
Development and Validation of a LC-MS/MS Technique for the Analysis of Short Chain Fatty Acids in Tissues and Biological Fluids without Derivatisation Using Isotope Labelled Internal Standards


Abstract
:1. Introduction
2. Results
2.1. Mass Spectrometry Conditions
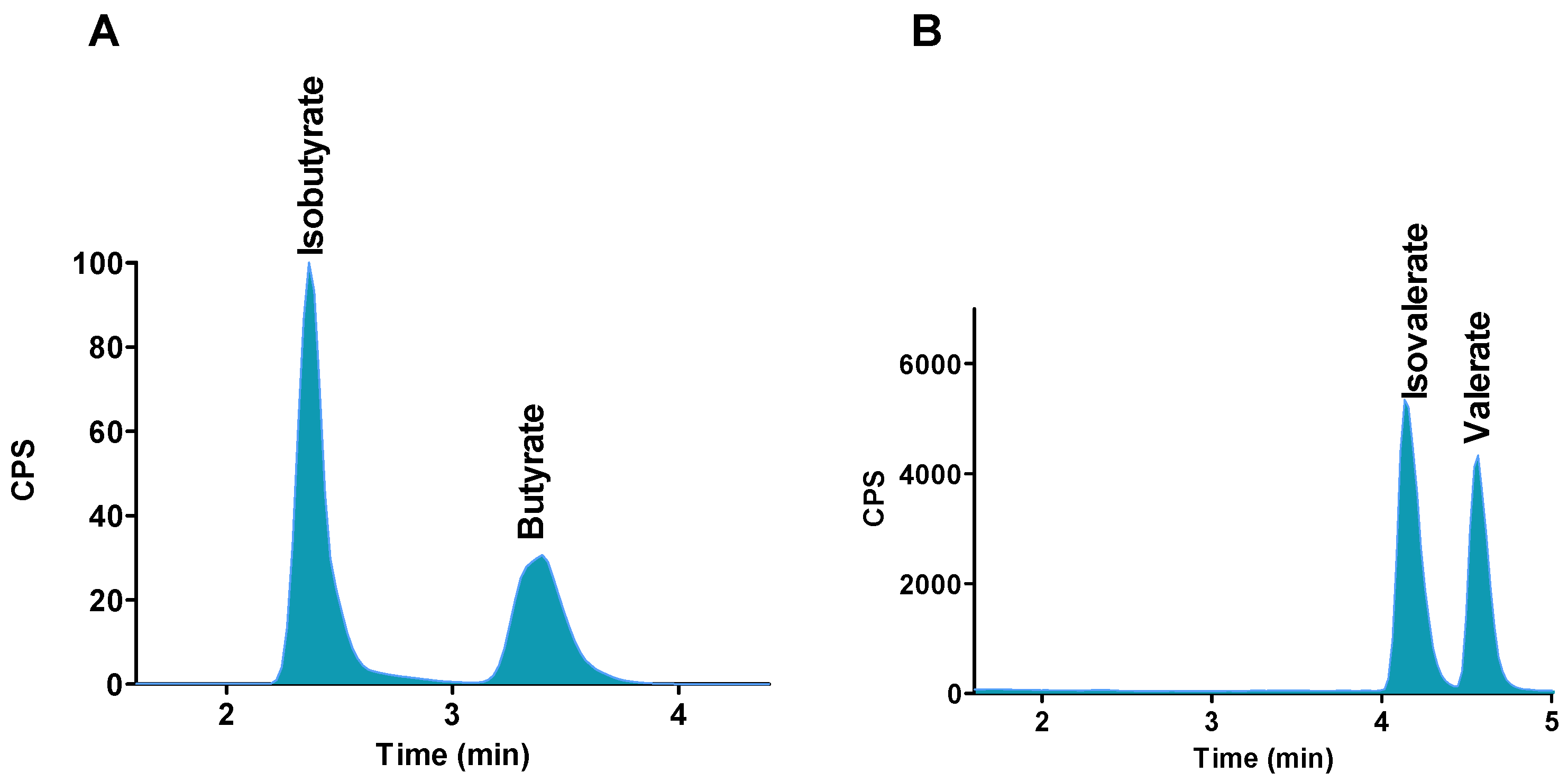
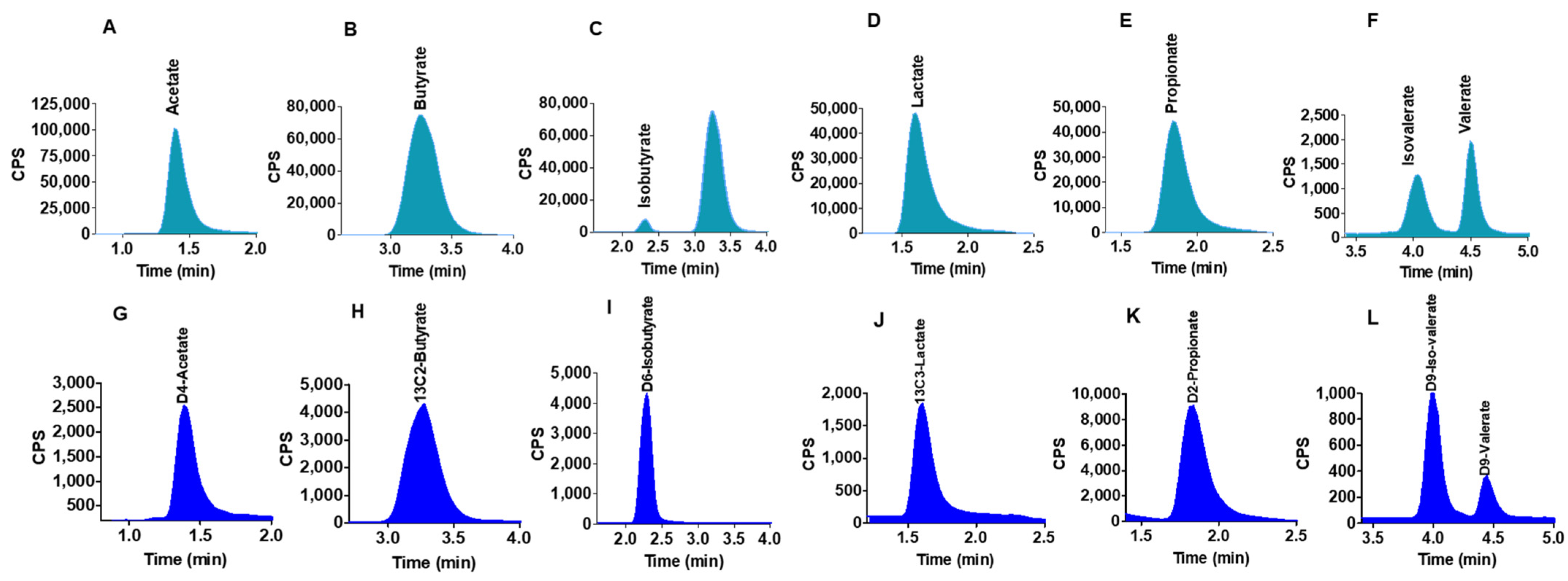
2.2. Chromatographic Separations
2.3. Method Performance
2.3.1. Linearity and Sensitivity
2.3.2. Precision and Accuracy
2.3.3. Carry-Over Effect
2.3.4. Recovery and Matrix Effect
2.3.5. Sample Stability
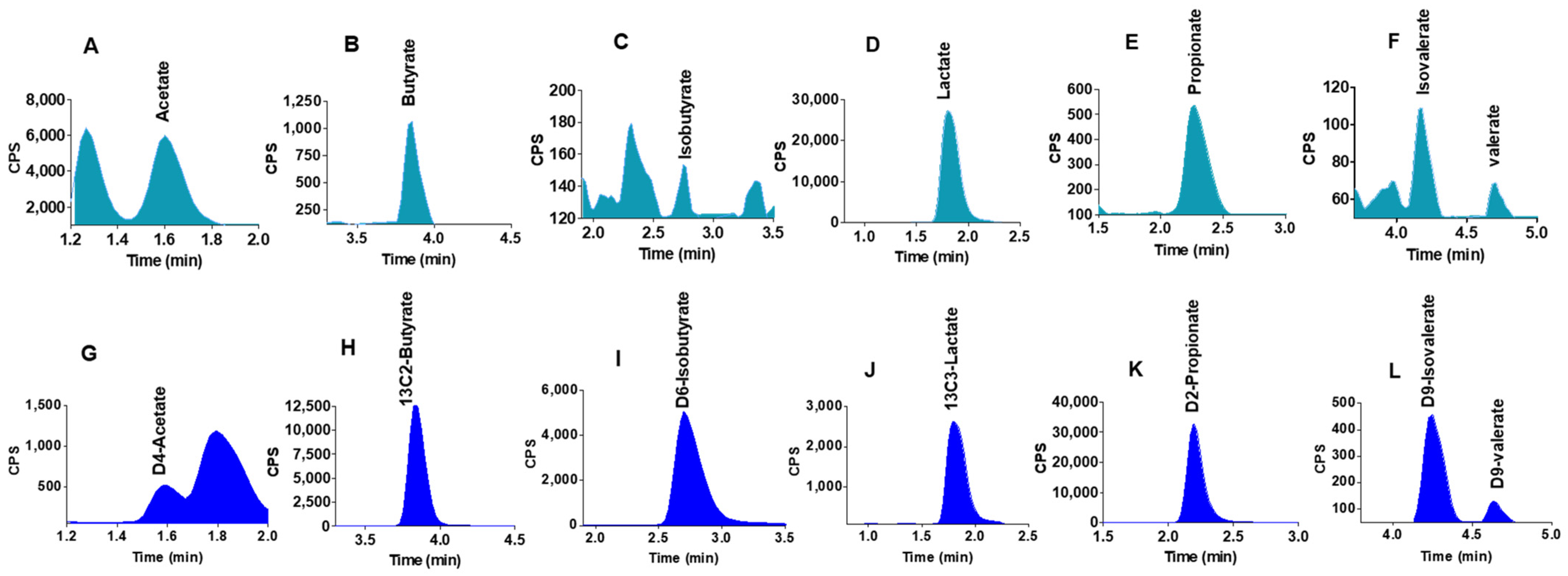
2.4. Quantification of SCFAs in Human Colon Model Fermentation Samples
2.5. Quantification of SCFAs in Mouse Liver Samples
3. Discussion
4. Materials and Methods
4.1. Chemical and Reagents
4.2. Colon Model Fermentation Media and Mouse Tissue Samples
4.2.1. Colon Model Study Participants
4.2.2. Animals and Sample Processing
4.2.3. Sample Processing
4.3. LC-MS/MS Analysis
4.4. Method Validation
4.4.1. Linearity
4.4.2. Sensitivity
4.4.3. Precision and Accuracy
4.4.4. Carry-Over Effect
4.4.5. Recovery and Matrix Effect (or Ion Suppression)
4.5. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Blaak, E.E.; Canfora, E.E.; Theis, S.; Frost, G.; Groen, A.K.; Mithieux, G.; Nauta, A.; Scott, K.; Stahl, B.; van Harsselaar, J.; et al. Short chain fatty acids in human gut and metabolic health. Benef Microbes 2020, 11, 411–455. [Google Scholar] [CrossRef] [PubMed]
- Holmes, Z.C.; Silverman, J.D.; Dressman, H.K.; Wei, Z.; Dallow, E.P.; Armstrong, S.C.; Seed, P.C.; Rawls, J.F.; David, L.A. Short-Chain Fatty Acid Production by Gut Microbiota from Children with Obesity Differs According to Prebiotic Choice and Bacterial Community Composition. mBio 2020, 11, e00914-20. [Google Scholar] [CrossRef] [PubMed]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.T.; Davis-Richardson, A.G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 2011, 6, e25792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Saez-Lara, M.J.; Gomez-Llorente, C.; Plaza-Diaz, J.; Gil, A. The role of probiotic lactic acid bacteria and bifidobacteria in the prevention and treatment of inflammatory bowel disease and other related diseases: A systematic review of randomized human clinical trials. BioMed Res. Int. 2015, 2015, 505878. [Google Scholar] [CrossRef]
