
A Novel Dual Organocatalyst for the Asymmetric Pinder Reaction and a Mechanistic Proposal Consistent with the Isoinversion Effect Thereof


Abstract
:
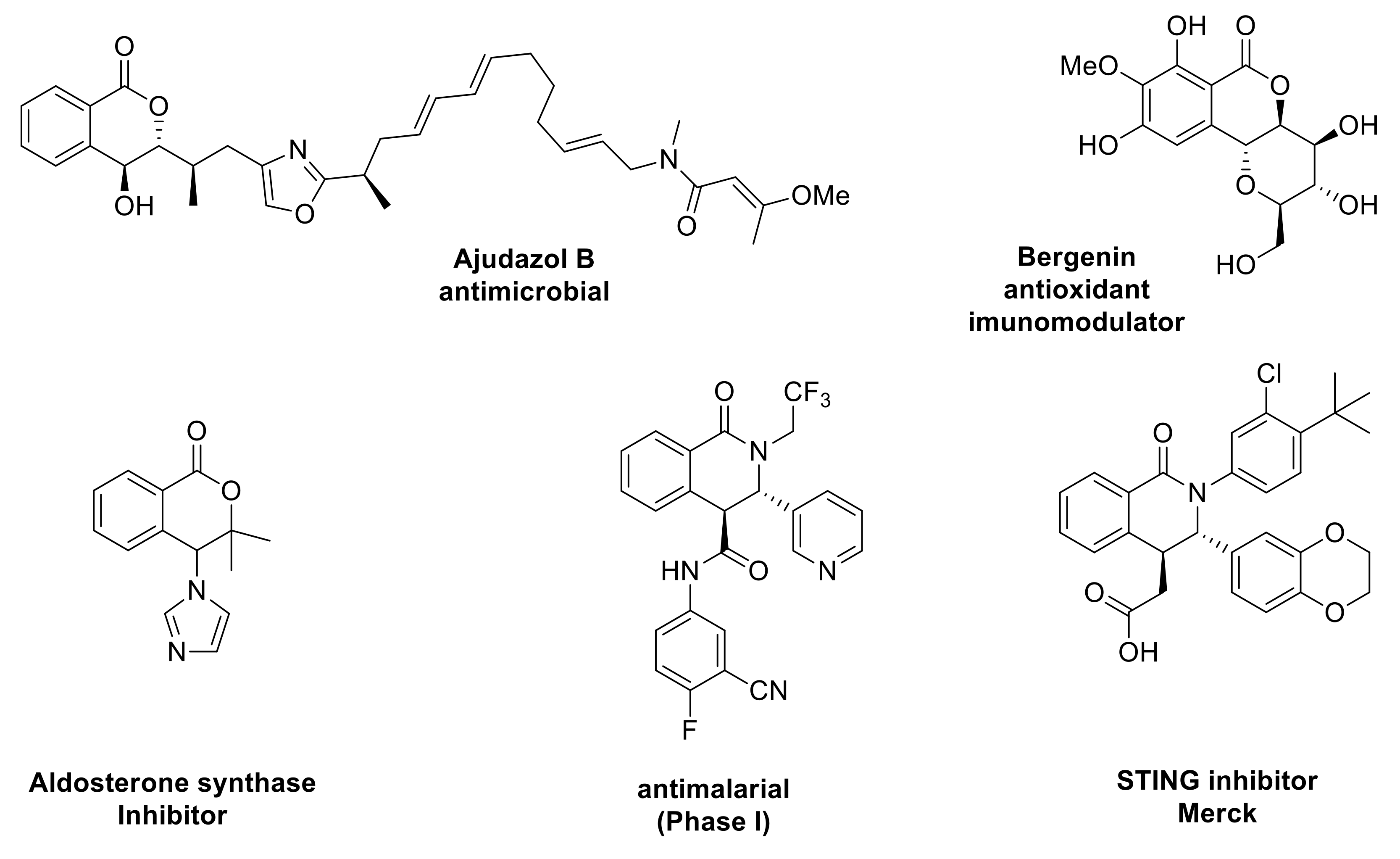
1. Introduction
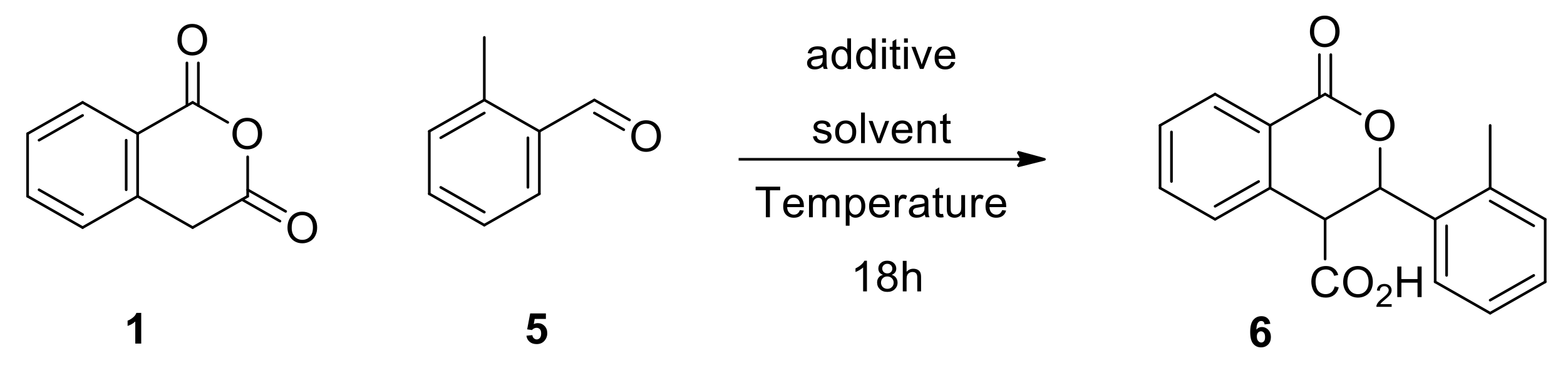
2. Results and Discussion
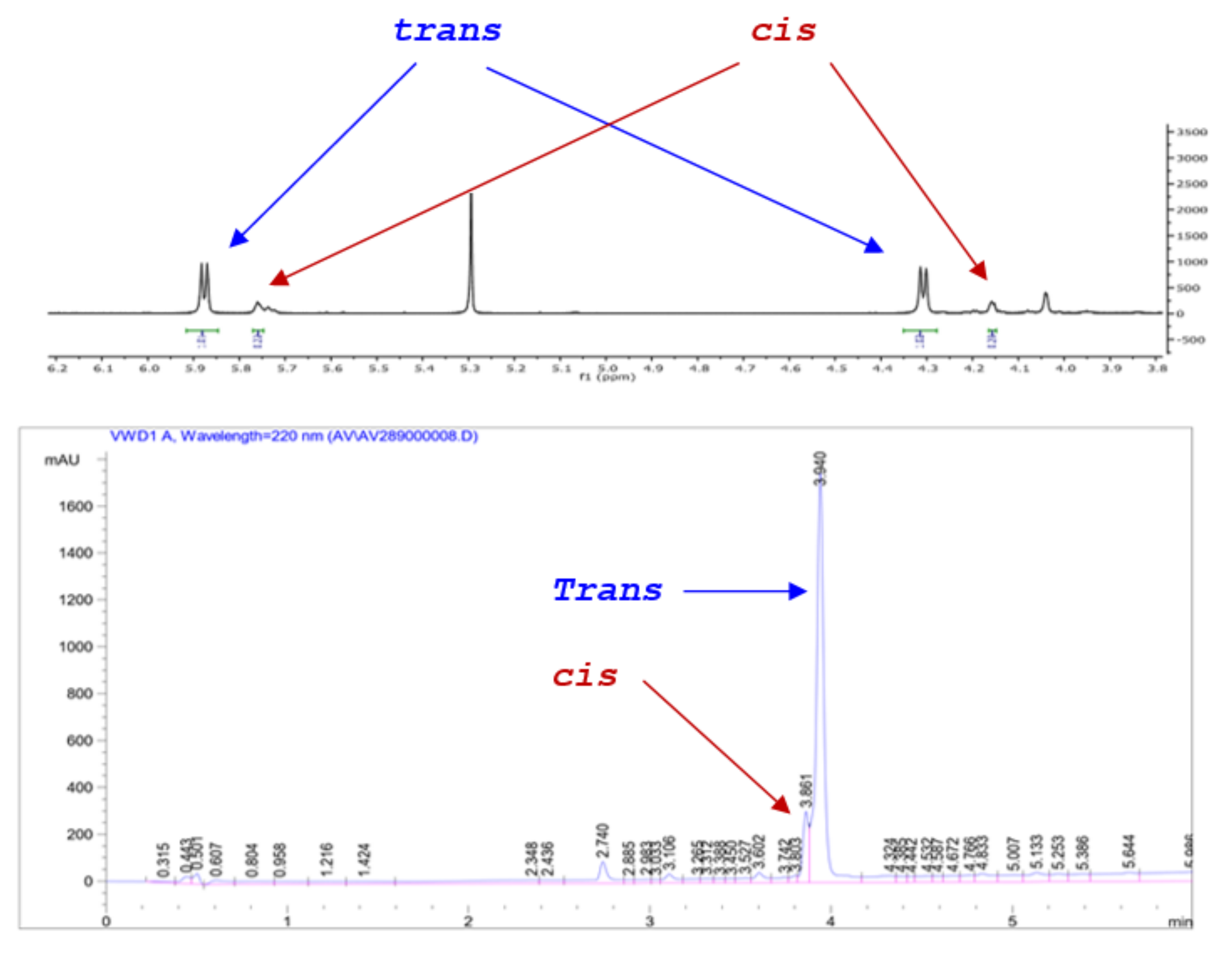
2.1. Exploring Conditions for Promoting and Suppressing the Racemic Reaction
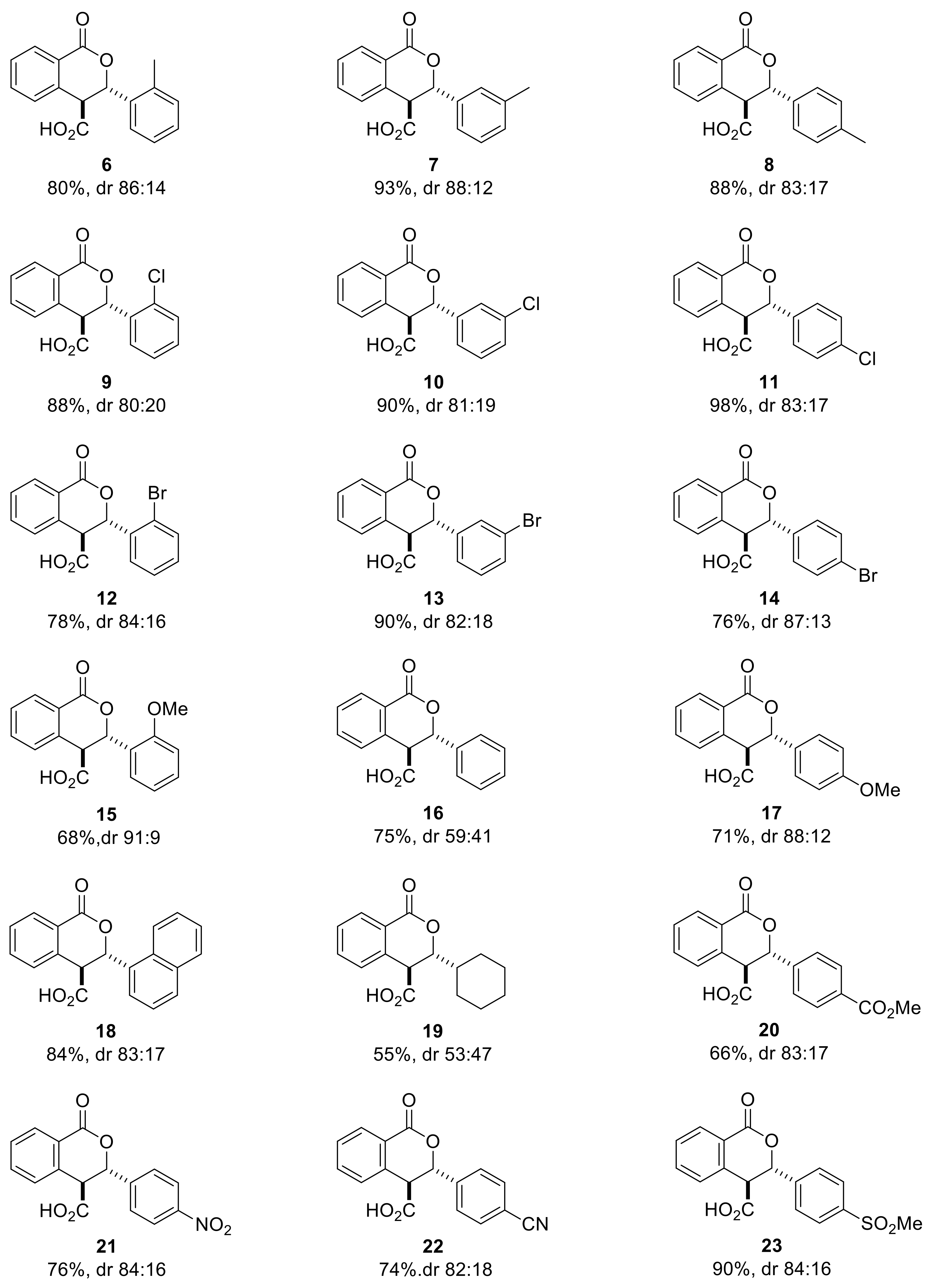
2.2. Exploring the Scope of the Aldehyde Substrate
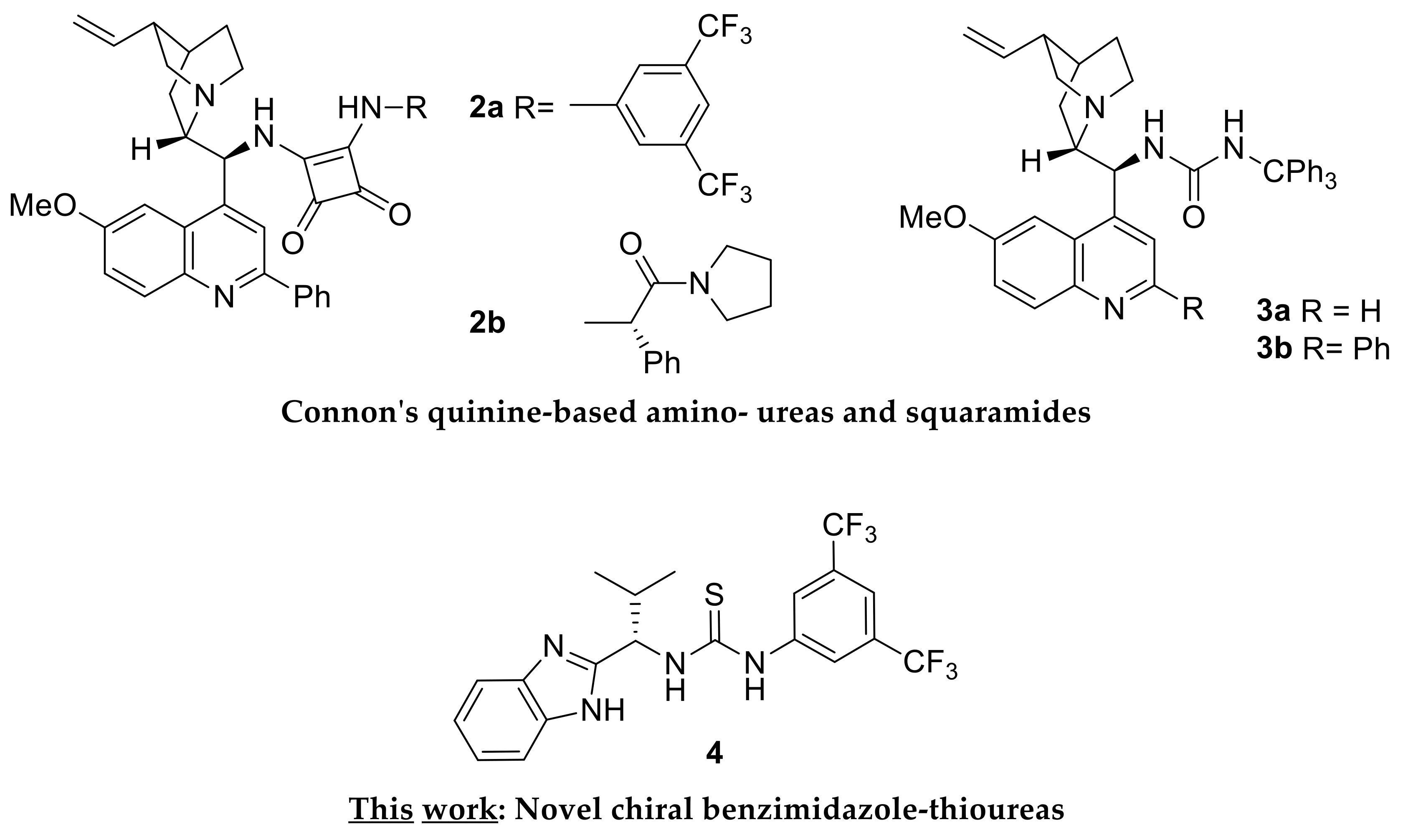
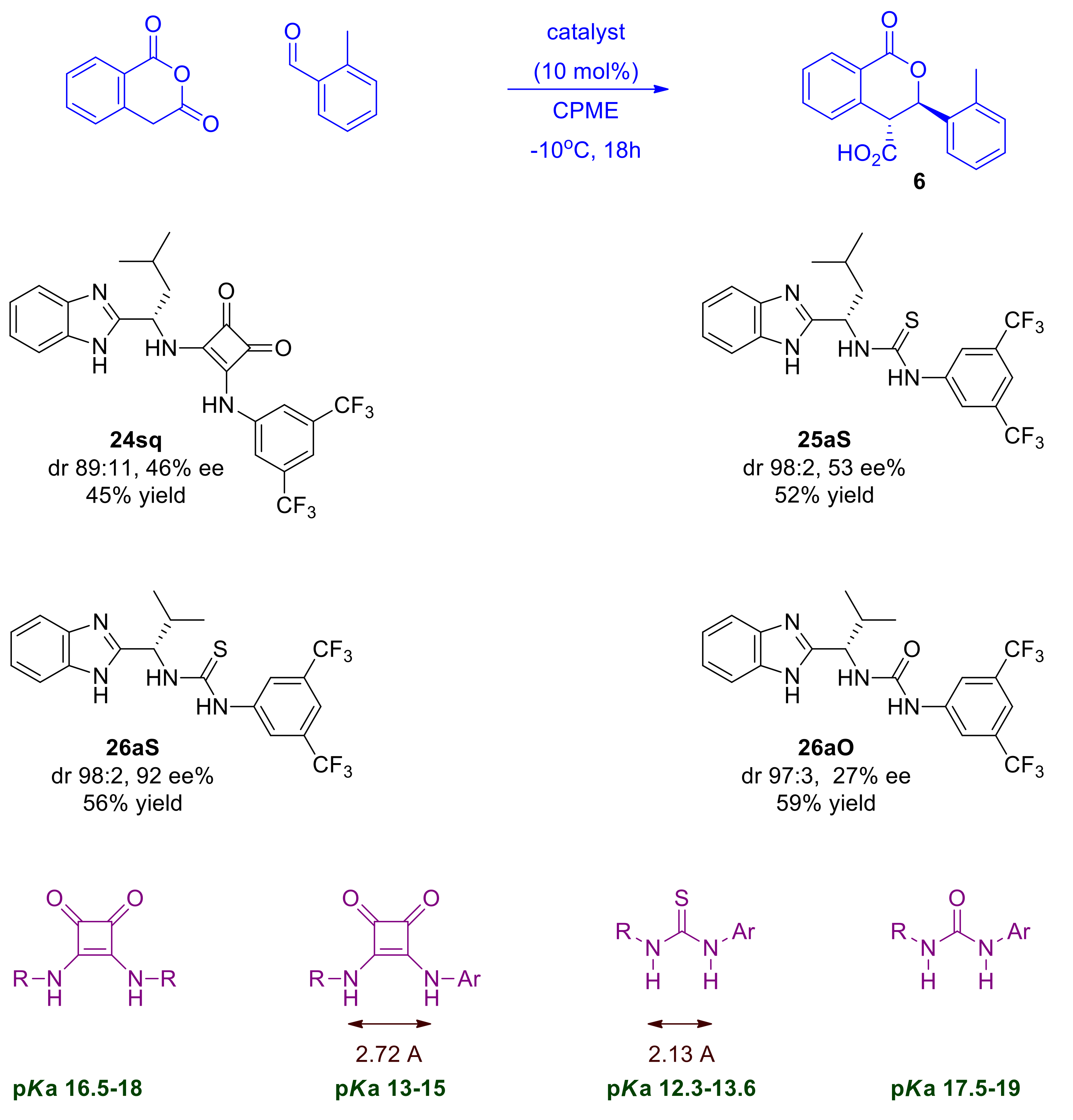
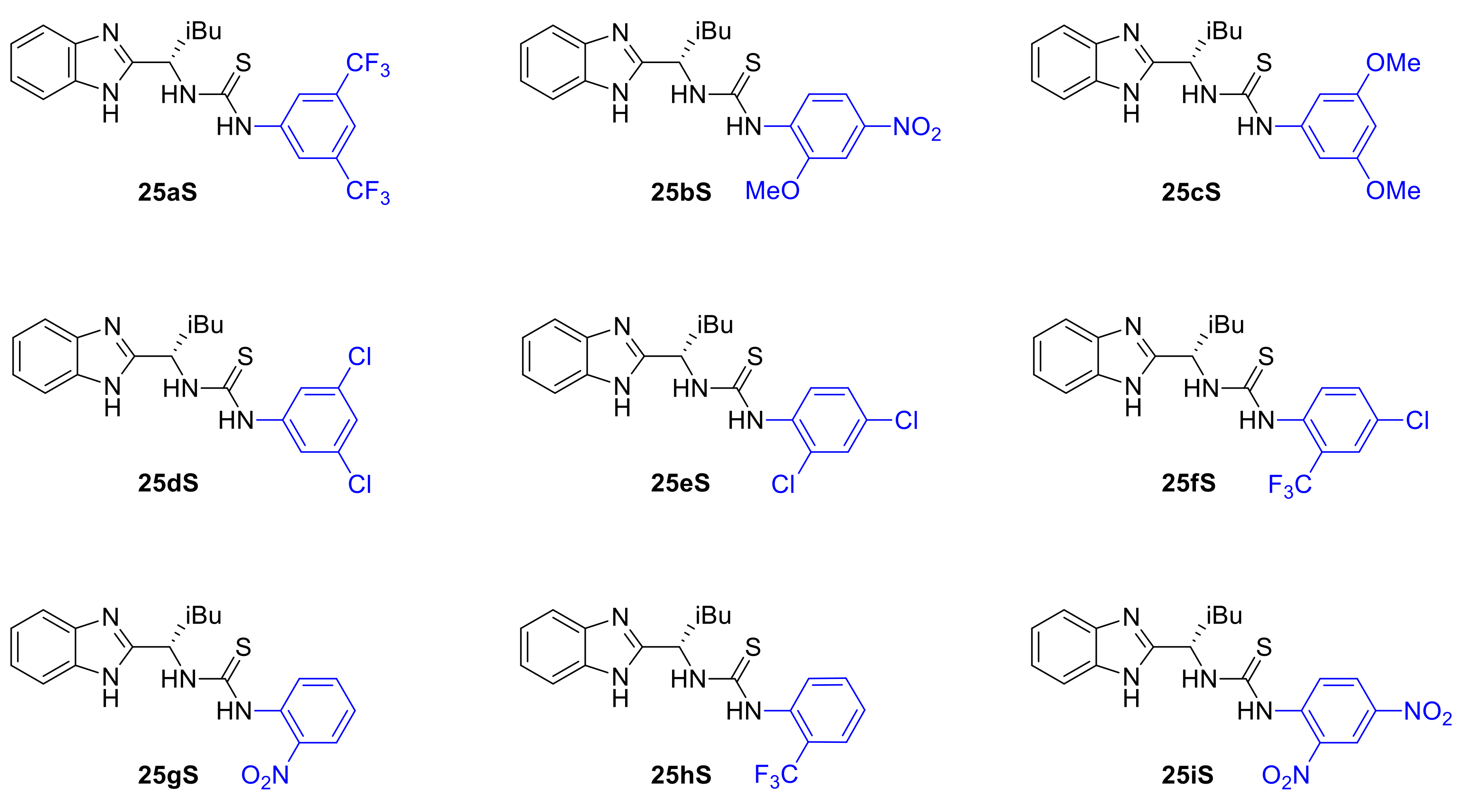
2.3. Design, Synthesis and Evaluation of Chiral Dual Organocatalysts
2.4. Optimization of Reaction Conditions for the Asymmetric Pinder Reaction
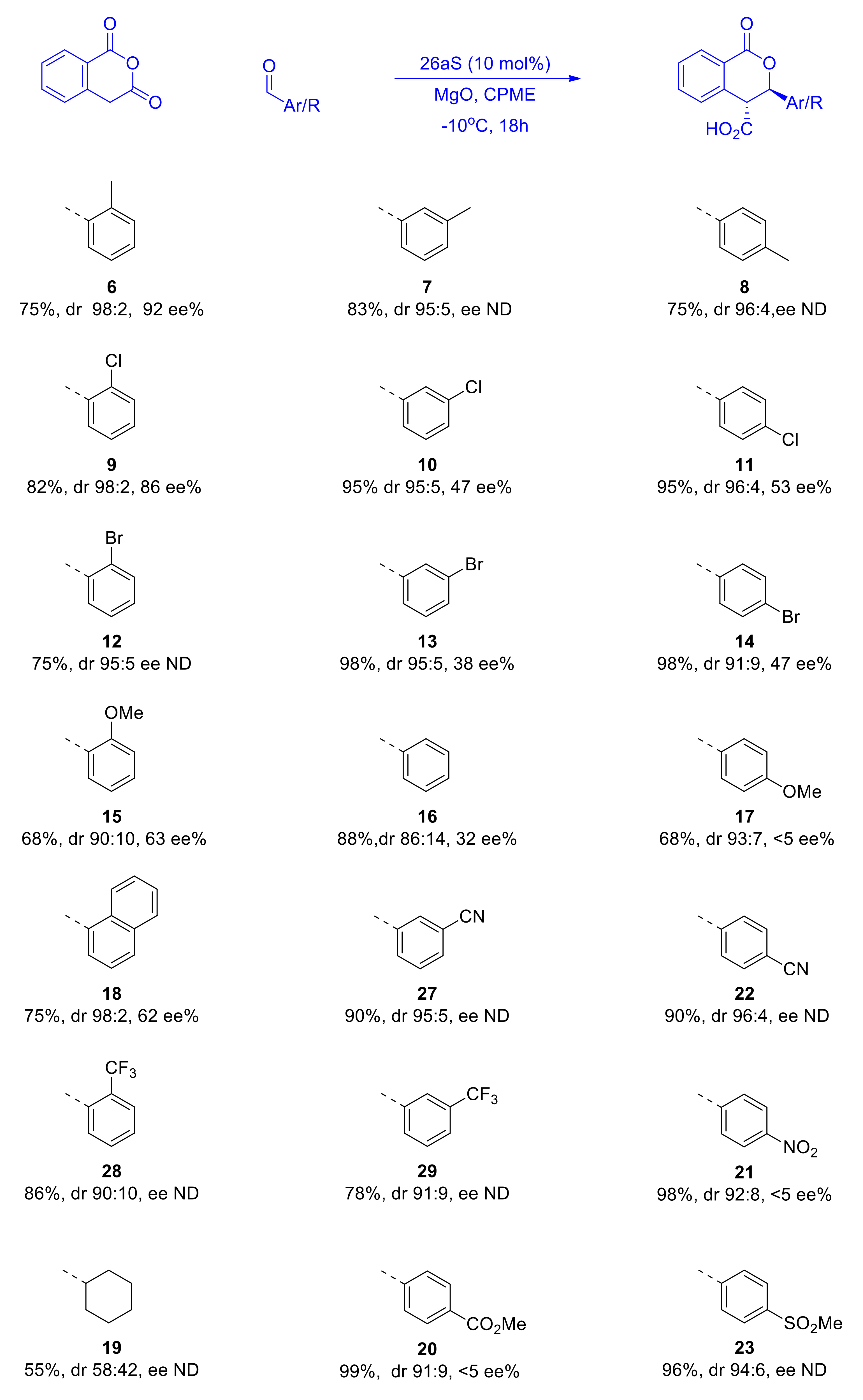
2.5. Substrate Scope in the Asymmetric Pinder Reaction Catalyzed by BIMAH-Thioureas
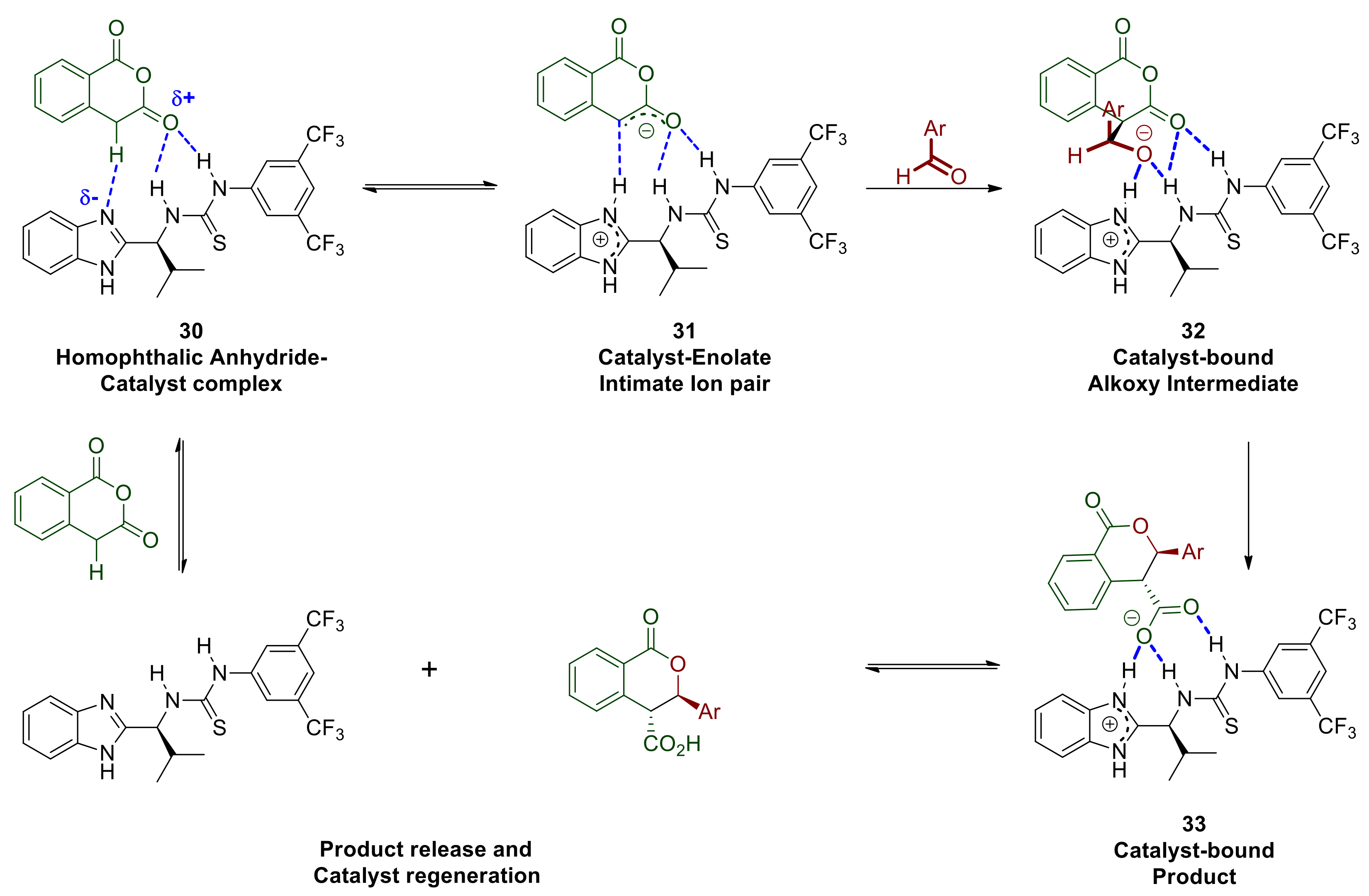
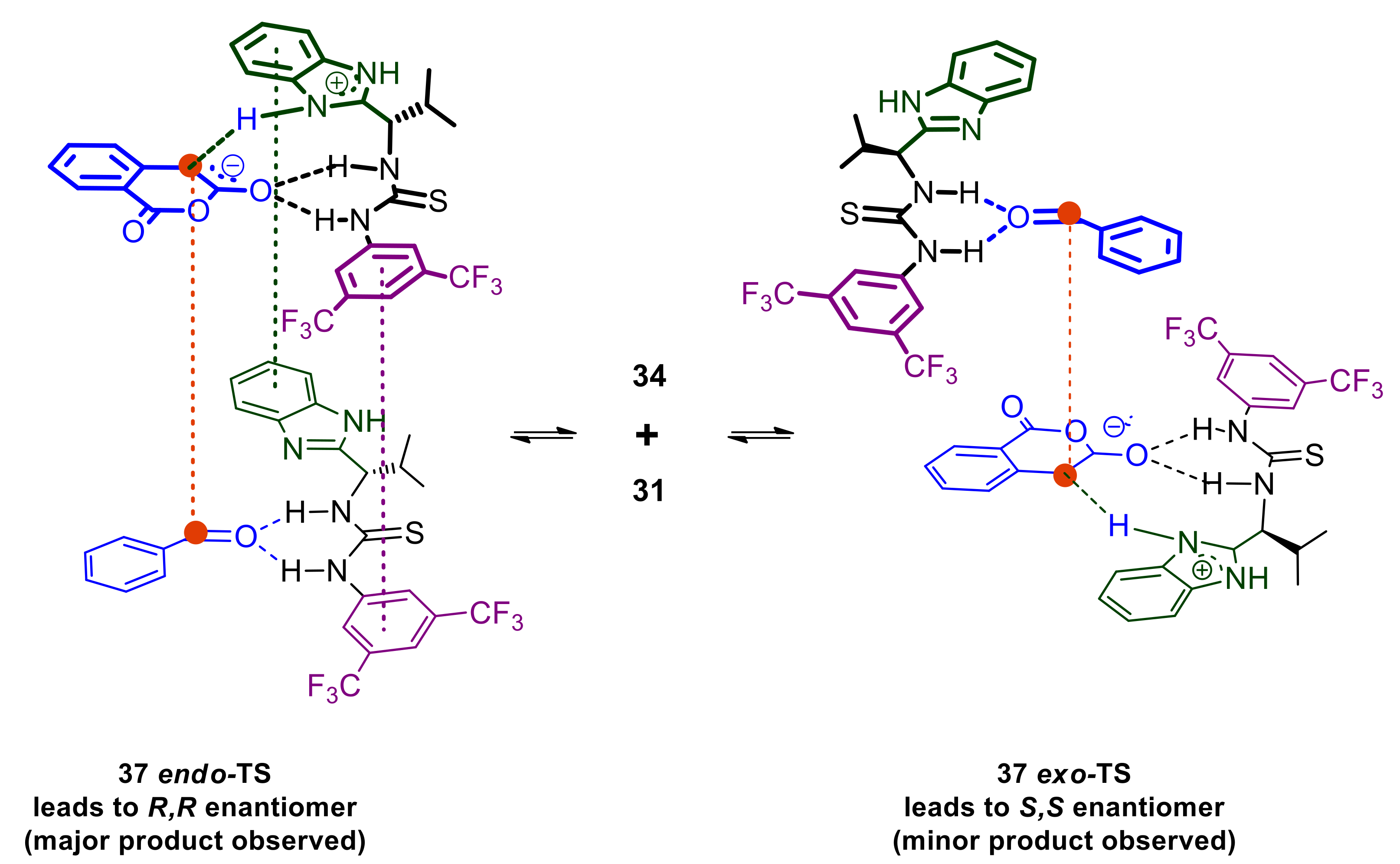
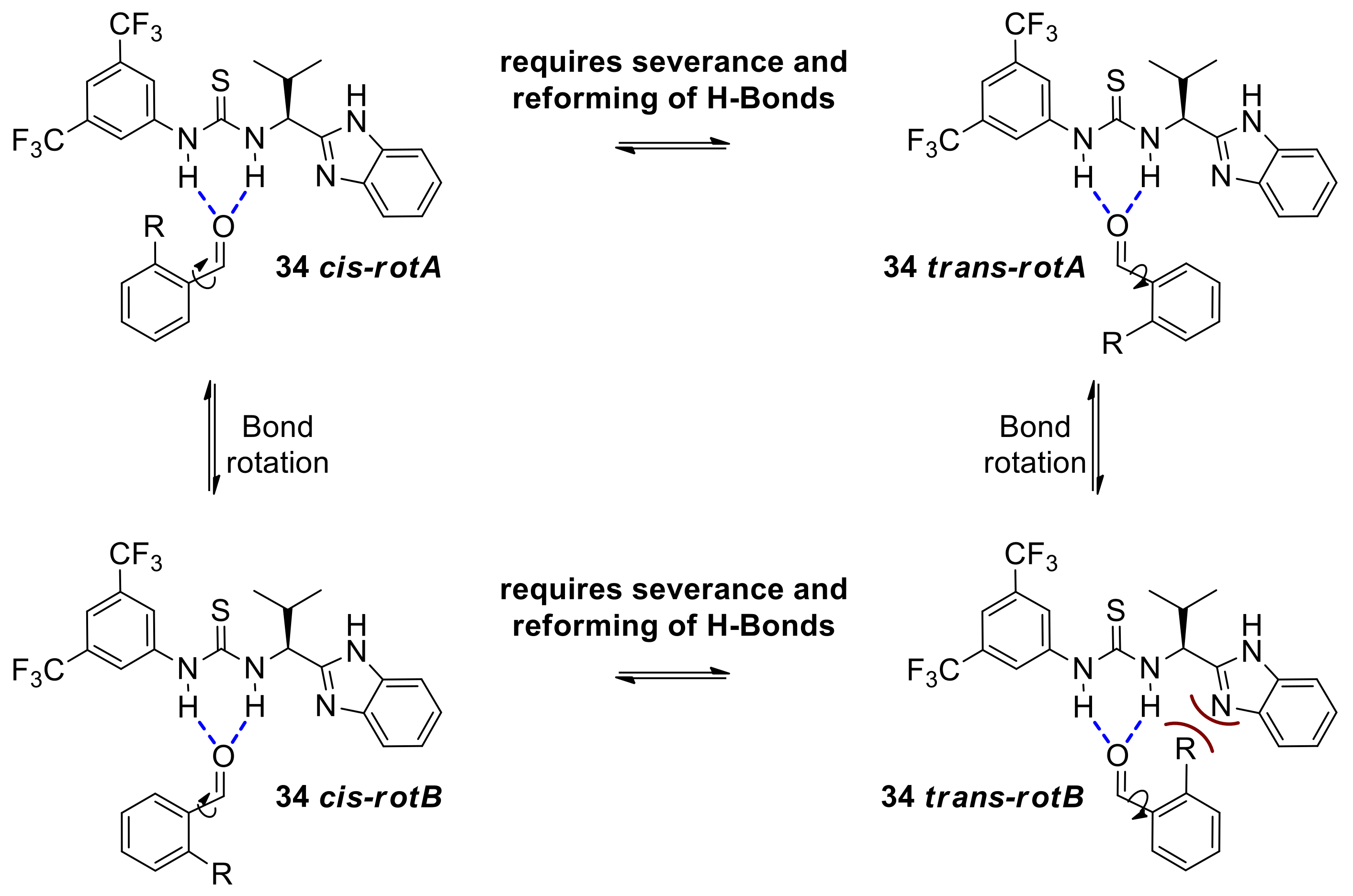
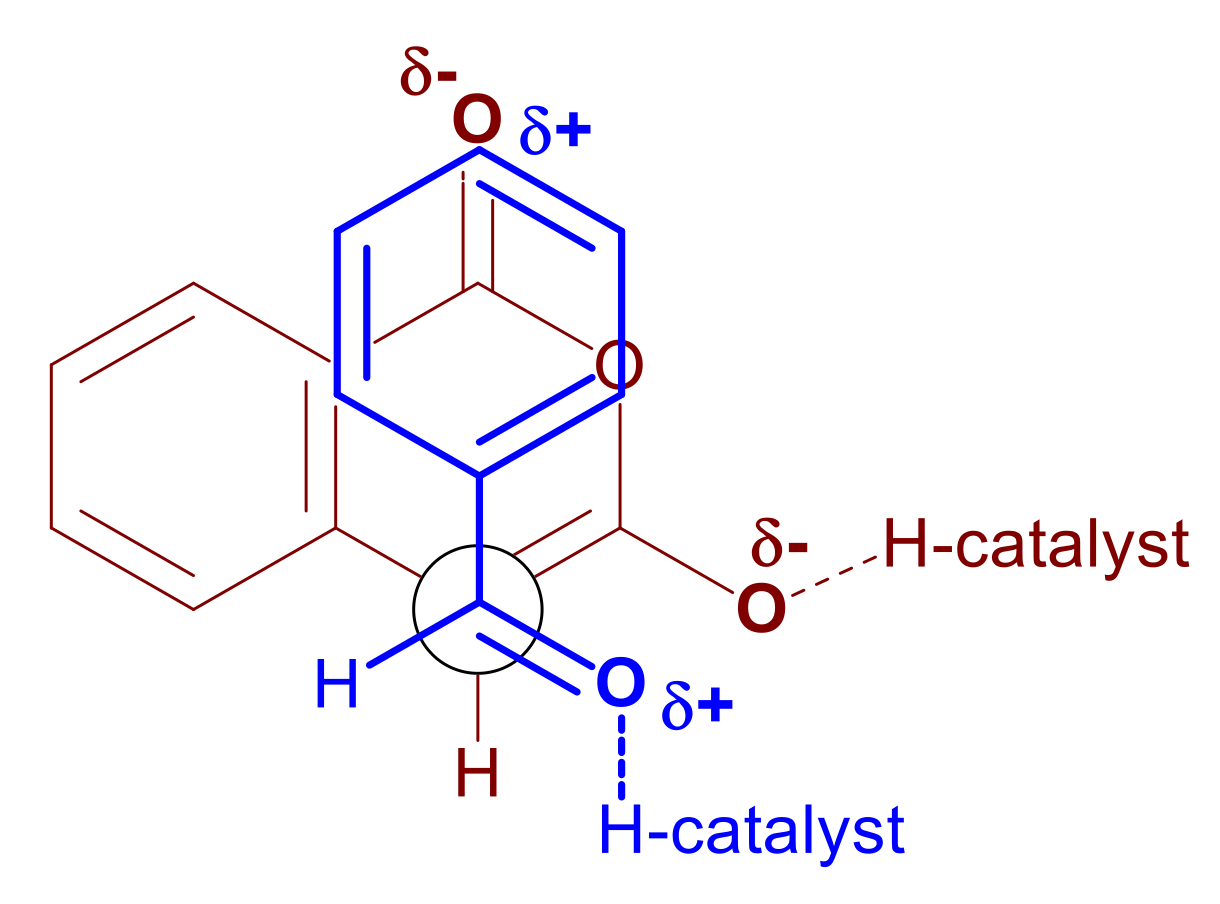
2.6. Mechanistic Considerations
- solvents of low polarity benefit the enantioselectivity of the process
- a mildly basic additive improves the reaction rate
- strongly electron donating or withdrawing groups on the aldehyde substrate lead to poor enantioselectivity
- the enantioselectivity of the catalyzed process is increased at higher catalyst loading and/or concentration
- the enantioselectivity-temperature relationship of the catalyzed process is governed by an inversion temperature
- the sterically congested ortho substituted benzaldehydes give higher enantioselectivity than their meta and para counterparts
- the diastereoselectivity is not correlated with enantioselectivity and is enhanced in the catalyzed process.
3. Materials and Methods
3.1. Chemistry Materials and Instrumentation
3.2. Experimental Procedures
3.2.1. Pinder Reaction Promoted by DIPEA – General Procedure A (Racemic Products)
3.2.2. Preparation of Methyl Esters: General Esterification Procedure Using TMS-DiazoMethane
3.2.3. Organocatalyzed Pinder Reaction: General Procedure B for Screening Chiral Catalysts and the Asymmetric Pinder Reaction with Various Substrates
3.2.4. Synthesis of Catalyst 24sq
