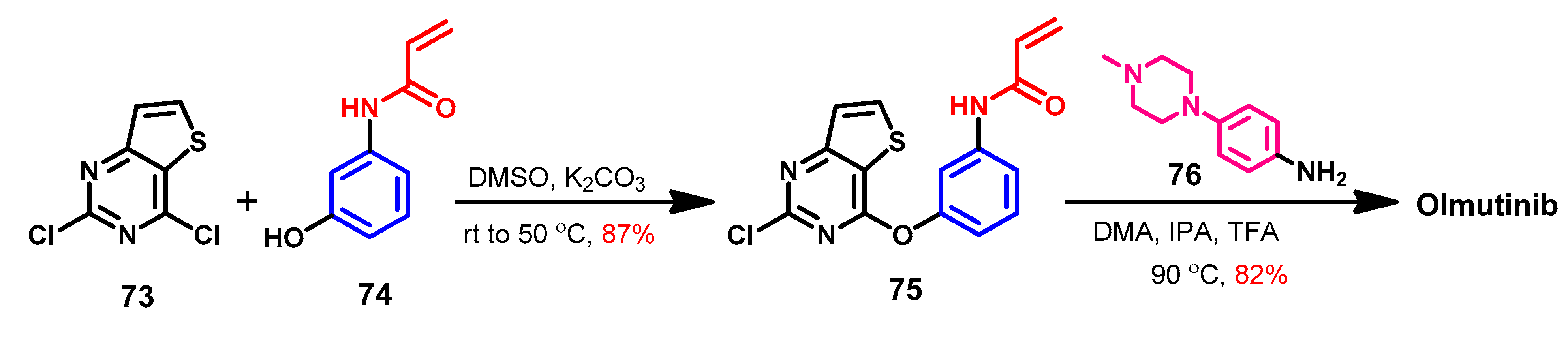
Approval History
Synthesis
Target Kinases
Crystal Structures and Binding Interactions
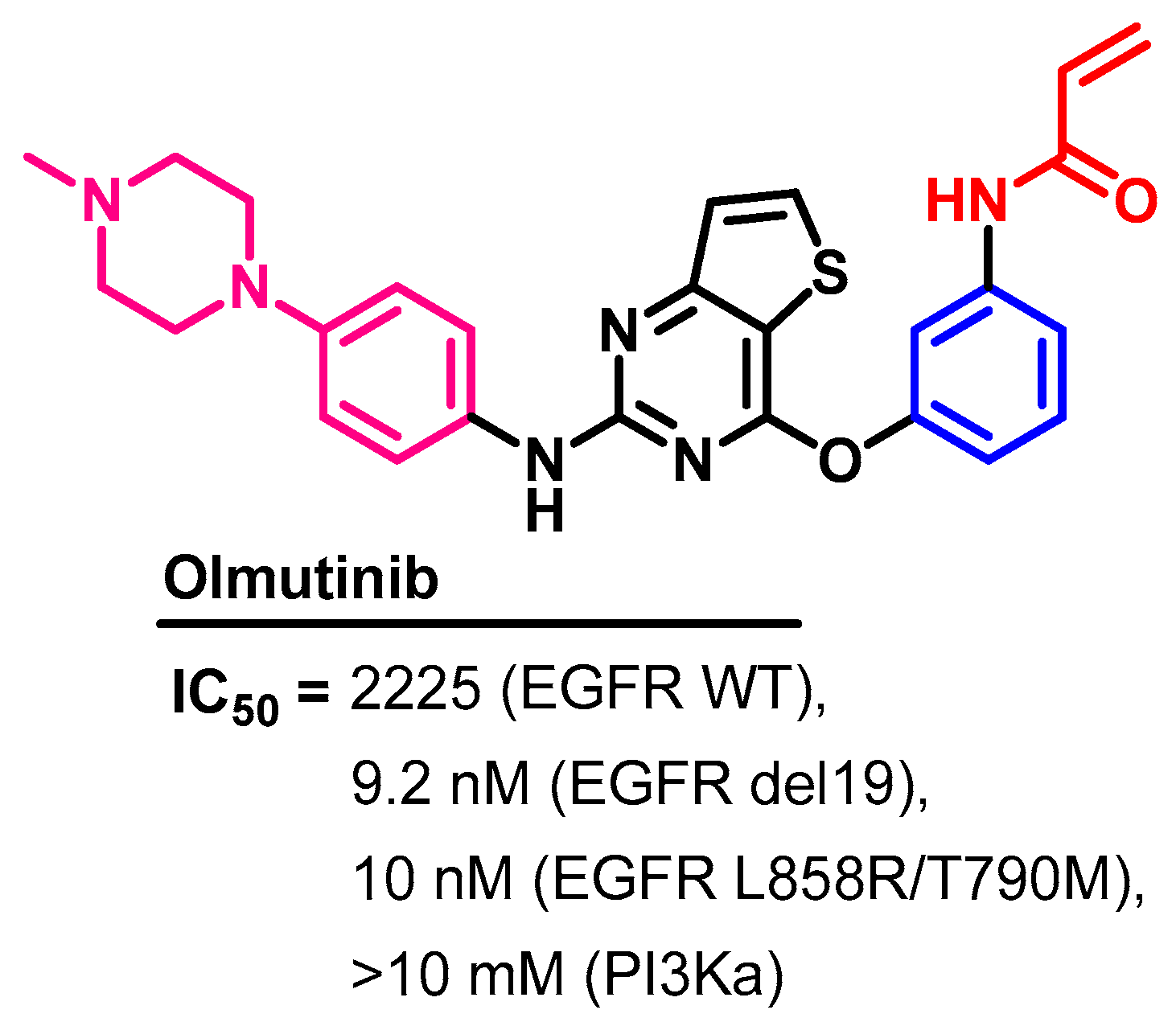
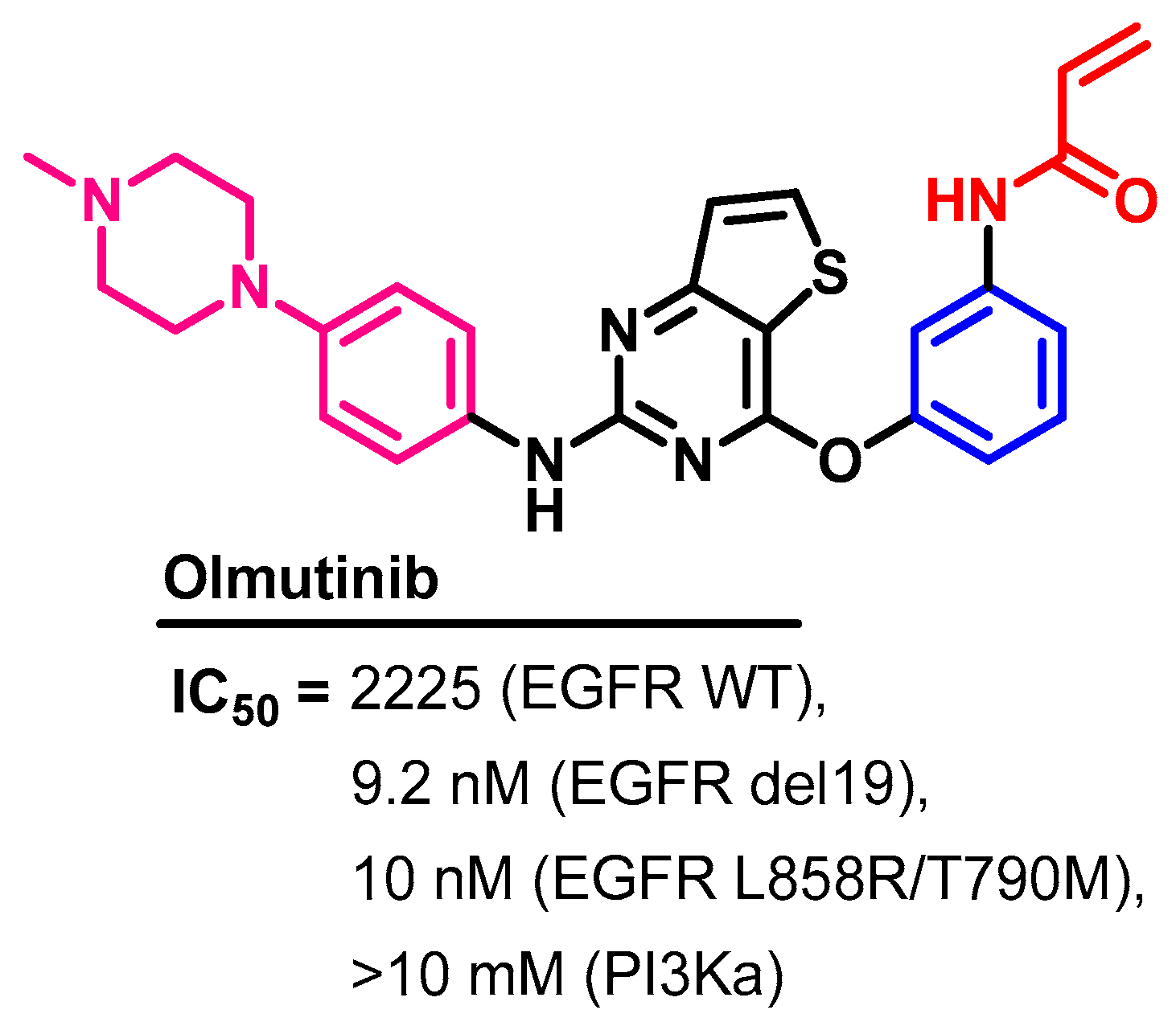
Biological Activity
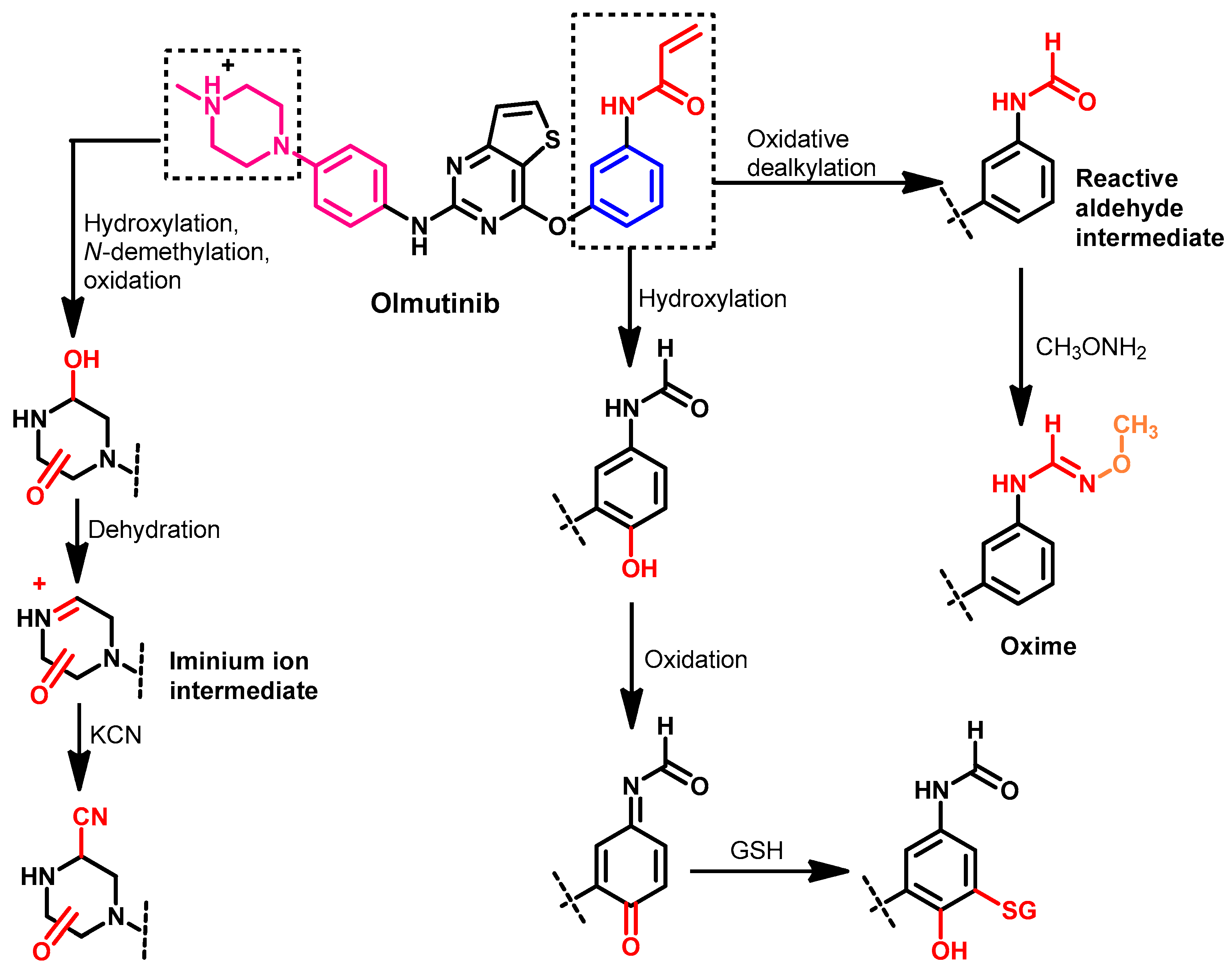
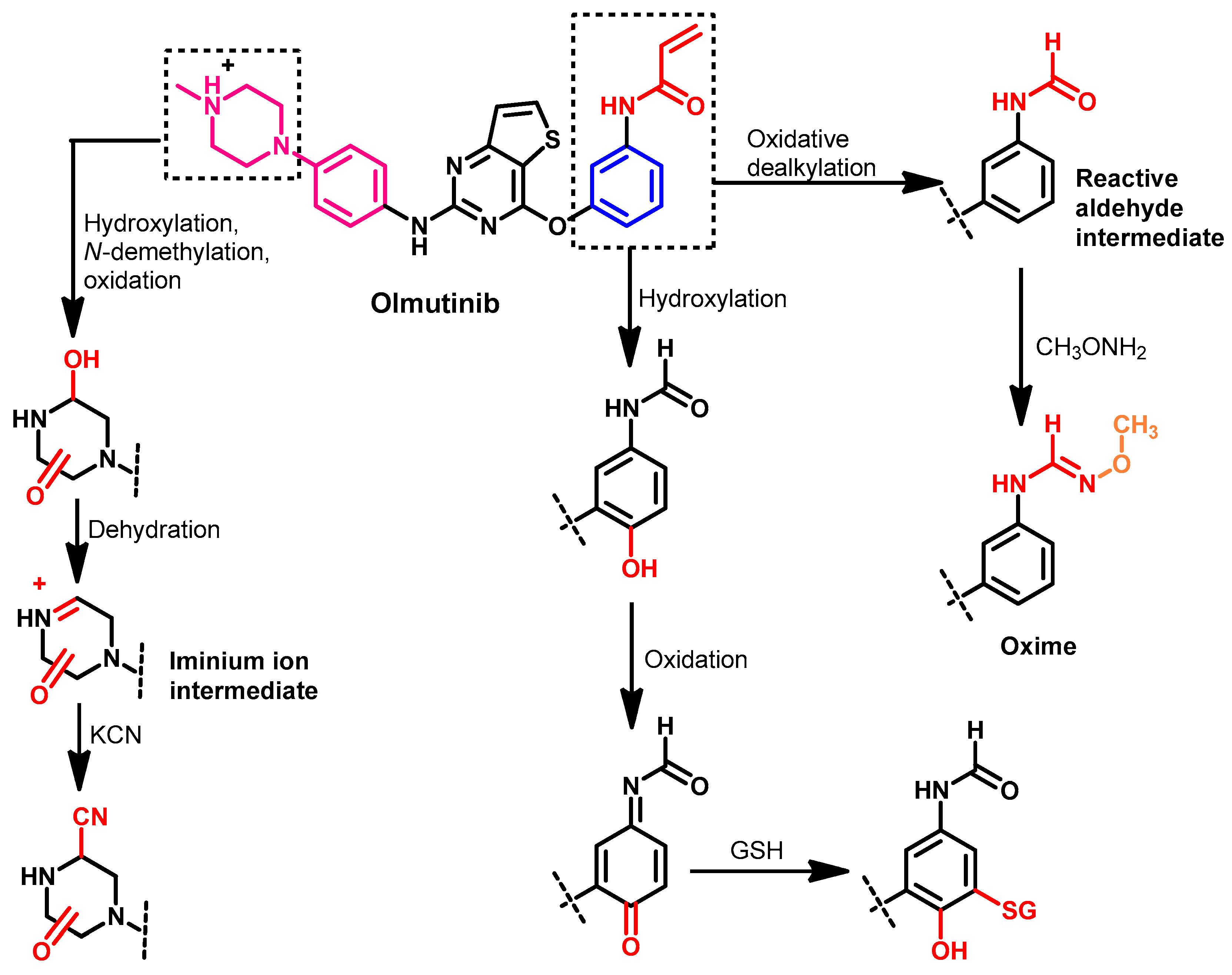
Metabolism
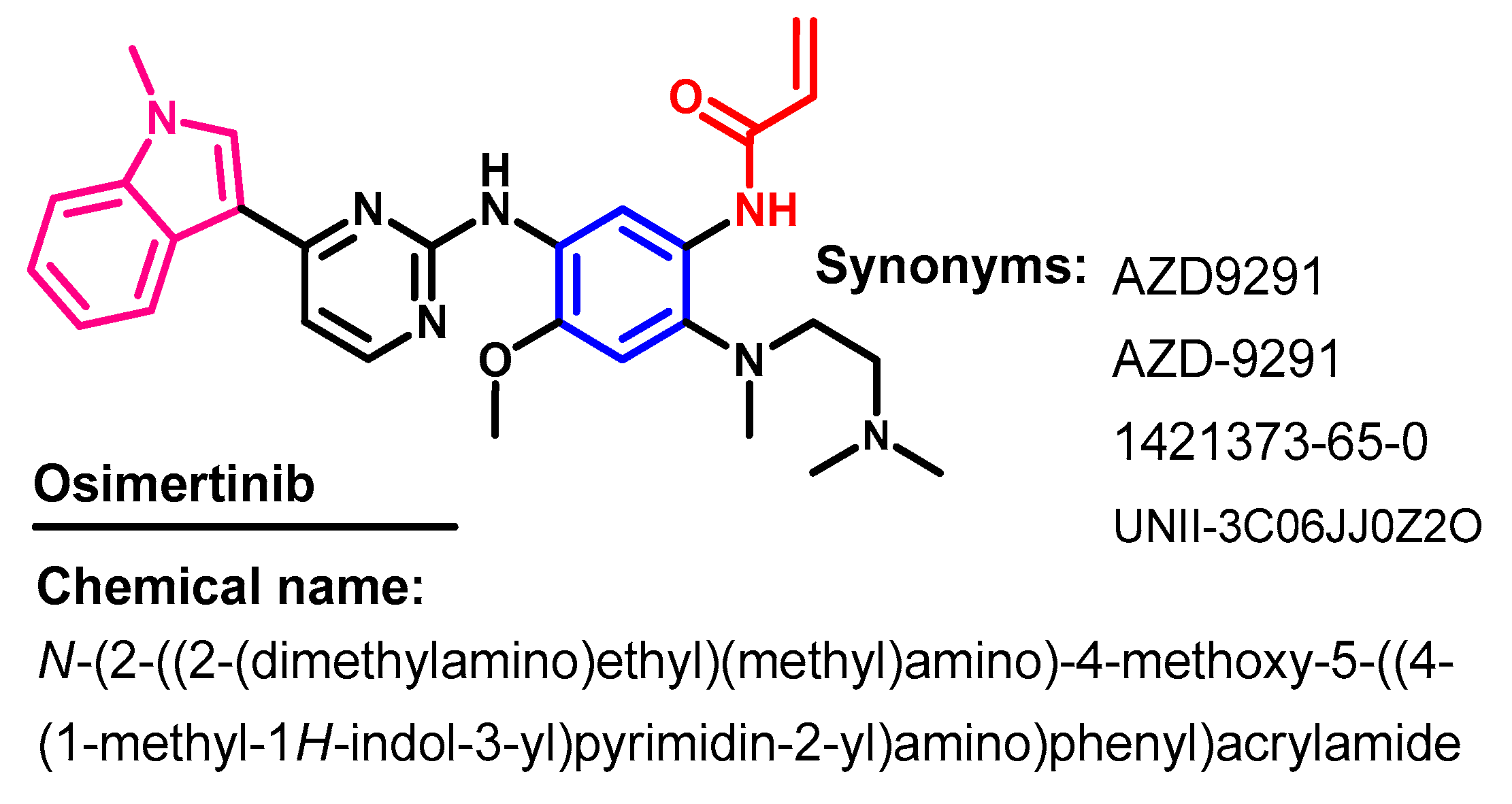
1.2.11. Osimertinib
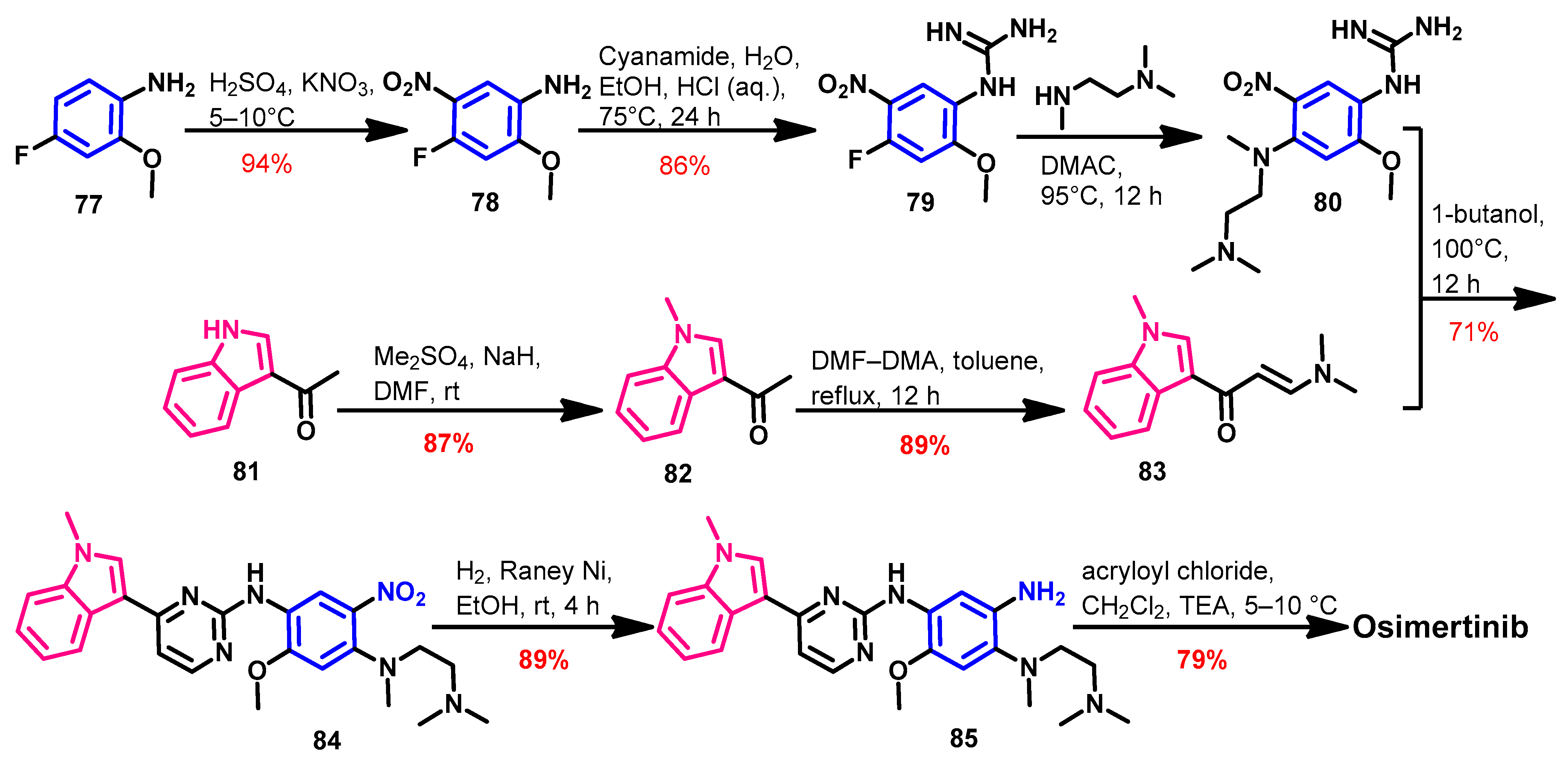
Approval History
Synthesis
Target Kinases
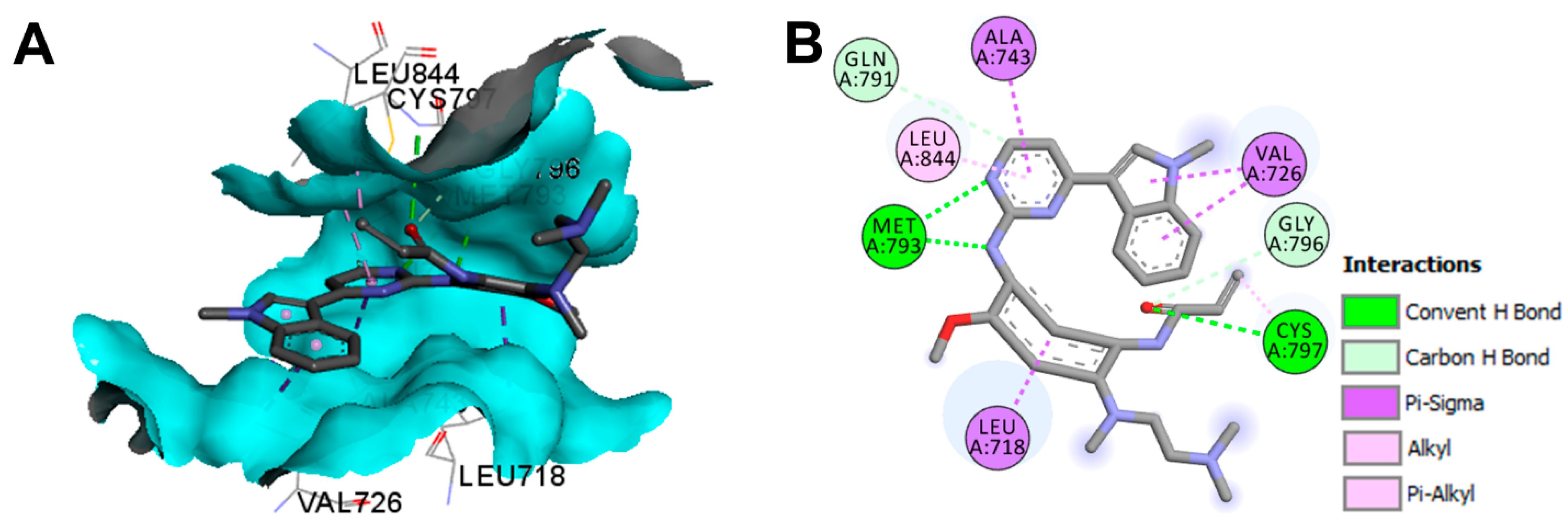
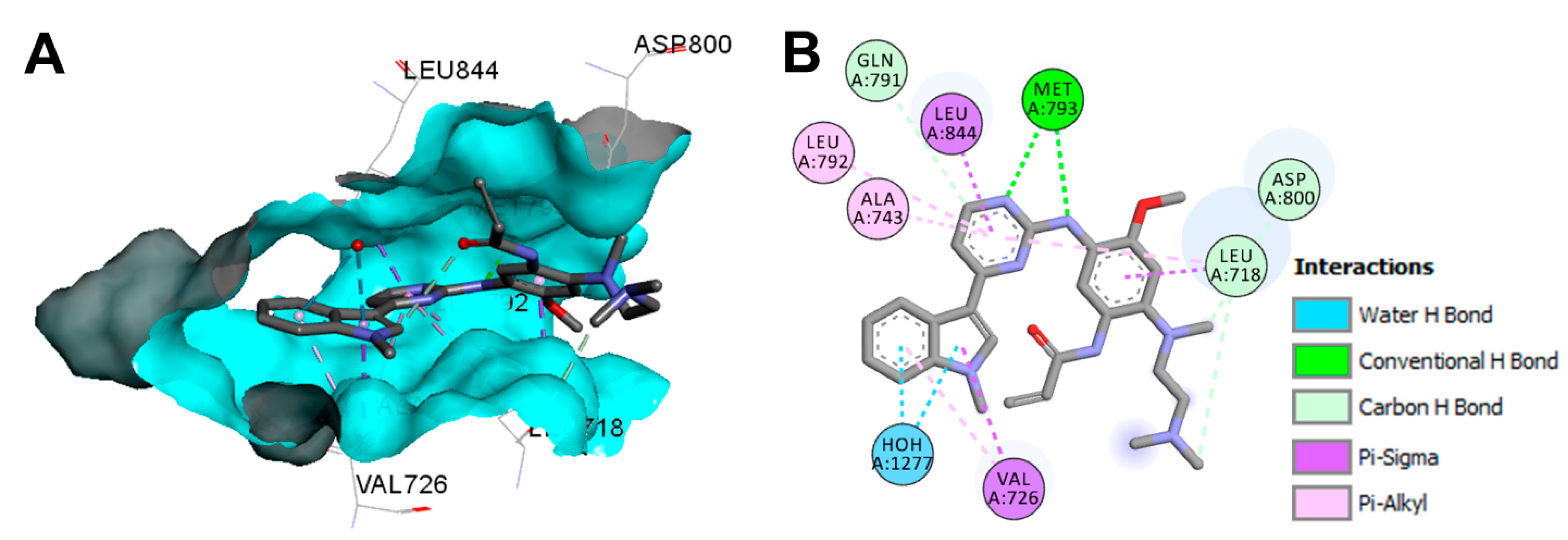
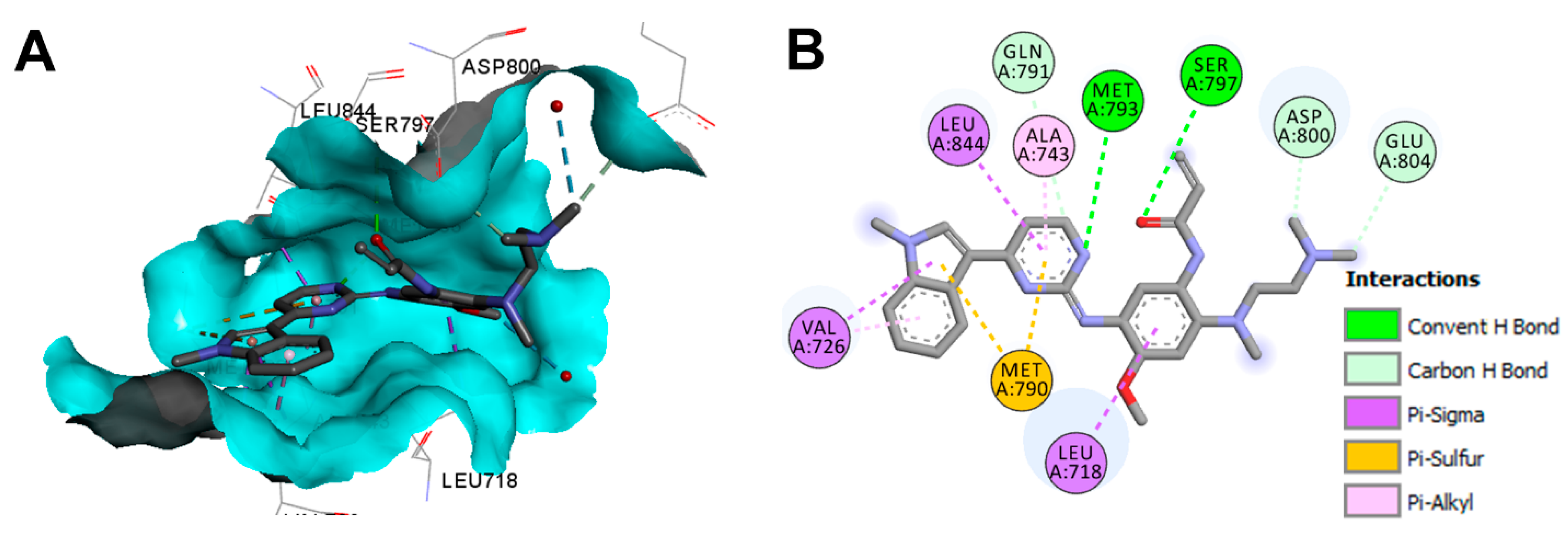
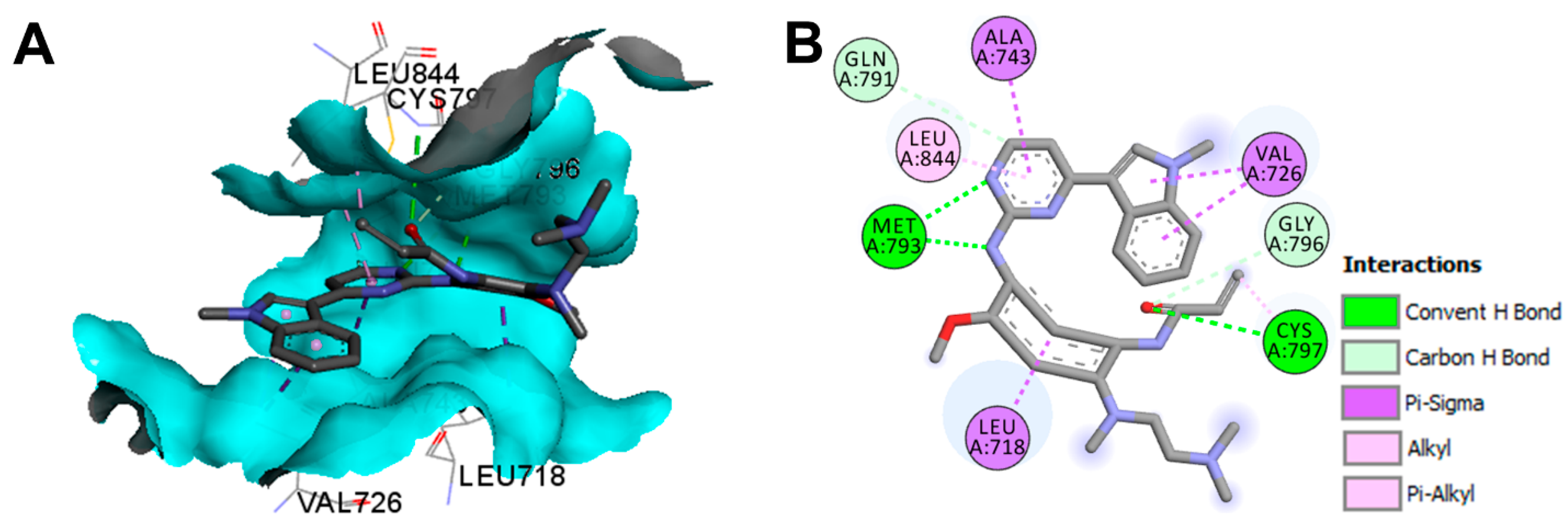
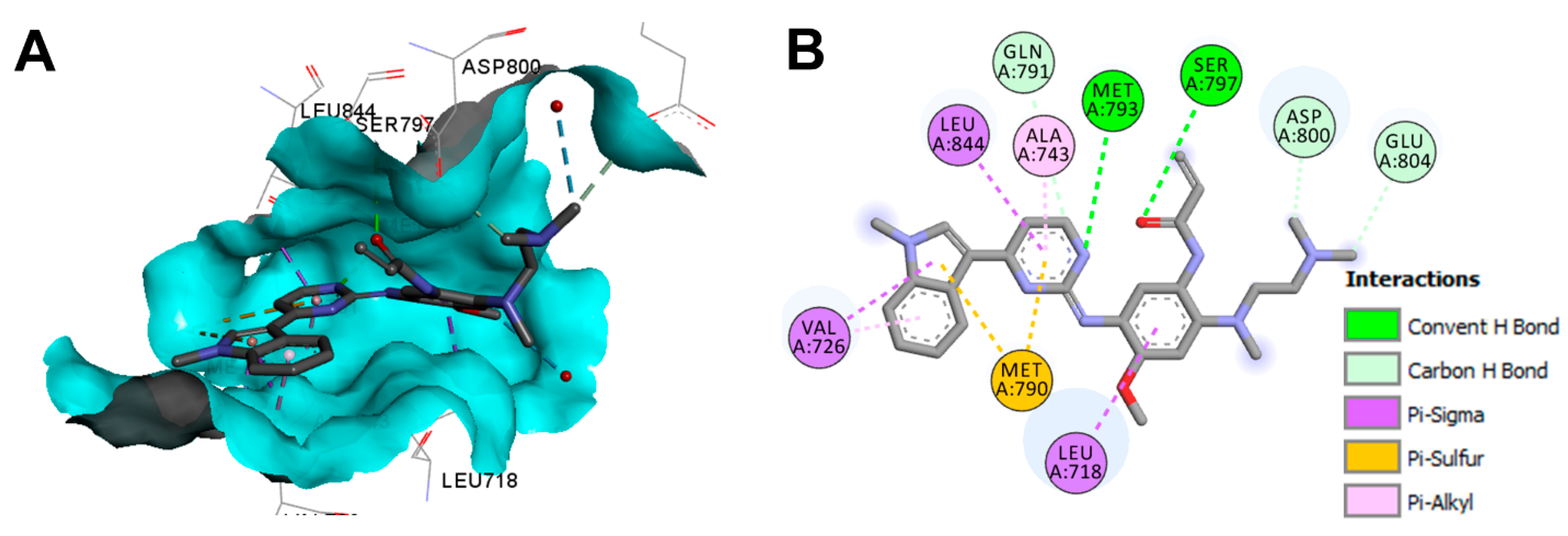
Crystal Structures and Binding Interactions
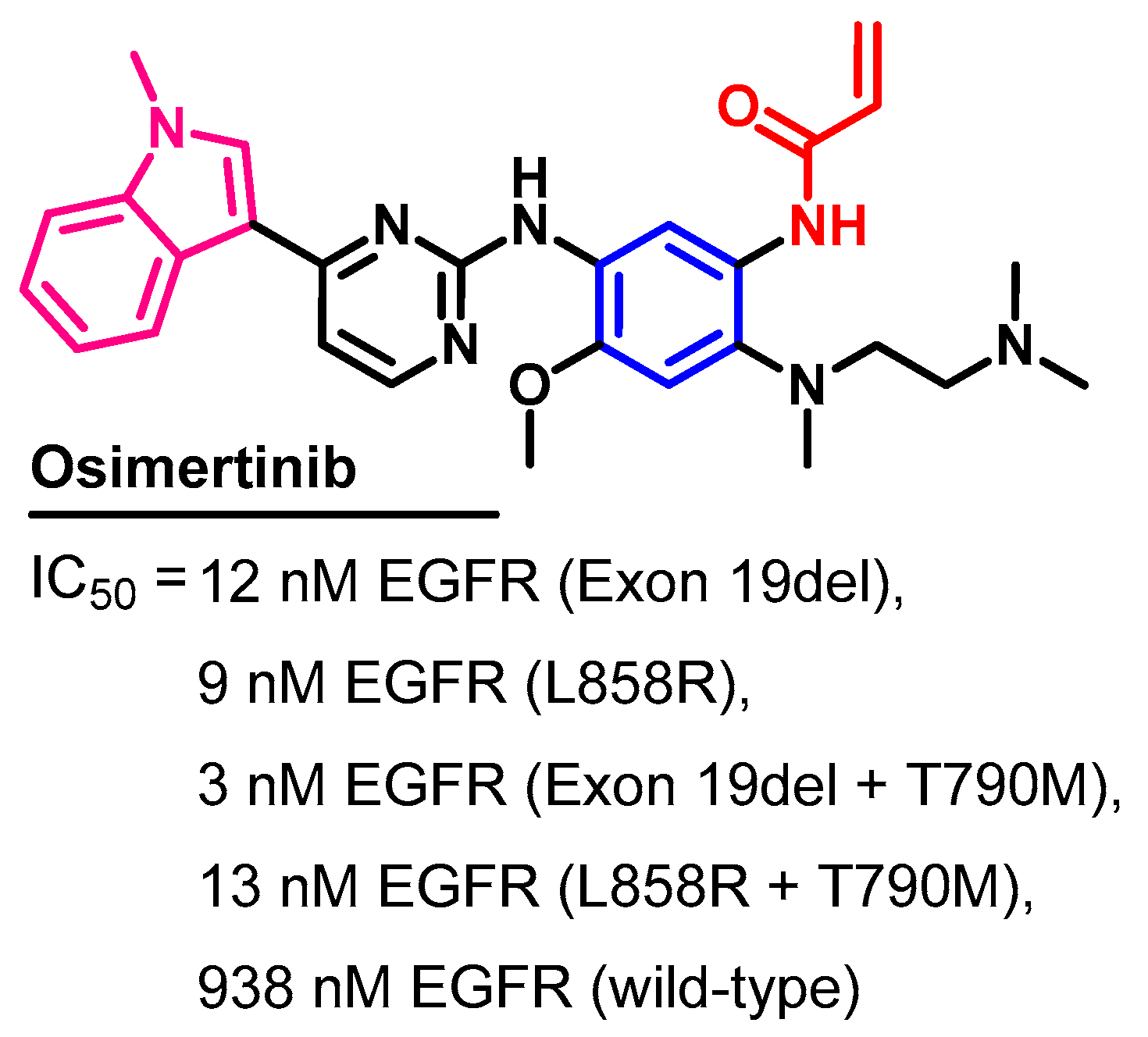
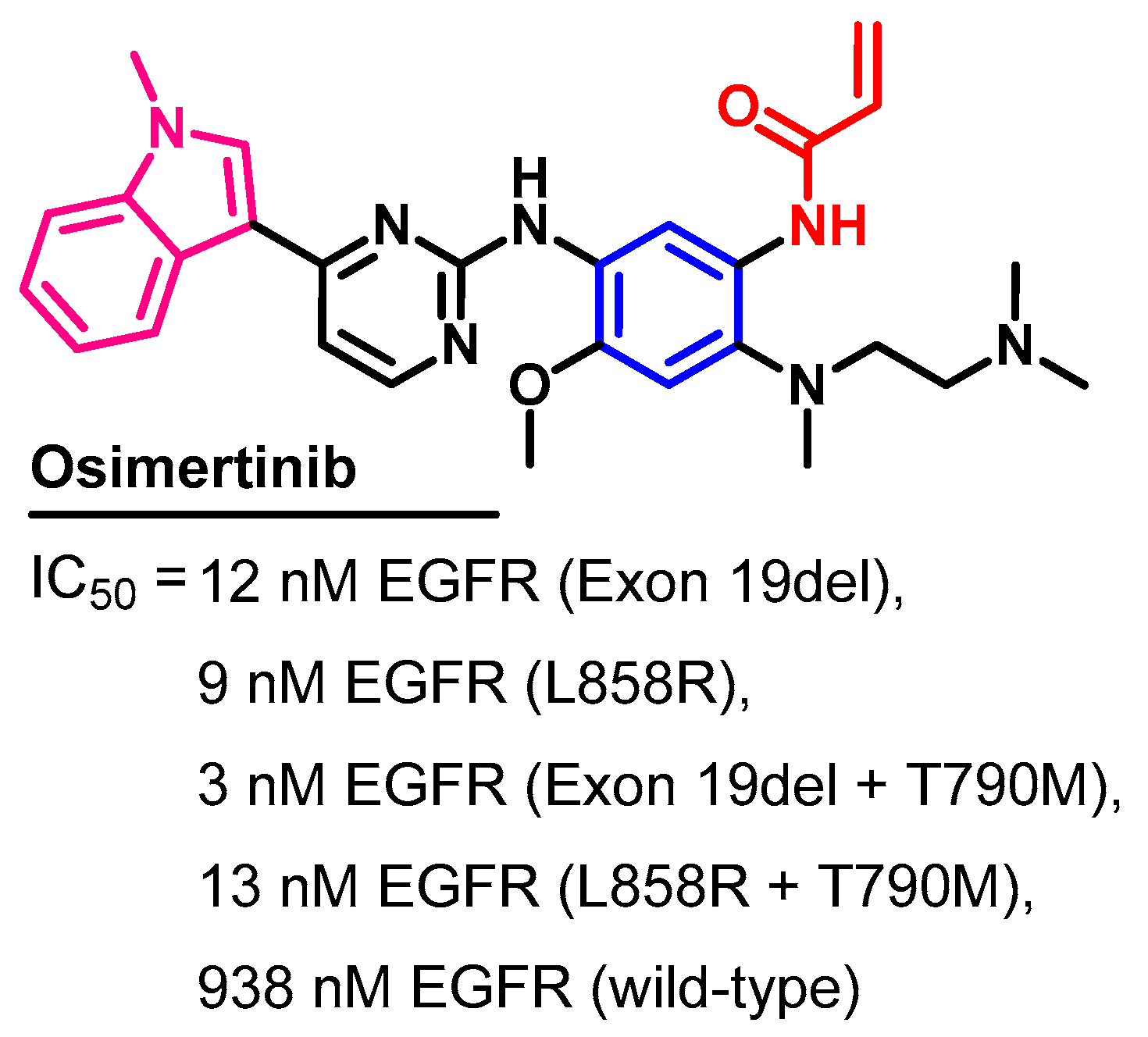
Biological Activity
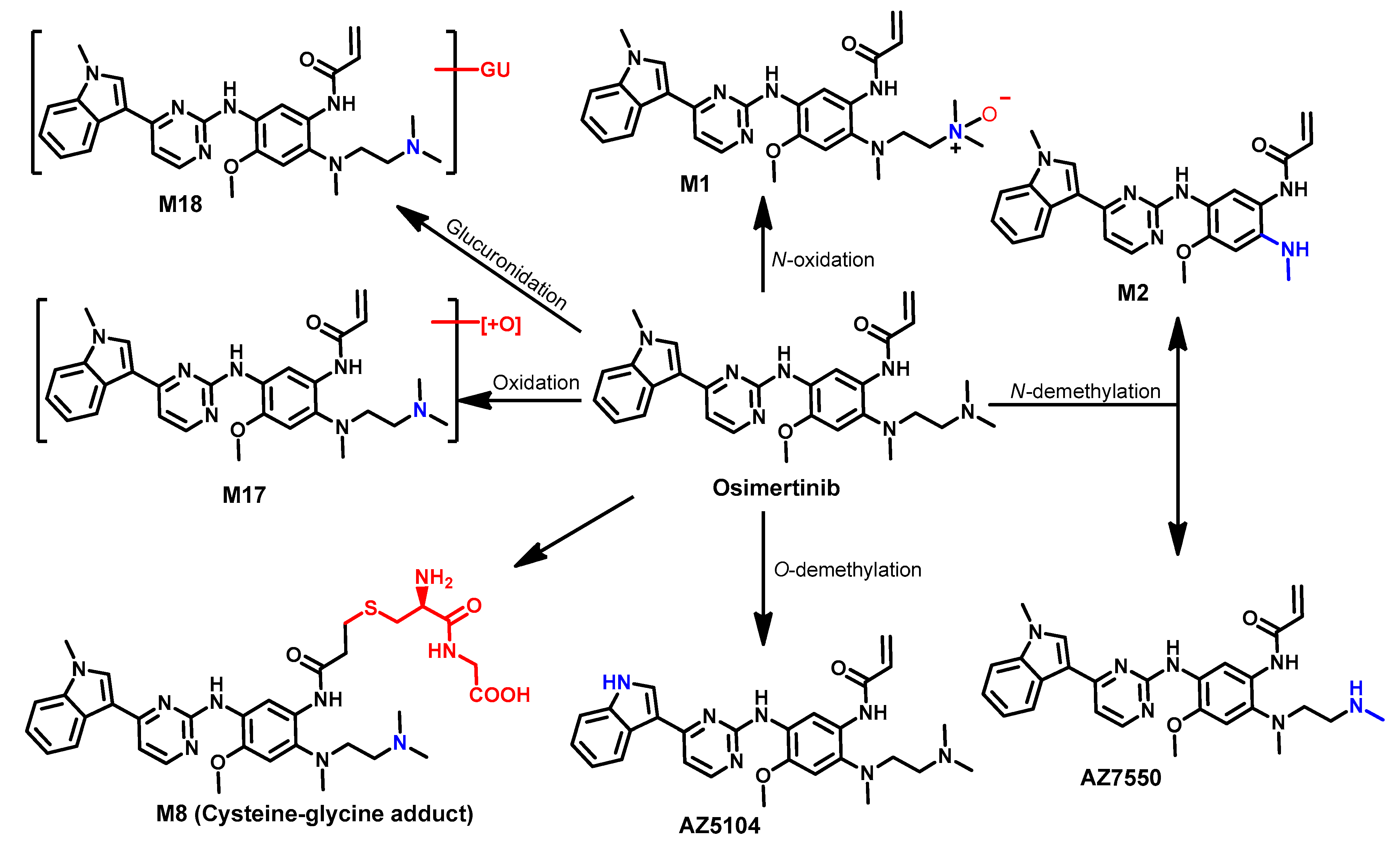
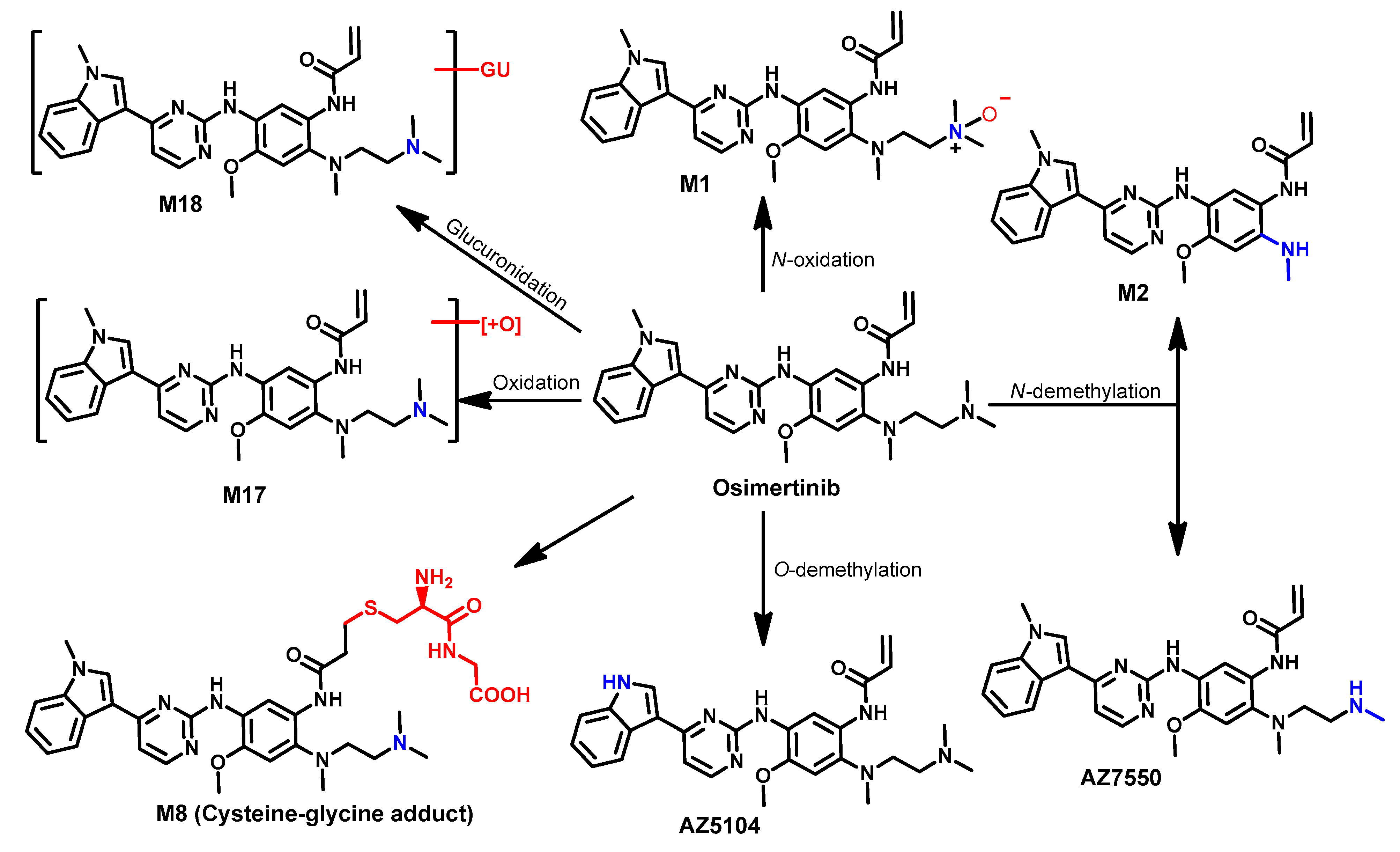
Metabolism
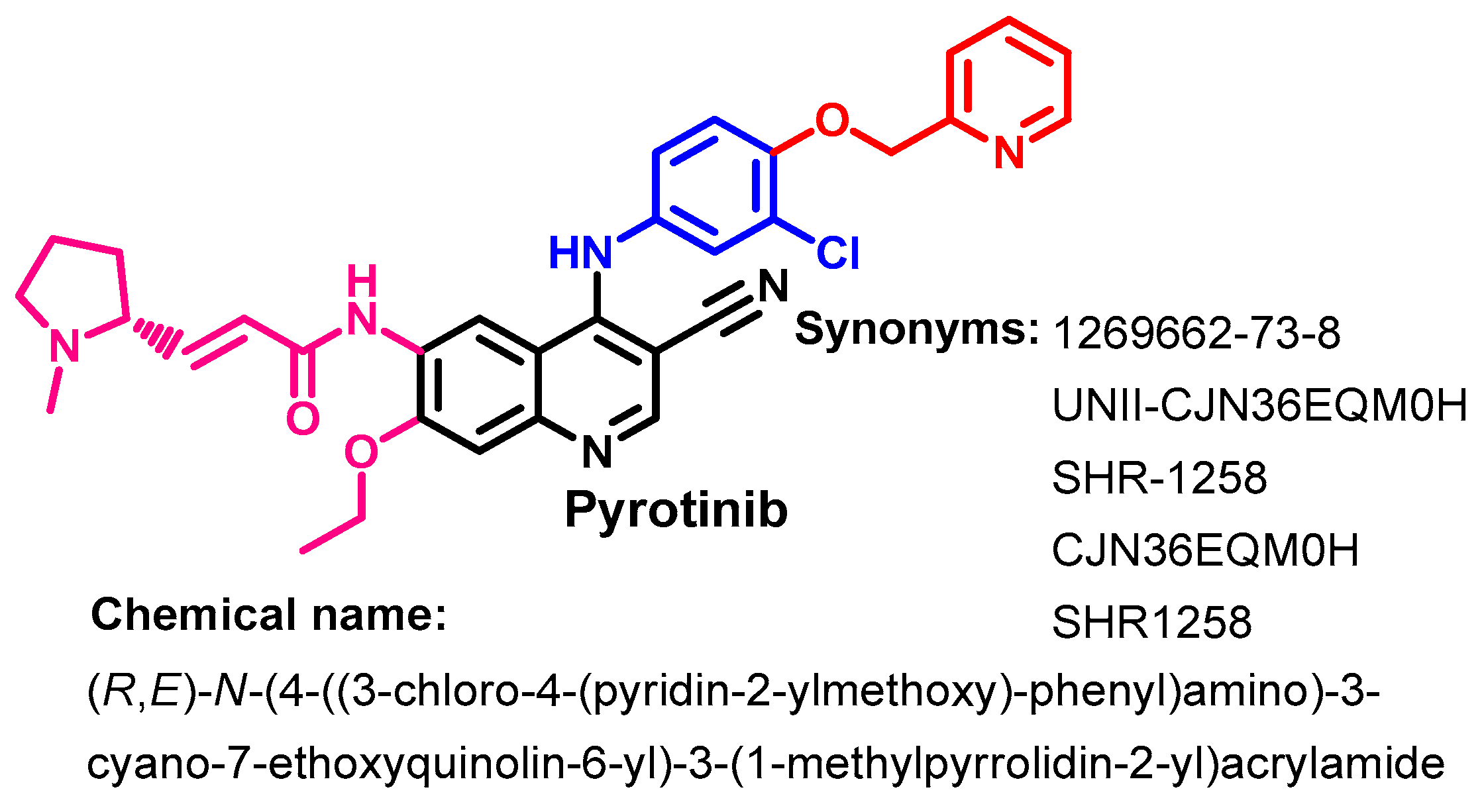
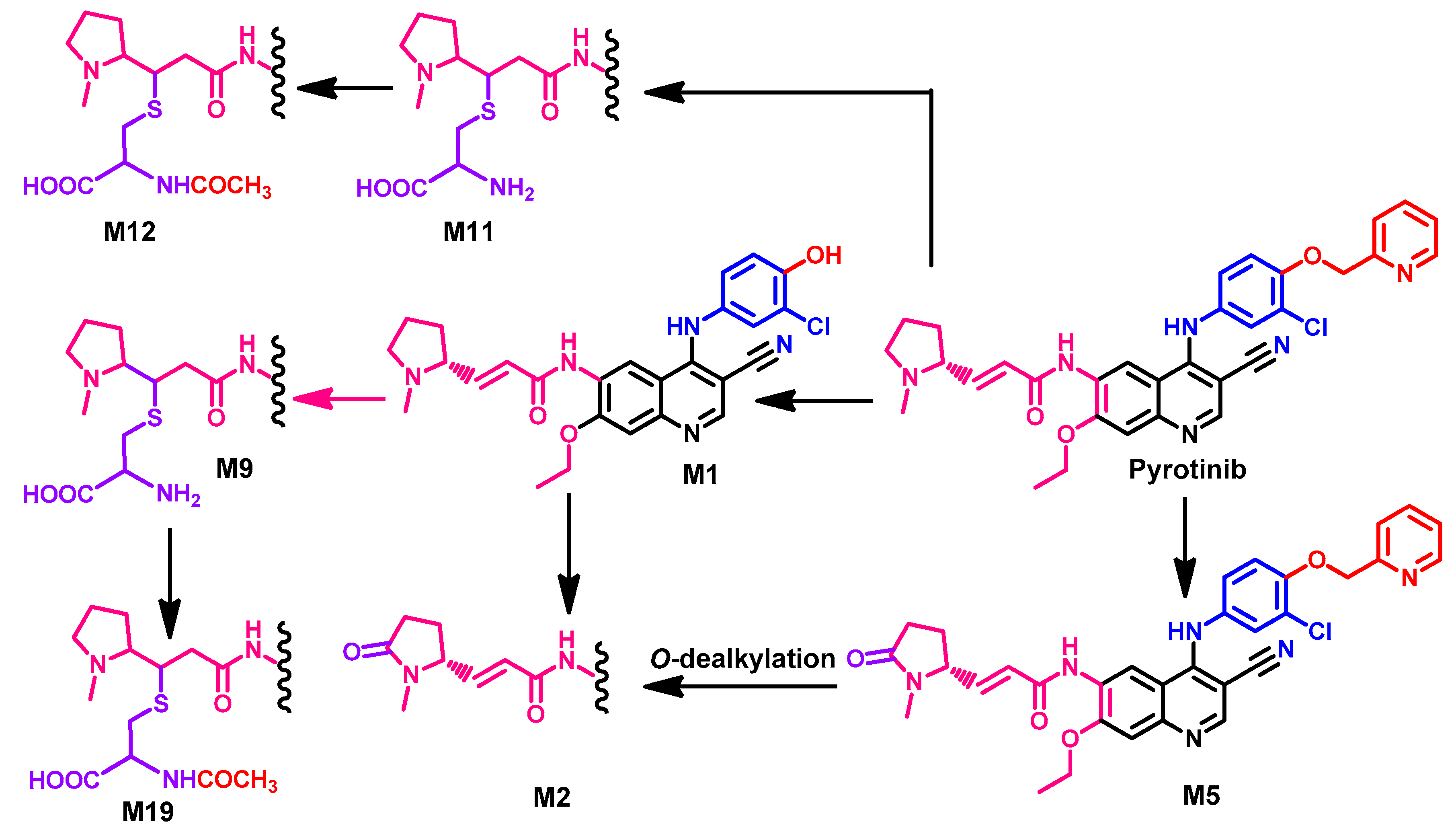
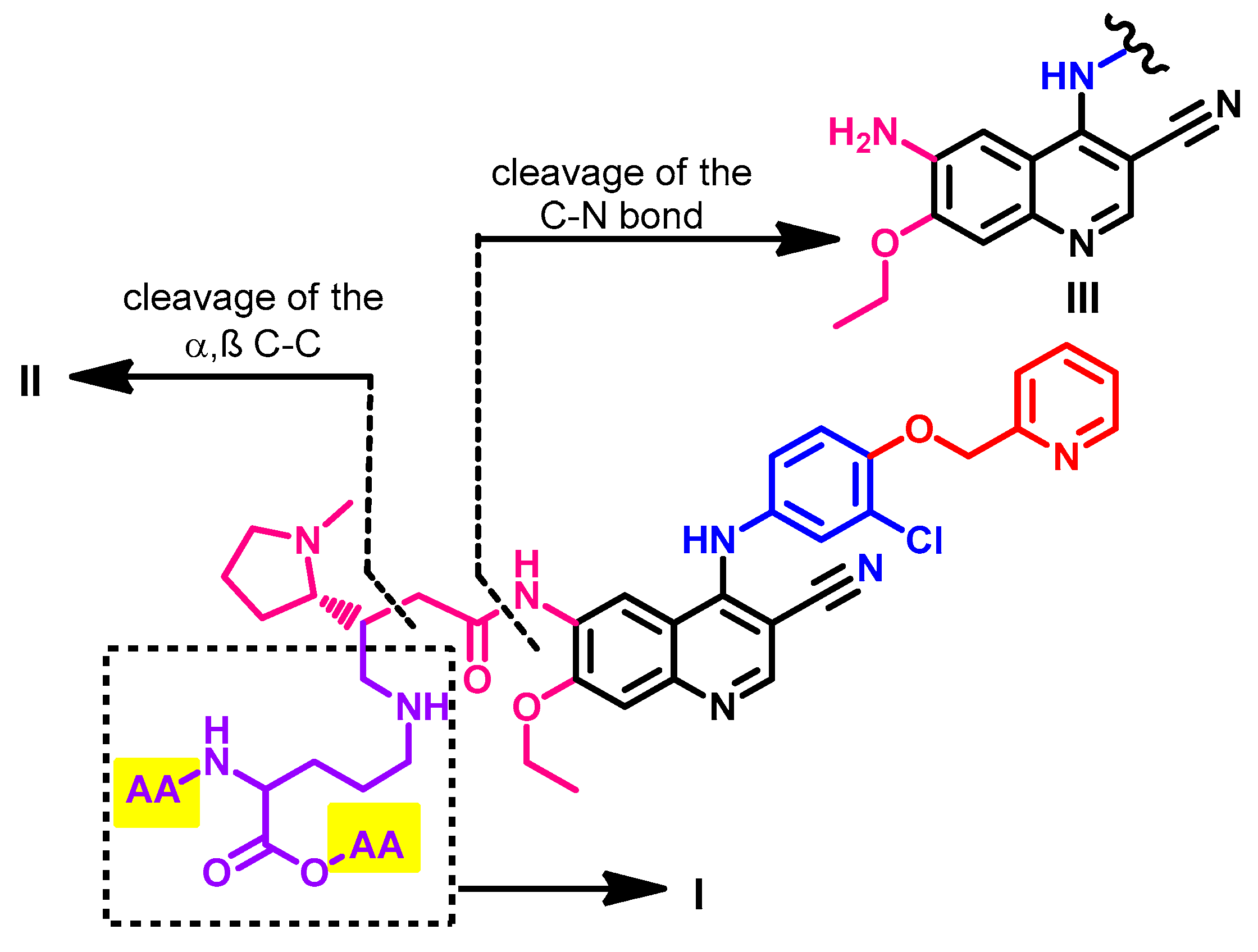
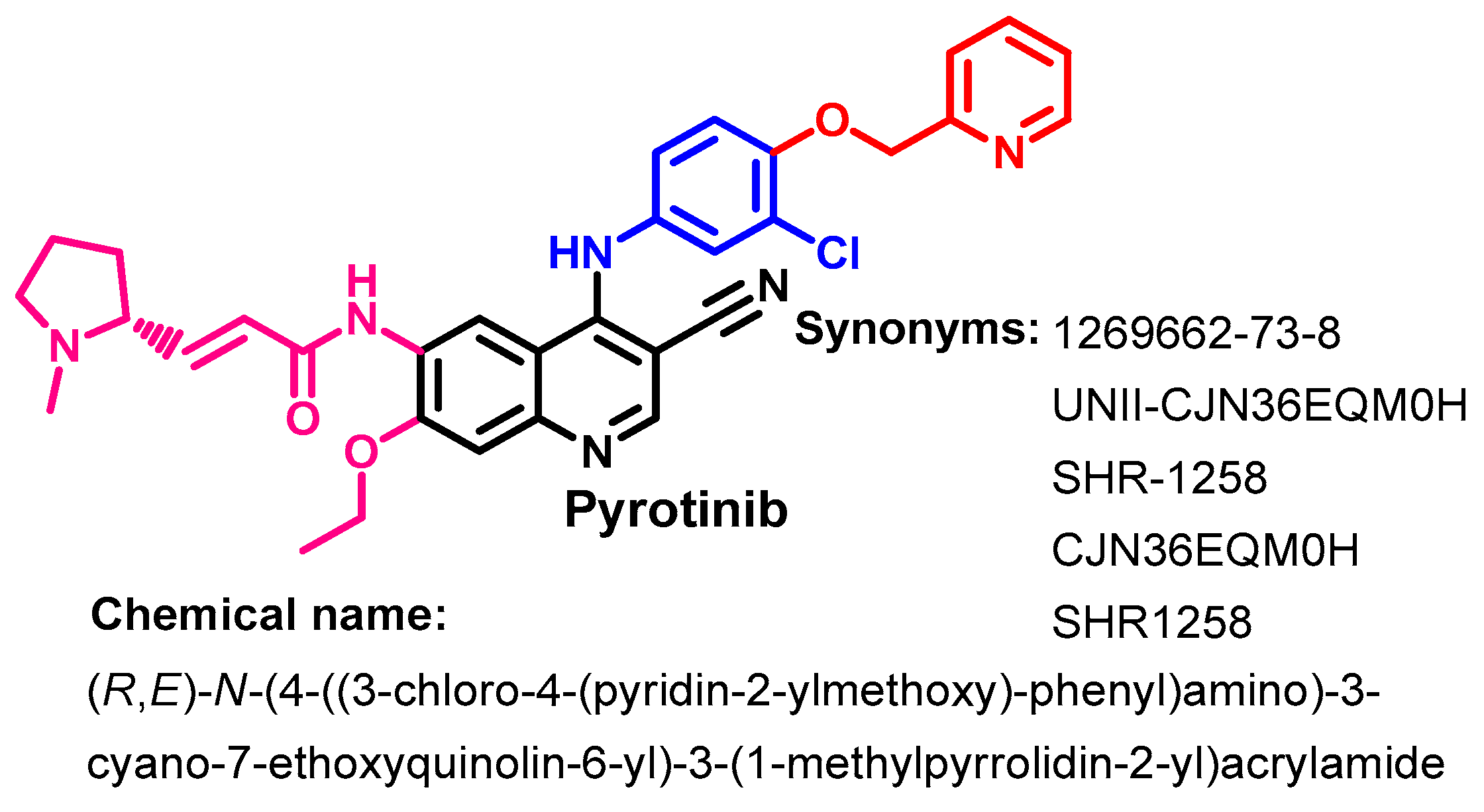
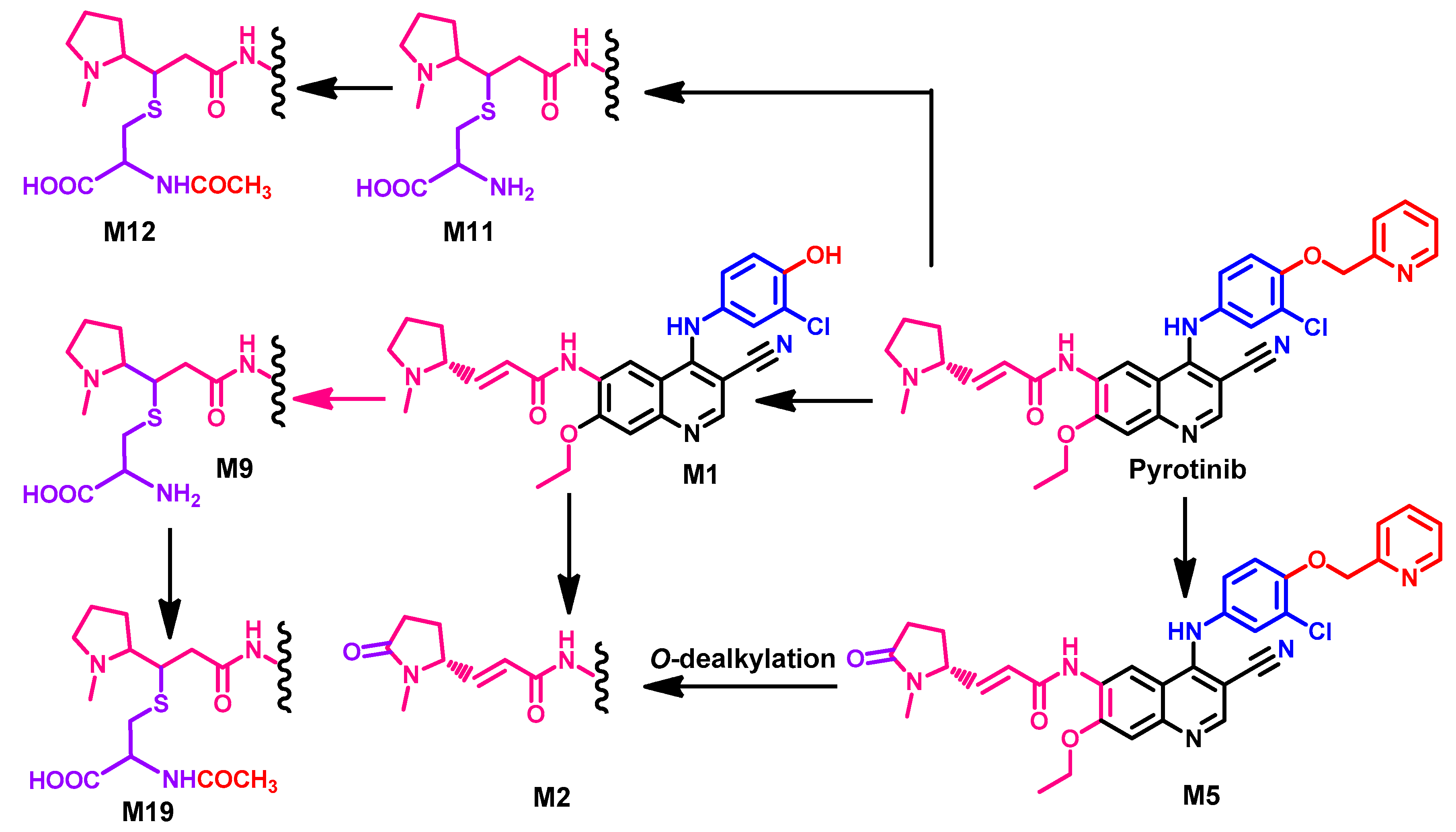
1.2.12. Pyrotinib
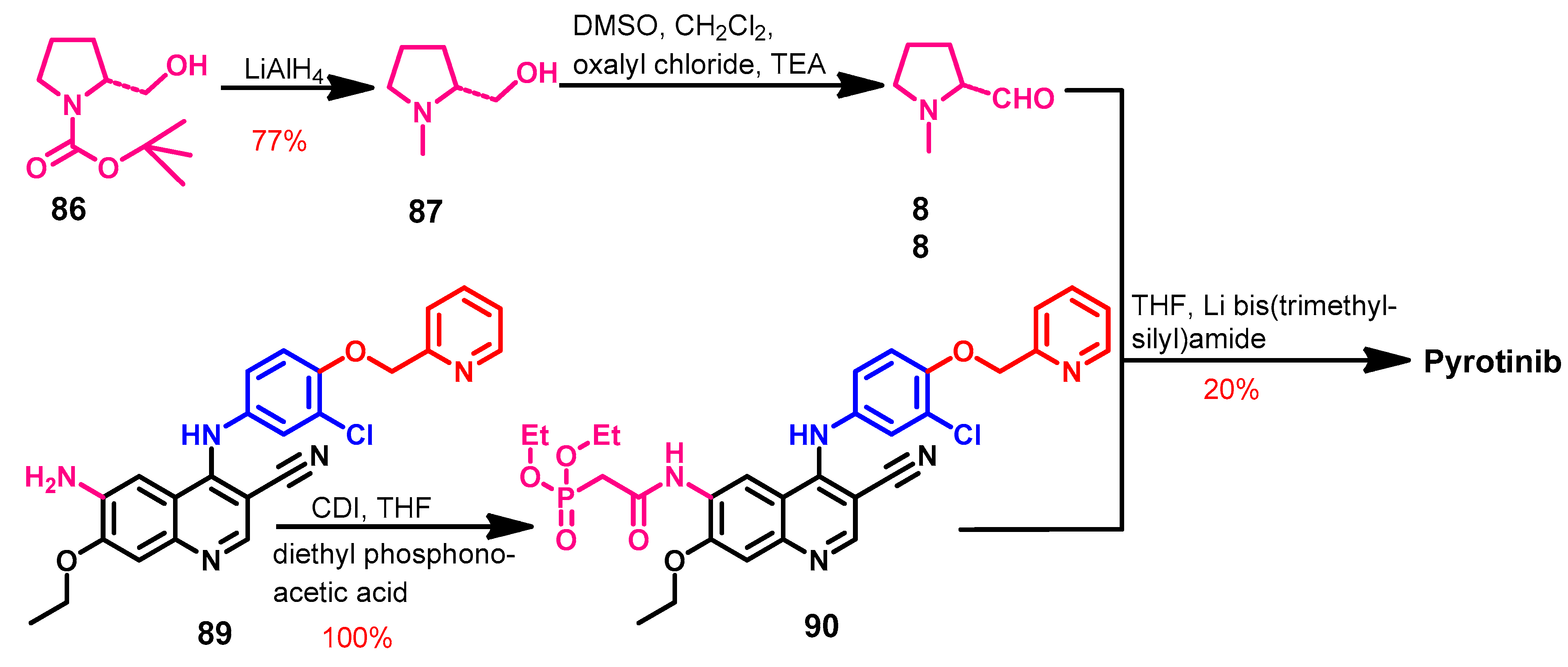
Approval History
Synthesis
Target Kinases
Crystal Structures and Binding Interactions
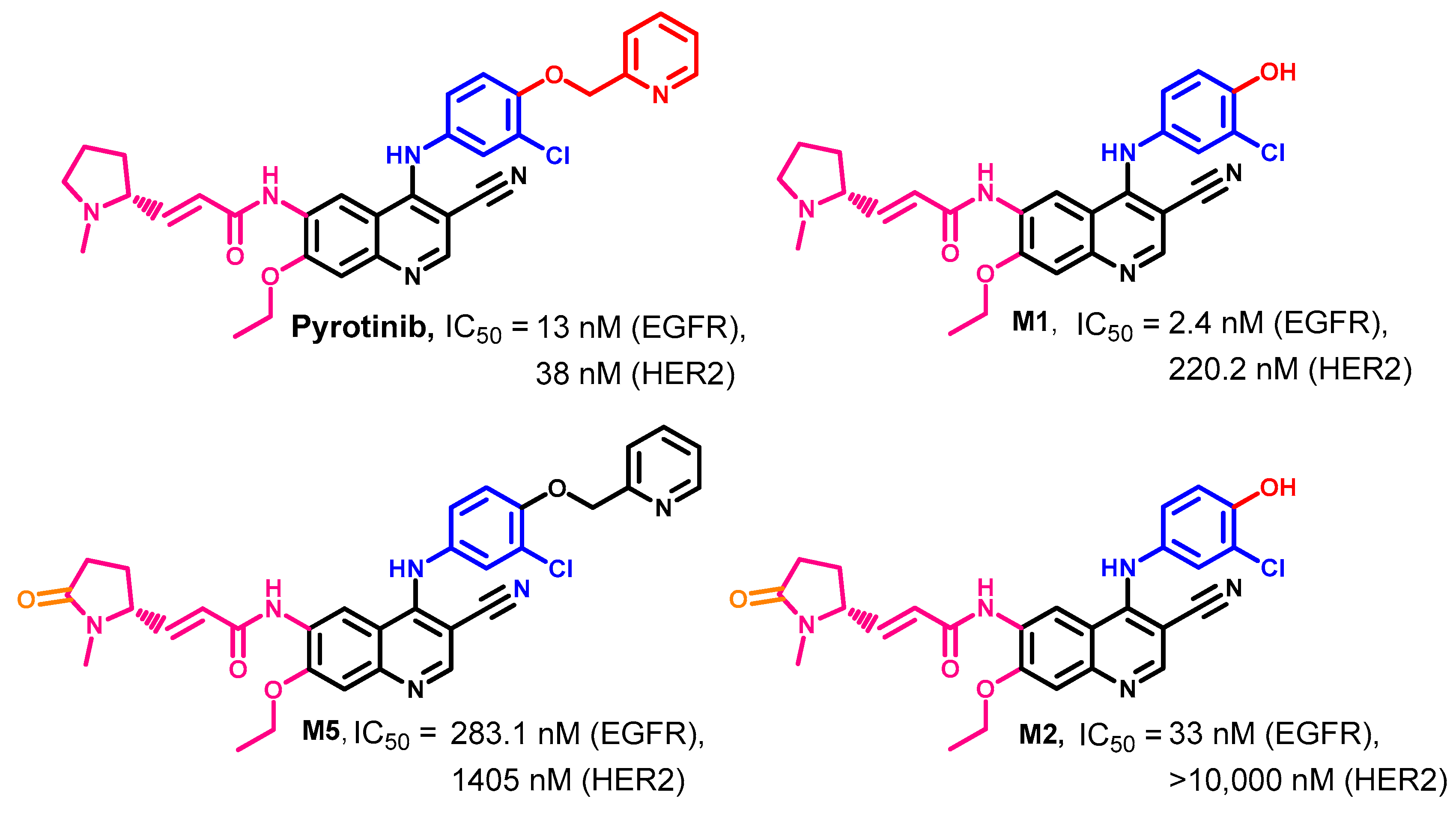
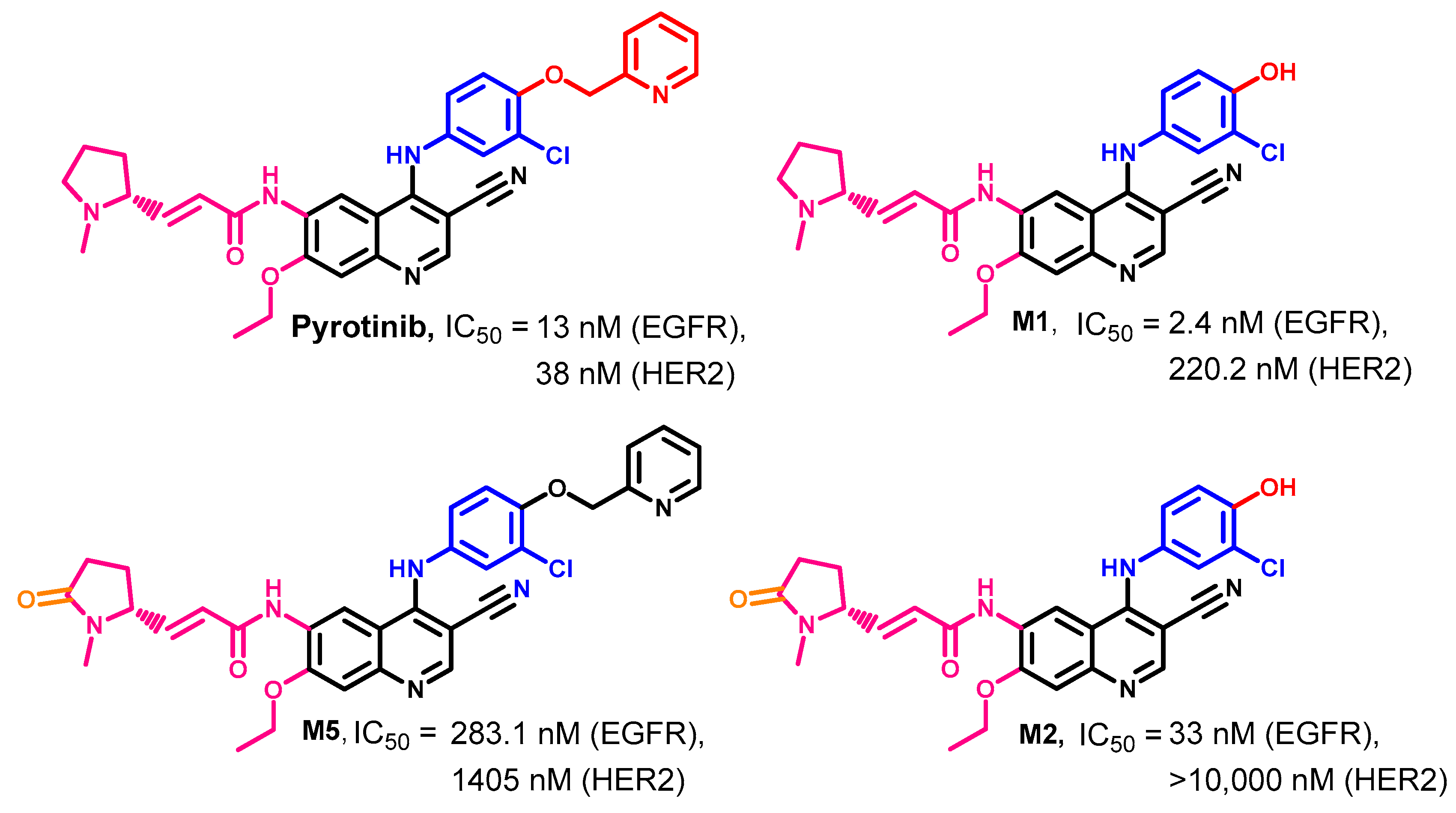
Biological Activity
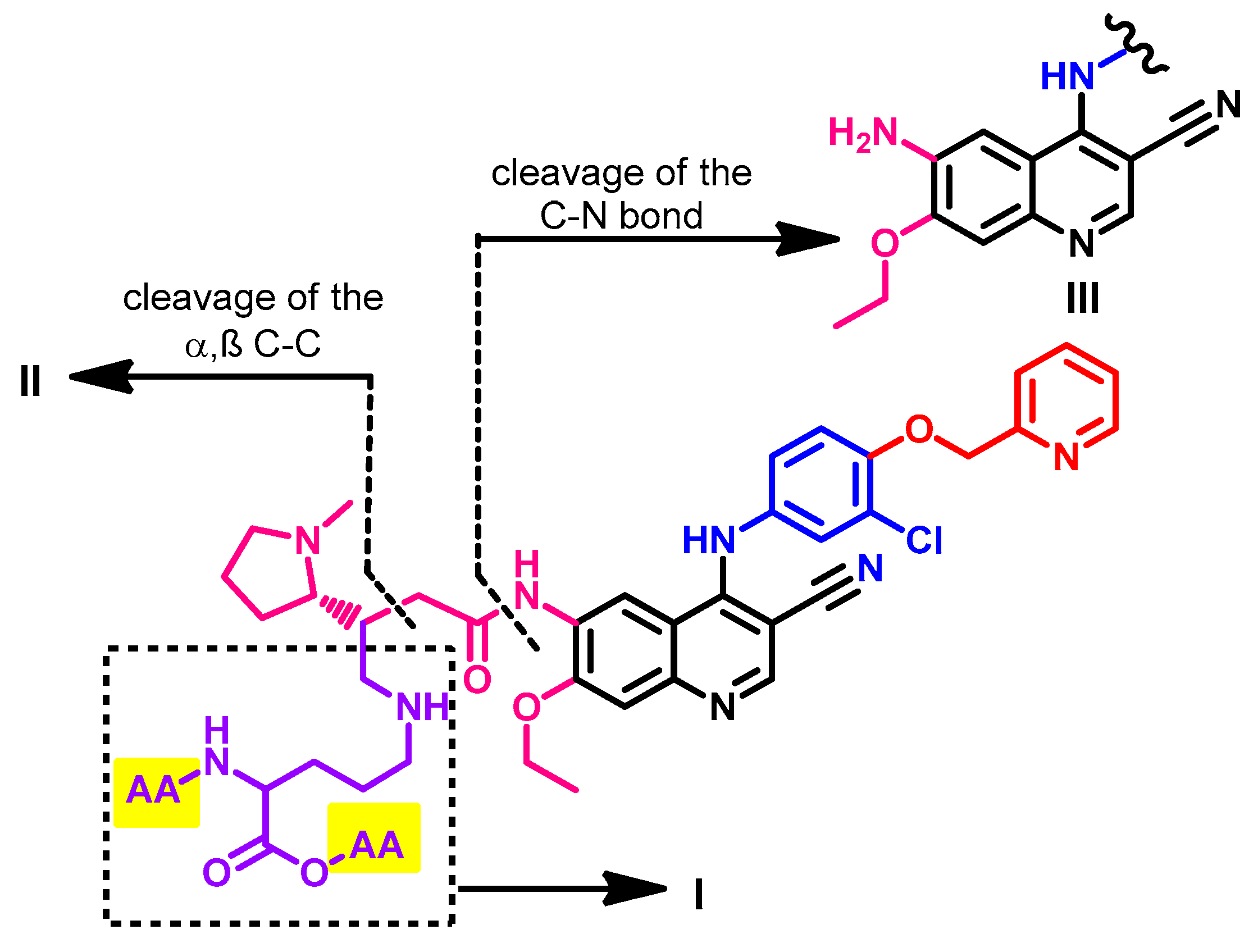
Metabolism
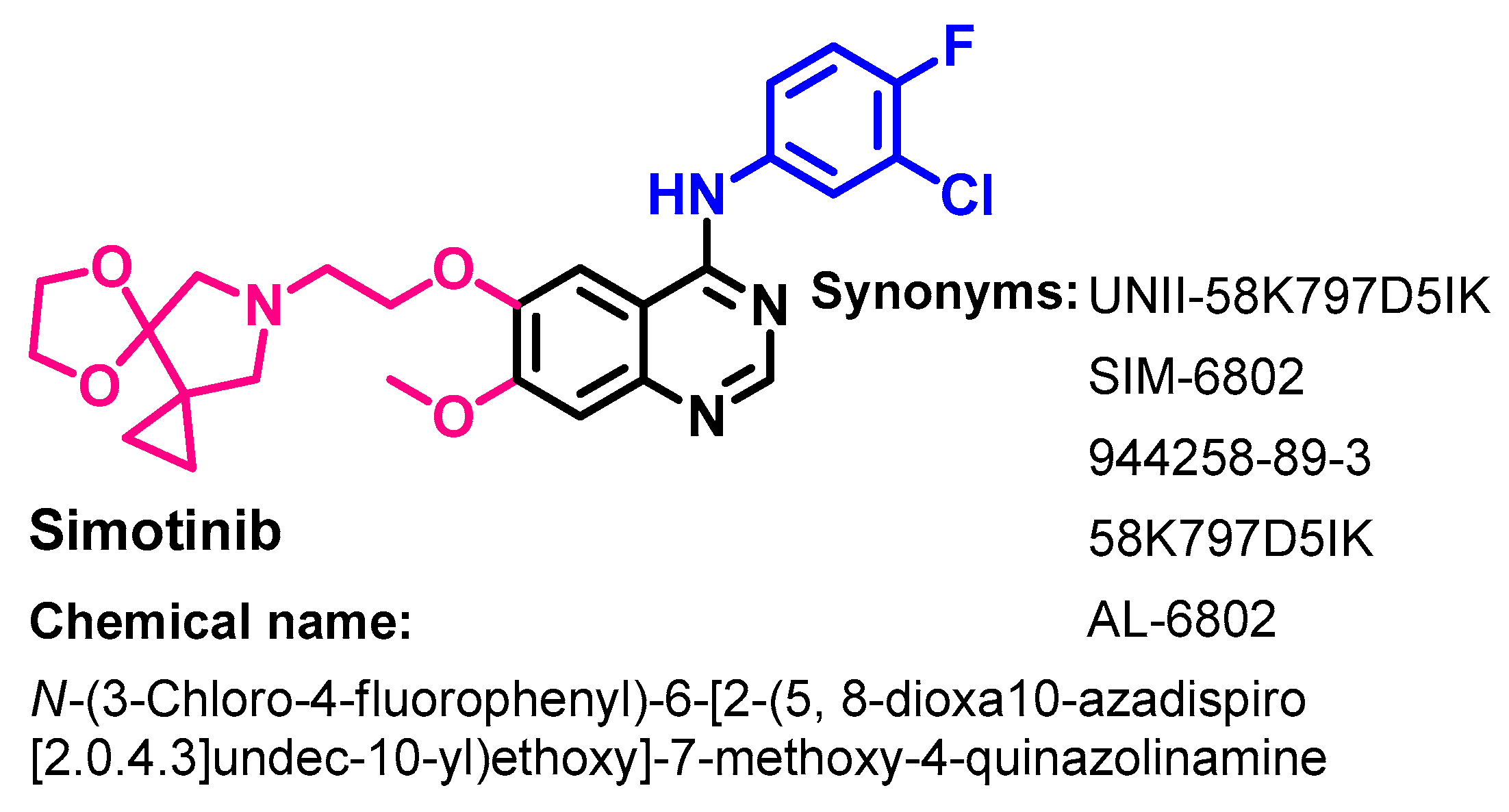
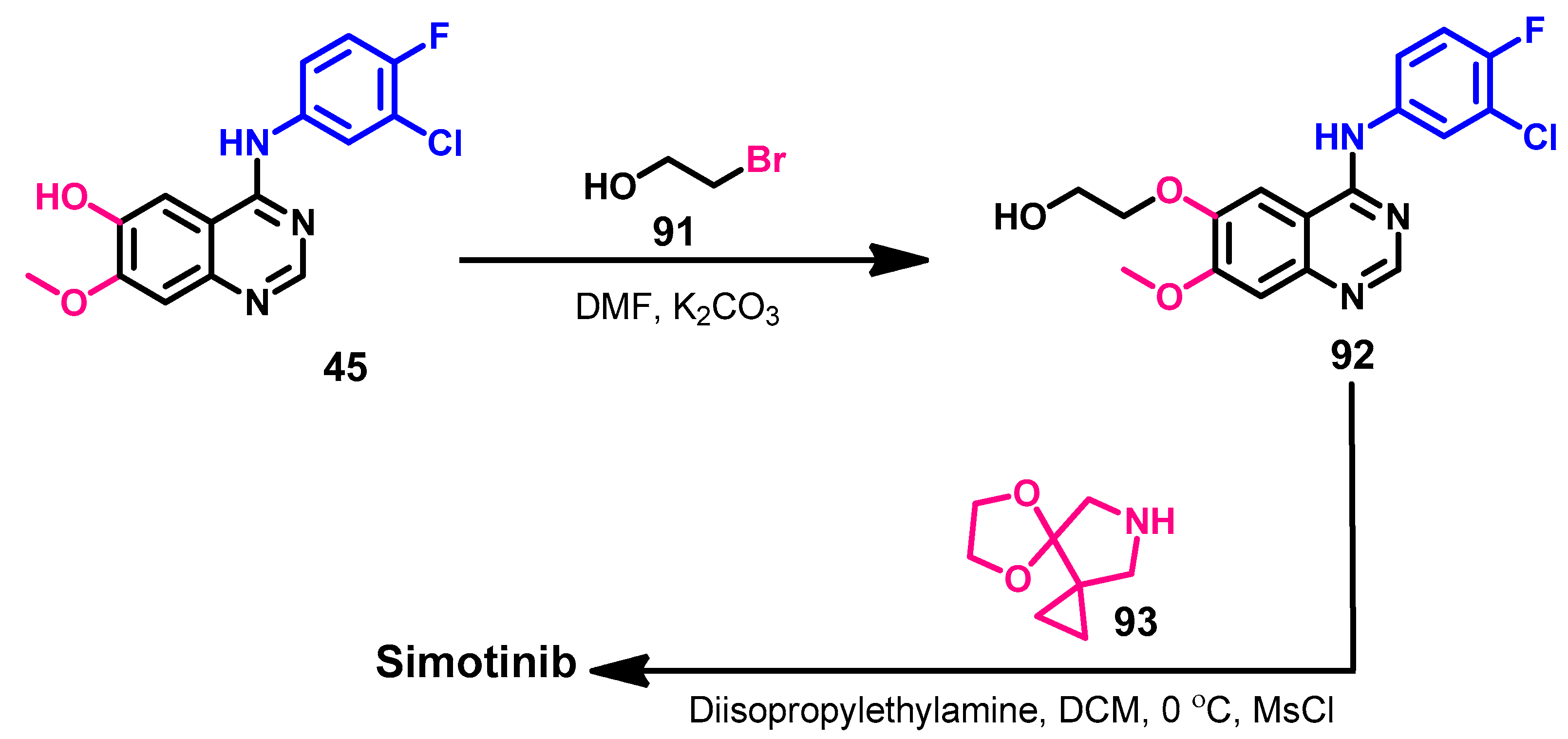
1.2.13. Simotinib
Approval History
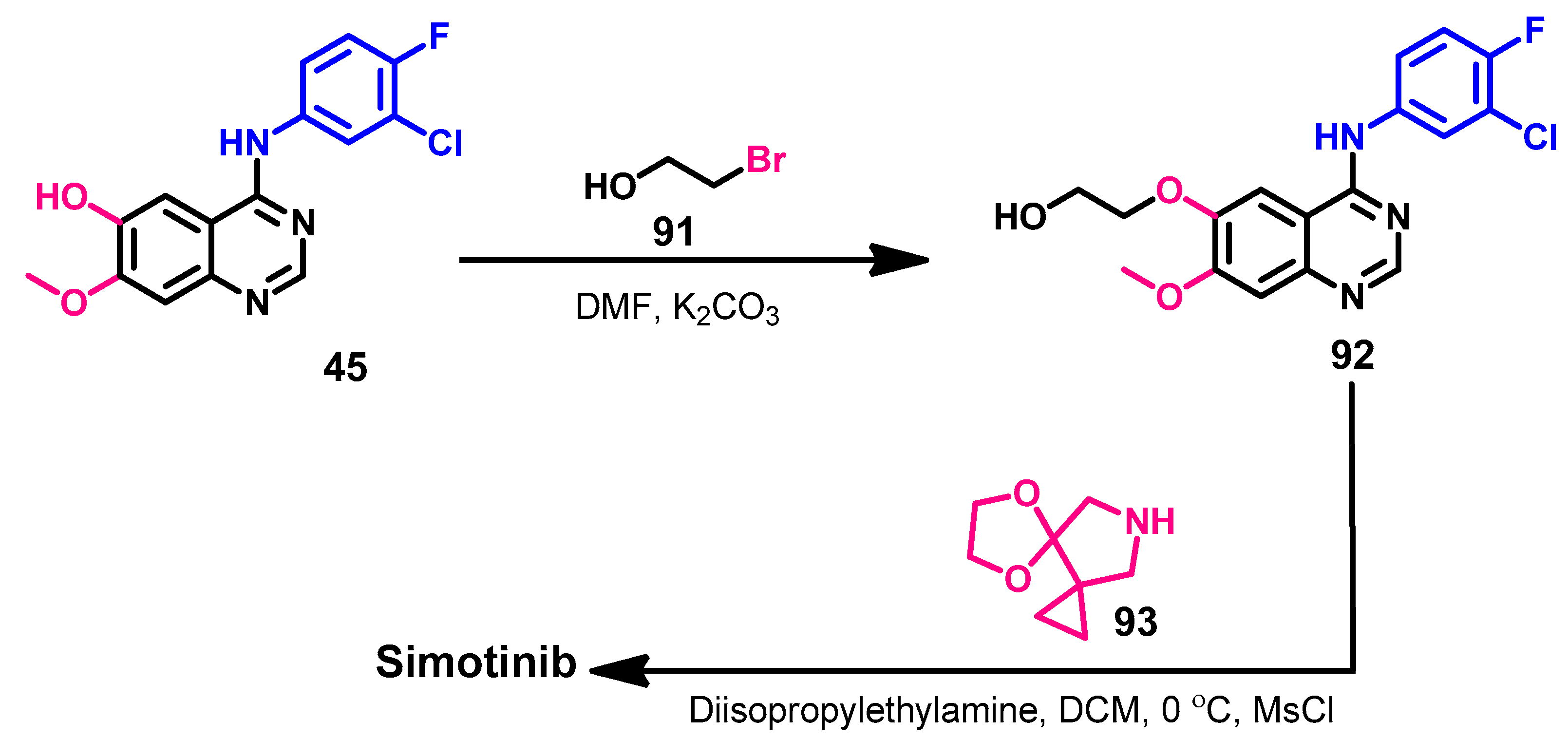
Synthesis
Target Kinases
Crystal Structures and Binding Interactions
Biological Activity
Metabolism
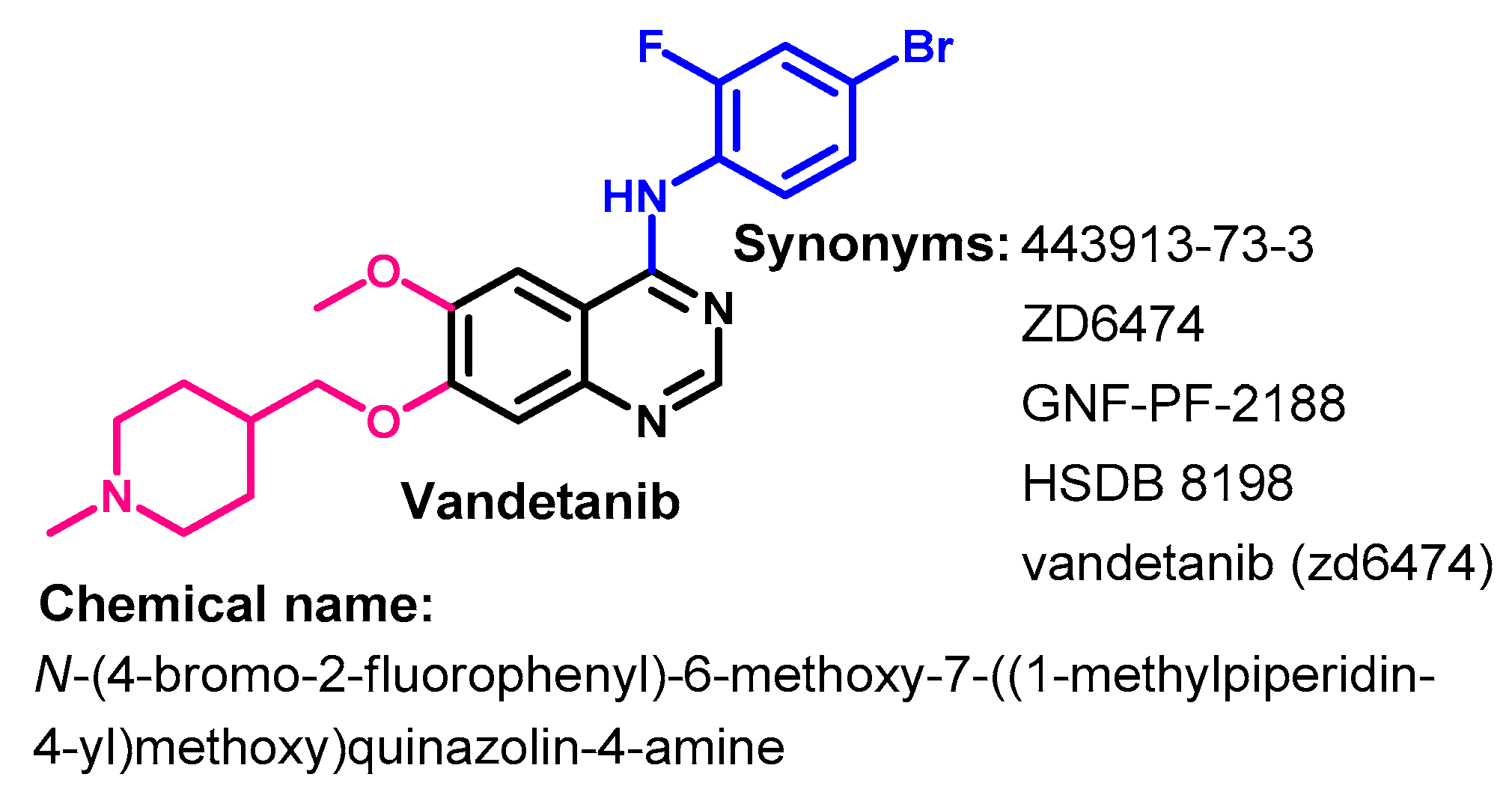
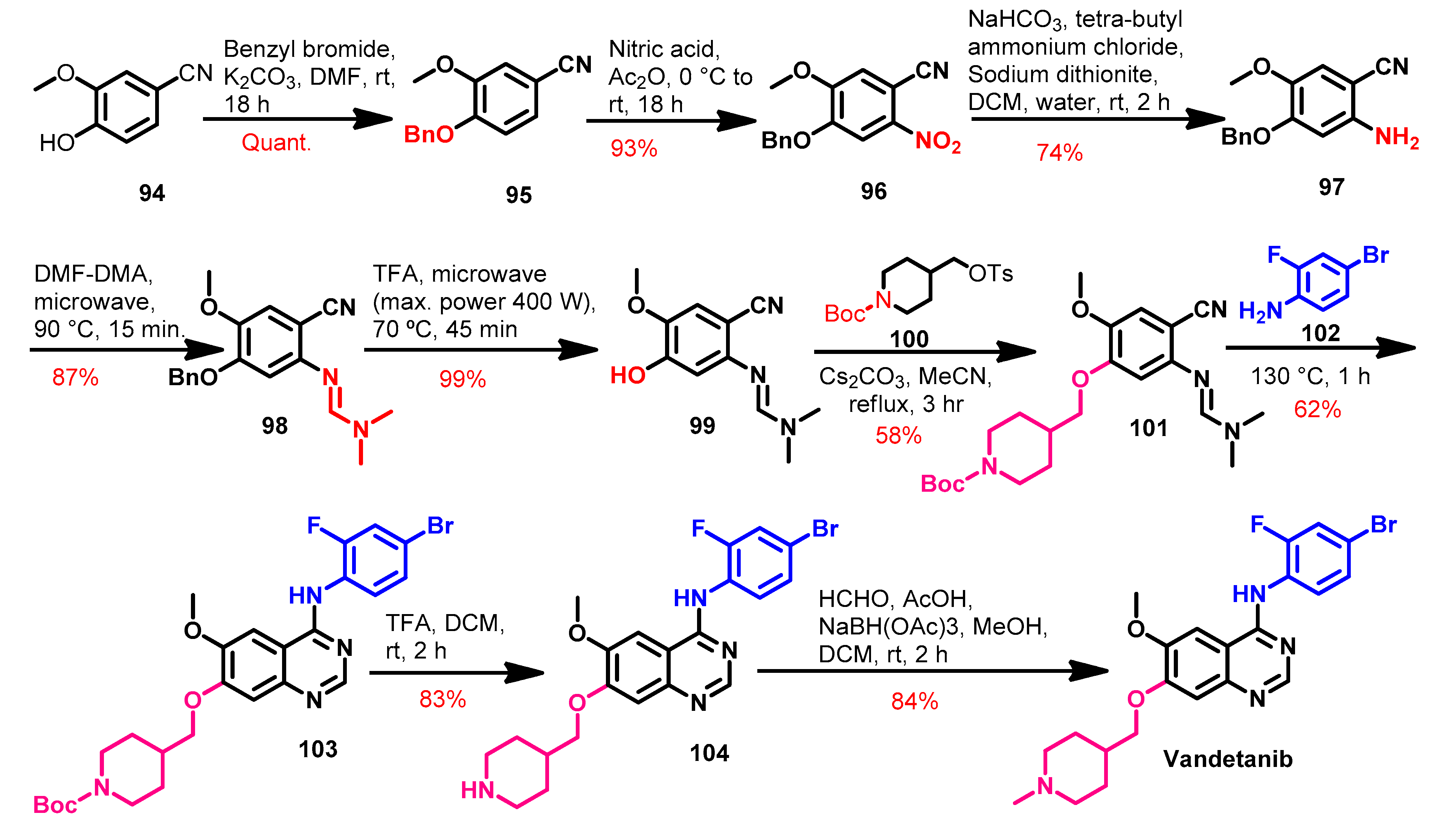
1.2.14. Vandetanib
Approval History
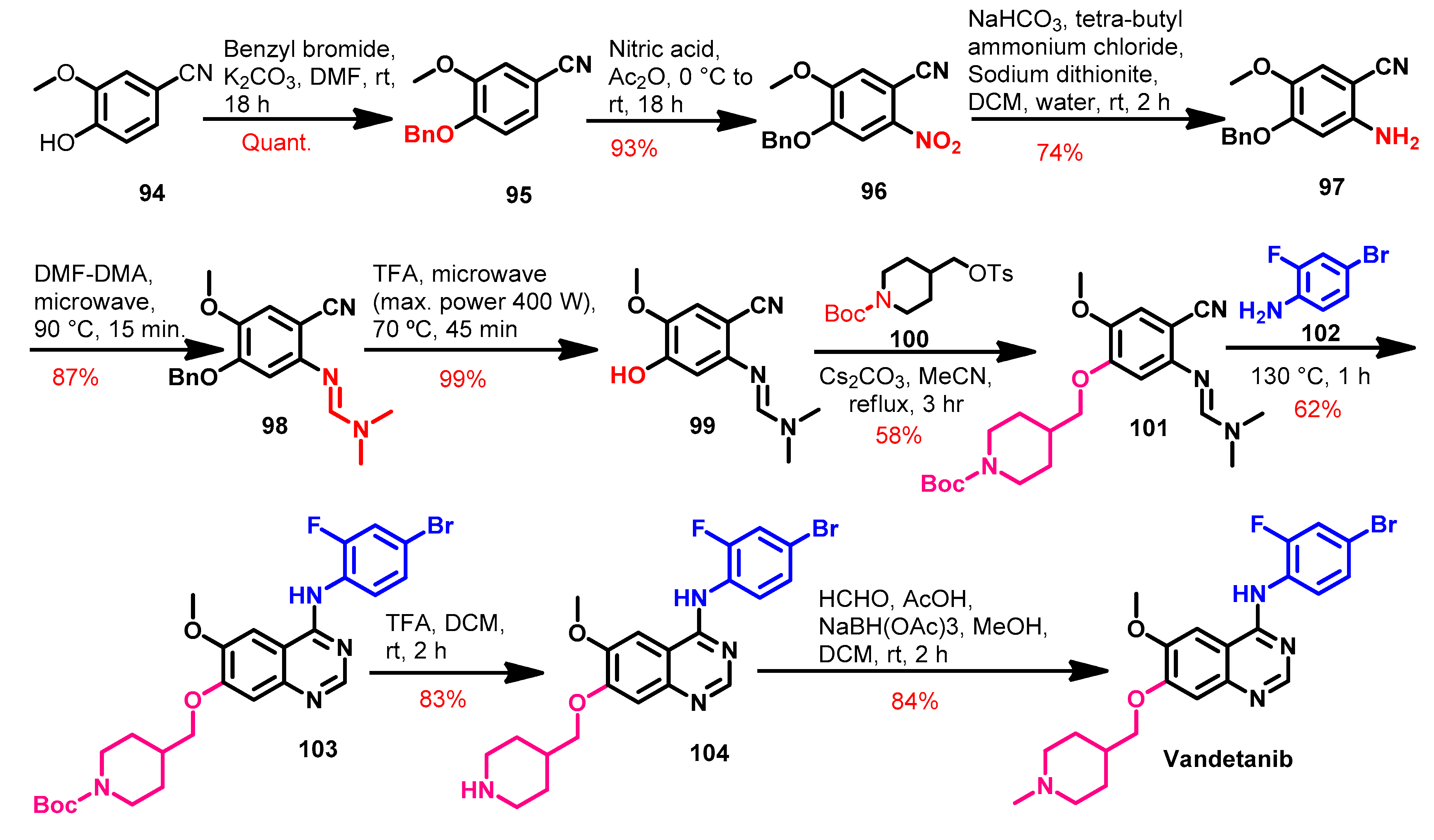
Synthesis
Target Kinases
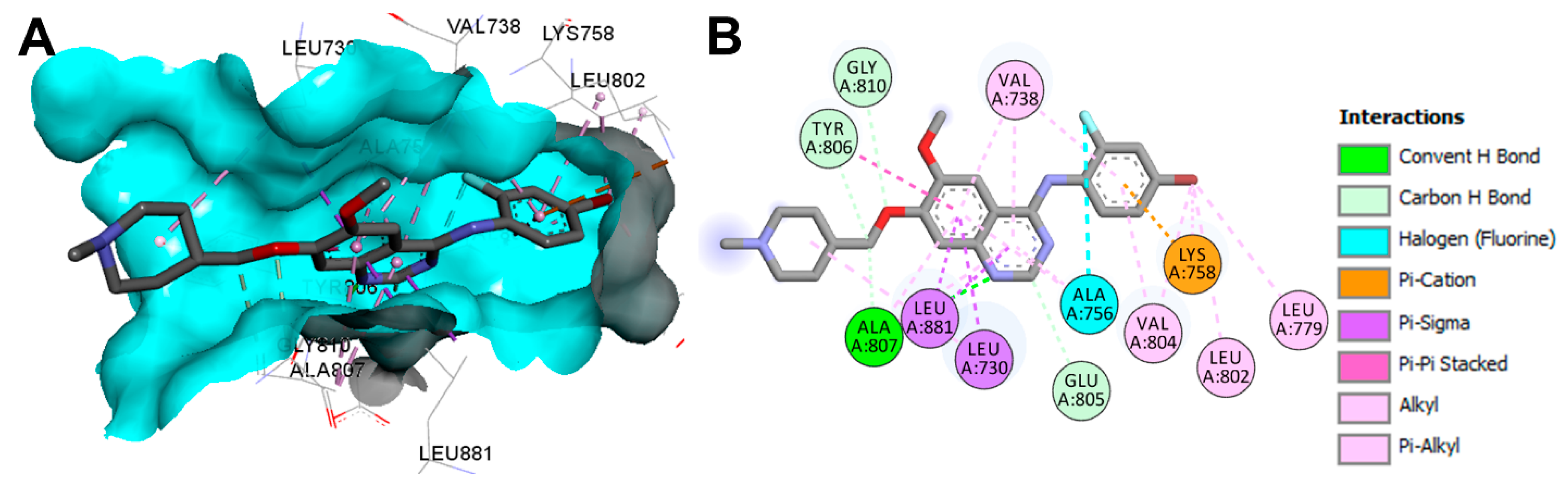
Crystal Structures and Binding Interactions
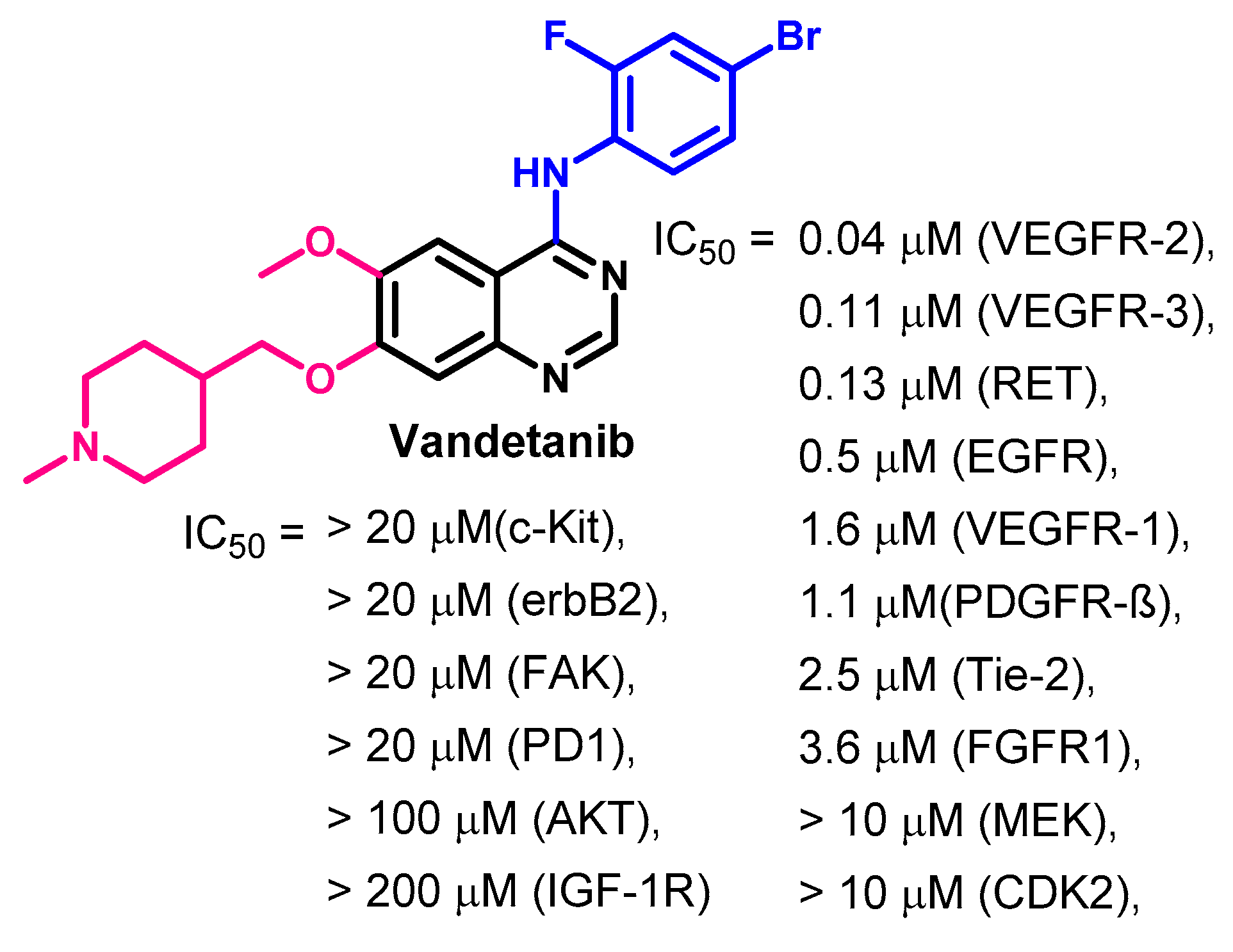
Biological Activity
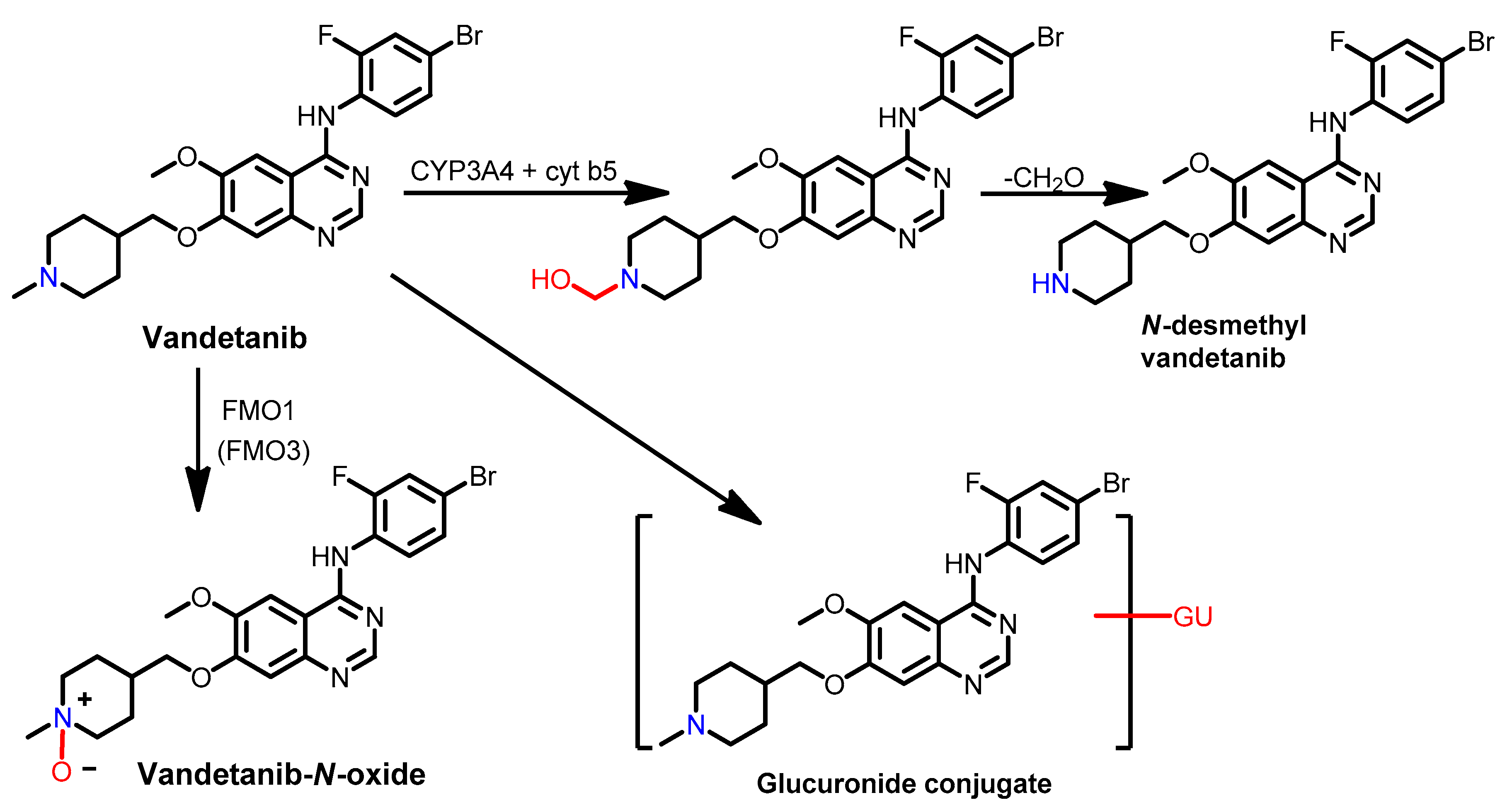
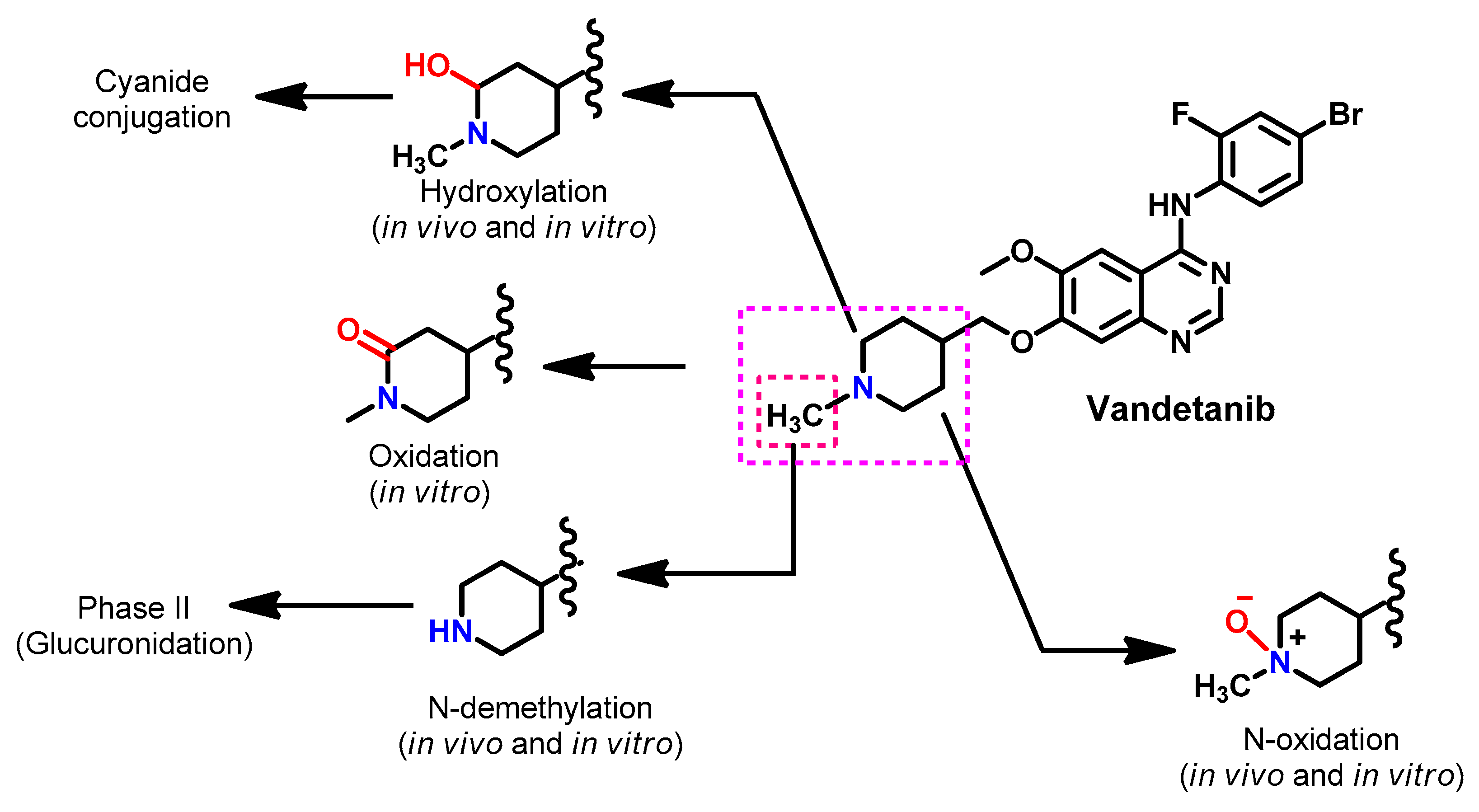
Metabolism
2. Conclusions
3. Perspective
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 21–26. [Google Scholar] [CrossRef]
- Lee, N.Y.; Hazlett, T.L.; Koland, J.G. Structure and dynamics of the epidermal growth factor receptor C-terminal phosphorylation domain. Protein Sci. 2006, 15, 1142–1152. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Fang, W.; Mu, L.; Tang, Y.; Gao, L.; Ren, S.; Cao, D.; Zhou, L.; Zhang, A.; Liu, D.; et al. Overexpression of wildtype EGFR is tumorigenic and denotes a therapeutic target in non-small cell lung cancer. Oncotarget 2016, 7, 3884–3896. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Vallbohmer, D.; Lenz, H.-J. Epidermal growth factor receptor as a target for chemotherapy. Clin. Colorectal Cancer 2005, 5 (Suppl. 1), S19–S27. [Google Scholar] [CrossRef]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef]
- Singh, D.; Attri, B.K.; Gill, R.K.; Bariwal, J. Review on EGFR Inhibitors: Critical Updates. Mini Rev. Med. Chem. 2016, 16, 1134–1166. [Google Scholar] [CrossRef]
- Soltan, O.M.; Shoman, M.E.; Abdel-Aziz, S.A.; Narumi, A.; Konno, H.; Abdel-Aziz, M. Molecular hybrids: A five-year survey on structures of multiple targeted hybrids of protein kinase inhibitors for cancer therapy. Eur. J. Med. Chem. 2021, 225, 113768. [Google Scholar] [CrossRef] [PubMed]
- Tay, R.Y.; Wong, R.; Hawkes, E.A. Treatment of metastatic colorectal cancer: Focus on panitumumab. Cancer Manag. Res. 2015, 7, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesnelle, K.M.; Boehm, A.L.; Grandis, J.R. STAT-mediated EGFR signaling in cancer. J. Cell. Biochem. 2007, 102, 311–319. [Google Scholar] [CrossRef]