- Rios-Covian, D.; González, S.; Nogacka, A.M.; Arboleya, S.; Salazar, N.; Gueimonde, M.; de los Reyes-Gavilán, C.G. An Overview on Fecal Branched Short-Chain Fatty Acids Along Human Life and as Related With Body Mass Index: Associated Dietary and Anthropometric Factors. Front. Microbiol. 2020, 11, 973. [Google Scholar] [CrossRef]
- Agus, A.; Clément, K.; Sokol, H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut 2021, 70, 1174–1182. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Liu, M.; Lan, Y.; Sun, H.; Mai, K.; Wan, M. Short-Chain Fatty Acids Promote Intracellular Bactericidal Activity in Head Kidney Macrophages From Turbot (Scophthalmus maximus L.) via Hypoxia Inducible Factor-1α. Front Immunol. 2020, 11, 615536. [Google Scholar] [CrossRef] [PubMed]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carretta, M.D.; Quiroga, J.; López, R.; Hidalgo, M.A.; Burgos, R.A. Participation of Short-Chain Fatty Acids and Their Receptors in Gut Inflammation and Colon Cancer. Front. Physiol. 2021, 12, 662739. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Wu, W.; Liu, Z.; Cong, Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J. Gastroenterol. 2017, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Primec, M.; Mičetić-Turk, D.; Langerholc, T. Analysis of short-chain fatty acids in human feces: A scoping review. Anal. Biochem. 2017, 526, 9–21. [Google Scholar] [CrossRef]
- Chai, L.; Luo, Q.; Cai, K.; Wang, K.; Xu, B. Reduced fecal short-chain fatty acids levels and the relationship with gut microbiota in IgA nephropathy. BMC Nephrol. 2021, 22, 209. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Nyman, M.; Jönsson, J.A. Rapid determination of short-chain fatty acids in colonic contents and faeces of humans and rats by acidified water-extraction and direct-injection gas chromatography. Biomed Chromatogr. 2006, 20, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Kotani, A.; Miyaguchi, Y.; Kohama, M.; Ohtsuka, T.; Shiratori, T.; Kusu, F. Determination of short-chain fatty acids in rat and human feces by high-performance liquid chromatography with electrochemical detection. Anal Sci. 2009, 25, 1007–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-Y.; Wang, C.; Guo, L.-X.; Zheng, Y.-F.; Hu, W.-H.; Dong, T.T.X.; Wang, T.-J.; Tsim, K.W.K. Simultaneous determination of short-chain fatty acids in human feces by HPLC with ultraviolet detection following chemical derivatization and solid-phase extraction segmental elution. J. Sep. Sci. 2019, 42, 2500–2509. [Google Scholar] [CrossRef] [PubMed]