3.2.5. Synthesis of Catalyst 26aS: General Procedure for Thiourea/BIMAH Organocatalysts
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Perkin, W.H.J.J. XI.—On the formation of coumarin and of cinnamic and of other analogous acids from the aromatic aldehydes. Chem. Soc. 1877, 31, 388–427. [Google Scholar] [CrossRef] [Green Version]
- Fittig, R.; Jayne, H.W. Ueber das Phenyl-Butyrolacton und die Phenyl-Paraconsäure. Justus Liebigs Ann. Chem. 1883, 216, 97. [Google Scholar]
- Jones, J.B.; Pinder, A.R.J. Some synthetical investigations in isocoumarin chemistry. Chem. Soc. 1958, 531, 2612–2618. [Google Scholar] [CrossRef]
- Yu, N.; Poulain, R.; Tartar, A.; Gesquiere, J.-C. Cycloaddition of homophthalic anhydrides with aldehydes and ketones: A route to 3,4-dihydroisocoumarin-4-carboxylic acid derivatives. Tetrahedron 1999, 55, 13735–13740. [Google Scholar] [CrossRef]
- Bogdanov, M.G.; Palamareva, M.D. cis/trans-Isochromanones. DMAP induced cycloaddition of homophthalic anhydride and aldehydes. Tetrahedron 2004, 60, 2525. [Google Scholar] [CrossRef]
- Castagnoli, N.J. Condensation of succinic anhydride with N-benzylidene-N-methylamine. Stereoselective synthesis of trans-and cis-1-methyl-4-carboxy-5-phenyl-2-pyrrolidinone. Org. Chem. 1969, 34, 3187. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, N.; Cushman, M.J. The condensation of succinic anhydrides with Schiff bases. Scope and mechanism. Org. Chem. 1971, 36, 3404. [Google Scholar] [CrossRef] [PubMed]
- Cushman, M.; Castagnoli, N.J. Synthesis of trans-3′-methylnicotine. Org. Chem. 1972, 37, 1268. [Google Scholar] [CrossRef]
- Cushman, M.; Castagnoli, N.J. Synthesis of pharmacologically active nitrogen analogs of the tetrahydrocannabinols. Org. Chem. 1974, 39, 1546. [Google Scholar] [CrossRef]
- Cushman, M.; Castagnoli, N.J. Novel approach to the synthesis of nitrogen analogs of the tetrahydrocannabinols. Org. Chem. 1973, 38, 440. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Z.; Levin, A.; Emge, T.J.; Rablen, P.R.; Floyd, D.M.; Knapp, S.J. N-Methylimidazole Promotes the Reaction of Homophthalic Anhydride with Imines. Org. Chem. 2014, 79, 7593–7599. [Google Scholar] [CrossRef]
