
Huperzine A and Its Neuroprotective Molecular Signaling in Alzheimer’s Disease


Abstract
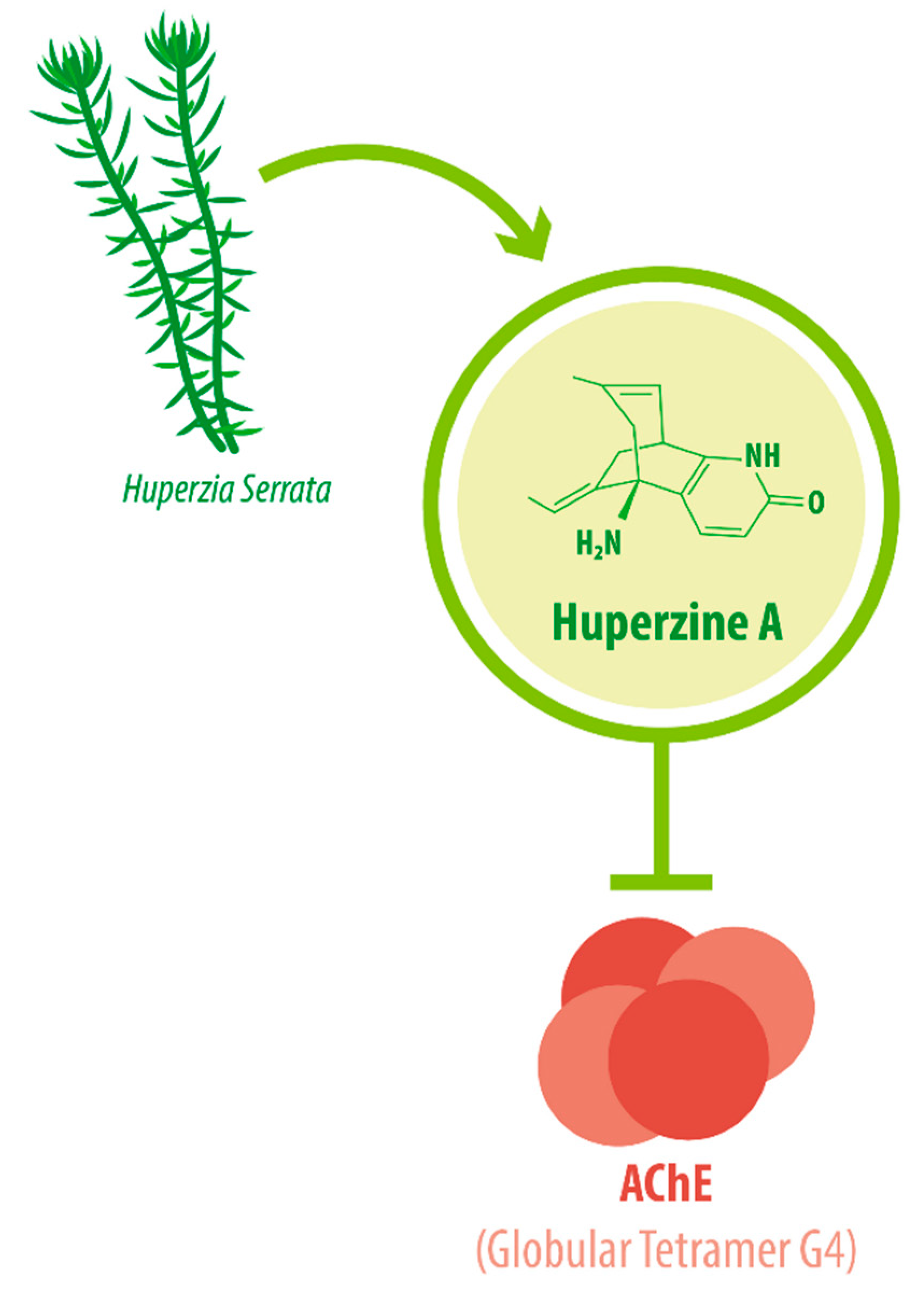
1. Introduction
2. HupA within the CNS: Modulation of Critical Pathological Conditions
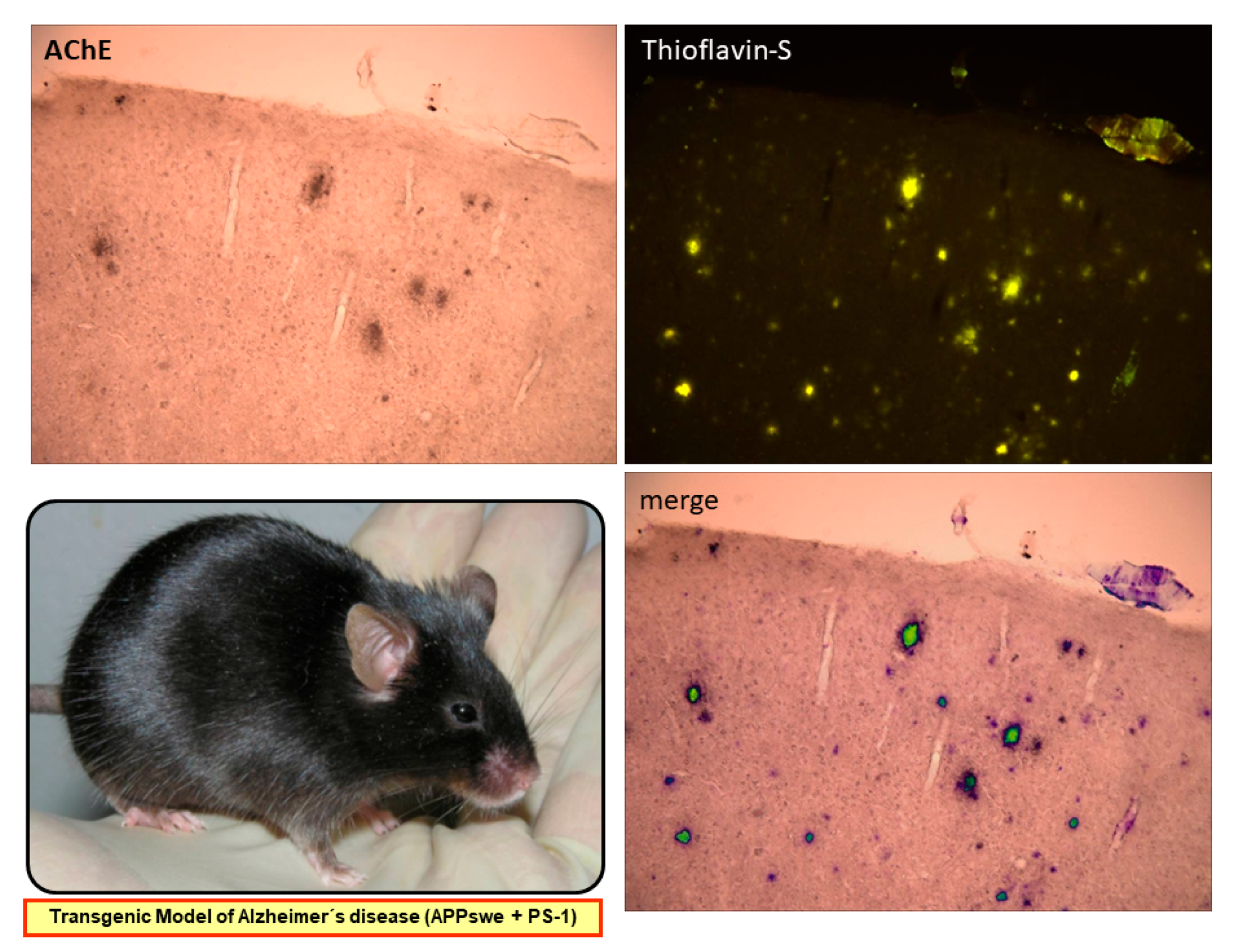
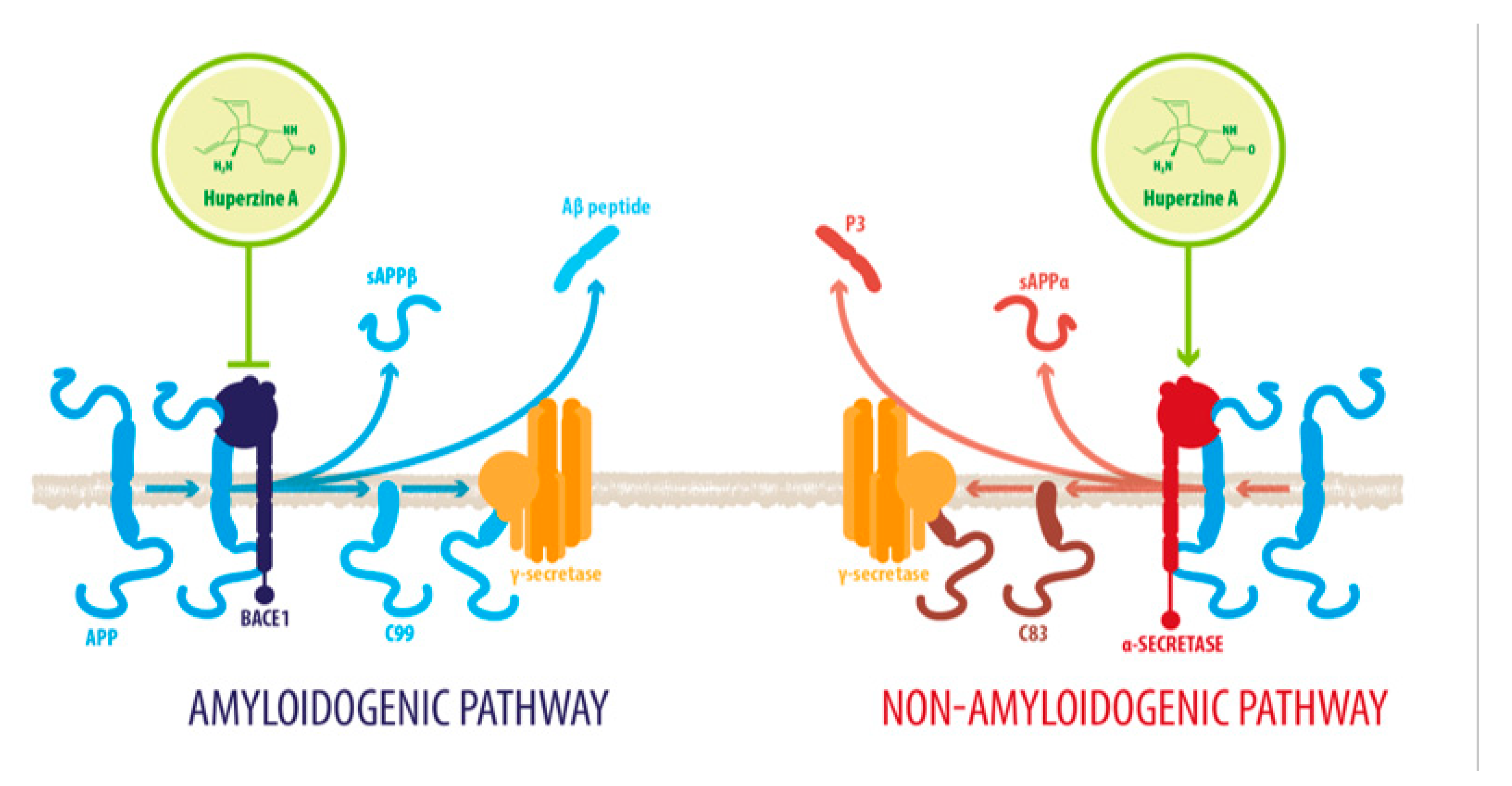
2.1. HupA Mediated Modulation of Amyloid-β PeptideToxicity
2.2. HupA on Dementia
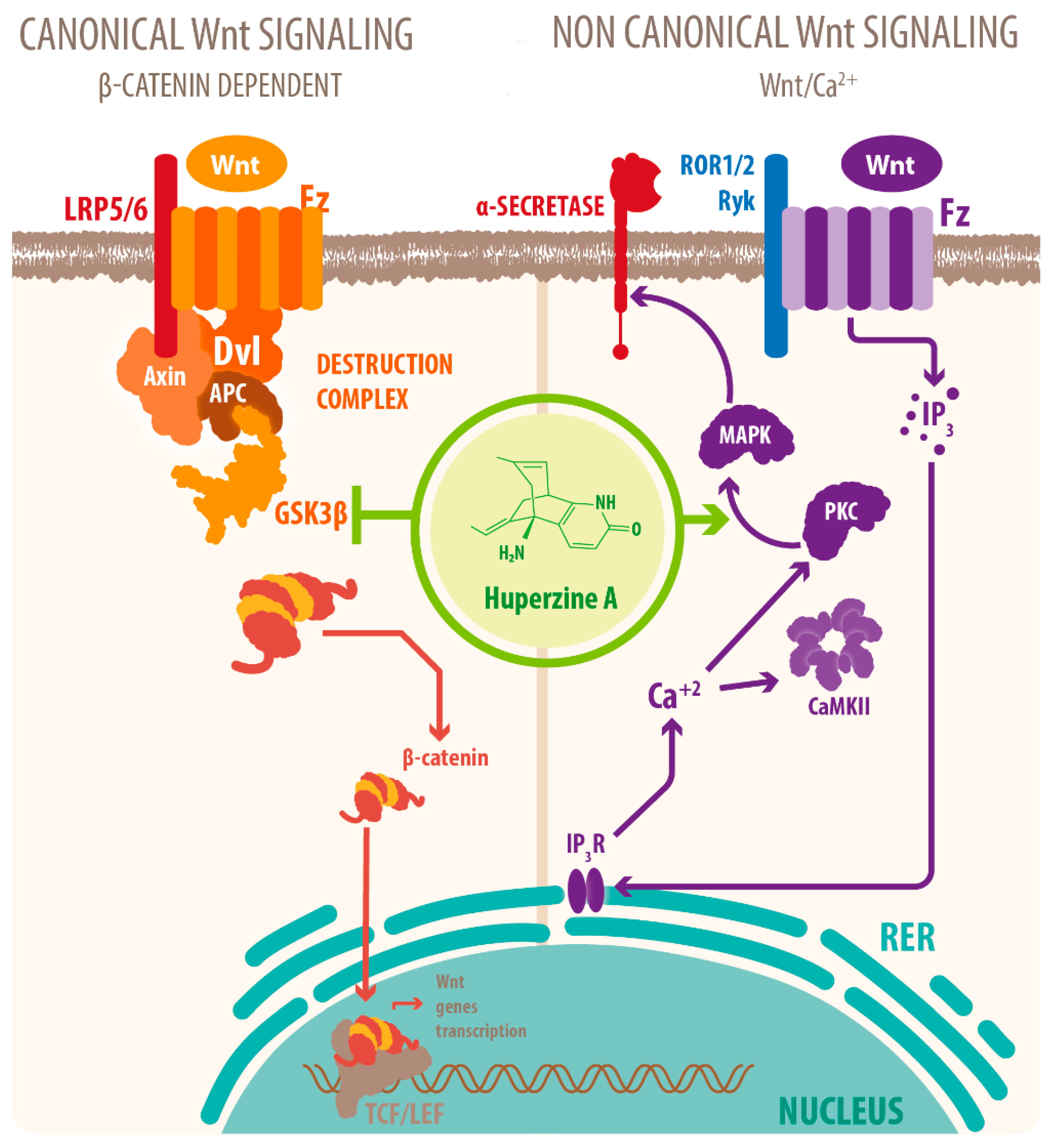
3. Neuroprotective Molecular Signaling of HupA against AD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Target | Effects of HupA Treatment | References |
|---|---|---|
| Inhibition of AChE | Reduced neuronal loss Decreased Aβ neurotoxicity]. Attenuated p53-mediated cell death Downregulated NF-kβ signaling Downregulated p65 translocation-related neurodegeneration [ | [9,37,39,46,49] |
| Activation of α7-nAChRs and α4β2-nAChRs | Decreased NF-κB signaling Increased GABAergic transmission Reduced pro-inflammatory cytokines | [31,46,47] |
| Upregulation of Neurotrophin | Reduced loss of cholinergic neurons | [52] |
| Activation of BDNF/TrkB | Improved neuronal survival through PI3K/TrkB/mTOR signaling Induced LTP and synaptic transmission | [48,51,52,54] |
| Upregulation of Bcl-2 and downregulation of Bax | Decreased apoptotic activity | [38] |
| Upregulation of Synaptotagmin | Improved synaptic vesicle exocytosis | [48] |
| Interaction with ChE-like domain of Neuroligin-1 | Decreased Aβ aggregation | [37,46,60] |
| Downregulation of activity of GSK-3β | Decreased tau phosphorylation and Aβ accumulation Increased neurogenesis and modulation of synaptic plasticity Increased nonamyloidogenic processing of APP] Downregulated NF-kβ signaling downregulated p65 expression-related neurodegeneration | [39,46,48,54] |
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AChE | Acetylcholinesterase |
| AChEI | Acetylcholinesterase Inhibitor |
| AD | Alzheimer´s Disease |
| ADAM10 | Disintegrin and Metalloproteinase Domain-containing Protein 10 |
| AIF | Apoptosis Inducing Factor |
| APP | Amyloid Precursor Protein |
| ATP | Adenosine Triphosphate |
| Aβ | Amyloid- β peptide |
| BACE1 | β-site of the APP-cleaving enzyme |
| Bax | Bcl-2-like protein 4 |
| BBB | Blood Brain Barrier |
| Bcl-2 | Bcl-2 B-cell lymphoma-2 |
| BDNF | Brain-Derived Neurotrophic Factor |
| CAT | Catalase |
| CNS | Central Nervous System |
| CREB | Cyclic Adenosine Monophosphate Responsive Element Binding Protein |
| CytoC | Cytochrome C |
| GSH-PX | Glutathione Peroxidase |
| GSK-3β | Glycogen Synthase Kinase-3β |
| HupA | Huperzine A |
| IL-6 | Interleukin-6 |
| IRE | Iron-Responsive Element |
| IRP-1 | Iron Regulatory Protein-1 |