- Tahara, Y.; Yamazaki, M.; Sukigara, H.; Motohashi, H.; Sasaki, H.; Miyakawa, H.; Haraguchi, A.; Ikeda, Y.; Fukuda, S.; Shibata, S. Gut Microbiota-Derived Short Chain Fatty Acids Induce Circadian Clock Entrainment in Mouse Peripheral Tissue. Sci. Rep. 2018, 8, 1395. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Ichikawa, T.; Inagaki, S.; Min, J.Z.; Fukushima, T.; Toyo’oka, T. Simple and practical derivatization procedure for enhanced detection of carboxylic acids in liquid chromatography–electrospray ionization-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2010, 52, 809–818. [Google Scholar] [CrossRef]
- Liebisch, G.; Ecker, J.; Roth, S.; Schweizer, S.; Öttl, V.; Schött, H.F.; Yoon, H.; Haller, D.; Holler, E.; Burkhardt, R.; et al. Quantification of Fecal Short Chain Fatty Acids by Liquid Chromatography Tandem Mass Spectrometry-Investigation of Pre-Analytic Stability. Biomolecules 2019, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Song, H.E.; Lee, H.Y.; Kim, S.J.; Back, S.H.; Yoo, H.J. A Facile Profiling Method of Short Chain Fatty Acids Using Liquid Chromatography-Mass Spectrometry. Metabolites 2019, 9, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, N.T.; Zhang, M.; Juraschek, S.P.; Miller, E.R.; Appel, L.J. Effects of high-fiber diets enriched with carbohydrate, protein, or unsaturated fat on circulating short chain fatty acids: Results from the OmniHeart randomized trial. Am. J. Clin. Nutr. 2020, 111, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Arora, I.; Stoll, M.L.; Li, Y.; Morrow, C.D.; Barnes, S.; Berryhill, T.F.; Li, S.; Tollefsbol, T.O. Nutritional combinatorial impact on the gut microbiota and plasma short-chain fatty acids levels in the prevention of mammary cancer in Her2/neu estrogen receptor-negative transgenic mice. PLoS ONE 2020, 15, e0234893. [Google Scholar] [CrossRef] [PubMed]