- Laws, S.W.; Moore, L.C.; Di Maso, M.J.; Nguyen, Q.N.N.; Tantillo, D.J.; Shaw, J.T. Diastereoselective Base-Catalyzed Formal [4 + 2] Cycloadditions of N-Sulfonyl Imines and Cyclic Anhydrides. Org. Lett. 2017, 19, 2466–2469. [Google Scholar] [CrossRef] [Green Version]
- González-López, M.; Shaw, J.T. Cyclic anhydrides in formal cycloadditions and multicomponent reactions. Chem. Rev. 2009, 109, 164–189. [Google Scholar] [CrossRef] [PubMed]
- Essig, S.; Bretzke, S.; Miller, R.; Menche, D.J. Menche. Am. Chem. Soc. 2012, 134, 19362. [Google Scholar] [CrossRef]
- Enomoto, M.; Kuwahara, S. Total synthesis of bacilosarcins A and B. Angew. Chem. Int. Ed. 2009, 48, 1144. [Google Scholar] [CrossRef]
- Guiguemde, W.A.; Shelat, A.A.; Bouck, D.; Duffy, S.; Crowther, G.J.; Davis, P.H.; Smithson, D.C.; Connelly, M.; Clark, J.; Zhu, F.; et al. Chemical genetics of Plasmodium falciparum. Nature 2010, 465, 311–315. [Google Scholar] [CrossRef]
- Floyd, D.M.; Stein, P.; Wang, Z.; Liu, J.; Castro, S.; Clark, J.A.; Connelly, M.; Zhu, F.; Holbrook, G.; Matheny, A.; et al. Hit-to-lead studies for the antimalarial tetrahydroisoquinolone carboxanilides. J. Med. Chem. 2016, 59, 7950–7962. [Google Scholar] [CrossRef] [Green Version]
- Siu, T.; Altman, M.D.; Baltus, G.A.; Childers, M.; Ellis, J.M.; Gunaydin, H.; Hatch, H.; Ho, T.; Jewell, J.; Lacey, B.M.; et al. Discovery of a novel cGAMP competitive ligand of the inactive form of STING. ACS Med. Chem. Lett. 2019, 10, 92–97. [Google Scholar] [CrossRef]
- Croston, G.E.; Olsson, R.; Currier, E.A.; Burstein, E.S.; Weiner, D.; Nash, N.; Severance, D.; Allenmark, S.G.; Thunberg, L.; Ma, J.N.; et al. Discovery of the first nonpeptide agonist of the GPR14/urotensin-II receptor: 3-(4-chlorophenyl)-3-(2-(dimethylamino) ethyl) isochroman-1-one (AC-7954). J. Med. Chem. 2002, 45, 4950. [Google Scholar] [CrossRef] [PubMed]
- Papillon, J.; Adams, C. Imidazoles as aldosterone synthase inhibitors. EP 2301931 A1, 14 December 2007. [Google Scholar]
- Cornaggia, C.; Manoni, F.; Torrente, E.; Tallon, S.; Connon, S.J. Michael Addition–Lactonization of Arylacetyl Phosphonate to β,γ-Unsaturated α-Keto Esters for the Synthesis of Chiral syn-3,4-Dihydropyranones and 5,6-Dihydropyranones. Org. Lett. 2012, 14, 1850–1853. [Google Scholar] [CrossRef] [PubMed]