| MAPK/ERK | Mitogen-activated Protein Kinases/Extracellular Signal-regulated Kinases |
| MIP-1α | Macrophage Inflammatory Protein-1α |
| mTOR | Mammalian Target of Rapamycin |
| NF-kB | Nuclear Factor Kappa-light-chain-enhancer of Activated B cells |
| NL1 | Neuroligin 1 |
| NMDA | N-methyl-D-aspartate |
| NMDAR | N-methyl-D-aspartate Receptor |
| PI3K | Phosphatidylinositol 3-Kinase |
| PKC | Protein Kinase C |
| PS1 | Presenilin 1 |
| ROS | Reactive Oxygen Species |
| SOD | Superoxide Dismutase |
| TBI | Transferrin-Bound Iron |
| TFR1 | Transferrin Receptor Protein 1 |
| TNF-α | Tumor Necrosis Factor-α |
| TrkB | Tropomyosin receptor kinase B |
| VaD | Vascular-associated Dementia |
| Wnt | Wingless-related Integration Site |
| WT | Wild-type |
References
- Howes, M.R.; Fang, R.; Houghton, P.J. Effect of Chinese Herbal Medicine on Alzheimer’s Disease. Int. Rev. Neurobiol. 2017, 135, 29–56. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.A.; Sarpong, R. Heathcock-inspired strategies for the synthesis of fawcettimine-type Lycopodium alkaloids. Chemistry 2014, 20, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.Y.; Chen, C.P.; Jinn, T.R. Traditional Chinese medicines and Alzheimer’s disease. Taiwan J. Obstet. Gynecol. 2011, 50, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wang, Y.; Tian, J.; Liu, J.P. Huperzine A for Alzheimer’s disease: A systematic review and meta-analysis of randomized clinical trials. PLoS ONE 2013, 8, e74916. [Google Scholar] [CrossRef]
- Nett, R.S.; Dho, Y.; Low, Y.; Sattely, E.S. A metabolic regulon reveals early and late acting enzymes in neuroactive Lycopodium alkaloid biosynthesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2102949118. [Google Scholar] [CrossRef]
- Callizot, N.; Campanari, M.L.; Rouvière, L.; Jacquemot, G.; Henriques, A.; Garayev, E.; Poindron, P. Huperzia serrata Extract ‘NSP01’ with Neuroprotective Effects-Potential Synergies of Huperzine A and Polyphenols. Front. Pharmacol. 2021, 12, 681532. [Google Scholar] [CrossRef]
- Kong, Y.R.; Tay, K.C.; Su, Y.X.; Wong, C.K.; Tan, W.N.; Khaw, K.Y. Potential of Naturally Derived Alkaloids as Multi-Targeted Therapeutic Agents for Neurodegenerative Diseases. Molecules 2021, 26, 728. [Google Scholar] [CrossRef]
- De La Garza, R.; Verrico, C.D.; Newton, T.F.; Mahoney, J.J.; Thompson-Lake, D.G. Safety and Preliminary Efficacy of the Acetylcholinesterase Inhibitor Huperzine A as a Treatment for Cocaine Use Disorder. Int. J. Neuropsychopharmacol. 2015, 19, pyv098. [Google Scholar] [CrossRef][Green Version]