- van Eijk, H.M.H.; Bloemen, J.G.; Dejong, C.H.C. Application of liquid chromatography–mass spectrometry to measure short chain fatty acids in blood. J. Chromatogr. B. 2009, 877, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Schäpe, S.S.; Krause, J.L.; Engelmann, B.; Fritz-Wallace, K.; Schattenberg, F.; Liu, Z.; Müller, S.; Jehmlich, N.; Rolle-Kampczyk, U.; Herberth, G.; et al. The Simplified Human Intestinal Microbiota (SIHUMIx) Shows High Structural and Functional Resistance against Changing Transit Times in In Vitro Bioreactors. Microorganisms 2019, 7, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderón-Pérez, L.; Gosalbes, M.J.; Yuste, S.; Valls, R.M.; Pedret, A.; Llauradó, E.; Jimenez-Hernandez, N.; Artacho, A.; Pla-Pagà, L.; Companys, J.; et al. Gut metagenomic and short chain fatty acids signature in hypertension: A cross-sectional study. Sci. Rep. 2020, 10, 6436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, M.A. Porous Graphitic Carbon-Based Metabolomics and Development of an Alternative Biomass-Derived Carbon Stationary Phase. Ph.D. Thesis, University of York, York, UK, 2013. [Google Scholar]
- Sargent, M. (Ed.) Guide to Achieving Reliable Quantitative LC-MS Measurements; RSC Analytical Methods Committee: Cambridge, UK, 2013. [Google Scholar]
- van de Merbel, N.C. Quantitative determination of endogenous compounds in biological samples using chromatographic techniques. TrAC Trends Anal. Chem. 2008, 27, 924–933. [Google Scholar] [CrossRef]
- Bao, X.; Wu, J.; Shuch, B.; LoRusso, P.; Bindra, R.S.; Li, J. Quantitative Profiling of Oncometabolites in Frozen and Formalin-Fixed Paraffin-Embedded Tissue Specimens by Liquid Chromatography Coupled with Tandem Mass Spectrometry. Sci Rep. 2019, 9, 11238. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wiegand, R.; LoRusso, P.; Li, J. A stable isotope-labeled internal standard is essential for correcting for the interindividual variability in the recovery of lapatinib from cancer patient plasma in quantitative LC-MS/MS analysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 941, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Kadhi, O.; Melchini, A.; Mithen, R.; Saha, S. Development of a LC-MS/MS Method for the Simultaneous Detection of Tricarboxylic Acid Cycle Intermediates in a Range of Biological Matrices. J. Anal. Methods Chem. 2017, 2017, 5391832. [Google Scholar] [CrossRef] [Green Version]