- Farid, U.; Aiello, M.L.; Connon, S.J. Highly Enantioselective Catalytic Kinetic Resolution of α-Branched Aldehydes through Formal Cycloaddition with Homophthalic Anhydrides. Chem. Eur. J. 2019, 25, 10074–10079. [Google Scholar] [CrossRef]
- Majee, D.; Jakkampudi, S.; Arman, H.D.; Zhao, J.C.-G. Enantioselective Synthesis of Cyclohexenol Derivatives from γ-Aryl-Substituted Enals via an Organocatalyzed Three-Component Reaction. Org. Lett. 2019, 21, 9166–9170. [Google Scholar] [CrossRef] [PubMed]
- Aiello, M.L.; Farid, U.; Trujillo, C.; Twamley, B.; Connon, S.J.J. Catalytic Asymmetric Cycloadditions between Aldehydes and Enolizable Anhydrides: Cis-Selective Dihydroisocoumarin Formation. Org. Chem. 2018, 83, 15499–15511. [Google Scholar] [CrossRef]
- Cornaggia, C.; Gundala, S.; Manoni, F.; Gopalasetty, N.; Connon, S.J. Catalytic formal cycloadditions between anhydrides and ketones: Excellent enantio and diastereocontrol, controllable decarboxylation and the formation of adjacent quaternary stereocentres +. Org. Biomol. Chem. 2016, 14, 3040–3046. [Google Scholar] [CrossRef]
- Collar, A.G.; Trujillo, C.; Lockett-Walters, B.; Twamley, B.; Connon, S.J. Catalytic Asymmetric γ-Lactam Synthesis from Enolisable Anhydrides and Imines. Chem. Eur J. 2019, 25, 7275–7279. [Google Scholar] [CrossRef]
- Cronin, S.A.; Gutiérrez, C.A.; Gundala, S.; Cornaggia, C.; Torrente, E.; Manoni, F.; Botte, A.; Twamley, B.; Connon, S.J. The first catalytic asymmetric cycloadditions of imines with an enolisable anhydride. Org. Biomol. Chem. 2016, 14, 6955–6959. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, C.L.; Hirschi, J.S.; Vetticatt, M.J.; Seidel, D. Catalytic enantioselective synthesis of lactams through formal [4+ 2] cycloaddition of imines with homophthalic anhydride. Angew. Chem. Int. Ed. Engl. 2017, 56, 2670–2674. [Google Scholar] [CrossRef]
- Li, Y.; Ding, K.; Sandoval, C.A. Hybrid NH2-Benzimidazole Ligands for Efficient Ru-Catalyzed Asymmetric Hydrogenation of Aryl Ketones. Org Lett. 2009, 11, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Jakab, G.; Tancon, C.; Zhang, Z.; Lippert, K.M.; Schreiner, P.M. (Thio) urea organocatalyst equilibrium acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef]
- Xiang, N.; Xin, L.; Zhen, W.; Jin-Pei, C. Applications of chiral squaramides: From asymmetric organocatalysis to biologically active compounds. Org. Lett. 2014, 16, 1786–1789. [Google Scholar] [CrossRef]
