Abstract
To search for novel androgen receptor (AR) modulators for the potential treatment of castration-resistant prostate cancer (CRPC), naturally occurring silibinin was sought after as a lead compound because it possesses a moderate potency towards AR-positive prostate cancer cells and its chemical scaffold is dissimilar to all currently marketed AR antagonists. On the basis of the structure–activity relationships that we have explored, this study aims to incorporate carbamoyl groups to the alcoholic hydroxyl groups of silibinin to improve its capability in selectively suppressing AR-positive prostate cancer cell proliferation together with water solubility. To this end, a feasible approach was developed to regioselectively introduce a carbamoyl group to the secondary alcoholic hydroxyl group at C-3 without causing the undesired oxidation at C2–C3, providing an avenue for achieving 3-O-carbamoyl-5,7,20-O-trimethylsilybins. The application of the synthetic method can be extended to the synthesis of 3-O-carbamoyl-3′,4′,5,7-O-tetramethyltaxifolins. The antiproliferative potency of 5,7,20-O-trimethylsilybin and its nine 3-carbamoyl derivatives were assessed in an AR-positive LNCaP prostate cancer cell line and two AR-null prostate cancer cell lines (PC-3 and DU145). Our preliminary bioassay data imply that 5,7,20-O-trimethylsilybin and four 3-O-carbamoyl-5,7,20-O-trimethylsilybins emerge as very promising lead compounds due to the fact that they can selectively suppress AR-positive LNCaP cell proliferation. The IC50 values of these five 5,7,20-O-trimethylsilybins against the LNCaP cells fall into the range of 0.11–0.83 µM, which exhibit up to 660 times greater in vitro antiproliferative potency than silibinin. Our findings suggest that carbamoylated 5,7,20-O-trimethylsilybins could serve as a natural product-based scaffold for new antiandrogens for lethal castration-resistant prostate cancer.
1. Introduction
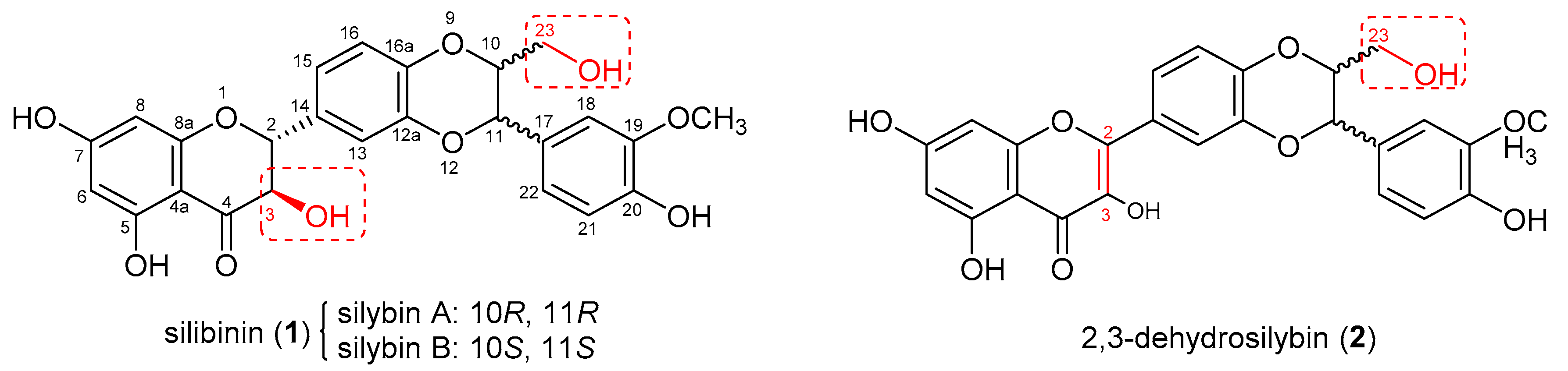
Castration-resistant prostate cancer (CRPC) is a lethal version of prostate cancer that continues to deprive the lives of over 30,000 American men per year [1]. The androgen receptor (AR)-regulated gene expression still holds the foremost impetus for the progression of CRPC [2]. In addition to acquiring drug resistance with the time of treatment, a considerable portion of patients with CRPC are primarily resistant to the current FDA-approved treatments targeting the AR-signaling axis as evidenced by their hazard ratios for the primary end points in the phase III trials [3]. Consequently, novel potential therapeutic strategies for CRPC, including poly(adenosine diphosphate (ADP)-ribose)polymerase inhibitors [4,5], androgen receptor degraders [6,7], and CAR-T cell therapy [8], are emerging. As part of our ongoing project to discover new AR modulators with dissimilar chemical scaffolds for the potential treatment of CRPC, this study picks up naturally occurring silibinin (1, also known as silybin, Figure 1) as a lead compound because it has a chemical scaffold completely different from all marketed AR antagonists and possesses moderate potency towards AR-positive prostate cancer cells. Silibinin (1) is the most abundant flavonolignan of silymarin, a well-known traditional European medicine and the crude extract from milk thistle (Silybum marianum) [9]. Its initial name is silybin because it was considered as a single compound in 1968 [9] and later silibinin was recommended as an alternative name to highlight the fact that it is a diastereomeric mixture of silybin A and silybin B [10]. Throughout this paper, silibinin is used to stand for the commercially available mixture of silybin A and silybin B. In addition to having appreciable potential in treating prostate cancer on the ground of its in vitro and in vivo experimental data [11,12,13], silibinin (1) acts as an anti-prostate cancer agent with mechanisms of action that are associated with the AR-signaling axis as summarized in our review article [14]. Specifically, silibinin can inhibit the secretion of prostate-specific antigens (PSA) [11,15,16,17], lower the AR level, prevent AR nuclear localization, and promote AR degradation [11,16,18]. Its non-toxic profiles, which have been corroborated by its long-term dietary application along with its clinical trial data, make it even more alluring as a lead compound [19]. However, its moderate potency, poor selectivity, and poor bioavailability pose significant impediments to clinical applications. Chemical manipulations have been verified by us and others to be a viable strategy to boost its potency [20,21,22,23,24] and to improve its pharmacokinetic profiles [25,26].
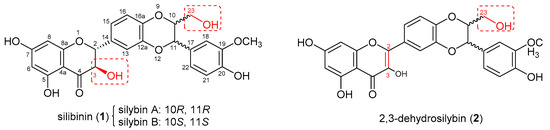
Figure 1.
Chemical structures of silibinin (1) and 2,3-dehydrosilybin (2).
The earlier studies on the structure–activity relationships of silibinin (1) and 2,3-dehydrosilybin (2) from our laboratories revealed that chemical modification on silibinin (1) led to significantly higher selectivity in the proliferation inhibition of AR-positive LNCaP cells vs. AR-null PC-3 and DU145 cells when compared with the same chemical modification on 2,3-dehydrosilybin (2) [20,21,22]. Our recent investigation suggests that modification of the alcoholic hydroxyl group at C-23 of 3,5,7,20-O-tetramethyl-2,3-dehydrosilybin resulted in appreciably higher selectivity in suppressing AR-positive LNCaP prostate cancer cell proliferation compared with modification of the phenolic hydroxyl groups [27]. These results prompted us to aim for the appropriately designed substituents at alcoholic hydroxyl groups at C-3 and C-23 for 5,7,20-O-trimethylsilybin (3) in hopes to enhance the antiproliferative potency and selectivity towards AR-positive prostate cancer cells. Structure manipulations on the alcoholic hydroxyl groups at C-3 and C-23 have been reported to yield silybin 3,23-bis-O-hemisuccinate and 23-phosphodiester silybin with improved pharmacokinetic profiles compared with silibinin [25,26]. However, these derivatives do not show significant improvement in antiproliferative potency against prostate cancer cells (both LNCaP and PC-3 cell lines). The carbamate-incorporated compounds have been proven to possess sufficient water solubility and improved biological activity [28,29]. The carbamate derivatives of 5,7,20-O-trimethylsilybin (3) have thus been designed to fine-tune the alcoholic hydroxyl groups at C-23 and C-3 in hopes to simultaneously increase the potency, selectivity, and aqueous solubility. In this paper, a regioselective synthesis of 3-O-carbamoyl-5,7,20-O-trimethylsilybins, along with the antiproliferative potency towards AR-containing and AR-null prostate cancer cell lines, are presented.
2. Results and Discussion
2.1. Synthesis
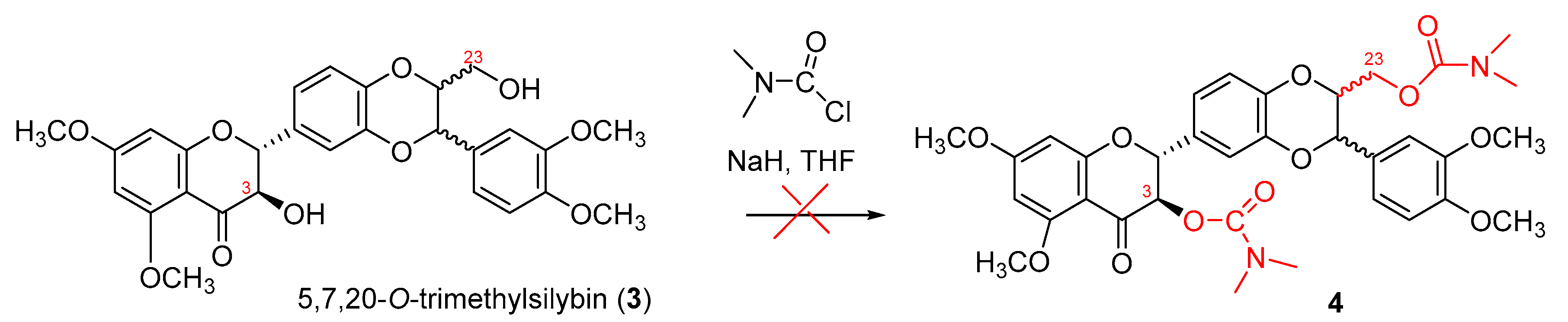
So far, no 3-O-carbamoyl derivative of flavanonol-based flavonolignans has yet been reported. The synthesis of 3-O-carbamoyl derivatives of flavanonol-based flavonolignans could be a challenge due to the fact that an oxidation at the C-2, C-3 position can readily occur under the basic conditions [30] and the typical bases employed for the carbamoylation of the alcoholic hydroxyl groups are NaH or KH [31]. Encouraged by the successful preparation of 7-O-benzylsilybin and 5,7,20-O-trimethylsilybin mediated by potassium carbonate under strictly anaerobic conditions [21], we initially attempted to synthesize 3,23-O-dicarbamoyl-5,7,20-O-trimethylsilybin (4) by treating 5,7,20-O-trimethylsilybin (3) [21] with N,N-dimethylcarbamoyl chloride using NaH as a base under strictly anaerobic conditions (Scheme 1). Unfortunately, the TLC plates from our several trials showed this reaction was very messy and did not yield the desired carbamate 4, suggesting the starting material 3 (5,7,20-O-trimethylsilybin) was decomposed under the strong basic conditions [32].
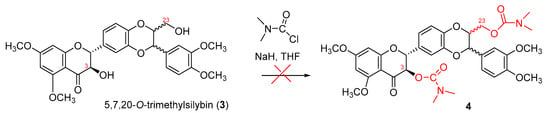
Scheme 1.
Original attempt to synthesize dicarbamoylsilybin 4.