- Ohba, T.; Yoshino, Y.; Ishisaka, M.; Abe, N.; Tsuruma, K.; Shimazawa, M.; Oyama, M.; Tabira, T.; Hara, H. Japanese Huperzia serrata extract and the constituent, huperzine A, ameliorate the scopolamine-induced cognitive impairment in mice. Biosci. Biotechnol. Biochem. 2015, 79, 1838–1844. [Google Scholar] [CrossRef]
- Damar, U.; Gersner, R.; Johnstone, J.T.; Schachter, S.; Rotenberg, A. Huperzine A as a neuroprotective and antiepileptic drug: A review of preclinical research. Expert Rev. Neurother. 2016, 16, 671–680. [Google Scholar] [CrossRef]
- Ferreira, A.; Rodrigues, M.; Fortuna, A.; Falcão, A.; Alves, G. Huperzine A from Huperzia serrata: A review of its sources, chemistry, pharmacology and toxicology. Phytochem. Rev. 2016, 15, 51–85. [Google Scholar] [CrossRef]
- Ashani, Y.; Grunwald, J.; Kronman, C.; Velan, B.; Shafferman, A. Role of tyrosine 337 in the binding of huperzine A to the active site of human acetylcholinesterase. Mol. Pharmacol. 1994, 45, 555–560. [Google Scholar]
- Pang, Y.P.; Kozikowski, A.P. Prediction of the binding sites of huperzine A in acetylcholinesterase by docking studies. J. Comput. Aided Mol. Des. 1994, 8, 669–681. [Google Scholar] [CrossRef]
- Raves, M.L.; Harel, M.; Pang, Y.P.; Silman, I.; Kozikowski, A.P.; Sussman, J.L. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (-)-huperzine A. Nat. Struct. Biol. 1997, 4, 57–63. [Google Scholar] [CrossRef]
- Tun, M.K.; Herzon, S.B. The pharmacology and therapeutic potential of (-)-huperzine A. Int. J. Exp. Pharmacol. 2012, 4, 113–123. [Google Scholar] [CrossRef]
- Colović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef]
- Geula, C.; Mesulam, M.M.; Saroff, D.M.; Wu, C.K. Relationship between plaques, tangles, and loss of cortical cholinergic fibers in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1998, 57, 63–75. [Google Scholar] [CrossRef]
- Reyes, A.E.; Chacón, M.A.; Dinamarca, M.C.; Cerpa, W.; Morgan, C.; Inestrosa, N.C. Acetylcholinesterase-Abeta complexes are more toxic than Abeta fibrils in rat hippocampus: Effect on rat beta-amyloid aggregation, laminin expression, reactive astrocytosis, and neuronal cell loss. Am. J. Pathol. 2004, 164, 2163–2174. [Google Scholar] [CrossRef]
- Fuentes, M.E.; Inestrosa, N.C. Characterization of a tetrameric G4 form of acetylcholinesterase from bovine brain: A comparison with the dimeric G2 form of the electric organ. Mol. Cell. Biochem. 1988, 81, 53–64. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alvarez, A.; Pérez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase accelerates assembly of amyloid-b-peptides into Alzheimer’s fibrils: Possible role of the peripheral site of the enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef]