- Hunter, T. The genesis of tyrosine phosphorylation. Cold Spring Harb. Perspect. Biol. 2014, 6, a020644. [Google Scholar] [CrossRef] [Green Version]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Fernandez, M.L.; Clarke, D.T.; Roberts, S.K.; Zanetti-Domingues, L.C.; Gervasio, F.L. Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and Its Relevance to Non-Small Cell Lung Cancer. Cells 2019, 8, 316. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Xie, L.; Bourne, P.E. Structural Insights into Characterizing Binding Sites in Epidermal Growth Factor Receptor Kinase Mutants. J. Chem. Inf. Model. 2019, 59, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, S.; Kukimoto-Niino, M.; Parker, L.; Handa, N.; Terada, T.; Fujimoto, T.; Terazawa, Y.; Wakiyama, M.; Sato, M.; Sano, S.; et al. Structural basis for the altered drug sensitivities of non-small cell lung cancer-associated mutants of human epidermal growth factor receptor. Oncogene 2013, 32, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassault Systems BIOVIA. Discovery Studio Visualizer, v16.1.0.15350; Dassault Systems: San Diego, CA, USA, 2016. [Google Scholar]
- Ghosh, S.; Liu, X.P.; Zheng, Y.; Uckun, F.M. Rational design of potent and selective EGFR tyrosine kinase inhibitors as anticancer agents. Curr. Cancer Drug Targets 2001, 1, 129–140. [Google Scholar] [CrossRef]
- Mohamed, F.A.M.; Gomaa, H.A.M.; Hendawy, O.M.; Ali, A.T.; Farghaly, H.S.; Gouda, A.M.; Abdelazeem, A.H.; Abdelrahman, M.H.; Trembleau, L.; Youssif, B.G.M. Design, synthesis, and biological evaluation of novel EGFR inhibitors containing 5-chloro-3-hydroxymethyl-indole-2-carboxamide scaffold with apoptotic antiproliferative activity. Bioorg. Chem. 2021, 112, 104960. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Gouda, A.M.; Abou-Ghadir, O.F.; Salem, O.I.A.; Ali, A.T.; Farghaly, H.S.; Abdelrahman, M.H.; Trembleau, L.; Abdu-Allah, H.H.M.; Youssif, B.G.M. Design and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAF(V600E) dual inhibitors. Bioorg. Chem. 2020, 104, 104260. [Google Scholar] [CrossRef]
- Johnson, J.R.; Bross, P.; Cohen, M.; Rothmann, M.; Chen, G.; Zajicek, A.; Gobburu, J.; Rahman, A.; Staten, A.; Pazdur, R. Approval summary: Imatinib mesylate capsules for treatment of adult patients with newly diagnosed philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1972–1979. [Google Scholar]
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; Pazdur, R. FDA drug approval summary: Gefitinib (ZD1839) (Iressa) tablets. Oncologist 2003, 8, 303–306. [Google Scholar] [CrossRef]
- Cohen, M.H.; Johnson, J.R.; Chen, Y.-F.; Sridhara, R.; Pazdur, R. FDA drug approval summary: Erlotinib (Tarceva) tablets. Oncologist 2005, 10, 461–466. [Google Scholar] [CrossRef]
- Ryan, Q.; Ibrahim, A.; Cohen, M.H.; Johnson, J.; Ko, C.; Sridhara, R.; Justice, R.; Pazdur, R. FDA drug approval summary: Lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 2008, 13, 1114–1119. [Google Scholar] [CrossRef]
- Akher, F.B.; Farrokhzadeh, A.; Soliman, M.E.S. Covalent vs. Non-Covalent Inhibition: Tackling Drug Resistance in EGFR—A Thorough Dynamic Perspective. Chem. Biodivers. 2019, 16, e1800518. [Google Scholar] [CrossRef]
- Hossam, M.; Lasheen, D.S.; Abouzid, K.A.M. Covalent EGFR Inhibitors: Binding Mechanisms, Synthetic Approaches, and Clinical Profiles. Arch. Pharm. (Weinh.) 2016, 349, 573–593. [Google Scholar] [CrossRef] [PubMed]
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748. [Google Scholar] [CrossRef] [PubMed]
- You, K.S.; Yi, Y.W.; Cho, J.; Park, J.-S.; Seong, Y.-S. Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 589. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.R.; Maity, A. Cellular responses to EGFR inhibitors and their relevance to cancer therapy. Cancer Lett. 2007, 254, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Stasi, I.; Cappuzzo, F. Second generation tyrosine kinase inhibitors for the treatment of metastatic non-small-cell lung cancer. Transl. Respir. Med. 2014, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Nagasaka, M.; Zhu, V.W.; Lim, S.M.; Greco, M.; Wu, F.; Ou, S.-H.I. Beyond Osimertinib: The Development of Third-Generation EGFR Tyrosine Kinase Inhibitors for Advanced EGFR+ NSCLC. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 740–763. [Google Scholar] [CrossRef]