- Lotti, C.; Rubert, J.; Fava, F.; Tuohy, K.; Mattivi, F.; Vrhovsek, U. Development of a fast and cost-effective gas chromatography-mass spectrometry method for the quantification of short-chain and medium-chain fatty acids in human biofluids. Anal. Bioanal. Chem. 2017, 409, 5555–5567. [Google Scholar] [CrossRef]
- Zeng, M.; Cao, H. Fast quantification of short chain fatty acids and ketone bodies by liquid chromatography-tandem mass spectrometry after facile derivatization coupled with liquid-liquid extraction. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1083, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.Y.; Kioh, D.Y.Q.; Yap, G.C.; Lee, B.W.; Chan, E.C.Y. A novel LCMSMS method for quantitative measurement of short-chain fatty acids in human stool derivatized with 12C- and 13C-labelled aniline. J. Pharm. Biomed. Anal. 2017, 138, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.Y.; Wang, C.Y.; Lee, C.H.; Kao, H.L.; Wu, W.K.; Kuo, C.H. Development of an Efficient and Sensitive Chemical Derivatization-Based LC-MS/MS Method for Quantifying Gut Microbiota-Derived Metabolites in Human Plasma and Its Application in Studying Cardiovascular Disease. J. Proteome Res. 2021, 20, 3508–3518. [Google Scholar] [CrossRef]
- Oldendorf, W.H. Carrier-mediated blood-brain barrier transport of short-chain monocarboxylic organic acids. Am. J. Physiol. 1973, 224, 1450–1453. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, C.; Colombo, J.-P.; Berüter, J. Short chain fatty acids in plasma and brain: Quantitative determination by gas chromatography. Clin. Chim. Acta 1979, 92, 153–159. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Baere, S.; Eeckhaut, V.; Steppe, M.; De Maesschalck, C.; De Backer, P.; Van Immerseel, F.; Croubels, S. Development of a HPLC–UV method for the quantitative determination of four short-chain fatty acids and lactic acid produced by intestinal bacteria during in vitro fermentation. J. Pharm. Biomed. Anal. 2013, 80, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Day-Walsh, P.; Shehata, E.; Saha, S.; Savva, G.M.; Nemeckova, B.; Speranza, J.; Kellingray, L.; Narbad, A.; Kroon, P.A. The use of an in-vitro batch fermentation (human colon) model for investigating mechanisms of TMA production from choline, l-carnitine and related precursors by the human gut microbiota. Eur. J. Nutr. 2021, 60, 3987–3999. [Google Scholar] [CrossRef]