- Held, F.E.; Tsogoeva, S.B. Asymmetric cycloaddition reactions catalyzed by bifunctional thiourea and squaramide organocatalysts: Recent advances. Catal. Sci. Technol. 2016, 6, 645–667. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, Y. Development of chiral thiourea catalysts and its application to asymmetric catalytic reactions. Chem. Pharm. Bull. 2010, 58, 593–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvin, T.; Yadav, R.; Choudhury, L.H. Recent applications of thiourea-based organocatalysts in asymmetric multicomponent reactions (AMCRs). Org. Biomol. Chem. 2020, 18, 5513–5532. [Google Scholar] [CrossRef] [PubMed]
- Bora, P.; Jakkampudi, S.; Parella, R.; Sakkani, N.; Dai, Q.; Bihani, M.; Arman, H.D.; Zhao, J.C. Diastereodivergent synthesis of 4-oxocyclohexanecarbaldehydes by using the modularly designed organocatalysts upon switching on their iminium catalysis. Chem. Commun. 2021, 57, 5334–5337. [Google Scholar] [CrossRef]
- Sato, K.; Umeno, T.; Ueda, A.; Kato, T.; Doi, M.; Tanaka, M. Asymmetric 1,4-Addition Reactions Catalyzed by N-Terminal Thiourea-Modified Helical l-Leu Peptide with Cyclic Amino Acids. Chem. Eur. J. 2021, 27, 11216–11220. [Google Scholar] [CrossRef]
- Buschmann, H.; Scharf, H.-D.; Hoffmann, N.; Esser, P. Enthalpy-and/or entropy-controlled asymmetric oxidation: Stereocontrolling factors in Mn–salen-catalyzed oxidation. Angew. Chem. Int. Ed. 1991, 30, 477–515. [Google Scholar] [CrossRef]
- Lin, C.; Hiraga, Y.; Masaki, K.; Iefuji, H.; Ohkata, K. Temperature-dependence of enantioselectivity and desymmetrization in the acetylation of 2-mono- and 2,2-di-substituted 1,3-propanediols by a novel lipase isolated from the yeast Cryptococcus spp. S-2. Biocatal. Biotransfor. 2006, 24, 390–395. [Google Scholar] [CrossRef]
- Carrée, F.; Gil, R.; Collin, J. Enantioselective Ring Opening of meso-Epoxides by Aromatic Amines Catalyzed by Lanthanide Iodo Binaphtholates. Org. Lett. 2005, 7, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Stone, G. Oxazaborolidine catalyzed borane reductions of ketones: A significant effect of temperature on selectivity. Tetrahedron Asymmetry 1994, 5, 465–472. [Google Scholar] [CrossRef]