- Zhang, C.; Leighl, N.B.; Wu, Y.-L.; Zhong, W.-Z. Emerging therapies for non-small cell lung cancer. J. Hematol. Oncol. 2019, 12, 45. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, S.K.; Biswal, B.K. Allosteric mutant-selective fourth-generation EGFR inhibitors as an efficient combination therapeutic in the treatment of non-small cell lung carcinoma. Drug Discov. Today 2021, 26, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef]
- Du, X.; Yang, B.; An, Q.; Assaraf, Y.G.; Cao, X.; Xia, J. Acquired resistance to third-generation EGFR-TKIs and emerging next-generation EGFR inhibitors. Innovations 2021, 2, 100103. [Google Scholar] [CrossRef]
- Piper-Vallillo, A.J.; Sequist, L.V.; Piotrowska, Z. Emerging Treatment Paradigms for EGFR-Mutant Lung Cancers Progressing on Osimertinib: A Review. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 2926–2936. [Google Scholar] [CrossRef]
- Dungo, R.T.; Keating, G.M. Afatinib: First global approval. Drugs 2013, 73, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Santos, E.S.; Hart, L. Advanced Squamous Cell Carcinoma of the Lung: Current Treatment Approaches and the Role of Afatinib. Onco. Targets. Ther. 2020, 13, 9305–9321. [Google Scholar] [CrossRef]
- Ricciuti, B.; Baglivo, S.; de Giglio, A.; Chiari, R. Afatinib in the first-line treatment of patients with non-small cell lung cancer: Clinical evidence and experience. Ther. Adv. Respir. Dis. 2018, 12, 1753466618808659. [Google Scholar] [CrossRef]
- Kovacevic, T.; Mesic, M.; Avdagic, A.; Zegarac, M. An alternative synthesis of the non-small cell lung carcinoma drug afatinib. Tetrahedron Lett. 2018, 59, 4180–4182. [Google Scholar] [CrossRef]
- Hirano, T.; Yasuda, H.; Tani, T.; Hamamoto, J.; Oashi, A.; Ishioka, K.; Arai, D.; Nukaga, S.; Miyawaki, M.; Kawada, I.; et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non-small-cell lung cancer. Oncotarget 2015, 6, 38789–38803. [Google Scholar] [CrossRef] [Green Version]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef]
- Summers, Y.; Graham, D.M. Afatinib, an Irreversible ErbB Family Blocker for the Treatment of Epidermal Growth Factor Receptor Mutation-Positive Non-Small Cell Lung Cancer. Eur. J. Oncol. Pharm. 2019, 2, e18. Available online: https://journals.lww.com/ejop/Fulltext/2019/09000/Afatinib,_an_irreversible_ErbB_family_blocker_for.4.aspx (accessed on 8 September 2021). [CrossRef]
- Fry, D.W. Mechanism of action of erbB tyrosine kinase inhibitors. Exp. Cell Res. 2003, 284, 131–139. [Google Scholar] [CrossRef]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef] [Green Version]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Xu, H.; Zhou, Z.; Tian, Y.; Cao, X.; Cheng, G.; Liu, Q. Blocking of the EGFR-STAT3 signaling pathway through afatinib treatment inhibited the intrahepatic cholangiocarcinoma. Exp. Ther. Med. 2018, 15, 4995–5000. [Google Scholar] [CrossRef] [PubMed]
- Modjtahedi, H.; Cho, B.C.; Michel, M.C.; Solca, F. A comprehensive review of the preclinical efficacy profile of the ErbB family blocker afatinib in cancer. Naunyn. Schmiedebergs. Arch. Pharmacol. 2014, 387, 505–521. [Google Scholar] [CrossRef] [Green Version]
- Slobbe, P.; Windhorst, A.D.; Stigter-van Walsum, M.; Schuit, R.C.; Smit, E.F.; Niessen, H.G.; Solca, F.; Stehle, G.; van Dongen, G.A.M.S.; Poot, A.J. Development of [18F]afatinib as new TKI-PET tracer for EGFR positive tumors. Nucl. Med. Biol. 2014, 41, 749–757. [Google Scholar] [CrossRef]