- Day, P.E.L.; Chambers, K.F.; Winterbone, M.S.; García-Blanco, T.; Vauzour, D.; Kroon, P.A. Validation of control genes and a standardised protocol for quantifying gene expression in the livers of C57BL/6 and ApoE−/− mice. Sci. Rep. 2018, 8, 8081. [Google Scholar] [CrossRef] [PubMed]
- García-Villalba, R.; Giménez-Bastida, J.A.; García-Conesa, M.T.; Tomás-Barberán, F.A.; Carlos Espín, J.; Larrosa, M. Alternative method for gas chromatography-mass spectrometry analysis of short-chain fatty acids in faecal samples. J. Sep. Sci. 2012, 35, 1906–1913. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Analyte | Retention Time (mins) | Precursor Ion (m/z) | Product Ion (m/z) | Collision Energy | Cell Accelerator Energy | Polarity |
|---|---|---|---|---|---|---|
| Acetate | 1.5 (2.3) * | 61.1 | 43 | 16 | 4 | Positive |
| D4-Acetate | 1.5 (2.3) * | 65.1 | 47 | 14 | 4 | Positive |
| Butyrate | 3.7 (4.9) * | 89.1 | 43.1 | 14 | 4 | Positive |
| 13C2-Butyrate | 3.7 (4.9) | 91.1 | 44 | 14 | 4 | Positive |
| Isobutyrate | 2.9 (4.9) * | 89.1 | 43.1 | 14 | 4 | Positive |
| D6-Isobutyrate | 2.9 (4.9) | 95 | 49 | 14 | 4 | Positive |
| Iso-Valerate | 4.2 (5.4) * | 103.1 | 43 | 14 | 4 | Positive |
| D9-Isovalerate | 4.2 (5.4) * | 112.2 | 50.2 | 18 | 4 | Positive |
| Lactate | 1.7 (2.0) * | 89 | 42.9 | 10 | 5 | Negative |
| 13C3-Lactate | 1.7 (2.0) * | 92 | 46 | 10 | 4 | Negative |
| Propionate | 2.3 (3.6) * | 75 | 29 | 18 | 4 | Positive |
| D2-Propionate | 2.3 (3.6) * | 77 | 31.1 | 14 | 4 | Positive |
| Valerate | 4.7 (5.4) * | 103.1 | 75 | 10 | 4 | Positive |
| D9-Valerate | 4.7 (5.4) * | 112.1 | 80 | 10 | 4 | Positive |
| Analyte | R2 | Precision (n = 6) (Intra-Day) R.S.D. % | Precision (n = 5) (Inter-Day) R.S.D. % | Accuracy (R.S.D. %) | LOD (mM) | LOQ (mM) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L | M | H | L | M | H | L | M | H | ||||
| Acetate | 0.998 | 11.3 | 3 | 2.9 | 19.3 | 4.1 | 6.1 | 98.2 | 103 | 96.6 | 0.003 | 0.009 |
| Butyrate | 0.999 | 4.6 | 2.4 | 3.6 | 16.7 | 5.4 | 4.5 | 120.4 | 102.1 | 99.8 | 0.001 | 0.003 |
| Isobutyrate | 0.999 | 2 | 1.7 | 2 | 10.2 | 7.4 | 2.3 | 107.8 | 107.9 | 102.6 | 0.001 | 0.003 |
| Isovalerate | 0.998 | 6 | 3.2 | 2.7 | 11.8 | 4.2 | 4.7 | 120 | 119.3 | 100.2 | 0.001 | 0.003 |