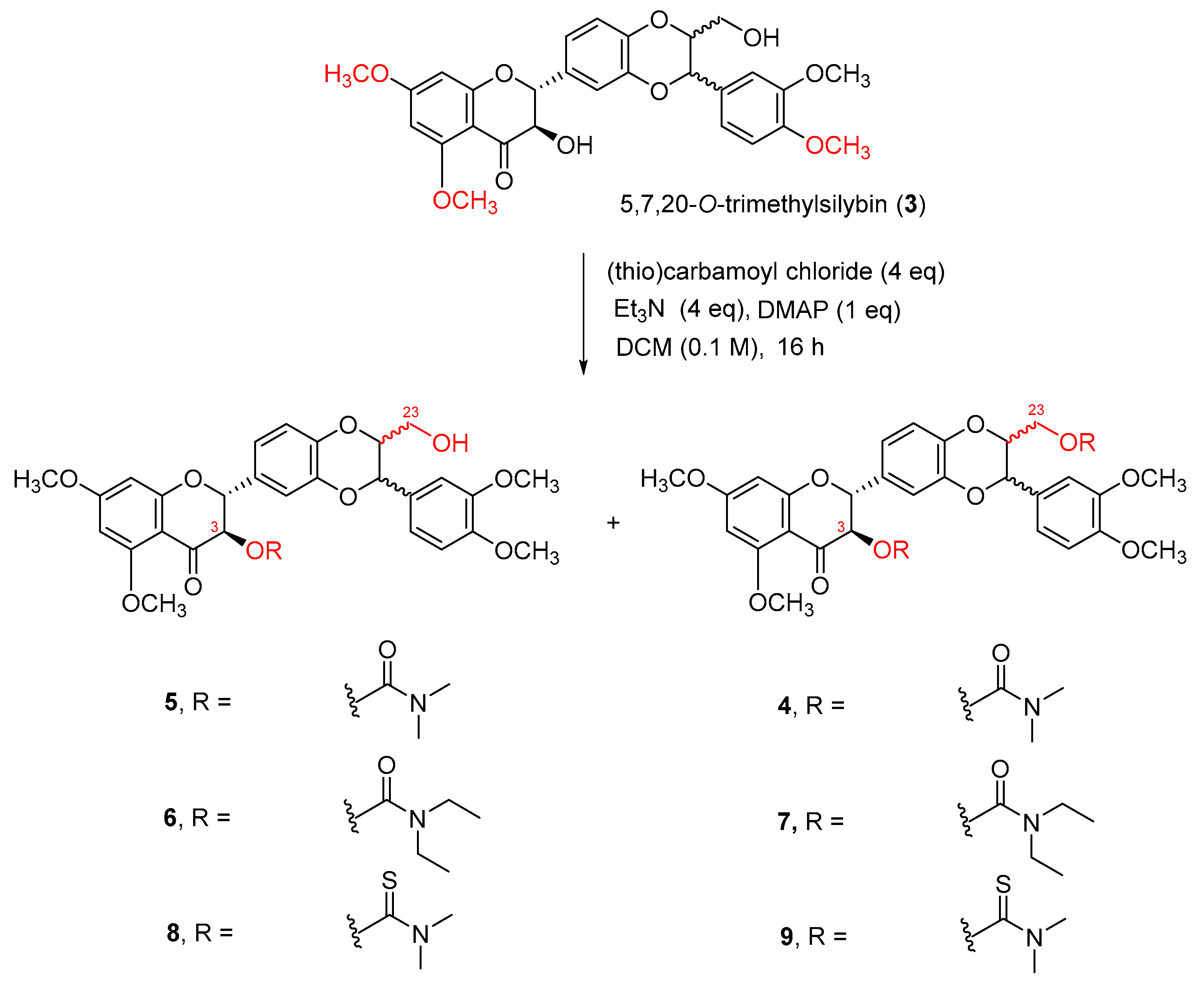
At this point, we revisited the literature and searched for weaker organic bases for this carbamoylation. It has been reported that carbamates could be achieved in good yield by refluxing alcohols with N,N-dimethylcarbamoyl chloride in pyridine [33]. These conditions are not appropriate for the synthesis of 3-O-carbamoylsilybin because heating silibinin at 80−90 °C in dry pyridine in the presence of air leads to the oxidation at C2–C3 [34]. A secondary benzylic alcohol was reported to be converted to the corresponding carbamate by treating it with a bulky carbamoyl chloride mediated by Et3N in DCM [35,36]. By integrating these two carbamoylation methods, we decided to prepare 3,23-O-dicarbamoyl-5,7,20-O-trimethylsilybin by treating 5,7,20-O-trimethylsilybin with N,N-dimethylcarbamoyl chloride (4 eq) in DCM (0.1 M) using triethylamine (4 eq) and 4-(N,N-dimethylamino)pyridine (DMAP,1 eq) at room temperature for 16 h (Scheme 2). Surprisingly, the carbamoylation regioselectively occurs at the secondary alcohol at C-3 in the presence of the primary alcohol at C-23 when treating 5,7,20-O-trimethylsilybin (3) with N,N-dimethylcarbamoyl chloride using triethylamine and DMAP as bases. This unexpected regioselective reaction has been repeated over forty times by two co-authors, and the structure of the 3-O-carbamoyl-5,7,20-O-trimethylsilybin (5) has been confirmed by 2D-NMR data and HRMS (see Structure Determination). Removal of DMAP resulted in no carbamoylation reaction, and the minimum amount of DMAP required for the completion of N,N-dimethylcarbamoylation of 5,7,20-O-trimethylsilybin (3) was 0.5 equivalents, suggesting an appropriate amount of DMAP is crucial for facilitating the carbamoylation.
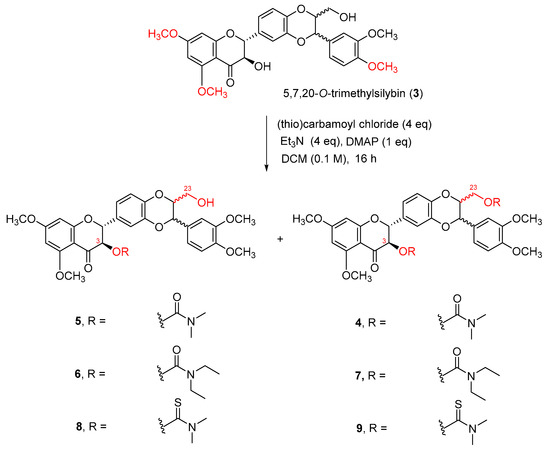
Scheme 2.
Carbamoylation of 5,7,20-O-trimethylsilybin.
In addition to obtaining the major 3-O-carbamoyl-5,7,20-O-trimethylsilybin (5) in 79% yield, a small amount of 5,7,20-O-trimethyl-3,23-O-di-(N,N-dimethylcarbamoyl)silybin (4) was also isolated in 13% yield (Table 1, entry 1). Adding more triethyl amine and DMAP, prolonging reaction time, or increasing reaction temperature can slightly increase the yields for 3,23-O-dicarbamoylsilbybin (4), but cannot promote the completion of carbamoylation at the primary alcoholic hydroxyl group at C-23 (Table 1, entries 4 and 7). This regioselective carbamoylation at the secondary alcoholic hydroxyl group at C-3 can be extended to other carbamoyl chlorides. A similar tendency was observed when reacting 5,7,20-O-trimethylsilybin (3) with N,N-diethylcarbamoyl chloride (Table 1, entries 5 and 8) or N,N-dimethylthiocarbamoyl chloride (Table 1, entries 6 and 9). It is worth noting that the bulky carbamoyl chlorides (i.e., N,N-diethylcarbamoyl chloride and N,N-dimethylthiocarbamoyl chloride) significantly decrease the conversion rates when compared with N,N-dimethylcarbamoyl chloride under the same set of carbamoylation conditions (Table 1).

Table 1.
Reaction conditions and yields for the carbamoylation of 5,7,20-O-trimethylsilybin (3).
2.2. Structure Determination of 3-O-Carbamoyl-5,7,20-O-trimethylsilybin 5
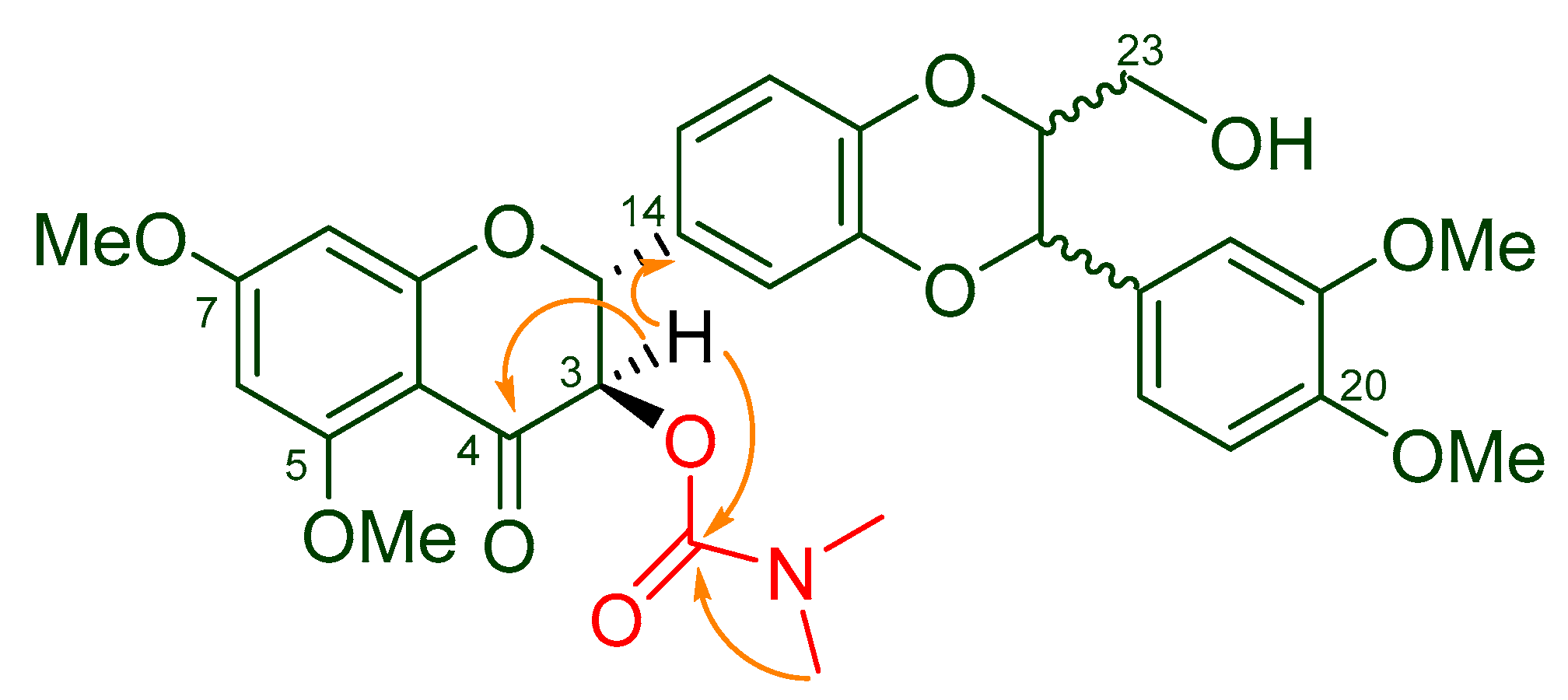
The structure of 5,7,20-O-trimethyl-3-O-(N,N-dimethylcarbamoyl)-silybin (5) was elucidated by interpreting its 1D- and 2D-NMR data (Table 2), as well as high resolution MS and IR data. The structure of 5 was characterized by the existence of one signal at 2.85 ppm representing 6 protons in its 1H NMR spectrum (Supplementary Materials) and at 36.75 (36.08) and 155.28 in its 13C NMR spectrum for an additional dimethylcarbamoyl group when compared with the starting material 5,7,20-O-trimethylsilybin (3), which was corroborated by the HRMS data. The dimethylcarbamoyl group in 5 was assigned to 3-OH based on the fact that the H-3 signal is downshifted from 4.42 ppm to 5.51 (5.49) ppm relative to 5,7,20-O-trimethylsilybin (3) [21]. The carbamoylation at 3-OH was further confirmed by the critical HMBC correlations from the signal at δH 5.51 (5.49) (H-3) to the signal at δC 155.28 (carbonyl carbon from the dimethylcarbamoyl group, Figure 2). It is worth noting that the starting material silibinin (1) (purchased from Fisher Scientific) was a nearly equimolar diastereoisomeric mixture of silybin A and silybin B. The carbamoyled derivatives of 5,7,20-O-trimethylsilybin are thus diastereoisomeric mixtures as well, and some NMR signals of these derivatives therefore appear as a pair (see Table 2 and Materials and Methods).