- Stopfer, P.; Marzin, K.; Narjes, H.; Gansser, D.; Shahidi, M.; Uttereuther-Fischer, M.; Ebner, T. Afatinib pharmacokinetics and metabolism after oral administration to healthy male volunteers. Cancer Chemother. Pharmacol. 2012, 69, 1051–1061. [Google Scholar] [CrossRef]
- Wind, S.; Schnell, D.; Ebner, T.; Freiwald, M.; Stopfer, P. Clinical Pharmacokinetics and Pharmacodynamics of Afatinib. Clin. Pharmacokinet. 2017, 56, 235–250. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef]
- Liang, X.; Yang, Q.; Wu, P.; He, C.; Yin, L.; Xu, F.; Yin, Z.; Yue, G.; Zou, Y.; Li, L.; et al. The synthesis review of the approved tyrosine kinase inhibitors for anticancer therapy in 2015-2020. Bioorg. Chem. 2021, 113, 105011. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.K.; Green, A.R. Me-too pharmaceutical products: History, definitions, examples, and relevance to drug shortages and essential medicines lists. Br. J. Clin. Pharmacol. 2020, 86, 2114–2122. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Camidge, D.R.; Yang, C.-T.; Zhou, J.; Guo, R.; Chiu, C.-H.; Chang, G.-C.; Shiah, H.-S.; Chen, Y.; Wang, C.-C.; et al. Safety, Efficacy, and Pharmacokinetics of Almonertinib (HS-10296) in Pretreated Patients With EGFR-Mutated Advanced NSCLC: A Multicenter, Open-label, Phase 1 Trial. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020, 15, 1907–1918. [Google Scholar] [CrossRef]
- Wu, C.-P.; Hung, T.-H.; Lusvarghi, S.; Chu, Y.-H.; Hsiao, S.-H.; Huang, Y.-H.; Chang, Y.-T.; Ambudkar, S.V. The third-generation EGFR inhibitor almonertinib (HS-10296) resensitizes ABCB1-overexpressing multidrug-resistant cancer cells to chemotherapeutic drugs. Biochem. Pharmacol. 2021, 188, 114516. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Xie, L.; Liu, W.; Zhang, L.; Zhou, S.; Wang, L.; Chen, J.; Li, H.; Zhao, Y.; Zhu, B.; et al. Absorption, metabolism, excretion, and safety of [(14)C]almonertinib in healthy Chinese subjects. Ann. Transl. Med. 2021, 9, 867. [Google Scholar] [CrossRef]
- Markham, A. Brigatinib: First Global Approval. Drugs 2017, 77, 1131–1135. [Google Scholar] [CrossRef]
- Huang, W.-S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2016, 59, 4948–4964. [Google Scholar] [CrossRef]
- Zhang, S.; Anjum, R.; Squillace, R.; Nadworny, S.; Zhou, T.; Keats, J.; Ning, Y.; Wardwell, S.D.; Miller, D.; Song, Y.; et al. The Potent ALK Inhibitor Brigatinib (AP26113) Overcomes Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in Preclinical Models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5527–5538. [Google Scholar] [CrossRef] [Green Version]
- Floch, N.; Finlay, M.R.V.; Bianco, A.; Bickerton, S.; Colclough, N.; Cross, D.A.; Cuomo, E.M.; Guerot, C.M.; Hargreaves, D.; Martin, M.J.; et al. Abstract 4451: Evaluation of the therapeutic potential of phosphine oxide pyrazole inhibitors in tumors harboring EGFR C797S mutation. Cancer Res. 2019, 79, 4451. [Google Scholar] [CrossRef]
- Bedi, S.; Khan, S.A.; AbuKhader, M.M.; Alam, P.; Siddiqui, N.A.; Husain, A. A comprehensive review on Brigatinib—A wonder drug for targeted cancer therapy in non-small cell lung cancer. Saudi Pharm. J. SPJ Off. Publ. Saudi Pharm. Soc. 2018, 26, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Kadi, A.A.; Attwa, M.W.; Darwish, H.W. LC-ESI-MS/MS reveals the formation of reactive intermediates in brigatinib metabolism: Elucidation of bioactivation pathways. RSC Adv. 2018, 8, 1182–1190. [Google Scholar] [CrossRef] [Green Version]
- Shirley, M. Dacomitinib: First Global Approval. Drugs 2018, 78, 1947–1953. [Google Scholar] [CrossRef]
- Flick, A.C.; Leverett, C.A.; Ding, H.X.; McInturff, E.; Fink, S.J.; Helal, C.J.; DeForest, J.C.; Morse, P.D.; Mahapatra, S.; O’Donnell, C.J. Synthetic Approaches to New Drugs Approved during 2018. J. Med. Chem. 2020, 63, 10652–10704. [Google Scholar] [CrossRef] [PubMed]
- Lavacchi, D.; Mazzoni, F.; Giaccone, G. Clinical evaluation of dacomitinib for the treatment of metastatic non-small cell lung cancer (NSCLC): Current perspectives. Drug Des. Devel. Ther. 2019, 13, 3187–3198. [Google Scholar] [CrossRef] [Green Version]