| Lactate | 0.999 | 2.6 | 2 | 1.7 | 9.5 | 7.6 | 2.2 | 120 | 104.9 | 98.6 | 0.001 | 0.003 |
| Propionate | 0.999 | 5.2 | 2 | 1.7 | 10 | 9 | 3.9 | 119.8 | 108.4 | 105.2 | 0.001 | 0.003 |
| Valerate | 0.998 | 8 | 4.4 | 3.9 | 14.7 | 8.4 | 5.3 | 116.3 | 111.4 | 92.9 | 0.001 | 0.003 |
| Sample Name | Acetate | Butyrate | Isobutyrate | Isovalerate | Lactate | Propionate | Valerate | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Recovery (%) | Matrix Effect (%) | Recovery (%) | Matrix Effect (%) | Recovery (%) | Matrix Effect (%) | Recovery (%) | Matrix Effect (%) | Recovery (%) | Matrix Effect (%) | Recovery (%) | Matrix Effect (%) | Recovery (%) | Matrix Effect (%) | |
| Brain | 69 | 0 | 89 | 5 | 82 | 0 | 92 | 22 | 47 | 22 | 79 | 0 | 93 | 57 |
| Faecal | 107 | 0 | 112 | 50 | 103 | 0 | 115 | 46 | 102 | 24 | 100 | 0 | 121 | 56 |
| Kidney | 89 | 17 | 97 | 33 | 96 | 0 | 107 | 34 | 96 | 37 | 104 | 0 | 109 | 53 |
| Liver | 81 | 32 | 96 | 53 | 97 | 0 | 115 | 65 | 64 | 38 | 95 | 0 | 105 | 75 |
| Milk | 95 | 34 | 79 | 11 | 76 | 0 | 112 | 0 | 97 | 0 | 92 | 0 | 109 | 0 |
| Muscle | 71 | 0 | 89 | 4 | 94 | 0 | 107 | 31 | 75 | 52 | 91 | 0 | 108 | 59 |
| Plasma | 96 | 17 | 89 | 0 | 80 | 0 | 107 | 0 | 64 | 0 | 95 | 0 | 109 | 0 |
| Spleen | 95 | 14 | 98 | 76 | 93 | 0 | 111 | 80 | 95 | 38 | 99 | 0 | 100 | 84 |
| Analytes Added | Measured Conc. (mM), Mean | Recovery (%) | CV (%) | Matrix Effect (%) |
|---|---|---|---|---|
| Acetate | ||||
| 0 | 1.97 | - | 5.79 | 43.2 |
| L (0.1 mM) | 2.07 | 98 | 0.33 | 42.7 |
| M (1.0 mM) | 3.10 | 113 | 0.02 | 42.5 |
| H (10 mM) | 11.6 | 97 | 1.88 | 37.7 |
| Butyrate | ||||
| 0 | 0.13 | - | - | 0 |
| L (0.1 mM) | 0.25 | 120 | 3.85 | 0 |
| M (1.0 mM) | 1.21 | 108 | 0.01 | 0 |
| H (10 mM) | 10.1 | 100 | 0.61 | 0 |
| Isobutyrate | ||||
| 0 | 0.05 | - | 2.04 | 2.50 |
| L (0.1 mM) | 0.17 | 116 | 3.64 | 11.21 |
| M (1.0 mM) | 1.12 | 107 | 0.02 | 9.47 |
| H (10 mM) | 9.83 | 98 | 1.60 | 14.98 |
| Isovalerate | ||||
| 0 | 0.01 | - | 6.25 | 18.44 |
| L (0.1 mM) | 0.12 | 109 | 5.09 | 17.52 |
| M (1.0 mM) | 1.01 | 100 | 0.04 | 16.57 |
| H (10 mM) | 8.85 | 88 | 1.39 | 18.41 |
| Lactate | ||||
| 0 | 0.16 | - | 5.90 | 23.98 |
| L (0.1 mM) | 0.28 | 117 | 1.96 | 29.43 |
| M (1.0 mM) | 1.21 | 105 | 0.02 | 32.08 |
| H (10 mM) | 9.61 | 95 | 0.87 | 22.78 |