- Alvarez, A.; Alarcón, R.; Opazo, C.; Campos, E.O.; Muñoz, F.J.; Calderón, F.H.; Dajas, F.; Gentry, M.K.; Doctor, B.P.; De Mello, F.G.; et al. Stable Complexes Involving Acetylcholinesterase and Amyloid-β Peptide Change the Biochemical Properties of the Enzyme and Increase the Neurotoxicity of Alzheimer’s Fibrils. J. Neurosci. 1998, 18, 3213–3223. [Google Scholar] [CrossRef]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. β-Amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar] [CrossRef]
- Zhang, H.Y. New insights into huperzine A for the treatment of Alzheimer’s disease. Acta Pharmacol. Sin. 2012, 33, 1170–1175. [Google Scholar] [CrossRef]
- Zhou, X.; Cui, G.; Tseng, H.H.; Lee, S.M.; Leung, G.P.; Chan, S.W.; Kwan, Y.W.; Hoi, M.P. Vascular Contributions to Cognitive Impairment and Treatments with Traditional Chinese Medicine. Evid. Based Complementary Altern. Med. 2016, 9627258. [Google Scholar] [CrossRef] [PubMed]
- Ha, G.T.; Wong, R.K.; Zhang, Y. Huperzine a as potential treatment of Alzheimer’s disease: An assessment on chemistry, pharmacology, and clinical studies. Chem. Biodivers. 2011, 8, 1189–1204. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Inestrosa, N.C. Wnt signaling loss accelerates the appearance of neuropathological hallmarks of Alzheimer’s disease in J20-APP transgenic and wild-type mice. J. Neurochem. 2018, 144, 443–465. [Google Scholar] [CrossRef]
- Rahman, M.; Bajgai, J.; Fadriquela, A.; Sharma, S.; Trinh, T.T.; Akter, R.; Lee, K.J. Therapeutic Potential of Natural Products in Treating Neurodegenerative Disorders and Their Future Prospects and Challenges. Molecules 2021, 26, 5327. [Google Scholar] [CrossRef]
- Xu, Z.Q.; Liang, X.M.; Juan, W.; Zhang, Y.F.; Zhu, C.X.; Jiang, X.J. Treatment with Huperzine A improves cognition in vascular dementia patients. Cell Biochem. Biophys. 2012, 62, 55–58. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Z.; Wu, J.; Chen, Y. Chemical Constituents of Plants from the Genus Phlegmariurus. Chem. Biodivers. 2016, 13, 269–274. [Google Scholar] [CrossRef]
- Orhan, I.E.; Orhan, G.; Gurkas, E. An overview on natural cholinesterase inhibitors—A multi-targeted drug class-and their mass production. Mini Rev. Med. Chem. 2011, 11, 836–842. [Google Scholar] [CrossRef]
- Chauhan, P.S.; Yadav, D. Dietary Nutrients and Prevention of Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2021. (E-pub Ahead of Print). [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Zheng, W.; Wang, T.; Xie, J.W.; Wang, S.L.; Zhao, B.L.; Teng, W.P.; Wang, Z.Y. Huperzine A activates Wnt/β-catenin signaling and enhances the nonamyloidogenic pathway in an Alzheimer transgenic mouse model. Neuropsychopharmacology 2011, 36, 1073–1089. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Lin, Z.X.; Wu, W.; Albert, W.N.; Zee, B.C.Y. Huperzine A in treatment of amyloid-β-associated neuropathology in a mouse model of Alzheimer disease: Abridged secondary publication. Hong Kong Med. J. 2020, 26 (Suppl.8), 34–37. [Google Scholar]