- Gajiwala, K.S.; Feng, J.; Ferre, R.; Ryan, K.; Brodsky, O.; Weinrich, S.; Kath, J.C.; Stewart, A. Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure 2013, 21, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ather, F.; Hamidi, H.; Fejzo, M.S.; Letrent, S.; Finn, R.S.; Kabbinavar, F.; Head, C.; Wong, S.G. Dacomitinib, an irreversible Pan-ErbB inhibitor significantly abrogates growth in head and neck cancer models that exhibit low response to cetuximab. PLoS ONE 2013, 8, e56112. [Google Scholar] [CrossRef] [Green Version]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S. Characterization of reactive intermediates formation in dacomitinib metabolism and bioactivation pathways elucidation by LC-MS/MS: In vitro phase I metabolic investigation. RSC Adv. 2018, 8, 38733–38744. [Google Scholar] [CrossRef] [Green Version]
- Kelley, R.K.; Ko, A.H. Erlotinib in the treatment of advanced pancreatic cancer. Biologics 2008, 2, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Barghi, L.; Aghanejad, A.; Valizadeh, H.; Barar, J.; Asgari, D. Modified synthesis of erlotinib hydrochloride. Adv. Pharm. Bull. 2012, 2, 119–122. [Google Scholar] [CrossRef]
- Chandregowda, V.; Rao, G.V.; Reddy, G.C. Improved Synthesis of Gefitinib and Erlotinib Hydrochloride- Anticancer Agents. Synth. Commun. 2007, 37, 3409–3415. [Google Scholar] [CrossRef]
- Knesl, P.; Röseling, D.; Jordis, U. Improved synthesis of substituted 6,7-dihydroxy-4-quinazolineamines: Tandutinib, erlotinib and gefitinib. Molecules 2006, 11, 286–297. [Google Scholar] [CrossRef] [Green Version]
- Asgari, D.; Aghanejad, A.; Mojarrad, J.S. An improved convergent approach for synthesis of erlotinib, a tyrosine kinase inhibitor, via a ring closure reaction of phenyl benzamidine intermediate. Bull. Korean Chem. Soc. 2011, 32, 909–914. [Google Scholar] [CrossRef] [Green Version]
- Kancha, R.K.; von Bubnoff, N.; Peschel, C.; Duyster, J. Functional analysis of epidermal growth factor receptor (EGFR) mutations and potential implications for EGFR targeted therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 460–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, D.; Yokota, K.; Gouda, M.; Narumi, Y.; Ohmoto, H.; Nishiwaki, E.; Akita, K.; Kirii, Y. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes Cells 2013, 18, 110–122. [Google Scholar] [CrossRef]
- Li, Z.; Xu, M.; Xing, S.; Ho, W.T.; Ishii, T.; Li, Q.; Fu, X.; Zhao, Z.J. Erlotinib effectively inhibits JAK2V617F activity and polycythemia vera cell growth. J. Biol. Chem. 2007, 282, 3428–3432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Liu, Y.; Lemmon, M.A.; Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 2012, 448, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Bart, A.G.; Scott, E.E. Structures of human cytochrome P450 1A1 with bergamottin and erlotinib reveal active-site modifications for binding of diverse ligands. J. Biol. Chem. 2018, 293, 19201–19210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, J.; Johnson, K.A.; Miao, Z.; Rakhit, A.; Pantze, M.P.; Hamilton, M.; Lum, B.L.; Prakash, C. Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab. Dispos. 2006, 34, 420–426. [Google Scholar] [CrossRef] [Green Version]
- Kazandjian, D.; Blumenthal, G.M.; Yuan, W.; He, K.; Keegan, P.; Pazdur, R. FDA Approval of Gefitinib for the Treatment of Patients with Metastatic EGFR Mutation-Positive Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1307–1312. [Google Scholar] [CrossRef] [Green Version]
- Chandregowda, V.; Rao, G.V.; Reddy, G.C. Convergent Approach for Commercial Synthesis of Gefitinib and Erlotinib. Org. Process Res. Dev. 2007, 11, 813–816. [Google Scholar] [CrossRef]