| Propionate | ||||
| 0 | 0.15 | - | 8.48 | 7.13 |
| L (0.1 mM) | 0.27 | 116 | 5.64 | 14.85 |
| M (1.0 mM) | 1.24 | 109 | 0.01 | 10.67 |
| H (10 mM) | 9.81 | 97 | 0.12 | 16.08 |
| Valerate | ||||
| 0 | 0.02 | - | 20.18 | 10.86 |
| L (0.1 mM) | 0.14 | 117 | 5.26 | 9.41 |
| M (1.0 mM) | 1.22 | 120 | 0.05 | 3.01 |
| H (10 mM) | 10.0 | 100 | 1.94 | 7.36 |
| Methodological Consideration | GC-MS | HPLC | LC-MS/MS | Current |
|---|---|---|---|---|
| Derivatization | Yes | No | Yes | No |
| Sensitivity | µM range | mM range | µM range | µM range |
| Instrument run time | Long | Long | Medium | short |
| Sample preparation | Laborious | Simple | Laborious | Simple |
| BSCFA detection | Yes | No | Yes | Yes |
| Lactate detection | No | Yes | No | Yes |
| Tested matrices | 4 | 3 | 3 | 9 |
| Sample preparation time | Long | Long | Long | Short (~10 min) |
| Instrument run time | 14–45 min | 45–75 min | 14–35 min | 10 min |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, S.; Day-Walsh, P.; Shehata, E.; Kroon, P.A. Development and Validation of a LC-MS/MS Technique for the Analysis of Short Chain Fatty Acids in Tissues and Biological Fluids without Derivatisation Using Isotope Labelled Internal Standards. Molecules 2021, 26, 6444. https://doi.org/10.3390/molecules26216444
Saha S, Day-Walsh P, Shehata E, Kroon PA. Development and Validation of a LC-MS/MS Technique for the Analysis of Short Chain Fatty Acids in Tissues and Biological Fluids without Derivatisation Using Isotope Labelled Internal Standards. Molecules. 2021; 26(21):6444. https://doi.org/10.3390/molecules26216444
Chicago/Turabian StyleSaha, Shikha, Priscilla Day-Walsh, Emad Shehata, and Paul Anthony Kroon. 2021. "Development and Validation of a LC-MS/MS Technique for the Analysis of Short Chain Fatty Acids in Tissues and Biological Fluids without Derivatisation Using Isotope Labelled Internal Standards" Molecules 26, no. 21: 6444. https://doi.org/10.3390/molecules26216444
APA StyleSaha, S., Day-Walsh, P., Shehata, E., & Kroon, P. A. (2021). Development and Validation of a LC-MS/MS Technique for the Analysis of Short Chain Fatty Acids in Tissues and Biological Fluids without Derivatisation Using Isotope Labelled Internal Standards. Molecules, 26(21), 6444. https://doi.org/10.3390/molecules26216444