Table 2.
NMR data for 5 (1H NMR: 300 MHz; 13C NMR: 75 MHz).
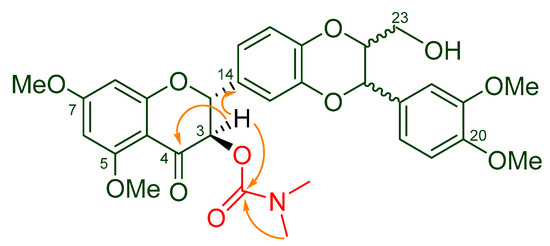
Figure 2.
Key HMBC correlations (from H to C) in 5.
2.3. Scope of the Reaction Approach
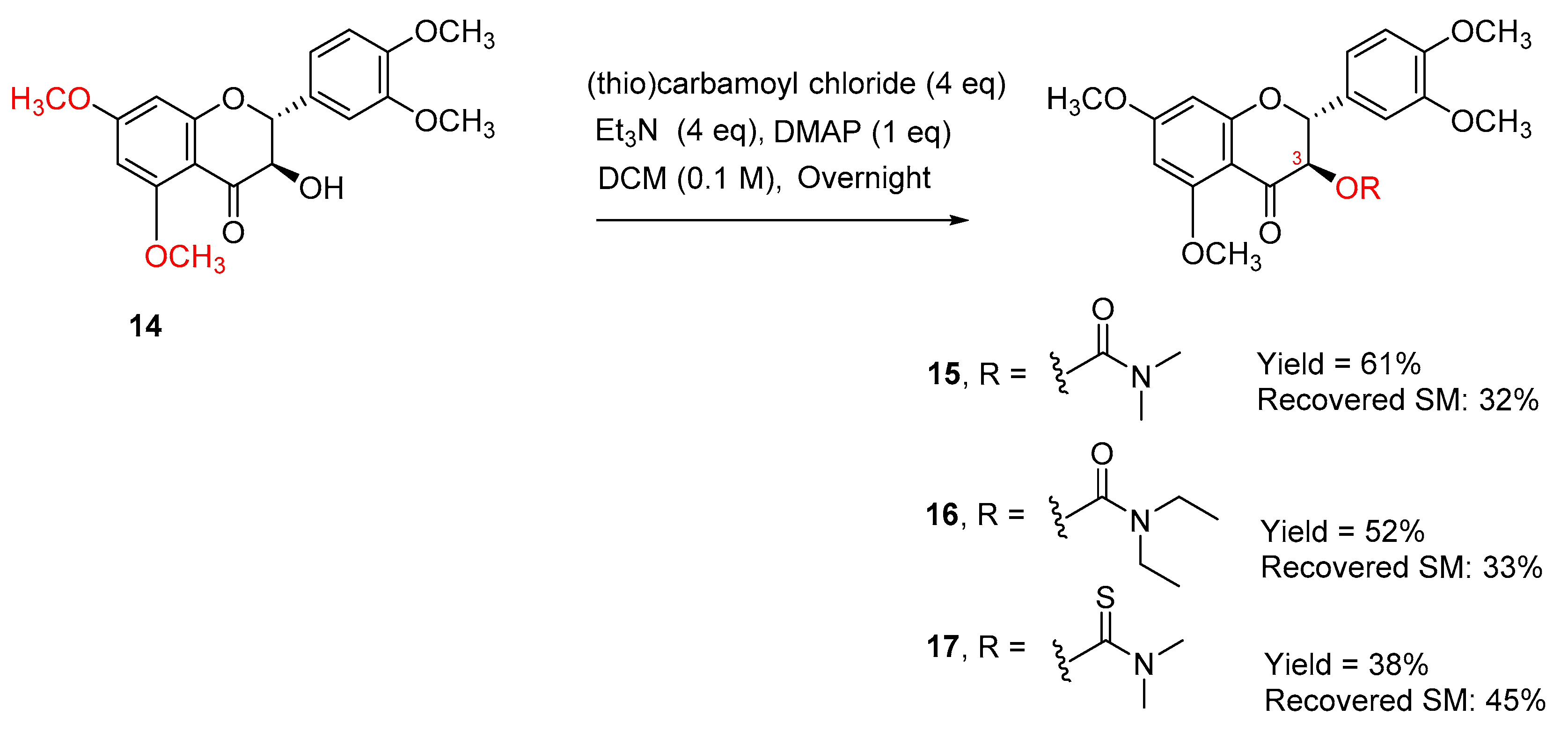
The selectivity may be caused by the hydrogen bonding between the hydrogen of the alpha-OH and the 4-carbonyl oxygen atom. To further confirm that the secondary 3-OH in 5,7,20-O-trimethylsilybin can be carbamoyled by using triethylamine and DMAP as bases, 5,7,20-O-trimethyl-23-O-TBS-silybin (10) was exposed to the same set of reaction conditions, resulting in three 3-O-carbamoyl-5,7,20-O-trimethyl-23-O-TBS-silybins (11, 12, and 13) in 45–67% yields together with the recovered starting material 10 (27–46%), as shown in Scheme 3. To further explore the applicability of the new carbamoylation method to flavanonols, the application of the same set of reaction conditions to 3′,4′,5,7-O-tetramethyltaxifolin (14) led to the formation of three desired taxifolin derivatives, 15, 16, and 17, in 38–61% yields along with the recovered starting material 14 (32–45%) (Scheme 4). This implies that the carbamoylation provides a feasible approach to not only 3-O-carbamoylated flavonolignans but also 3-O-carbamoylated flavanonols.
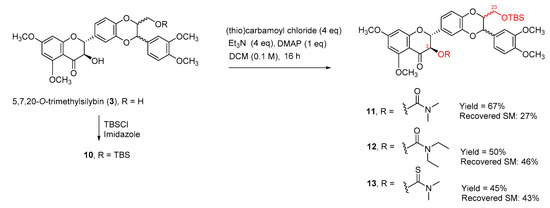
Scheme 3.
Synthesis of 3-O-carbamoyl-23-O-TBS-5,7,20-trimethylsilybins.
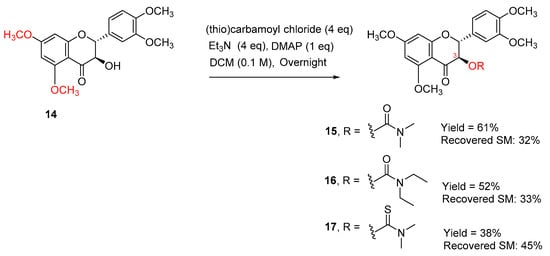
Scheme 4.
Synthesis of 3-O-carbamoyltaxifolins.
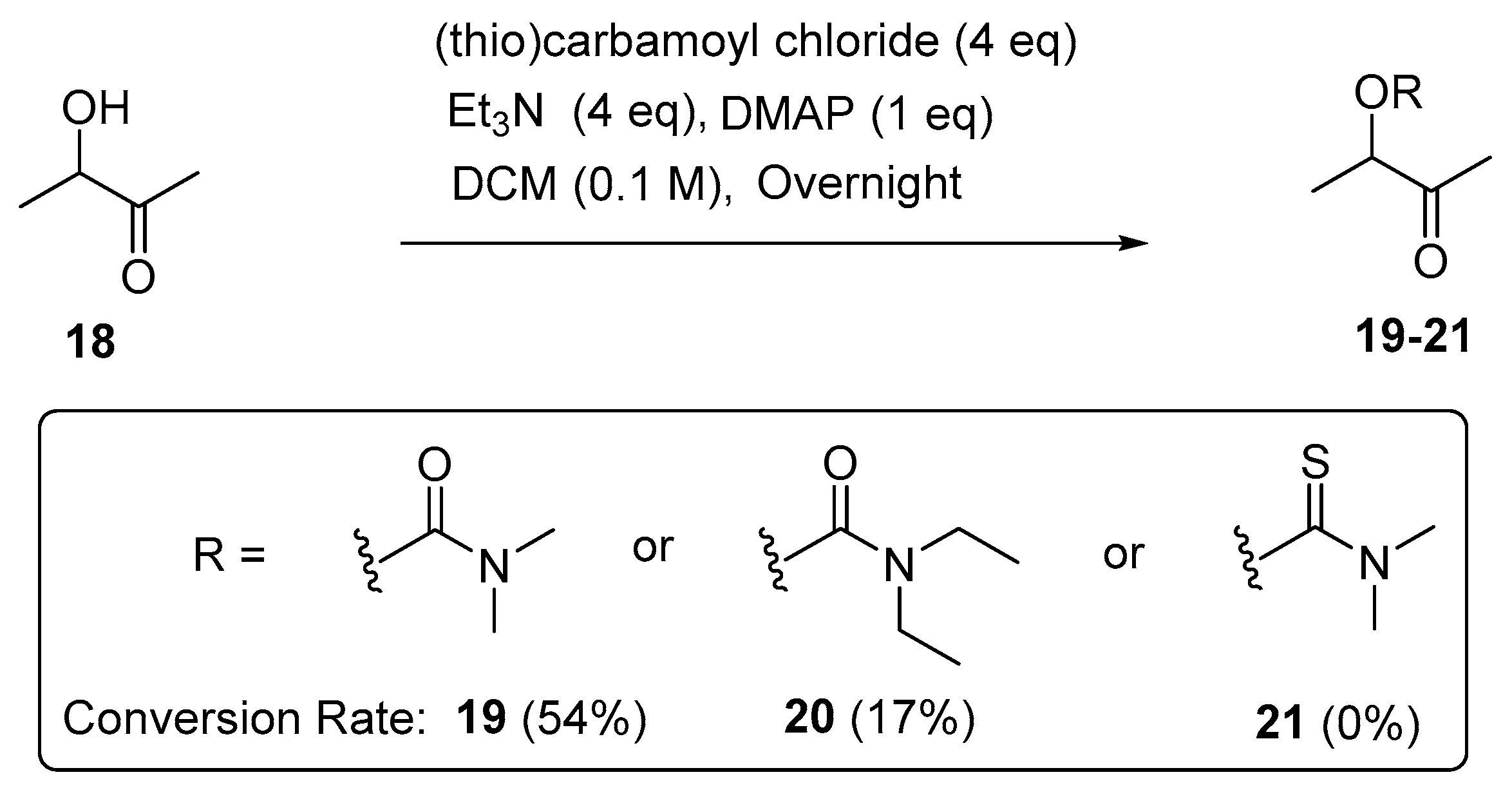
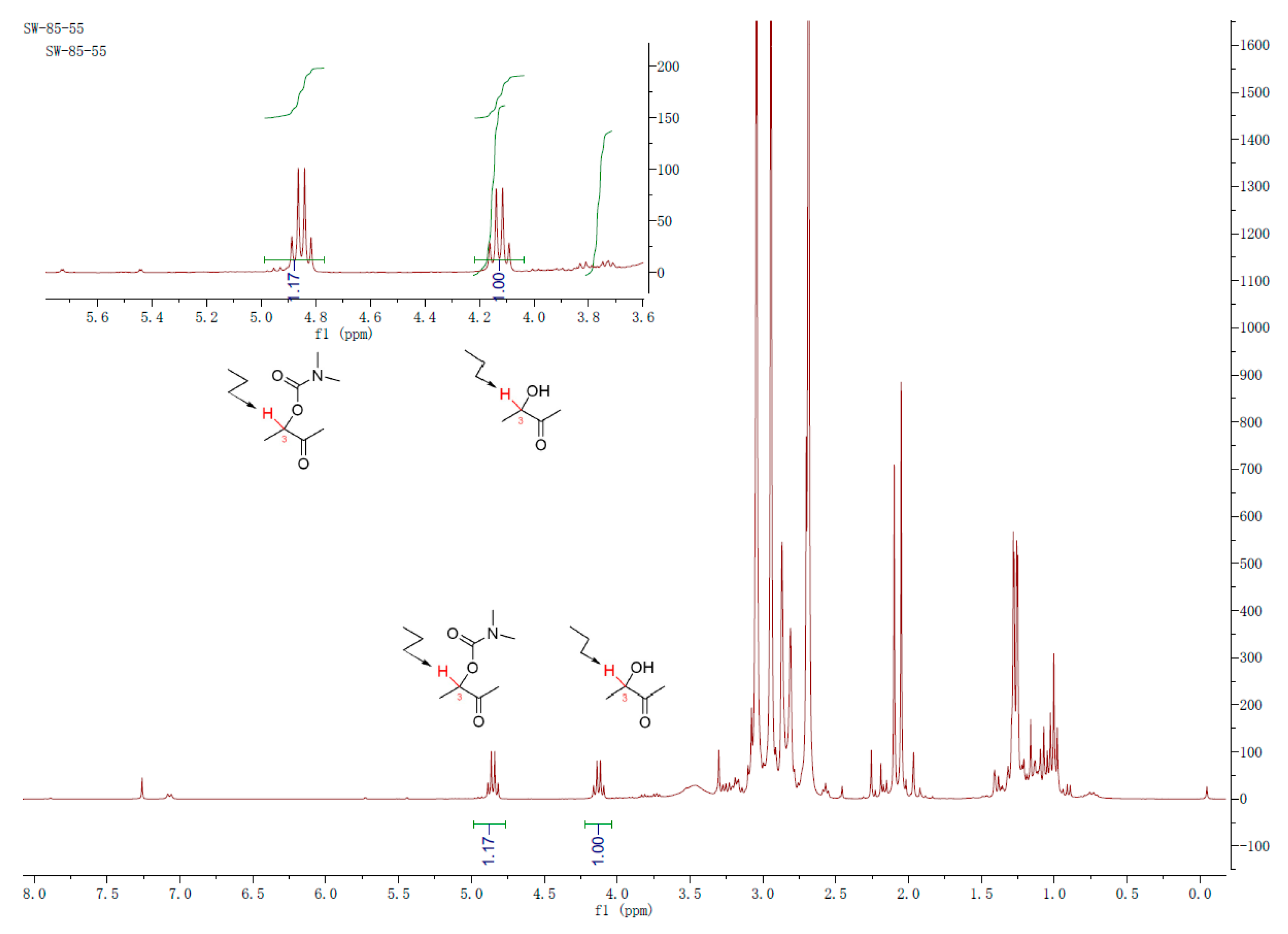
The applicability of the method was also evaluated in the open-chain α-hydroxyl ketones. Acyclic 3-hydroxy-2-butanone (18) was selected and treated with the respective (thio)carbamoyl chloride (4 eq) in the presence of triethylamine (4 eq) and DMPA (1 eq) in DCM (0.1 M). The 1H NMR analysis (Figure 3) of the characteristic H-3 in the crude product indicated that 54% and 17% of 3-hydroxy-2-butanone (18) can be converted to the corresponding N,N-dimethylcarbamoyl product (19) and N,N-diethylcarbamoyl product (20), respectively (Scheme 5). The overall conversion rates for the open-chain α-hydroxyl ketones are lower than those for the cyclic α-hydroxyl ketones, and no carbamoylation reaction of 3-hydroxy-2-butanone with N,N-dimethylthiocarbamoyl halide under the above-mentioned conditions was observed.
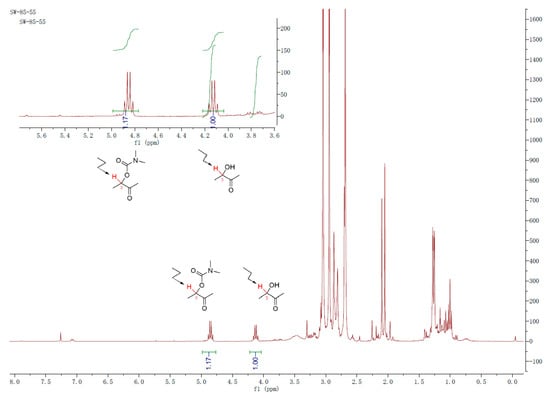
Figure 3.
1H NMR spectrum for the crude product from reaction of α-hydroxyl ketones with N,N-dimethylcarbamoyl chloride.
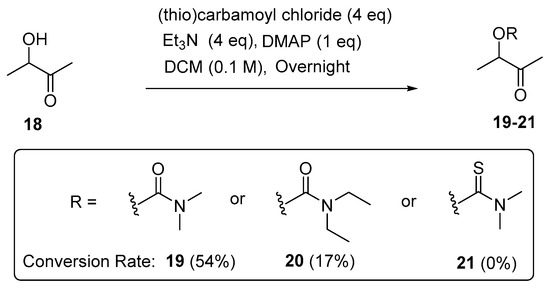
Scheme 5.
Carbamoylation of open-chain α-hydroxyl ketones.
2.4. Preliminary Anti-Proliferative Activity towards AR-Positive and AR-Null Prostate Cancer Cell Lines
The preliminary in vitro antiproliferative potency of 5,7,20-O-trimethylsilybin (3) and its nine carbamoyled derivatives towards the AR-positive LNCaP prostate cancer cell line was assessed using the WST-1 cell proliferation assay according to the procedure described in the Materials and Methods section. The AR-null DU145 and PC-3 prostate cancer cell models were used to evaluate the inhibitory selectivity towards AR-positive cells over AR-null ones. Enzalutamide, a current FDA-approved second-generation AR antagonist for CRPC, and silibinin were used as positive controls for comparison. The IC50 values were calculated based on the dose-response curves and summarized in Table 3. The IC50 values for 5,7,20-O-trimethylsilybin (3) and four derivatives (4, 5, 6, and 8) against the LNCaP cells are 0.11–0.83 µM, which exhibit up to 660 times greater in vitro antiproliferative potency than silibinin. All these 5,7,20-O-trimethylsilybins are also significantly more potent than enzalutamide in the AR-positive LNCaP cell model. These five 5,7,20-O-trimethylsilybins are clearly more potent in suppressing cell proliferation of AR-positive LNCaP prostate cancer cells than AR-negative PC-3 and DU145 cells by comparing their IC50 values.