- Xu, J.; Wei, T.; Zhang, Q.J. Effect of Temperature on the Enantioselectivity in the Oxazaborolidine-Catalyzed Asymmetric Reduction of Ketones. Noncatalytic Borane Reduction, a Nonneglectable Factor in the Reduction System. Org. Chem. 2003, 68, 10146–10151. [Google Scholar] [CrossRef]
- Santhi, V.; Rao, J.M. Asymmetric reduction of prochiral ketones using in situ generated oxazaborolidines derived from amino alcohols of (1R)-camphor as catalysts. Tetrahedron Asymmetry 2000, 11, 3553–3560. [Google Scholar] [CrossRef]
- Corey, E.J.; Bakshi, R.K.; Shibata, S.J. Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. Am. Chem. Soc. 1987, 109, 5551–5553. [Google Scholar] [CrossRef]
- Corey, E.J.; Bakshi, R.K.; Shibata, S.; Chen, C.; Singh, V.K.J. A stable and easily prepared catalyst for the enantioselective. Am. Chem. Soc. 1987, 109, 7925–7926. [Google Scholar] [CrossRef]
- Garzan, A.; Jaganathan, A.; Salehi Marzijarani, N.; Yousefi, R.; Whitehead, D.C.; Jackson, J.E.; Borhan, B. Solvent-Dependent Enantiodivergence in the Chlorocyclization of Unsaturated Carbamates. Chem. Eur. J. 2013, 19, 9015–9021. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, Y.; Du, D.-M.; Xu, J.J. Notable and obvious ketene substituent-dependent effect of temperature on the stereoselectivity in the Staudinger reaction. Org. Chem. 2007, 72, 990–997. [Google Scholar] [CrossRef]
- Matusmoto, A.; Fujiwara, S.; Hiyoshi, Y.; Zawatzky, K.; Makarov, A.A.; Welch, C.J.; Soai, K. Unusual reversal of enantioselectivity in the asymmetric autocatalysis of pyrimidyl alkanol triggered by chiral aromatic alkanols and amines. Org. Biomol. Chem. 2017, 15, 555–558. [Google Scholar] [CrossRef] [Green Version]
- Hale, K.J.; Ridd, J.H. A reassessment of the isoniversion relationship. J. Chem. Soc. 1995, 2, 1601–1605. [Google Scholar] [CrossRef]
- Cainelli, G.; Galletti, P.; Giacomini, D. Solvent effects on stereoselectivity: More than just an environment. Chem. Soc. Rev. 2009, 38, 990–1001. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Promoter | Solvent (10 vol) | Temperature °C | Conversion at 18 h (a/a% of 6 by HPLC) | dr (trans:cis) |
|---|---|---|---|---|---|
| 1 | DIPEA (1 eq) | ΤΒΜΕ | −10 | 55 | 88:12 |
| 2 | 0 | 74 | 85:15 | ||
| 3 | 20 | 82 | 85:15 | ||
| 4 | THF | 20 | 94 | 86:14 | |