- Huang, X.T.; Qian, Z.M.; He, X.; Gong, Q.; Wu, K.C.; Jiang, L.R.; Lu, L.N.; Zhu, Z.J.; Zhang, H.Y.; Yung, W.H.; et al. Reducing iron in the brain: A novel pharmacologic mechanism of huperzine A in the treatment of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1045–1054. [Google Scholar] [CrossRef]
- Yang, L.; Ye, C.Y.; Huang, X.T.; Tang, X.C.; Zhang, H.Y. Decreased accumulation of subcellular amyloid-β with improved mitochondrial function mediates the neuroprotective effect of huperzine A. J. Alzheimer’s Dis. 2012, 31, 131–142. [Google Scholar] [CrossRef]
- Lei, Y.; Yang, L.; Ye, C.Y.; Qin, M.Y.; Yang, H.Y.; Jiang, H.L.; Tang, X.C.; Zhang, H.Y. Involvement of Intracellular and Mitochondrial Aβ in the Ameliorative Effects of Huperzine A against Oligomeric Aβ42-Induced Injury in Primary Rat Neurons. PLoS ONE 2015, 10, e0128366. [Google Scholar] [CrossRef]
- Tao, Y.; Fang, L.; Yang, Y.; Jiang, H.; Yang, H.; Zhang, H.; Zhou, H. Quantitative proteomic analysis reveals the neuroprotective effects of huperzine A for amyloid beta treated neuroblastoma N2a cells. Proteomics 2013, 13, 1314–1324. [Google Scholar] [CrossRef]
- Zhu, N.; Lin, J.; Wang, K.; Wei, M.; Chen, Q.; Wang, Y. Huperzine A protects neural stem cells against Aβ-induced apoptosis in a neural stem cells and microglia co-culture system. Int. J. Clin. Exp. Pathol. 2015, 8, 6425–6433. [Google Scholar]
- Xie, L.; Jiang, C.; Wang, Z.; Yi, X.; Gong, Y.; Chen, Y.; Fu, Y. Effect of Huperzine A on Aβ-induced p65 of astrocyte in vitro. Biosci. Biotechnol. Biochem. 2016, 80, 2334–2337. [Google Scholar] [CrossRef]
- Rafii, M.S.; Walsh, S.; Little, J.T.; Behan, K.; Reynolds, B.; Ward, C.; Jin, S.; Thomas, R.; Aisen, P.S. A phase II trial of huperzine A in mild to moderate Alzheimer disease. Neurology 2011, 76, 1389–1394. [Google Scholar] [CrossRef]
- Xing, S.H.; Zhu, C.X.; Zhang, R.; An, L. Huperzine a in the treatment of Alzheimer’s disease and vascular dementia: A meta-analysis. Evid. Based Complementary Altern. Med. 2014, 363985. [Google Scholar] [CrossRef]
- Gul, A.; Bakht, J.; Mehmood, F. Huperzine-A response to cognitive impairment and task switching deficits in patients with Alzheimer’s disease. J. Chin. Med Assoc. 2019, 82, 40–43. [Google Scholar] [CrossRef]
- Xu, S.S.; Gao, Z.X.; Weng, Z.; Du, Z.M.; Xu, W.A.; Yang, J.S.; Zhang, M.L.; Tong, Z.H.; Fang, Y.S.; Chai, X.S.; et al. Efficacy of tablet huperzine-A on memory, cognition, and behavior in Alzheimer’s disease. Zhongguo Yao Li Xue Bao 1995, 16, 391–395. [Google Scholar]