Table 3.
Seventy-two hours of antiproliferative activity of carbamoyled 5,7,20-O-trimethylsilybins.
On the grounds of our preliminary bioassay data, 5,7,20-O-trimethylsilybin (3) and its four 3-substituted derivatives (4, 5, 6, and 8) emerge as very promising lead compounds due to the fact that they can selectively suppress AR-positive LNCaP cell proliferation with IC50 values of 0.11−0.83 µM and have more efficacy than the current FDA-approved second-generation AR antagonist enzalutamide (Table 3). Our findings suggest that 3-O-substituted-5,7,20-O-trimethylsilybin may serve as a natural product-based scaffold for new antiandrogens for lethal castration-resistant prostate cancer.
3. Materials and Methods
3.1. General Procedures
IR spectra were recorded on a Nicolet Nexus 470 FTIR spectrophotometer (Waltham, MA, USA). HRMS were obtained on an Orbitrap mass spectrometer with electrospray ionization (ESI). NMR spectra were obtained on a Bruker Fourier 300 spectrometer (Billerica, MA, USA) in CDCl3. The chemical shifts are given in ppm referenced to the respective solvent peak, and coupling constants are reported in Hz. The value of the central peak of the solvent was calibrated as δ = 7.26 ppm for the 1H NMR spectrum and as δ = 77.16 ppm for the 13C NMR spectrum, respectively. THF and dichloromethane were purified by the PureSolv MD 7 Solvent Purification System from Innovative Technologies (MB-SPS-800) (Herndon, VA, USA). All other reagents and solvents were purchased from commercial sources and were used without further purification. Silica gel column chromatography was performed using silica gel (32–63 µm). Preparative thin-layer chromatography (PTLC) separations were carried out on thin-layer chromatography plates loaded with silica gel 60 GF254 (EMD Millipore Corporation) (Berlington, MA, USA). 5,7,20-O-Trimethylsilybin (3) was synthesized from silibinin (>98%, purchased from Fisher Scientific (Portland, OR, USA)) using the procedure previously described by us [21]. The HPLC purity analyses were performed on an Agilent Hewlett-Packard 1100 series HPLC DAD system (Santa Clara, CA, USA) using a 5 µM C18 reversed phase column (4.6 × 250 mm) and a diode array detector. Solvent A was methanol and solvent B was 5% methanol in DI water. All testing samples were run for 30 min of 35−100% A in B, with a 20 min gradient. The flow rate was 1 mL/min.
3.2. General Procedure for the Synthesis of Carbamoyled Derivatives of 5,7,20-Trimethylsilybin
Triethylamine (56 µL, 0.4 mmoL) and 4-(N,N-dimethylamino)pyridine (12 mg, 0.1 mmol) were sequentially added to a solution of 5,7,20-O-trimethylsilybin (3, 52.5 mg, 0.1 mmol) in DCM (1 mL, 0.1 M). The subsequent mixture was stirred for 10 min at room temperature, to which the respective (thio)carbamoyl chloride (0.4 mmol) was added. The reaction was allowed to proceed at room temperature with stirring overnight under argon prior to being quenched with brine (50 mL). The resulting mixture was extracted with ethyl acetate (30 mL × 3), the combined organic extracts were dried over anhydrous sodium sulfate, and the organic solvents were removed. The crude product was subjected to PTLC purification eluting with DCM:MeOH (95:5, v/v) to afford the respective 3-carbomoyled derivative and 3,23-dicarbomoyled derivative. Their physical and spectral data are summarized below.
3.2.1. 5,7,20-O-Trimethyl-3-O-(N,N-dimethylcarbamoyl)silybin (5)
Yield, 79%; white solid; m.p. 113−115 °C. 1H NMR (300 MHz, CDCl3) and 13C NMR (75 MHz, CDCl3): see Table 2. HRMS (ESI): m/z calculated for C31H34NO11 [M+H]+: 596.2132. Found: 596.2128. IR (film) νmax: 3361, 2943, 2833, 1689, 1610, 1572, 1509 cm−1. HPLC purity 100% (two very close signals were observed for the diastereomers).