- Kang, S.K.; Lee, S.W.; Woo, D.; Sim, J.; Suh, Y.-G. Practical and efficient synthesis of gefitinib through selective O-alkylation: A novel concept for a transient protection group. Synth. Commun. 2017, 47, 1990–1998. [Google Scholar] [CrossRef]
- Brehmer, D.; Greff, Z.; Godl, K.; Blencke, S.; Kurtenbach, A.; Weber, M.; Müller, S.; Klebl, B.; Cotten, M.; Kéri, G.; et al. Cellular targets of gefitinib. Cancer Res. 2005, 65, 379–382. [Google Scholar]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohbayashi, N.; Murayama, K.; Kato-Murayama, M.; Kukimoto-Niino, M.; Uejima, T.; Matsuda, T.; Ohsawa, N.; Yokoyoma, S.; Nojima, H.; Shirouzu, M. Structural Basis for the Inhibition of Cyclin G-Associated Kinase by Gefitinib. ChemistryOpen 2018, 7, 721–727. [Google Scholar] [CrossRef]
- Wang, M.; Chang, A.Y.C. Molecular mechanism of action and potential biomarkers of growth inhibition of synergistic combination of afatinib and dasatinib against gefitinib-resistant non-small cell lung cancer cells. Oncotarget 2018, 9, 16533–16546. [Google Scholar] [CrossRef] [Green Version]
- Festuccia, C.; Muzi, P.; Millimaggi, D.; Biordi, L.; Gravina, G.L.; Speca, S.; Angelucci, A.; Dolo, V.; Vicentini, C.; Bologna, M. Molecular aspects of gefitinib antiproliferative and pro-apoptotic effects in PTEN-positive and PTEN-negative prostate cancer cell lines. Endocr. Relat. Cancer 2005, 12, 983–998. [Google Scholar] [CrossRef]
- Ono, M.; Hirata, A.; Kometani, T.; Miyagawa, M.; Ueda, S.; Kinoshita, H.; Fujii, T.; Kuwano, M. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol. Cancer Ther. 2004, 3, 465–472. [Google Scholar]
- Li, J.; Zhao, M.; He, P.; Hidalgo, M.; Baker, S.D. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 3731–3737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfieri, R.R.; Galetti, M.; Tramonti, S.; Andreoli, R.; Mozzoni, P.; Cavazzoni, A.; Bonelli, M.; Fumarola, C.; la Monica, S.; Galvani, E.; et al. Metabolism of the EGFR tyrosin kinase inhibitor gefitinib by cytochrome P450 1A1 enzyme in EGFR-wild type non small cell lung cancer cell lines. Mol. Cancer 2011, 10, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mckillop, D.; McCormick, A.D.; Millar, A.; Miles, G.S.; Phillips, P.J.; Hutchison, M. Cytochrome P450-dependent metabolism of gefitinib. Xenobiotica 2005, 35, 39–50. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, J.; Zhou, S.; Yu, L.; Han, F.; Ling, R.; Ling, J. Tentative identification of gefitinib metabolites in non-small-cell lung cancer patient plasma using ultra-performance liquid chromatography coupled with triple quadrupole time-of-flight mass spectrometry. PLoS ONE 2020, 15, e0236523. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.X.; Liu, K.K.-C.; Sakya, S.M.; Flick, A.C.; O’Donnell, C.J. Synthetic approaches to the 2011 new drugs. Bioorg. Med. Chem. 2013, 21, 2795–2825. [Google Scholar] [CrossRef]
- Mao, L.; Sun, G.; Zhao, J.; Xu, G.; Yuan, M.; Li, Y.-M. Design synthesis and antitumor activity of icotinib derivatives. Bioorg. Chem. 2020, 105, 104421. [Google Scholar] [CrossRef]
- Lu, X.; Wang, C.; Li, X.; Gu, P.; Jia, L.; Zhang, L. Synthesis and preliminary evaluation of (18)F-icotinib for EGFR-targeted PET imaging of lung cancer. Bioorg. Med. Chem. 2019, 27, 545–551. [Google Scholar] [CrossRef]
- Tan, F.; Shen, X.; Wang, D.; Xie, G.; Zhang, X.; Ding, L.; Hu, Y.; He, W.; Wang, Y.; Wang, Y. Icotinib (BPI-2009H), a novel EGFR tyrosine kinase inhibitor, displays potent efficacy in preclinical studies. Lung Cancer 2012, 76, 177–182. [Google Scholar] [CrossRef]
- Mao, L.-F.; Wang, Y.-W.; Zhao, J.; Xu, G.-Q.; Yao, X.-J.; Li, Y.-M. Discovery of Icotinib-1,2,3-Triazole Derivatives as IDO1 Inhibitors. Front. Pharmacol. 2020, 11, 579024. [Google Scholar] [CrossRef]