- Tsai, S. Huperzine-A, a versatile herb, for the treatment of Alzheimer’s disease. Crit. Care Med. 2019, 82, 750–751. [Google Scholar] [CrossRef]
- Ghassab-Abdollahi, N.; Mobasseri, K.; Dehghani Ahmadabad, A.; Nadrian, H.; Mirghafourvand, M. The effects of Huperzine A on dementia and mild cognitive impairment: An overview of systematic reviews. Phytother. Res. 2021, 35, 4971–4987. [Google Scholar] [CrossRef]
- Damar, U.; Gersner, R.; Johnstone, J.T.; Schachter, S.; Rotenberg, A. Huperzine A: A promising anticonvulsant, disease modifying, and memory enhancing treatment option in Alzheimer’s disease. Med. Hypotheses 2017, 99, 57–62. [Google Scholar] [CrossRef]
- Südhof, T.C. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 2013, 80, 675–690. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, X.C.; Zhang, H.Y. Huperzine A alleviates synaptic deficits and modulates amyloidogenic and nonamyloidogenic pathways in APPswe/PS1dE9 transgenic mice. J. Neurosci. Res. 2012, 90, 508–517. [Google Scholar] [CrossRef]
- Ma, T.; Gong, K.; Yan, Y.; Zhang, L.; Tang, P.; Zhang, X.; Gong, Y. Huperzine A promotes hippocampal neurogenesis in vitro and in vivo. Brain Res. 2013, 1506, 35–43. [Google Scholar] [CrossRef]
- Mao, X.Y.; Zhou, H.H.; Li, X.; Liu, Z.Q. Huperzine A Alleviates Oxidative Glutamate Toxicity in Hippocampal HT22 Cells via Activating BDNF/TrkB-Dependent PI3K/Akt/mTOR Signaling Pathway. Cell. Mol. Neurobiol. 2016, 36, 915–925. [Google Scholar] [CrossRef]
- Kerr, F.; Bjedov, I.; Sofola-Adesakin, O. Molecular Mechanisms of Lithium Action: Switching the Light on Multiple Targets for Dementia Using Animal Models. Front. Mol. Neurosci. 2018, 11, 297. [Google Scholar] [CrossRef]
- Qian, Z.M.; Ke, Y. Huperzine A: Is it an Effective Disease-Modifying Drug for Alzheimer’s Disease? Front. Aging Neurosci. 2014, 6, 216. [Google Scholar] [CrossRef]
- Rahman, M.H.; Bajgai, J.; Fadriquela, A.; Sharma, S.; Trinh Thi, T.; Akter, R.; Goh, S.H.; Kim, C.-S.; Lee, K.-J. Redox Effects of Molecular Hydrogen and Its Therapeutic Efficacy in the Treatment of Neurodegenerative Diseases. Processes 2021, 9, 308. [Google Scholar] [CrossRef]
- Du, Y.; Liang, H.; Zhang, L.; Fu, F. Administration of Huperzine A exerts antidepressant-like activity in a rat model of post-stroke depression. Pharmacol. Biochem. Behav. 2017, 158, 32–38. [Google Scholar] [CrossRef]
- Mak, S.; Li, W.; Fu, H.; Luo, J.; Cui, W.; Hu, S.; Pang, Y.; Carlier, P.R.; Tsim, K.W.; Pi, R.; et al. Promising tacrine/huperzine A-based dimeric acetylcholinesterase inhibitors for neurodegenerative disorders: From relieving symptoms to modifying diseases through multitarget. J. Neurochem. 2021, 158, 1381–1393. [Google Scholar] [CrossRef]