3.2.2. 5,7,20-O-Trimethyl-3,23-O-di(N,N-dimethylcarbamoyl)silybin (4)
Yield, 13%; colorless syrup. 1H NMR (300 MHz, CDCl3) δ 7.15 (d, J = 1.2 Hz, 1H, H-13), 6.09–7.01 (overlapped, 2H, H-15 and H-16), 6.94 (dt, J = 8.4, 1.5 Hz, 1H, H-22), 6.90–6.87 (overlapped, 2H, H-18 and H-22), 6.11 (d, J = 2.1 Hz, 1H, H-6), 6.09 (d, J = 2.4 Hz, 1H, H-8), 5.53 (5.51) (d, J = 11.7 Hz, 1H, H-3), 5.31 (5.30) (d, J = 11.7 Hz, 1H, H-2), 4.91 (4.90) (d, J = 7.8 Hz, 1H, H-11), 4.34 (dd, J = 11.9, 3.0 Hz, 1H, H-23), 4.27 (ddd, J = 7.8, 4.2, 3.0 Hz, 1H, H-10), 3.97 (dd, J = 11.9, 4.2 Hz, 1H, H-23), 3.91 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 2.88 (s, 12H, 2 × N(CH3)2). 13C NMR (75 MHz, CDCl3) δ 186.41 (C-4), 166.48, 164.24, 162.48, 155.90, 155.34, 149.87, 149.57 (149.51), 143.98, 143.86 (143.83), 129.63 (129.52), 128.34, 121.05, 120.22, 117.31 (117.21), 116.50 (116.39), 111.43 (111.35), 110.18 (110.02), 104.38 (104.34), 93.48, 81.03 (80.94), 76.87, 76.12, 74.75 (74.62), 63.94, 56.19 (56.13), 56.10, 56.05 (56.00), 55.77 (55.72), 36.79, 36.14. HRMS (ESI): m/z calculated for C34H39N2O12 [M+H]+: 667.2503. Found: 667.2498. IR (film) νmax: 2935, 2839, 1701, 1608, 1573, 1539, 1508 cm−1. HPLC purity 96.9% (only one signal was observed for the diastereomers).
3.2.3. 3-O-(N,N-Diethylcarbamoyl)-5,7,20-O-trimethylsilybin (6)
Yield, 34%; slight yellow solid; 114–116 °C. 1H NMR (300 MHz, CDCl3) δ 7.16 (7.13) (d, J = 1.5 Hz, 1H, H-13), 7.03–6.92 (overlapped, 4H, H-15, H-16, H-18, H-22), 6.89 (d, J = 8.1 Hz, 1H, H-21), 6.11 (d, J = 2.1 Hz, 1H, H-6), 6.08 (d, J = 2.1 Hz, 1H, H-8), 5.54 (d, J = 11.7 Hz, 1H, H-3), 5.30 (5.28) (d, J = 11.7 Hz, 1H, H-2), 4.95 (4.94) (d, J = 8.1 Hz, 1H, H-11), 4.02 (ddd, J = 8.1, 5.4, 3.6 Hz, 1H, H-10), 3.91 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 3.79 (3.78) (s, 3H, OCH3), 3.82−3.77 (overlapped, 1H, H-23), 3.53 (dd, J = 12.3, 3.6 Hz, 1H, H-23), 3.33–3.10 (m, 4H, 2 × CH2CH3), 1.11–0.97 (m, 6H, 2 × CH2CH3). 13C NMR (75 MHz, CDCl3) δ 186.47, 166.38, 164.18, 162.41, 154.51 (154.44), 149.72, 149.49 (149.43), 143.99 (143.92), 129.66 (129.58), 128.61 (128.58), 120.92 (120.89), 120.22 (120.18), 117.12 (117.00), 116.55, 111.33 (111.27), 110.19, 110.12 (110.07), 104.42 (104.38), 93.52 (93.47), 93.44 (93.39), 81.13 (81.05), 78.37, 76.34 (76.24), 74.31, 61.70, 56.21 (56.15), 56.07, 56.01 (55.96), 55.75 (55.69), 42.40 (41.55), 14.00 (13.44). HRMS (ESI): m/z calculated for C33H38NO11 [M+H]+: 624.2445. Found: 624.2443. IR (film) νmax: 3468, 2964, 2933, 2838, 1686, 1607, 1571, 1508 cm−1. HPLC purity 97.8% (two very close signals were observed for the diastereomers).
3.2.4. 3,23-O-Di(N,N-diethylcarbamoyl)-5,7,20-O-trimethylsilybin (7)
Yield, 9%; colorless wax. 1H NMR (300 MHz, CDCl3) δ 7.17 (7.13) (d, J = 1.8 Hz, 1H, H-13), 7.01–6.87 (overlapped, 5H, H-15, H-16, H-18, H-21, H-22), 6.11 (d, J = 2.1 Hz, 1H, aromatic H-6), 6.09 (d, J = 2.1 Hz, 1H, H-8), 5.55 (d, J = 11.7 Hz, 1H, H-3), 5.30 (5.29) (d, J = 11.7 Hz, 1H, H-2), 4.89 (4.87) (d, J = 7.2 Hz, 1H, H-11), 4.32 (dd, J = 12.0, 5.1 Hz, 1H, H-23), 4.25 (ddd, J = 7.2, 5.1, 4.5 Hz, 1H, H-10), 3.98 (dd, J = 12.0, 4.5 Hz, 1H, H-23), 3.90 (3.88) (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 3.80 (3.79) (s, 3H, OCH3), 3.34–3.12 (m, 8H, 4 × CH2CH3), 1.18–0.96 (m, 12H, 4 × CH2CH3). 13C NMR (75 MHz, CDCl3) δ 186.43, 166.35, 164.17, 162.42, 155.30, 154.50 (154.45), 149.83, 149.54 (149.47), 143.96 (143.93), 143.80 (143.77), 129.58 (129.51), 128.32 (128.30), 121.14 (120.97), 120.16, 117.25 (117.12), 116.57 (116.43), 111.37 (111.29), 110.08 (109.93), 104.43 (104.39), 93.52 (93.43), 93.43 (93.35), 81.15 (81.04), 76.15, 74.33, 69.59, 63.46, 56.29 (56.14), 56.08 (56.03), 55.94 (55.82), 55.69 (55.63), 42.05, 41.48, 14.18, 13.54. HRMS (ESI): m/z calculated for C38H47N2O12 [M+H]+: 723.3129. Found: 723.3129. IR (film) νmax: 2971, 2933, 1698, 1609, 1574, 1509 cm−1. HPLC purity 100% (only one signal was observed for the diastereomers).
3.2.5. 5,7,20-O-Trimethyl-3-O-(N,N-dimethylthiocarbamoyl)silybin (8)
Yield, 25%; slight yellow solid; 124–125 °C. 1H NMR (300 MHz, CDCl3) δ 7.17 (7.16) (d, J = 1.8 Hz, 1H, H-13), 7.08 (7.07) (dd, J = 8.4, 1.8 Hz, 1H, H-15), 7.02–6.98 (overlapped, 2H, H-16 and H-22), 6.94 (6.93) (d, J = 2.1 Hz, 1H, H-18), 6.90 (d, J = 8.1 Hz, 1H, H-21), 6.61 (6.57) (d, J = 11.6 Hz, 1H, H-3), 6.12 (6.11) (d, J = 2.3 Hz, 1H, H-6), 6.09 (d, J = 2.3 Hz, 1H, H-8), 5.37 (5.36) (d, J = 11.6 Hz, 1H, H-2), 4.97 (d, J = 8.4 Hz, 1H, H-11), 4.05 (ddd, J = 8.4, 3.9, 3.0 Hz, 1H, H-10), 3.91 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 3.80 (dd, J = 12.6, 3.0 Hz, 1H, H-23), 3.54 (dd, J = 12.6, 3.9 Hz, 1H, H-23), 3.33 (s, 3H, NCH3), 3.10 (3.09) (s, 3H, NCH3). 13C NMR (75 MHz, CDCl3) δ 187.46, 187.42, 166.49, 163.95, 162.40, 149.76, 149.50 (149.46), 144.17 (144.13), 144.00 (143.90), 128.99 (128.92), 128.55 (128.52), 121.26 (121.04), 120.25, 117.24, 116.96, 111.35, 110.23, 104.36, 93.56 (93.50), 93.46 (93.39), 81.06, 79.31 (79.06), 78.45 (78.30), 76.30, 61.73, 56.14, 55.99, 55.83, 55.70, 43.39, 38.33. HRMS (ESI): m/z calculated for C31H34NO10S [M+H]+: 612.1904. Found: 612.1920. IR (film) νmax: 3481, 2929, 2838, 1684, 1653, 1607, 1571, 1539, 1507 cm−1. HPLC purity 100% (only one signal was observed for the diastereomers).
3.2.6. 5,7,20-O-Trimethyl-3,23-O-(N,N-dimethylthiocarbamoyl)silybin (9)
Yield, 7%; colorless wax. 1H NMR (300 MHz, CDCl3) δ 7.17 (7.16) (d, J = 1.8 Hz, 1H, H-13), 7.07 (d, J = 8.1 Hz, 1H, H-16), 6.99 (dd, J = 8.4, 2.1 Hz, 1H, H-22), 6.93 (dd, J = 8.1, 1.8 Hz, 1H, H-15), 6.90–6.87 (overlapped, 2H, H-18 and H-21), 6.60 (6.56) (d, J = 11.6 Hz, 1H, H-3), 6.12 (d, J = 2.7 Hz, 1H, H-8), 6.09 (d, J = 2.7 Hz, 1H, H-6), 5.37 (d, J = 11.6 Hz, 1H, H-2), 4.92 (4.90) (d, J = 7.8 Hz, 1H, H-11), 4.67 (dd, J = 11.4, 2.4 Hz, 1H, H-23), 4.40–4.30 (overlapped, 2H, H-10, H-23), 3.89 (s, 6H, 2 × OCH3), 3.86 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 3.34 (s, 6H, 2 × NCH3), 3.11 (3.10, 3.08) (s, 6H, 2 × NCH3). 13C NMR (75 MHz, CDCl3) δ 187.69, 187.60, 187.49, 166.56, 164.06, 162.48, 150.00, 149.64 (149.61), 144.01 (143.98), 143.85 (143.77), 129.10 (129.01), 128.15, 121.34 (121.25), 120.30 (120.25), 117.39, 116.88, 111.46, 110.14 (110.03), 104.40, 93.51, 93.49, 81.09, 79.06, 76.87, 75.84, 69.60, 56.22, 56.15, 56.08, 55.81 (55.75), 43.36, 43.01, 38.30, 38.00. HRMS (ESI): m/z calculated for C34H39N2O10S2 [M+H]+: 699.2046. Found: 699.2050. IR (film) νmax: 2937, 1688, 1606, 1672, 1508 cm−1. HPLC purity 97.9% (only one signal was observed for the diastereomers).
3.3. Synthesis of 23-O-TBS-5,7,20-O-Trimethylsilybin (10)
Imidazole (36 mg, 0.53 mmol) and TBSCl (57 mg, 0.38 mmol) were added to a solution of 5,7,20-O-trimethylsilybin (3, 200 mg, 0.31 mmol, 1.0 eq) in DCM (3.1 mL). The reaction was stirred at room temperature for 2 h prior to being quenched with saturated aqueous solution of ammonium chloride (20 mL). The subsequent mixture was extracted with ethyl acetate (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate. Two PTLC plates were used to purify the crude product eluting with DCM:MeOH (97:3, v/v) to yield 10 as a white foam-like solid in 76% yield; 87−89 °C. 1H NMR (300 MHz, CDCl3) δ 7.22 (7.18) (d, J = 1.8 Hz, 1H, H-13), 7.11–6.97 (overlapped, 4H, H-15, H-16, H-18, H-22), 6.89 (d, J = 8.1 Hz, 1H, H-21), 6.11 (d, J = 2.1 Hz, 1H, H-8), 6.10 (d, J = 2.1 Hz, 1H, H-6), 5.02 (d, J = 8.1 Hz, 1H, H-11), 4.94 (d, J = 12.2 Hz, 1H, H-2), 4.45 (4.43) (d, J = 12.2 Hz, 1H, H-3), 3.96 (ddd, J = 8.1, 4.2, 2.4 Hz, 1H, H-10), 3.90 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 3.89–3.81 (overlapped, 1H, H-23), 3.80 (3.79) (s, 3H, OCH3), 3.55 (dd, J = 11.7, 2.7 Hz, 1H, H-23), 0.90 (s, 9H, TBS), 0.07 (0.06) (s, 6H, TBS). 13C NMR (75 MHz, CDCl3) δ 190.97, 167.12, 165.06, 162.24, 149.46, 149.19, 144.76 (144.69), 144.05 (143.97), 129.40 (129.35), 129.21, 121.32 (120.86), 120.19, 117.37 (117.21), 116.67 (116.33), 111.18, 110.45, 103.01, 93.79, 93.43, 83.12, 78.67, 76.17, 72.73 (72.64), 62.40, 56.42, 56.07, 55.92, 55.77, 25.94, 18.41, −4.95, −5.34. HRMS (ESI): m/z calculated for C24H43O10Si [M+H]+: 639.2626. Found: 639.2623. IR (film) νmax: 3446, 2951, 2929, 2882, 2854, 1716, 1675, 1606, 1573, 1540, 1507 cm−1.