| 5 | NMI (2 eq) | TBME | −10 | 35 | 88:12 |
| 6 | 0 | 37 | 87:13 | ||
| 7 | 20 | 54 | 87:13 | ||
| 8 | - | −10 | 0 | - | |
| 9 | 0 | 0 | - | ||
| 10 | CPME | −10 | 0 | - | |
| 11 | 0 | 0 | - |
| Entry | Catalyst | Conversion (a/a% of 6 by HPLC) | ee% | dr (trans:cis) |
|---|---|---|---|---|
| 1 | 25aS | 52 | 56 | 98:2 |
| 2 | 25bS | 51 | 18 | 98:2 |
| 3 | 25cS | 32 | 29 | 96:4 |
| 4 | 25dS | 8 | - | - |
| 5 | 25eS | 15 | - | - |
| 6 | 25fS | 18 | - | - |
| 7 | 25gS | 28 | - | 97:3 |
| 8 | 25hS | 62 | 18 | 98:2 |
| 9 | 25iS | 10 | - | - |
| Entry | Reaction Time | Solvent | Concentration | Conversion (a/a% of 6 by HPLC) | ee% | dr (trans:cis) |
|---|---|---|---|---|---|---|
| 1 | 54 h | THF | 0.16 M | 80 | 60 | 98:2 |
| 2 | 54 h | TBME | 0.16 M | 58 | 86 | 98:2 |
| 3 | 36 h | 2-MeTHF | 0.16 M | 21 | - | - |
| 4 | 18 h | CPME | 0.16 M | 56 | 92 | 98:2 |
| 5 | 36 h | CPME | 0.16 M | 72 | 92 | 98:2 |
| 6 | 54 h | CPME | 0.08 M | 70 | 76 | 98:2 |
| 7 | 54 h | CPME | 0.04 M | 57 | 69 | 98:2 |
| Entry | Temperature (°C) | Conversion at 18 h (a/a% of 6 by HPLC) | ee% | dr (trans:cis) |
|---|---|---|---|---|
| 1 | 25 | 92 | 24 | 97:3 |
| 2 | 0 | 87 | 78 | 98:2 |
| 3 | −10 | 56 | 92 | 98:2 |
| 4 | −15 | 51 | 76 | 98:2 |
| 5 | −20 | 47 | 66 | 98:2 |
| Entry | MgO (eq) | Conversion (a/a% of 6 by HPLC at 36 h) | ee% | |
|---|---|---|---|---|
| Without Catalyst | 10 mol% of 26aS | |||
| 1 | none | 0 | 56 | 90 |
| 2 | 0.5 | 4 | 65 | 88 |
| 3 | 1 | 6 | 78 | 89 |
| 4 | 4 | 7 | 82 | 78 |
| Substrate | Substituent | Column Type | HPLC Settings | Retention Times |
|---|---|---|---|---|
| 6-acid | ο-Me | Chiralcel OD-H (4.6 mm × 25 cm) | Hexane/EtOH: 80/20, 1.0 mL mL/min, 25 °C, UV detection at 220 nm. | 6.1 min 6.5 min |
| 7-ester | m-Me | Chiralcel OD-H (4.6 mm × 25 cm) | Hexane/IPA: 90/10, 1.0 mL/min, 40 °C, UV detection at 220 nm. | 11.66 min 12.53 min |
| 8-acid | p-Me | Chiralcel OD-H (4.6 mm × 25 cm) | Hexane/ EtOH: 95/5, 1.0 mL/min, 40 °C, UV detection at 220 nm. | 21.33 min 22.79 min |
| 9-acid | o-Cl | YMC Amylose SA S-5 (250 × 4.6 mml.D.) | Hexane/IPA: 80/20 1.0 mL/min, 25 °C, UV detection at 220 nm. | 10.50 min 11.53 min |
| 10-ester | m-Cl | Chiralcel IA-H (4.6 mm × 25 cm) | Hexane/IPA: 80/20, 0.6 mL/min, 25 °C, UV detection 254 nm | 14.04 min 15.44 min |