- Carvajal, F.J.; Inestrosa, N.C. Interactions of AChE with Aβ Aggregates in Alzheimer’s Brain: Therapeutic Relevance of IDN 5706. Front. Mol. Neurosci. 2011, 4, 19. [Google Scholar] [CrossRef]
- Rees, T.; Hammond, P.I.; Soreq, H.; Younkin, S.; Brimijoin, S. Acetylcholinesterase promotes beta-amyloid plaques in cerebral cortex. Neurobiol. Aging 2003, 24, 777–787. [Google Scholar] [CrossRef]
- Leone, P.; Comoletti, D.; Taylor, P.; Bourne, Y.; Marchot, P. Structure-function relationships of the alpha/beta-hydrolase fold domain of neuroligin: A comparison with acetylcholinesterase. Chem. Biol. Interact. 2010, 187, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Scholl, F.G.; Scheiffele, P. Making connections: Cholinesterase-domain proteins in the CNS. Trends Neurosci. 2003, 26, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Dinamarca, M.C.; Sagal, J.P.; Quintanilla, R.A.; Godoy, J.A.; Arrázola, M.S.; Inestrosa, N.C. Amyloid-beta-Acetylcholinesterase complexes potentiate neurodegenerative changes induced by the Abeta peptide. Implications for the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2010, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Dinamarca, M.C.; Di Luca, M.; Godoy, J.A.; Inestrosa, N.C. The soluble extracellular fragment of neuroligin-1 targets Aβ oligomers to the postsynaptic region of excitatory synapses. Biochem. Biophys. Res. Commun. 2015, 466, 66–71. [Google Scholar] [CrossRef]
- Nhan, H.S.; Chiang, K.; Koo, E.H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol. 2015, 129, 1–19. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Arenas, E. Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 2010, 11, 77–86. [Google Scholar] [CrossRef]
- Oliva, C.A.; Montecinos-Oliva, C.; Inestrosa, N.C. Wnt Signaling in the Central Nervous System: New Insights in Health and Disease. Prog. Mol. Biol. Transl. Sci. 2018, 153, 81–130. [Google Scholar] [CrossRef]
- Varela-Nallar, L.; Inestrosa, N.C. Wnt signaling in the regulation of adult hippocampal neurogenesis. Front. Cell. Neurosci. 2013, 7, 100. [Google Scholar] [CrossRef]
- Menet, R.; Lecordier, S.; ElAli, A. Wnt Pathway: An Emerging Player in Vascular and Traumatic Mediated Brain Injuries. Front. Physiol. 2020, 11, 565667. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Burgos, P.V.; Inestrosa, N.C. Inhibition of Wnt signaling induces amyloidogenic processing of amyloid precursor protein and the production and aggregation of Amyloid-β (Aβ)(42) peptides. J. Neurochem. 2016, 139, 1175–1191. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signaling in the nervous system and in Alzheimer’s disease. J. Mol. Cell Biol. 2014, 6, 64–74. [Google Scholar] [CrossRef]