3.4. General Procedure for the Synthesis of Carbamoyled Derivatives of 23-O-TBS-5,7,20-O-Trimethylsilybin
Triethylamine (56 µL, 0.4 mmol) and 4-dimethylaminopyridine (12 mg, 0.1 mmol) were sequentially added to a solution of 23-O-TBS-5,7,20-O-trimethylsilybin (10) (63.8 mg, 0.1 mmol) in DCM (1 mL, 0.1 M). The subsequent mixture was stirred for 10 min at room temperature, to which the respective (thio)carbamoyl chloride (0.4 mmol) was added. The reaction was allowed to proceed at room temperature overnight under argon prior to being quenched with brine (50 mL). The resulting mixture was extracted with ethyl acetate (30 mL × 3), the combined organic extracts were dried over anhydrous sodium sulfate, and the organic solvents were removed. The crude product was subjected to PTLC purification eluting with DCM/MeOH (95:5, v/v) to afford the respective 3-carbomoyled derivative. Their physical and spectral data are summarized below.
3.4.1. 23-O-TBS-5,7,20-O-Trimethyl-3-O-(N,N-dimethylcarbamoyl)silybin (11)
Yield, 67%; white solid; 81−82 °C. 1H NMR (300 MHz, CDCl3) δ 7.15 (7.11) (d, J = 1.5 Hz, 1H, H-13), 7.01–6.93 (overlapped, 4H, H-15, H-16, H18, H-22), 6.87 (d, J = 8.1 Hz, 1H, H-21), 6.10 (6.09) (d, J = 2.3 Hz, 1H, H-8), 6.08 (d, J = 2.3 Hz, 1H, H-6), 5.53 (5.51) (d, J = 11.7 Hz, 1H, H-3), 5.30 (5.29) (d, J = 11.7 Hz, 1H, H-2), 5.00 (4.99) (d, J = 8.1 Hz, 1H, H-11), 3.97 (ddd, J = 8.1, 3.0, 2.3 Hz, 1H, H-10), 3.91 (s, 3H, OCH3), 3.91 (3.87) (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 3.79 (3.78) (s, 3H, OCH3), 3.85 (dd, J = 11.7, 2.3 Hz, 1H, H-23), 3.55 (dd, J = 11.7, 3.0 Hz, 1H, H-23), 2.87 (s, 6H, 2 × NCH3), 0.89 (s, 9H, TBS), 0.06 (s, 3H, TBS), 0.057 (0.070) (s, 3H, TBS). 13C NMR (75 MHz, CDCl3) δ 186.44, 166.44, 164.20, 162.41, 155.30, 149.67, 149.44 (149.38), 149.15 (149.10), 144.54, 144.04 (144.00), 143.96 (143.90), 129.60 (129.47), 129.09 (129.05), 129.01 (128.89), 128.55 (128.52), 121.00 (120.80), 120.20, 117.13 (117.03), 116.55 (116.46), 111.25 (111.19), 110.15 (110.00), 104.30, 93.52 (93.42), 93.42 (93.35), 80.99 (80.90), 78.46 (78.28), 76.26, 74.69 (74.54), 61.68, 56.19 (56.10), 56.04, 55.95, 55.79 (55.66), 36.79, 36.12, 25.95 (25.89), 25.76 (25.70), 18.38 (18.05), −3.47 (−3.56), −5.30 (−5.39). HRMS (ESI): m/z calculated for C37H48NO11Si [M+H]+: 710.2997. Found: 710.3006. IR (film) νmax: 2928, 2855, 1713, 1693, 1607, 1573, 1508 cm−1. HPLC purity 94.2% (only one signal was observed for the diastereomers).
3.4.2. 23-O-TBS-3-O-(N,N-diethylcarbamoyl)-5,7,20-O-trimethylsilybin (12)
Yield, 50%; white solid; 79−81 °C. 1H NMR (300 MHz, CDCl3) δ 7.15 (7.11) (d, J = 1.8 Hz, 1H, H-13), 7.02–6.93 (overlapped, 4H, H-15, H-16, H-18, H-22), 6.89 (d, J = 8.1 Hz, 1H, H-21), 6.11 (6.10 (d, J = 2.3 Hz, 1H, H-8), 6.09 (6.08) (d, J = 2.3 Hz, 1H, H-6), 5.57 (5.55) (d, J = 11.9 Hz, 1H, H-3), 5.29 (5.28) (d, J = 11.9 Hz, 1H, H-2), 5.00 (4.98) (d, J = 7.8 Hz, 1H, H-11), 3.97 (ddd, J = 7.8, 3.0, 2.1 Hz, 1H, H-10), 3.89 (s, 3H, OCH3), 3.89 (3.88) (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 3.91−3.82 (overlapped, 1H, H-23), 3.79 (3.78) (s, 3H, OCH3), 3.55 (dd, J = 11.7, 3.0 Hz, 1H, H-23), 3.30–3.09 (m, 4H, 2 × CH2CH3), 1.04 (br.s, 6H, 2 × CH2CH3), 0.89 (s, 9H, TBS), 0.06 (s, 6H, TBS). 13C NMR (75 MHz, CDCl3) δ 186.53, 166.30, 164.20, 162.37, 154.46 (154.40), 149.40, 149.15 (149.09), 144.48, 143.89 (143.85), 129.13 (129.09), 129.03 (128.94), 120.91 (120.78), 120.14 (120.08), 117.04 (116.88), 116.54 (116.36), 111.12 (111.07), 110.43 (110.27), 104.42 (104.38), 93.43, 93.36, 81.25 (81.15), 78.66, 76.09 (76.02), 74.35 (74.27), 62.35, 56.19 (56.17), 56.02 (55.99), 55.94, 55.70 (55.67), 42.15 (41.49), 25.90, 18.37, 13.89 (13.43), −5.07, −5.34. HRMS (ESI): m/z calculated for C39H52NO11Si [M+H]+: 738.3310. Found: 738.3329. IR (film) νmax: 2930, 2854, 1693, 1607, 1573, 1508 cm−1. HPLC purity 100% (only one signal was observed for the diastereomers).
3.4.3. 23-O-TBS-5,7,20-O-Trimethyl-3-O-(N,N-dimethylthiocarbamoyl)silybin (13)
This derivative was achieved by PTLC purification using EtOAc/hexane (7:3, v/v) followed by hexane/EtOAc (1:1, v/v). Yield, 45%; slight yellow solid; 83–85 °C. 1H NMR (300 MHz, CDCl3) δ 7.16 (7.13) (d, J = 2.1 Hz, 1H, H-13), 7.09–6.95 (overlapped, 4H, H-15, H-16, H-18, H-22), 6.89 (d, J = 8.4 Hz, 1H, H-21), 6.60 (6.57) (d, J = 11.6 Hz, 1H, H-3), 6.12 (6.11) (d, J = 2.3 Hz, 1H, H-8), 6.09 (d, J = 2.3 Hz, 1H, H-6), 5.36 (d, J = 11.6 Hz, 1H, H-2), 5.01 (5.00) (d, J = 7.8 Hz, 1H, H-11), 3.98 (ddd, J = 7.8, 4.2, 2.7 Hz, 1H, H-10), 3.90 (s, 3H, OCH3), 3.90 (3.89) (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 3.90–3.82 (overlapped, 1H, H-23), 3.80 (s, 3H, OCH3), 3.55 (dd, J = 11.7, 3.0 Hz, 1H, H-23), 3.34 (s, 3H, NCH3), 3.10 (s, 3H, NCH3), 0.90 (s, 9H, TBS), 0.06 (s, 3H, TBS), 0.06 (0.05) (s, 3H, TBS). 13C NMR (75 MHz, CDCl3) δ 187.53, 187.49, 166.46, 164.02, 162.40, 149.49, 149.17, 144.74, 143.98, 129.14 (129.09), 128.42 (128.33), 121.05, 120.20, 117.29 (117.17), 116.93 (116.80), 111.18 (111.15), 110.53 (110.38), 104.37, 93.48, 93.46, 81.17, 78.68 (78.64), 77.4, 76.22 (76.16), 62.41, 56.20, 56.06, 56.02, 55.79, 43.37, 38.33, 25.98, 18.44, −5.00, −5.29. HRMS (ESI): m/z calculated for C37H48NO10SSi [M+H]+: 726.2769. Found: 726.2796. IR (film) νmax: 2929, 2854, 1690, 1607, 1573, 1508 cm−1. HPLC purity 96.7% (only one signal was observed for the diastereomers).
3.5. Synthesis of Tetramethyltaxifolin (14)
A reaction flask was charged with taxifolin (20 mg, 0.066 mmol) and potassium carbonate (82 mg, 0.59 mmol), which was vacuumed three times under argon before the addition of anhydrous acetone (0.47 mL, 0.14 M). Dimethylsulfate (75 µL, 0.79 mmol) was then added into the reaction flask through a long needle. The reaction solution was refluxed overnight and cooled down to room temperature before the aqueous solution of ammonium chloride (20 mL) was added to quench the reaction. The subsequent mixture was extracted with ethyl acetate (10 mL × 3), the combined organic extracts were dried over anhydrous sodium sulfate, and the organic solvents were removed under vacuum. The crude product was purified over preparative TLC plate eluting with ethyl acetate/hexane (5:1, v/v) and ethyl acetate/hexane (7:3, v/v), sequentially, to furnish the desired 14 as a slight yellow solid in 86% yield; 149–151 °C; in [37], 165–166 °C; in [38], 153–155 °C. 1H NMR (300 MHz, CDCl3) δ 7.11 (dd, J = 8.3, 1.9 Hz, 1H, H-6′), 7.07 (d, J = 1.9 Hz, 1H, H-2′), 6.93 (d, J = 8.3 Hz, 1H, H-5′), 6.13 (d, J = 2.1 Hz, 1H, H-6), 6.11 (d, J = 2.1 Hz, 1H, H-8), 4.97 (d, J = 12.3 Hz, 1H, H-2), 4.45 (d, J = 12.3 Hz, 1H, H-3), 3.93 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.82 (s, 3H, OCH3). 13C NMR (75 MHz, CDCl3) δ 190.93, 167.13, 165.06, 162.25, 149.91, 149.28, 129.02, 120.51, 111.23, 110.39, 103.02, 93.80, 93.43, 83.42, 72.72, 56.32, 56.11, 56.02, 55.89. HRMS (ESI): m/z calculated for C19H21O7 [M+H]+: 361.1287. Found: 361.1281. IR (film) νmax: 3446, 2939, 2838, 1673, 1606, 1572, 1516 cm−1. The NMR data are mostly consistent with those reported in the literature [39]. Only one exception is that the chemical shift for H-5′ was reported as 7.08 ppm in the literature [39] rather than at 6.93 ppm, without showing the original 1H NMR spectrum. Our chemical shift at H-5′ is closer to the value reported in another work [40].