- Wang, D.-S.; Patel, A.; Shukla, S.; Zhang, Y.-K.; Wang, Y.-J.; Kathawala, R.J.; Robey, R.W.; Zhang, L.; Yang, D.-H.; Talele, T.T.; et al. Icotinib antagonizes ABCG2-mediated multidrug resistance, but not the pemetrexed resistance mediated by thymidylate synthase and ABCG2. Oncotarget 2014, 5, 4529–4542. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Yao, Y.; Zhou, J.; Zhao, Q. Effects of icotinib, a novel epidermal growth factor receptor tyrosine kinase inhibitor, in EGFR-mutated non-small cell lung cancer. Oncol. Rep. 2012, 27, 2066–2072. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, D.; Zheng, X.; Zhao, Q.; Jiang, J.; Hu, P. Relative contributions of the major human CYP450 to the metabolism of icotinib and its implication in prediction of drug–drug interaction between icotinib and CYP3A4 inhibitors/inducers using physiologically based pharmacokinetic modeling. Expert Opin. Drug Metab. Toxicol. 2015, 11, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, K.; Ma, L.; Li, Z.; Wang, J.; Zhang, Y.; Lu, C.; Zhu, M.; Zhuang, X. Metabolic Pathway of Icotinib In Vitro: The Differential Roles of CYP3A4, CYP3A5, and CYP1A2 on Potential Pharmacokinetic Drug-Drug Interaction. J. Pharm. Sci. 2018, 107, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Opdam, F.L.; Guchelaar, H.-J.; Beijnen, J.H.; Schellens, J.H.M. Lapatinib for advanced or metastatic breast cancer. Oncologist 2012, 17, 536–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, G.; Guo, J.; McClure, M.; Mitchell, M.; Salaun, M.-C.; Whitehead, A. Synthesis of Lapatinib via direct regioselective arylation of furfural. Tetrahedron Lett. 2014, 55, 6007–6010. [Google Scholar] [CrossRef]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Knight, W.B.; Mullin, R.J.; et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001, 1, 85–94. [Google Scholar] [PubMed]
- Kim, H.-P.; Yoon, Y.-K.; Kim, J.-W.; Han, S.-W.; Hur, H.-S.; Park, J.; Lee, J.-H.; Oh, D.-Y.; Im, S.-A.; Bang, Y.-J.; et al. Lapatinib, a dual EGFR and HER2 tyrosine kinase inhibitor, downregulates thymidylate synthase by inhibiting the nuclear translocation of EGFR and HER2. PLoS ONE 2009, 4, e5933. [Google Scholar] [CrossRef] [Green Version]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Tarrant, M.K.; Choi, S.H.; Sathyamurthy, A.; Bose, R.; Banjade, S.; Pal, A.; Bornmann, W.G.; Lemmon, M.A.; Cole, P.A.; et al. Mechanism of activation and inhibition of the HER4/ErbB4 kinase. Structure 2008, 16, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Konecny, G.E.; Pegram, M.D.; Venkatesan, N.; Finn, R.; Yang, G.; Rahmeh, M.; Untch, M.; Rusnak, D.W.; Spehar, G.; Mullin, R.J.; et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006, 66, 1630–1639. [Google Scholar] [CrossRef] [Green Version]
- Wainberg, Z.A.; Anghel, A.; Desai, A.J.; Ayala, R.; Luo, T.; Safran, B.; Fejzo, M.S.; Hecht, J.R.; Slamon, D.J.; Finn, R.S. Lapatinib, a dual EGFR and HER2 kinase inhibitor, selectively inhibits HER2-amplified human gastric cancer cells and is synergistic with trastuzumab in vitro and in vivo. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 1509–1519. [Google Scholar] [CrossRef] [Green Version]
- Collins, D.M.; Conlon, N.T.; Kannan, S.; Verma, C.S.; Eli, L.D.; Lalani, A.S.; Crown, J. Preclinical Characteristics of the Irreversible Pan-HER Kinase Inhibitor Neratinib Compared with Lapatinib: Implications for the Treatment of HER2-Positive and HER2-Mutated Breast Cancer. Cancers 2019, 11, 737. [Google Scholar] [CrossRef] [Green Version]
- Castellino, S.; O’Mara, M.; Koch, K.; Borts, D.J.; Bowers, G.D.; MacLauchlin, C. Human metabolism of lapatinib, a dual kinase inhibitor: Implications for hepatotoxicity. Drug Metab. Dispos. 2012, 40, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Nunes, P.; Zhang, Z.; Zhang, C.; Carvalho, I.; Benard, F.; Lin, K.-S. Facile Synthesis of 18F-Labeled Lapatinib for Imaging with Positron Emission Tomography. J. Nucl. Med. 2018, 59, 1055. Available online: http://jnm.snmjournals.org/content/59/supplement_1/1055.abstract (accessed on 8 September 2021).