- Lu, H.; Jiang, M.; Lu, L.; Zheng, G.; Dong, Q. Ultrastructural mitochondria changes in perihematomal brain and neuroprotective effects of Huperzine A after acute intracerebral hemorrhage. Neuropsychiatr. Dis. Treat. 2015, 11, 2649–2657. [Google Scholar] [CrossRef]
- Yang, X.; Wei, H.M.; Hu, G.Y.; Zhao, J.; Long, L.-N.; Li, C.-J.; Zhao, Z.-J.; Zeng, H.-K.; Nie, H. Combining antioxidant astaxantin and cholinesterase inhibitor huperzine A boosts neuroprotection. Mol. Med. Rep. 2020, 21, 1043–1050. [Google Scholar] [CrossRef]
- Báez-Pagán, C.A.; Delgado-Vélez, M.; Lasalde-Dominicci, J.A. Activation of the Macrophage α7 Nicotinic Acetylcholine Receptor and Control of Inflammation. J. Neuroimmune Pharmacol. 2015, 10, 468–476. [Google Scholar] [CrossRef]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef]






| Mechanism | Molecular Target | Effects of HupA Treatment | References |
|---|---|---|---|
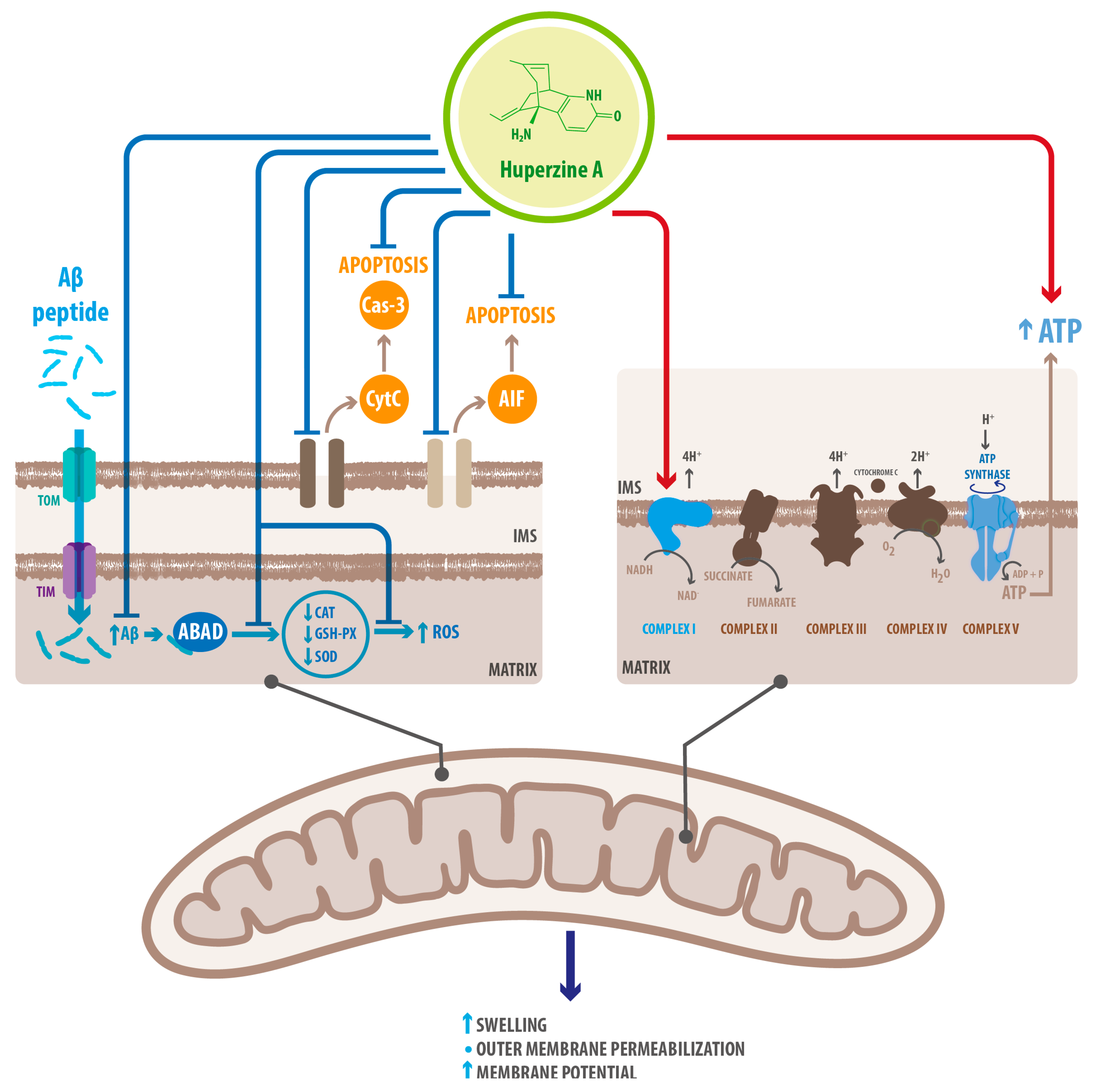
| Mitochondrial protection | Upregulation of oxidative phosphorylation proteins | Recovered energetic homeostasis | [52] |
| Inhibition of cytoplasmic translocation of CytoC | Prevented mitochondria-mediated apoptosis | [69] | |
| Downregulation of AIF | Prevented cas3-independent apoptosis | [52] | |
| Inhibition of Caspase-3 cleavage | Prevented cas-3 dependent apoptosis | [52] | |
| Upregulation of GSH-PX SOD CAT | Decreased ROS accumulation Decreased oxidative stress | [52] | |
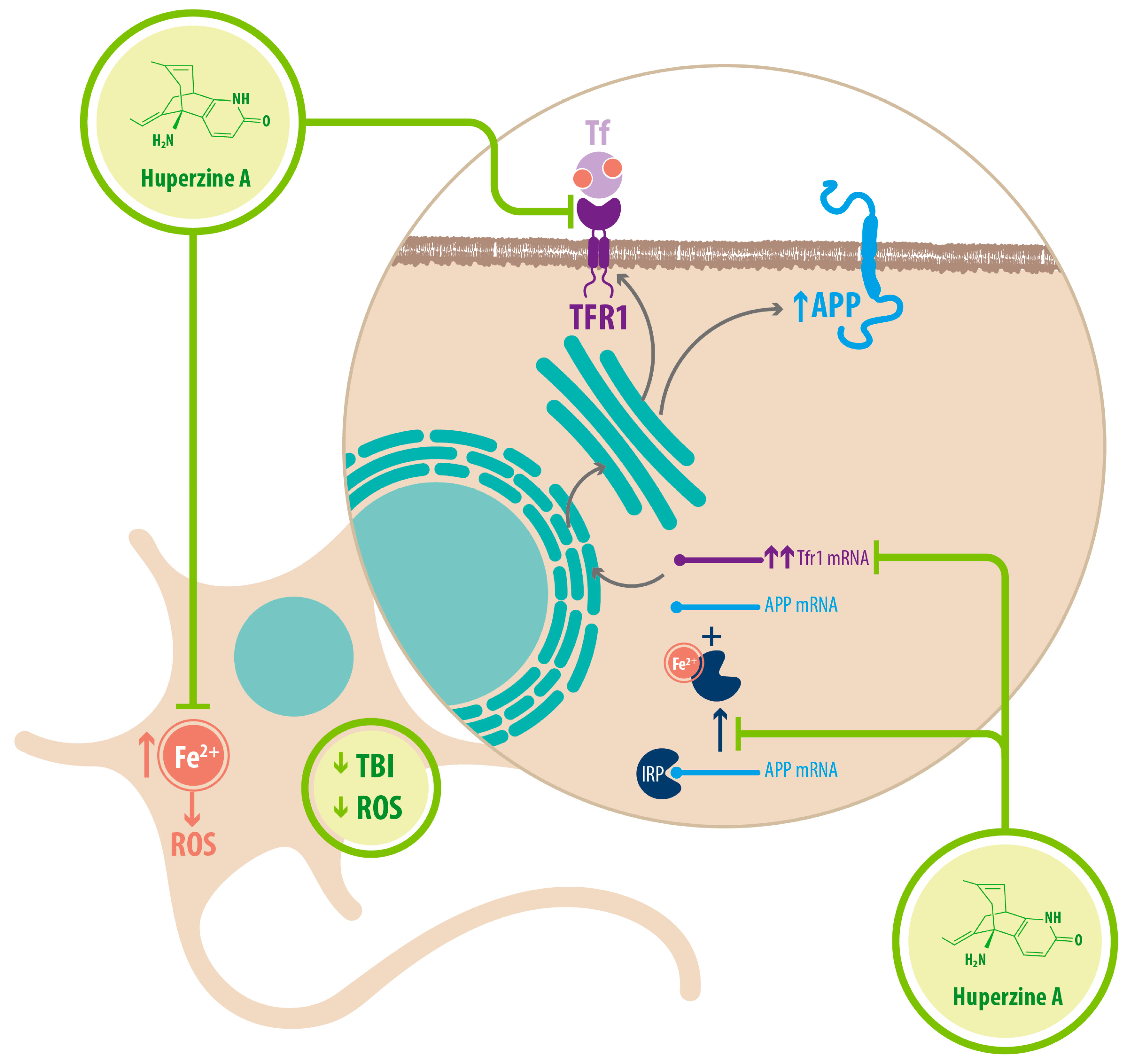
| Fe2+ homeostasis | Chelator/ Reducer of Fe2+ | Increased nonamyloidogenic processing of APP Downregulated expression of APP through IRP-1 | [34] |
| Downregulation of TFR1 | Decreased Fe2+ uptake in neurons | [34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friedli, M.J.; Inestrosa, N.C. Huperzine A and Its Neuroprotective Molecular Signaling in Alzheimer’s Disease. Molecules 2021, 26, 6531. https://doi.org/10.3390/molecules26216531
Friedli MJ, Inestrosa NC. Huperzine A and Its Neuroprotective Molecular Signaling in Alzheimer’s Disease. Molecules. 2021; 26(21):6531. https://doi.org/10.3390/molecules26216531
Chicago/Turabian StyleFriedli, María Jesús, and Nibaldo C. Inestrosa. 2021. "Huperzine A and Its Neuroprotective Molecular Signaling in Alzheimer’s Disease" Molecules 26, no. 21: 6531. https://doi.org/10.3390/molecules26216531
APA StyleFriedli, M. J., & Inestrosa, N. C. (2021). Huperzine A and Its Neuroprotective Molecular Signaling in Alzheimer’s Disease. Molecules, 26(21), 6531. https://doi.org/10.3390/molecules26216531