3.6. General Procedure for the Synthesis of 3-Carbamoyled Derivatives of Tetramethyltaxifolin
Triethylamine (56 µL, 0.4 mmoL) and 4-dimethylaminopyridine (12 mg, 0.1 mmol) were sequentially added to a solution of tetramethyltoxifolin (14) (36 mg, 0.1 mmol) in DCM (1 mL, 0.1 M). The subsequent mixture was stirred for 10 min at room temperature, to which the respective (thio)carbamoyl chloride (0.4 mmol) was added. The reaction was allowed to proceed at room temperature with stirring overnight under argon prior to being quenched with brine (50 mL). The resulting mixture was extracted with ethyl acetate (30 mL × 3), the combined organic extracts were dried over anhydrous sodium sulfate, and the organic solvents were removed. The crude product was subjected to PTLC purification eluting with hexane/EtOAc (3:7, v/v) to afford the respective 3-carbomoyled derivative. Their physical and spectral data are summarized below.
3.6.1. 3′,4′,5,7-O-Tetramethyl-3-O-(N,N-dimethylcarbamoyl)taxifolin (15)
Yield, 61%; white solid; 153–155 °C. 1H NMR (300 MHz, CDCl3) δ 7.04 (dd, J = 8.4, 1.8 Hz, 1H, H-6′), 7.03 (s, 1H, H-2′), 6.89 (d, J = 8.4 Hz, 1H, H-5′), 6.12 (d, J = 2.1 Hz, 1H, H-8), 6.10 (d, J = 2.1 Hz, 1H, H-6), 5.63 (d, J = 12.3 Hz, 1H, H-3), 5.32 (d, J = 12.3 Hz, 1H, H-2), 3.91 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 2.86 (s, 6H, 2 × NCH3). 13C NMR (75 MHz, CDCl3) δ 186.69, 166.47, 164.34, 162.51, 155.27, 149.84, 149.17, 128.46, 120.74, 111.08, 110.42, 104.47, 93.59, 93.51, 81.51, 74.34, 56.19, 56.08, 56.05, 55.78, 36.76, 36.16. HRMS (ESI): m/z calculated for C22H26NO8 [M+H]+: 432.1659. Found: 432.1657. IR (film) νmax: 2937, 2839, 1708, 1694, 1607, 1595, 1572, 1517 cm−1.
3.6.2. 3-O-(N,N-Diethylcarbamoyl)-3′,4′,5,7-O-tetramethyltaxifolin (16)
Yield, 52%; colorless syrup. 1H NMR (300 MHz, CDCl3) δ 7.06 (d, J = 8.1, 2.1 Hz, 1H, H-6′), 7.03 (s, 1H, H-2′), 6.88 (d, J = 8.1 Hz, 1H, H-5′), 6.13 (d, J = 2.1 Hz, 1H, H-6 or H-8), 6.11 (d, J = 2.4 Hz, 1H, H-8 or H-6), 5.69 (d, J = 12.2 Hz, 1H, H-3), 5.31 (d, J = 12.2 Hz, 1H, H-2), 3.91 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 3.30–3.06 (m, 4H, 2 × CH2CH3), 1.02 (brs, 6H, 2 × CH2CH3). 13C NMR (75 MHz, CDCl3) δ 186.81, 166.37, 164.31, 162.47, 154.46, 149.80, 149.12, 128.45, 120.87, 111.02, 110.41, 104.53, 93.55, 93.50, 81.64, 73.91, 56.32, 56.13, 55.99, 55.71, 42.40, 41.63, 13.96, 13.47. HRMS (ESI): m/z calculated for C24H30NO8 [M+H]+: 460.1972. Found: 460.1970. IR (film) νmax: 2965, 2934, 2839, 1697, 1595, 1571, 1518 cm−1.
3.6.3. 3′,4′,5,7-O-Tetramethyl-3-O-(N,N-dimethylthiocarbamoyl)taxifolin (17)
Yield, 38%; white solid; 152–154 °C. 1H NMR (300 MHz, CDCl3) δ 7.15 (d, J = 1.8 Hz, 1H, H-2′), 7.06 (dd, J = 8.1, 1.8 Hz, 1H, H-6′), 6.88 (d, J = 8.1 Hz, 1H, H-5′), 6.79 (d, J = 12.0 Hz, 1H, H-3), 6.12 (d, J = 2.4 Hz, 1H, H-8), 6.10 (d, J = 2.4 Hz, 1H, H-6), 5.36 (d, J = 12.0 Hz, 1H, H-2), 3.92 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 3.31 (s, 3H, NCH3), 3.06 (s, 3H, NCH3). 13C NMR (75 MHz, CDCl3) δ 187.53, 187.49, 166.47, 164.10, 162.41, 149.99, 149.21, 127.67, 121.47, 110.94, 110.81, 104.42, 93.59, 93.50, 81.56, 78.74, 56.30, 56.15, 55.94, 55.88, 43.41, 38.31. HRMS (ESI): m/z calculated for C22H26NO7S [M+H]+: 448.1430. Found: 448.1431. IR (film) νmax: 2937, 2895, 2838, 1687, 1607, 1572, 1518 cm−1.
3.7. Cell Culture
The LNCaP, DU145, and PC-3 prostate cancer cell lines were originally purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). The PC-3 and LNCaP cell lines were routinely cultured in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin. Cultures were maintained in a high humidity environment supplemented with 5% carbon dioxide at a temperature of 37 °C. The DU-145 prostate cancer cells were routinely cultured in Eagle’s Minimum Essential Medium (EMEM) supplemented with 10% FBS and 1% penicillin/streptomycin.
3.8. WST-1 Cell Proliferation Assay
The LNCaP, DU-145, or PC-3 prostate cancer cells were plated in 96-well plates at a density of 3200 in each well in 200 µL of culture medium. The cells were then treated with positive reference, or synthesized mimics separately at different doses for 3 days, while equal treatment volumes of DMSO were used as the vehicle control. The cells were cultured in a CO2 incubator at 37 °C for three days. An amount of 10 µL of the premixed WST-1 cell proliferation reagent (Clontech) was added to each well. After mixing gently for 1 min on an orbital shaker, the cells were incubated for an additional 3 h at 37 °C. To ensure homogeneous distribution of color, it was important to mix gently on an orbital shaker for 1 min. The absorbance of each well was measured using a microplate-reader (Synergy HT, BioTek) at a wavelength of 430 nm. The IC50 value was the concentration of each compound that inhibits cell proliferation by 50% under the experimental conditions and was the average from triplicate determinations that were reproducible and statistically significant. For calculating the IC50 values, a linear proliferative inhibition was made based on at least five dosages for each compound.
3.9. Statistical Analysis
All data are represented as the mean ± standard deviation (S.D.) for the number of experiments indicated. Other differences between treated and control groups were analyzed using the Student’s t-test. A p-value < 0.05 was considered statistically significant.
4. Conclusions
Collectively, the regioselective carbamoylation at the secondary alcoholic hydroxyl group at C-3 of flavanonols and flavanonol-type flavonolignans was developed for the first time. This approach avoids the oxidation of the C2–C3, offering a practical approach to the synthesis of 3-O-carbamoyl derivatives of flavanonols and flavanonol-type flavonolignans. Our preliminary WST cell proliferation assay data exhibit that 5,7,20-O-trimethylsilybin and four 3-O-carbamoyl-5,7,20-O-trimethylsilybins bear high selectivity and promising potency towards the AR-positive LNCaP prostate cancer cell line. These findings advocate that 3-O-substituted-5,7,20-O-trimethylsilybin may serve as a natural product-based scaffold for new antiandrogens for lethal castration-resistant prostate cancer. Further evaluation of their (including their optically pure versions) promising potency and selectivity in other AR-positive prostate cancer cell lines, as well as their AR modulating capability, are in progress and will be reported in due course.
Supplementary Materials
The following are available online. Figures S1–S5, S8, S9, S12, S13, S16, S17, S20, S21, S24, S25, S28, S29, S31, S32, S34, S35, S38, S39, S42, S43, S45, S46, S48, S49, S51 and S52: NMR spectra, Figures S6, S10, S14, S18, S22, S26, S30, S33, S36, S40, S44, S47, S50, & S53: High resolution mass spectra, Figures S7, S11, S15, S19, S23, S27, S33, S37 and S41: HPLC chromatograms, Figure S54: The concentration-effect curves.
Author Contributions
Conceptualization and design: Q.-H.C.; experiments and data analysis: S.W., G.C., Q.Z., G.W. and Q.-H.C.; funding acquisition, Q.-H.C. and G.W. All authors have read and agreed to the published version of the manuscript.
Funding
We are grateful to The California State University Program for Education and Research in Biotechnology (CSUPERB) for (i) the Graduate Student Research Restart award (to S. Wu) and the COVID-19 Research Recovery Microgrant (to Q. Chen). This work was also partially supported by the Scholarly and Creative Activity Award from the College of Science and Mathematics at California State University Fresno (Q. Chen) and the NIH RCMI program at Xavier University of Louisiana through Grant 2G12MD007595-064 (G. Wang).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We thank the Department of Chemistry and Biochemistry and the College of Science and Mathematics at CSU-Fresno for all administrative support. Special thanks go to Douglas Kliewer from the Department of Chemistry and Biochemistry at CSU-Fresno for assisting us in performing HPLC purify analysis.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Sample Availability
Samples of the compounds 4–17 are available from the authors.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Elshan, N.G.R.D.; Rettig, M.; Jung, M.E. Molecules targeting the androgen receptor (AR) signaling axis beyond the AR-Ligand binding domain. Med. Res. Rev. 2018, 39, 910–960. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, P.; Rivera, A.; Muthima, K.; Olveda, N.; Muchalski, H.; Chen, Q.-H. Second-generation androgen receptor antagonists as hormonal therapeutics for three forms of prostate cancer. Molecules 2020, 25, 2448. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA damage repair gene alteration and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: Analysis from the phase II TRITON2 study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.-Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef]