| 11-ester | p-Cl | Chiralcel IA-H (4.6 mm × 25 cm) | Hexane/IPA: 85/15, 0.6 mL/min, 25 °C, UV detection at 280 nm | 21.82 min 24.24 min |
| 12-ester | o-Br | Chiralcel OD-H (4.6 mm × 25cm) | Hexane/IPA: 75/25, 0.8 mL/min, 18 °C, UV detection at 220 nm. | 7.9 min 8.5 min |
| 13-ester | m-Br | Chiralcel IA-H (4.6 mm × 25 cm) | Hexane/IPA: 90/10, 1.0 mL min, 25oC, UV detection at 210 nm. | 17.90 min 20.11 min |
| 14-ester | p-Br | Chiralcel IA-H (4.6 mm × 25 cm) | Hexane/IPA: 95/5, 0.6 mL/min, 25 °C, UV detection at 230 nm. | 56.38 min 63.67 min |
| 15-ester | o-MeO | Chiralcel IA-H (4.6 mm × 25 cm) | Hexane/IPA: 85/15, 0.6 mL/min, 25 °C, UV detection at 230 nm | 28.21 min 30.68 min |
| 16-ester | H | Chiralcel IA-H (4.6 mm × 25 cm) | Hexane/IPA: 85/15, 0.6 mL/min, 25 °C, UV detection at 254 nm. | 18.83 min 21.10 min |
| 17-ester | p-MeO | Chiralcel IA-H (4.6 mm × 25 cm) | Hexane /IPA: 85/15, 0.6 mL/min, 25 °C, UV detection at 230 nm. | 17.87 min 22.00 min |
| 21-ester | p-NO2 | Chiralcel IA-H (4.6 mm × 25 cm) | Hexane/IPA:80/20, 0.6 mL/min, 25 °C, UV detection at 254 nm. | 13.00 min 14.77 min |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moschona, F.; Vagena, A.; Vidali, V.P.; Rassias, G. A Novel Dual Organocatalyst for the Asymmetric Pinder Reaction and a Mechanistic Proposal Consistent with the Isoinversion Effect Thereof. Molecules 2021, 26, 6398. https://doi.org/10.3390/molecules26216398
Moschona F, Vagena A, Vidali VP, Rassias G. A Novel Dual Organocatalyst for the Asymmetric Pinder Reaction and a Mechanistic Proposal Consistent with the Isoinversion Effect Thereof. Molecules. 2021; 26(21):6398. https://doi.org/10.3390/molecules26216398
Chicago/Turabian StyleMoschona, Fotini, Athena Vagena, Veroniki P. Vidali, and Gerasimos Rassias. 2021. "A Novel Dual Organocatalyst for the Asymmetric Pinder Reaction and a Mechanistic Proposal Consistent with the Isoinversion Effect Thereof" Molecules 26, no. 21: 6398. https://doi.org/10.3390/molecules26216398
APA StyleMoschona, F., Vagena, A., Vidali, V. P., & Rassias, G. (2021). A Novel Dual Organocatalyst for the Asymmetric Pinder Reaction and a Mechanistic Proposal Consistent with the Isoinversion Effect Thereof. Molecules, 26(21), 6398. https://doi.org/10.3390/molecules26216398