- Saleem, A.; Searle, G.E.; Kenny, L.M.; Huiban, M.; Kozlowski, K.; Waldman, A.D.; Woodley, L.; Palmieri, C.; Lowdell, C.; Kaneko, T.; et al. Lapatinib access into normal brain and brain metastases in patients with Her-2 overexpressing breast cancer. Ejnmmi Res. 2015, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeks, E.D. Neratinib: First Global Approval. Drugs 2017, 77, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Jackisch, C.; Barcenas, C.H.; Bartsch, R.; di Palma, J.; Glück, S.; Harbeck, N.; Macedo, G.; O’Shaughnessy, J.; Pistilli, B.; Ruiz-Borrego, M.; et al. Optimal Strategies for Successful Initiation of Neratinib in Patients with HER2-Positive Breast Cancer. Clin. Breast Cancer 2021, 21, e575–e583. [Google Scholar] [CrossRef]
- Tsou, H.-R.; Overbeek-Klumpers, E.G.; Hallett, W.A.; Reich, M.F.; Floyd, M.B.; Johnson, B.D.; Michalak, R.S.; Nilakantan, R.; Discafani, C.; Golas, J.; et al. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J. Med. Chem. 2005, 48, 1107–1131. [Google Scholar] [CrossRef]
- Gu, N.; Yang, J.; Wang, P.; Li, L.; Chen, Y.; Ji, M. The Wittig–Horner reaction for the synthesis of neratinib. Res. Chem. Intermed. 2013, 39, 3105–3110. [Google Scholar] [CrossRef]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [CrossRef] [Green Version]
- Kulukian, A.; Lee, P.; Taylor, J.; Rosler, R.; de Vries, P.; Watson, D.; Forero-Torres, A.; Peterson, S. Preclinical Activity of HER2-Selective Tyrosine Kinase Inhibitor Tucatinib as a Single Agent or in Combination with Trastuzumab or Docetaxel in Solid Tumor Models. Mol. Cancer Ther. 2020, 19, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Sogabe, S.; Kawakita, Y.; Igaki, S.; Iwata, H.; Miki, H.; Cary, D.R.; Takagi, T.; Takagi, S.; Ohta, Y.; Ishikawa, T. Structure-Based Approach for the Discovery of Pyrrolo[3,2-d]pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors. ACS Med. Chem. Lett. 2013, 4, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Li, S.; Wu, Y.; Yan, X.; Zhu, Y.-M.; Huang, J.-H.; Chen, Z. Metabolic profiles of neratinib in rat by using ultra-high-performance liquid chromatography coupled with diode array detector and Q-Exactive Orbitrap tandem mass spectrometry, Biomed. Chromatography 2018, 32, e4272. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Olmutinib: First Global Approval. Drugs 2016, 76, 1153–1157. [Google Scholar] [CrossRef]
- Flick, A.C.; Ding, H.X.; Leverett, C.A.; Fink, S.J.; O’Donnell, C.J. Synthetic Approaches to New Drugs Approved During 2016. J. Med. Chem. 2018, 61, 7004–7031. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.S.; Cho, B.C.; Soo, R.A. Next-generation epidermal growth factor receptor tyrosine kinase inhibitors in epidermal growth factor receptor -mutant non-small cell lung cancer. Lung Cancer 2016, 93, 59–68. [Google Scholar] [CrossRef]
- Hu, X.; Tang, S.; Yang, F.; Zheng, P.; Xu, S.; Pan, Q.; Zhu, W. Design, Synthesis, and Antitumor Activity of Olmutinib Derivatives Containing Acrylamide Moiety. Molecules 2021, 26, 3041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Fan, Y.-F.; Cai, C.-Y.; Wang, J.-Q.; Teng, Q.-X.; Lei, Z.-N.; Zeng, L.; Gupta, P.; Chen, Z.-S. Olmutinib (BI1482694/HM61713), a Novel Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor, Reverses ABCG2-Mediated Multidrug Resistance in Cancer Cells. Front. Pharmacol. 2018, 9, 1097. [Google Scholar] [CrossRef] [Green Version]
- Noh, Y.S.; Yoon, S.; Kim, S.R.; Lee, K.-T.; Jang, I.-J. A safety, pharmacokinetic, pharmacogenomic and population pharmacokinetic analysis of the third-generation EGFR TKI, olmutinib (HM61713), after single oral administration in healthy volunteers. Basic Clin. Pharmacol. Toxicol. 2019, 125, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S. Detection and characterization of olmutinib reactive metabolites by LC–MS/MS: Elucidation of bioactivation pathways. J. Sep. Sci. 2020, 43, 708–718. [Google Scholar] [CrossRef]
- Greig, S.L. Osimertinib: First Global Approval. Drugs 2016, 76, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Finlay, M.R.V.; Anderton, M.; Ashton, S.; Ballard, P.; Bethel, P.A.; Box, M.R.; Bradbury, R.H.; Brown, S.J.; Butterworth, S.; Campbell, A.; et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 2014, 57, 8249–8267. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, X.; Wang, F.; Mao, Y.; Wang, H. New and Convergent Synthesis of Osimertinib. J. Heterocycl. Chem. 2017, 54, 2898–2901. [Google Scholar] [CrossRef]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Zhang, X.; Wang, Z.; Zhang, X.; Zhao, L.; Fu, W.; Liang, X.; Zhang, Z.; Wang, Y. SH-1028, an Irreversible Third-Generation EGFR TKI, Overcomes T790M-Mediated Resistance in Non-Small Cell Lung Cancer. Front. Pharmacol. 2021, 12, 983. [Google Scholar] [CrossRef]
- Yosaatmadja, Y.; Silva, S.; Dickson, J.M.; Patterson, A.V.; Smaill, J.B.; Flanagan, J.U.; McKeage, M.J.; Squire, C.J. Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J. Struct. Biol. 2015, 192, 539–544. [Google Scholar] [CrossRef]
- Yan, X.-E.; Ayaz, P.; Zhu, S.-J.; Zhao, P.; Liang, L.; Zhang, C.H.; Wu, Y.-C.; Li, J.-L.; Choi, H.G.; Huang, X.; et al. Structural Basis of AZD9291 Selectivity for EGFR T790M. J. Med. Chem. 2020, 63, 8502–8511. [Google Scholar] [CrossRef] [PubMed]
- Kashima, K.; Kawauchi, H.; Tanimura, H.; Tachibana, Y.; Chiba, T.; Torizawa, T.; Sakamoto, H. CH7233163 Overcomes Osimertinib-Resistant EGFR-Del19/T790M/C797S Mutation. Mol. Cancer Ther. 2020, 19, 2288–2297. [Google Scholar] [CrossRef]
- Gao, H.; Yang, Z.; Yang, X.; Rao, Y. Synthesis and evaluation of osimertinib derivatives as potent EGFR inhibitors. Bioorg. Med. Chem. 2017, 25, 4553–4559. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, P.A.; Cantarini, M.V.; Collier, J.; Frewer, P.; Martin, S.; Pickup, K.; Ballard, P. Metabolic Disposition of Osimertinib in Rats, Dogs, and Humans: Insights into a Drug Designed to Bind Covalently to a Cysteine Residue of Epidermal Growth Factor Receptor. Drug Metab. Dispos. 2016, 44, 1201–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, H.A. Pyrotinib: First Global Approval. Drugs 2018, 78, 1751–1755. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, C.; Wan, H.; Zhang, G.; Feng, J.; Zhang, L.; Chen, X.; Zhong, D.; Lou, L.; Tao, W.; et al. Discovery and development of pyrotinib: A novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor with favorable safety profiles for the treatment of breast cancer. Eur. J. Pharm. Sci. 2017, 110, 51–61. [Google Scholar] [CrossRef]
- Su, B.; Huang, T.; Jin, Y.; Yin, H.; Qiu, H.; Yuan, X. Apatinib exhibits synergistic effect with pyrotinib and reverses acquired pyrotinib resistance in HER2-positive gastric cancer via stem cell factor/c-kit signaling and its downstream pathways. Gastric Cancer 2021, 24, 352–367. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, L.; Zhang, G.; Wan, H.; Yang, C.; Diao, X.; Chen, X.; Zhang, L.; Zhong, D. Metabolic characterization of pyrotinib in humans by ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1033–1034, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Liu, X.-Y.; Ma, S.; Zhang, H.; Yu, S.; Zhang, Y.-F.; Chen, M.-X.; Zhu, X.-Y.; Liu, Y.; Yi, L.; et al. Metabolism and disposition of pyrotinib in healthy male volunteers: Covalent binding with human plasma protein. Acta Pharmacol. Sin. 2019, 40, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Kwapiszewski, R.; Pawlak, S.D.; Adamkiewicz, K. Anti-EGFR Agents: Current Status, Forecasts and Future Directions. Target. Oncol. 2016, 11, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.P.; Moorpark, C.A. Spiro Compounds and Methods of Use. U.S. Patent US 2007/0167470 A1, 19 July 2007. [Google Scholar]
- Hu, X.-S.; Han, X.-H.; Yang, S.; Li, N.; Wang, L.; Song, Y.-Y.; Mu, H.; Shi, Y.-K. Safety, tolerability, and pharmacokinetics of simotinib, a novel specific EGFR tyrosine kinase inhibitor, in patients with advanced non-small cell lung cancer: Results of a phase Ib trial. Cancer Manag. Res. 2019, 11, 4449–4459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Shen, C.; Chen, Y.; Yan, H.; Cheng, Z.; Zhu, Q. Simotinib as a modulator of P-glycoprotein: Substrate, inhibitor, or inducer? Anticancer Drugs. 2016, 27, 300–311. [Google Scholar] [CrossRef]
- Li, N.; Han, X.; Du, P.; Song, Y.; Hu, X.; Yang, S.; Shi, Y. Development and validation of a UPLC-MS/MS assay for the quantification of simotinib in human plasma. Anal. Bioanal. Chem. 2014, 406, 1799–1805. [Google Scholar] [CrossRef]
- Han, X.; Li, N.; Hu, X.; Song, Y.; Zhang, Y.; Shi, Y. Clinical pharmacokinetics of simotinib, an oral EGFR tyrosine kinase inhibitor, in patients with advanced NSCLC patients. J. Clin. Oncol. 2015, 33, e13575. [Google Scholar] [CrossRef]
- Zhu, Q.; Liu, Z.; Li, P.; Cheng, Z. Drug interaction studies reveal that simotinib upregulates intestinal absorption by increasing the paracellular permeability of intestinal epithelial cells. Drug Metab. Pharmacokinet. 2014, 29, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Commander, H.; Whiteside, G.; Perry, C. Vandetanib: First global approval. Drugs 2011, 71, 1355–1365. [Google Scholar] [CrossRef]
- Chau, N.G.; Haddad, R.I. Vandetanib for the Treatment of Medullary Thyroid Cancer. Clin. Cancer Res. 2013, 19, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Yuan, X.-H.; Wang, S.-Q.; Zhao, W.; Chen, X.-B.; Yu, B. FDA-approved pyrimidine-fused bicyclic heterocycles for cancer therapy: Synthesis and clinical application. Eur. J. Med. Chem. 2021, 214, 113218. [Google Scholar] [CrossRef] [PubMed]
- Brocklesby, K.L.; Waby, J.S.; Cawthorne, C.; Smith, G. An alternative synthesis of Vandetanib (CaprelsaTM) via a microwave accelerated Dimroth rearrangement. Tetrahedron Lett. 2017, 58, 1467–1469. [Google Scholar] [CrossRef]
- Wedge, S.R.; Ogilvie, D.J.; Dukes, M.; Kendrew, J.; Chester, R.; Jackson, J.A.; Boffey, S.J.; Valentine, P.J.; Curwen, J.O.; Musgrove, H.L.; et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002, 62, 4645–4655. [Google Scholar]
- Carlomagno, F.; Vitagliano, D.; Guida, T.; Ciardiello, F.; Tortora, G.; Vecchio, G.; Ryan, A.J.; Fontanini, G.; Fusco, A.; Santoro, M. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002, 62, 7284–7290. [Google Scholar] [PubMed]
- Knowles, P.P.; Murray-Rust, J.; Kjaer, S.; Scott, R.P.; Hanrahan, S.; Santoro, M.; Ibáñez, C.F.; McDonald, N.Q. Structure and chemical inhibition of the RET tyrosine kinase domain. J. Biol. Chem. 2006, 281, 33577–33587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, F.; Koh, Y.; Koizumi, F.; Tamura, T.; Saijo, N.; Nishio, K. Anticancer effects of ZD6474, a VEGF receptor tyrosine kinase inhibitor, in gefitinib (“Iressa”)-sensitive and resistant xenograft models. Cancer Sci. 2004, 95, 984–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, D.; Ojima, H.; Kokubu, A.; Ochiya, T.; Kasai, S.; Hirohashi, S.; Shibata, T. Vandetanib (ZD6474), an inhibitor of VEGFR and EGFR signalling, as a novel molecular-targeted therapy against cholangiocarcinoma. Br. J. Cancer 2009, 100, 1257–1266. [Google Scholar] [CrossRef]
- Sarkar, S.; Mazumdar, A.; Dash, R.; Sarkar, D.; Fisher, P.B.; Mandal, M. ZD6474, a dual tyrosine kinase inhibitor of EGFR and VEGFR-2, inhibits MAPK/ERK and AKT/PI3-K and induces apoptosis in breast cancer cells. Cancer Biol. Ther. 2010, 9, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.; Oliver, S.; Kennedy, S.-J.; Partridge, E.; Hutchison, M.; Clarke, D.; Giles, P. Pharmacokinetics of vandetanib: Three phase I studies in healthy subjects. Clin. Ther. 2012, 34, 221–237. [Google Scholar] [CrossRef]
- Indra, R.; Pompach, P.; Martínek, V.; Takácsová, P.; Vavrová, K.; Heger, Z.; Adam, V.; Eckschlager, T.; Kopečková, K.; Arlt, V.M.; et al. Identification of Human Enzymes Oxidizing the Anti-Thyroid-Cancer Drug Vandetanib and Explanation of the High Efficiency of Cytochrome P450 3A4 in its Oxidation. Int. J. Mol. Sci. 2019, 20, 3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwa, M.W.; Kadi, A.A.; Darwish, H.W.; Amer, S.M.; Al-shakliah, N.S. Identification and characterization of in vivo, in vitro and reactive metabolites of vandetanib using LC–ESI–MS/MS. Chem. Cent. J. 2018, 12, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karachaliou, N.; Fernandez-Bruno, M.; Bracht, J.W.P.; Rosell, R. EGFR first- and second-generation TKIs—there is still place for them in EGFR -mutant NSCLC patients. Transl. Cancer Res. 2018, 8 (Suppl. 1), S24–S47. Available online: https://tcr.amegroups.com/article/view/24920 (accessed on 8 September 2021). [CrossRef]
- Barnet, M.B.; O’Toole, S.; Horvath, L.G.; Selinger, C.; Yu, B.; Ng, C.C.; Boyer, M.; Cooper, W.A.; Kao, S. EGFR-Co-Mutated Advanced NSCLC and Response to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2017, 12, 585–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]

























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abourehab, M.A.S.; Alqahtani, A.M.; Youssif, B.G.M.; Gouda, A.M. Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules 2021, 26, 6677. https://doi.org/10.3390/molecules26216677
Abourehab MAS, Alqahtani AM, Youssif BGM, Gouda AM. Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules. 2021; 26(21):6677. https://doi.org/10.3390/molecules26216677
Chicago/Turabian StyleAbourehab, Mohammed A. S., Alaa M. Alqahtani, Bahaa G. M. Youssif, and Ahmed M. Gouda. 2021. "Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism" Molecules 26, no. 21: 6677. https://doi.org/10.3390/molecules26216677
APA StyleAbourehab, M. A. S., Alqahtani, A. M., Youssif, B. G. M., & Gouda, A. M. (2021). Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules, 26(21), 6677. https://doi.org/10.3390/molecules26216677