- Sayegh, N.; Swami, U.; Agarwal, N. Recent advances in the management of metastatic prostate cancer. J. Clin. Oncol. 2020, 38 (Suppl. 15), 3500. [Google Scholar]
- Schepisi, G.; Cursano, M.C.; Casadei, C.; Menna, C.; Altavilla, A.C.L.; Cerchione, C.; Paganelli, G.; Santini, D.G.T.; Martinelli, G.; De Giorgi, U. CAR-T cell therapy: A potential new strategy against prostate cancer. Immunother. Cancer 2019, 7, 258. [Google Scholar] [CrossRef] [PubMed]
- Pelter, A.; Haensel, R. The structure of silybin (Silybum substance E6), the first flavonolignan. Tetrahedron Lett. 1968, 9, 2911–2916. [Google Scholar] [CrossRef]
- Kroll, D.J.; Shaw, H.S.; Oberlies, N.H. Milk thistle nomenclature: Why it matters in cancer research and pharmacokinetic studies. Integr. Cancer Ther. 2007, 6, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Davis-Searles, P.R.; Nakanishi, Y.; Kim, N.-C.; Graf, T.N.; Oberlies, N.H.; Wani, M.C.; Wall, M.E.; Agarwal, R.; Kroll, D.J. Milk thistle and prostate cancer: Differential effects of pure flavonolignans from Silybum marianum on antiproliferative end points in human prostate carcinoma cells. Cancer Res. 2005, 65, 4448–4457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.P.; Dhanalakshmi, S.; Tyagi, A.K.; Chan, D.C.F.; Agarwal, C.; Agarwal, R. Dietary feeding of silibinin inhibits advance human prostate carcinoma growth in athymic nude mice and increases plasma insulin-like growth factor-binding protein-3 levels. Cancer Res. 2002, 62, 3063–3069. [Google Scholar] [PubMed]
- Raina, K.; Blouin, M.-J.; Singh, R.P.; Majeed, N.; Deep, G.; Varghese, L.; Glodé, L.M.; Greenberg, N.M.; Hwang, D.; Cohen, P.; et al. Dietary feeding of silibinin inhibits prostate tumor growth and progression in transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2007, 67, 11083–11091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vue, B.; Chen, Q.-H. The potential of flavonolignans in prostate cancer management. Curr. Med. Chem. 2016, 23, 3925–3950. [Google Scholar] [CrossRef] [PubMed]
- Thelen, P.; Wuttke, W.; Jarry, H.; Grzmil, M.; Ringert, R.H. Inhibition of telomerase activity and secretion of prostate specific antigen by silibinin in prostate cancer cells. J. Urol. 2004, 171, 1934–1938. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, J.-S.; Young, C.Y. Silymarin inhibits function of the androgen receptor by reducing nuclear localization of the receptor in the human prostate cancer cell line LNCaP. Carcinogenesis 2001, 22, 1399–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zi, X.; Agarwal, R. Silibinin decreases prostate-specific antigen with cell growth inhibition via G1 arrest, leading to differentiation of prostate carcinoma cells: Implications for prostate cancer intervention. Proc. Natl. Acad. Sci. USA. 1999, 96, 7490–7495. [Google Scholar] [CrossRef] [Green Version]
- Deep, G.; Gangar, S.C.; Oberlies, N.H.; Kroll, D.J.; Agarwal, R. Isosilybin A induces apoptosis in human prostate cancer cells via targeting Akt, NF-κB, and androgen receptor signaling. Mol. Carcinog. 2010, 49, 902–912. [Google Scholar] [CrossRef] [Green Version]
- Flaig, T.W.; Gustafson, D.L.; Su, L.J.; Zirrolli, J.A.; Crighton, F.; Harrison, G.S.; Pierson, A.S.; Agarwal, R.; Glodé, L.M. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Investig. New Drugs 2007, 25, 139–146. [Google Scholar] [CrossRef]
- Vue, B.; Zhang, S.; Zhang, X.; Parisis, K.; Zhang, Q.; Zheng, S.; Wang, G.; Chen, Q.-H. Silibinin derivatives as anti-prostate cancer agents: Synthesis and cell-based evaluations. Eur. J. Med. Chem. 2016, 109, 36–46. [Google Scholar] [PubMed] [Green Version]
- Vue, B.; Zhang, S.; Vignau, A.; Chen, G.; Zhang, X.; Diaz, W.; Zhang, Q.; Zheng, S.; Wang, G.; Chen, Q.-H. O-aminoalkyl-O-trimethyl-2,3-dehydrosilybins: Synthesis and in vitro effects towards prostate cancer cells. Molecules 2018, 23, 3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Vue, B.; Huang, M.; Zhang, X.; Lee, T.; Zhang, Q.; Zheng, S.; Wang, G.; Chen, Q.-H. 3-O-Alkyl-2,3-dehydrosilibinins: Two synthetic approaches and in vitro effects toward prostate cancer cells. Bioorg. Med. Chem. Lett. 2016, 26, 3226–3231. [Google Scholar] [CrossRef] [Green Version]
- Sy-Cordero, A.A.; Graf, T.N.; Runyon, S.P.; Wani, M.C.; Kroll, D.J.; Agarwal, R.; Brantley, S.J.; Paine, M.F.; Polyak, S.J.; Oberlies, N.H. Enhanced bioactivity of silybin B methylation products. Bioorg. Med. Chem. 2013, 21, 742–747. [Google Scholar] [PubMed] [Green Version]
- Agarwal, C.; Wadhwa, R.; Deep, G.; Biedermann, D.; Gažák, R.; Křen, V.; Agarwal, R. Anti-cancer efficacy of silybin derivatives—A structure-activity relationship. PLoS ONE 2013, 8, e60074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanucci, V.; Agarwal, C.; Agarwal, R.; Pannecouque, C.; Luliano, M.; Tommaso, G.D.; Caruso, T.; Fabio, G.D.; Zarrelli, A. Silibinin phosphodiester glyco-conjugates: Synthesis, redox behaviour and biological investigations. Bioorg. Chem. 2018, 77, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.X.; Huang, K.X.; Li, H.B.; Gong, J.X.; Wang, F.; Feng, Y.B.; Tao, Q.F.; Wu, Y.H.; Li, X.K.; Wu, X.M.; et al. Design, synthesis, and examination of neuron protective properties of alkenylated and amidated dehydro-silybin derivatives. J. Med. Chem. 2009, 52, 7732–7752. [Google Scholar] [CrossRef]
- Jiang, Z.; Sekhon, A.; Oka, Y.; Chen, G.; Alrubati, N.; Kaur, J.; Orozco, A.; Zhang, Q.; Wang, G.; Chen, Q.-H. 23-O-Substituted-2,3-dehydrosilybins selectively suppress androgen receptor-positive LNCaP prostate cancer cell proliferation. Nat. Prod. Commun. 2020, 15, 1934578X20922326. [Google Scholar] [CrossRef]
- Simoni, D.; Invidiata, F.P.; Eleopra, M.; Marchetti, P.; Rondanin, R.; Baruchello, R.; Grisolia, G.; Tripathi, A.; Kellogg, G.E.; Durrant, D.; et al. Design, synthesis and biological evaluation of novel stilbene-based antitumor agents. Bioorg. Med. Chem. 2009, 17, 512–522. [Google Scholar] [CrossRef]
- Chassaing, C.; Berger, M.; Heckeroth, A.; Ilg, T.; Jaeger, M.; Kern, C.; Schmid, K.; Uphoff, M. Highly water-soluble prodrugs of anthelmintic benzimidazole carbamates: Synthesis, pharmacodynamics, and pharmacokinetics. J. Med. Chem. 2008, 51, 1111–1114. [Google Scholar] [CrossRef]
- Biedermann, D.; Vavrikova, E.; Cvak, L.; Kren, V. Chemistry of silybin. Nat. Prod. Rep. 2014, 31, 1138–1157. [Google Scholar] [CrossRef] [PubMed]
- Fiaud, J.-C.; Legros, J.-Y. Substrate leaving group control of the enantioselectivity in the palladium-catalyzed asymmetric allylic substitution of 4-alkyl-1-vinylcyclohexyl derivatives. J. Org. Chem. 1990, 55, 4840–4846. [Google Scholar] [CrossRef]
- Gazak, R.; Trouillas, P.; Biedermann, D.; Fuksova, K.; Marhol, P.; Kuzma, M.; Kren, V. Base-catalyzed oxidation of silybin and isosilybin into 2,3-dehydro derivatives. Tetrahedron Lett. 2013, 54, 315–317. [Google Scholar] [CrossRef]
- Atkinson, R.F.; Balko, T.W.; Westman, T.R.; Sypniewski, G.C.; Carmody, M.A.; Pauler, C.T.; Schade, C.L.; Coulter, D.E.; Pham, H.T.; Barea, F. Formation of olefins in the pyrolysis of N,N-disubstituted carbamates. J. Org. Chem. 1981, 46, 2804–2806. [Google Scholar] [CrossRef]
- Gazak, R.; Svobodova, A.; Psotova, J.; Sedmera, P.; Prikrylova, V.; Walterova, D.; Kren, V. Oxidized derivatives of silybin and their antiradical and antioxidant activity. Bioorg. Med. Chem. 2004, 12, 5677–5687. [Google Scholar] [CrossRef]
- Zhao, H.; Tong, M.; Wang, H.; Xu, S. Transition-metal-free synthesis of 1,1-diboronate esters with a fully substituted benzylic center via diborylation of lithiated carbamates. Org. Biomol. Chem. 2017, 15, 3418–3422. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Sun, X.; Potter, K.; Cao, Y.; Zakharov, L.N.; Blakemore, P.R. Stereospecific synthesis of alkenes by eliminative cross-coupling of enantioenriched sp(3) -hybridized carbenoids. Angew. Chem. Int. Ed. Engl. 2016, 55, 12285–12289. [Google Scholar] [CrossRef] [PubMed]
- Bernini, R.; Crisante, F.; Gentili, P.; Morana, F.; Pierini, M.; Piras, M. Chemoselective C-4 aerobic oxidation of catechin derivatives catalyzed by the trametes villosa laccase/1-hydroxybenzotriazole system: Synthetic and mechanistic aspects. J. Org. Chem. 2011, 76, 820–832. [Google Scholar] [CrossRef]
- Porter, L.J.; Foo, L.Y. Leucocyanidin: Synthesis and properties of (2R,3S,4R)-(+)-3,4,5,7,3’,4’-hexahydroxyflavan. Phytochemistry 1980, 21, 2947–2952. [Google Scholar] [CrossRef]
- Taniguchi, T.; Monde, K. Exciton chirality method in vibrational circular dichroism. J. Am. Chem. Soc. 2012, 134, 3695–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiehlmann, E.; Slade, P.W. Methylation of dihydroquercetin acetates: Synthesis of 5-O-methyldihydroquercetin. J. Nat. Prod. 2003, 66, 1562–1566. [Google Scholar] [CrossRef]
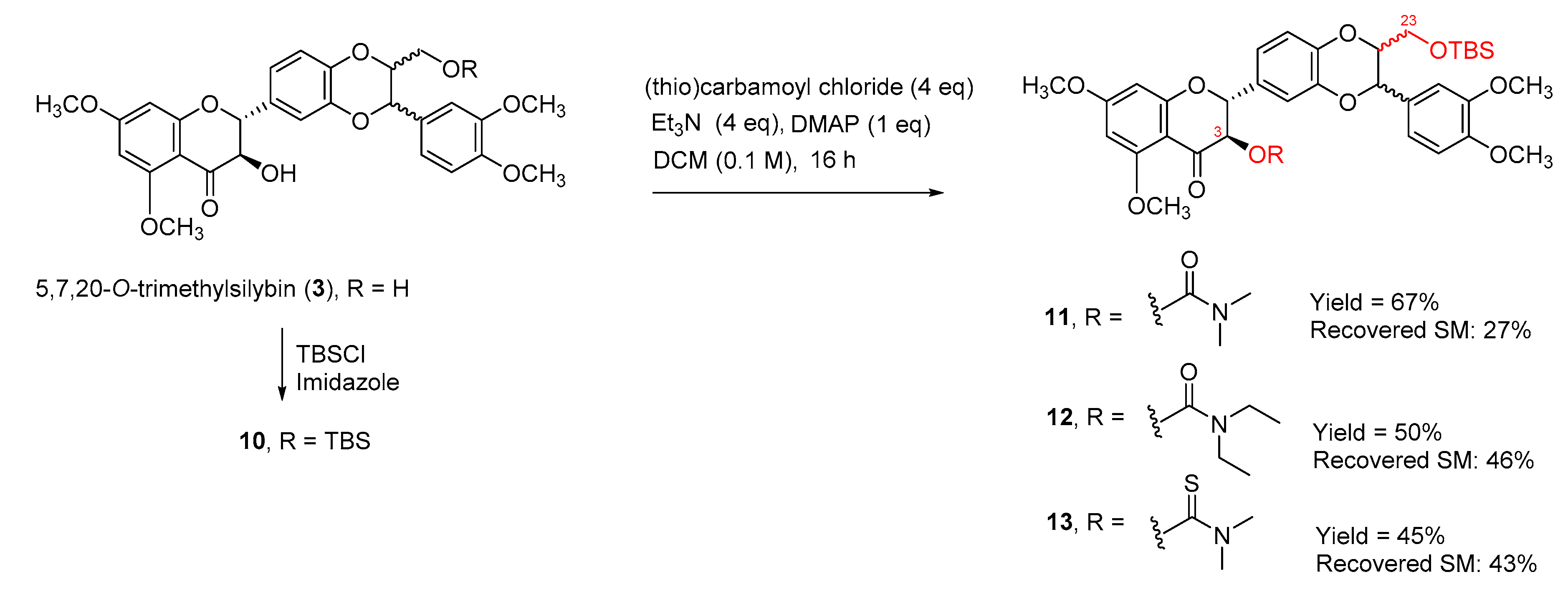
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).