3.2. Experimental Procedures and Compound Data
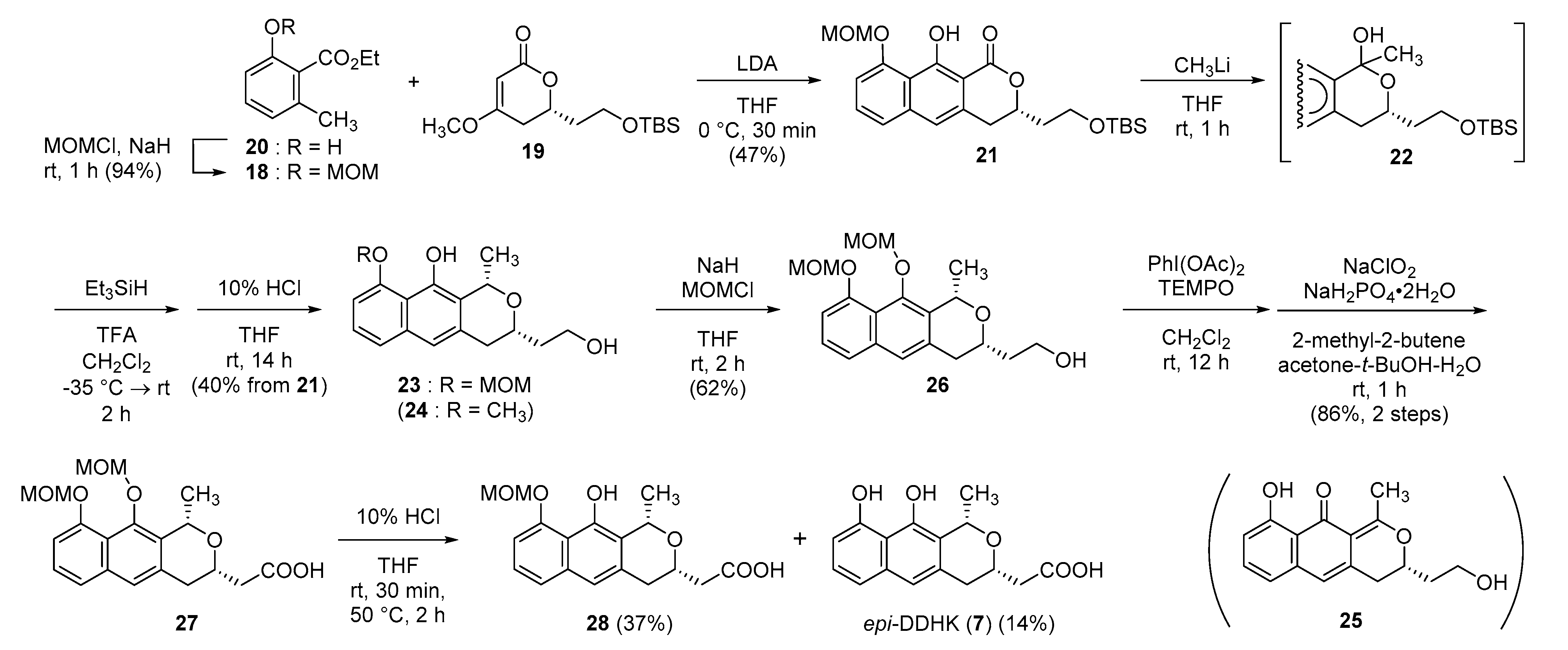
Ethyl 2-(methoxymethoxy)-6-methylbenzoate (18). To a mixture of NaH (55%, 487 mg, 11.2 mmol) in THF (3.0 mL), a solution of 20 (1.00 g, 5.55 mmol) in THF (7.0 mL) was added at 0 °C. The mixture was stirred at 0 °C for 30 min. MOMCl (0.55 mL, 7.24 mmol) was added at 0 °C, and the whole was stirred at rt for 1 h. Saturated aqueous NaHCO3 (10 mL) was added, and the whole was partitioned with AcOEt (10 mL) and H2O (3 mL). The aqueous layer was extracted with AcOEt (3 mL × 10 mL). Combined organic phase was washed with brine (1 mL × 10 mL) and was dried over Na2SO4. Solvent was evaporated in vacuo and the residue was purified by column chromatography (CC) (hexane–AcOEt = 95:5–82:18) to give 18 as a colorless oil (1.17 g, 94%). IR (ATR) νmax cm−1: 1725 (C=O). 1H-NMR (400 MHz, CDCl3) δ: 1.38 (3H, t, J = 7.3 Hz, CO2CH2CH3), 2.31 (3H, s, C6-CH3), 3.45 (3H, s, OCH3), 4.40 (2H, q, J = 7.3 Hz, CO2CH2), 5.18 (2H, s, OCH2O), 6.85 (1H, d, J = 7.8 Hz, C3-H), 6.99 (1H, d, J = 8.2 Hz, C5-H), 7.22 (1H, dd, J = 8.2, 7.8 Hz, C4-H). 13C-NMR (125 MHz, CDCl3) δ:14.2, 19.1, 56.0, 61.0, 94.5, 112.1, 123.4, 125.0, 130.0, 136.2, 153.7, 168.1. HRESIMS m/z 247.0940 (calcd for C12H16NaO4: 247.0946).
(S)-3-{2-[(tert-Butyldimethylsilyl)oxy]ethyl}-10-hydroxy-9-(methoxymethoxy)-3,4-dihydro-1H-benzo[g]isochromen-1-one (21). To a solution of diisopropylamine (0.74 mL, 5.28 mmol) in THF (12 mL), BuLi (1.63 M in hexane, 3.25 mL, 5.30 mmol) was added at −78 °C, and the mixture was stirred at −78 °C for 20 min. A solution of 18 (398 mg, 1.77 mmol) in THF (1.7 mL) was added at −78 °C, and the whole was stirred at −78 °C for 15 min. A solution of 19 (504 mg, 1.76 mmol) in THF (1.7 mL) was added at −78 °C, and the mixture was stirred at 0 °C for 30 min. Saturated aqueous NH4Cl (15 mL) was added at 0 °C, and the whole was partitioned with AcOEt (15 mL) and H2O (5 mL). Aqueous layer was extracted with AcOEt (2 mL × 15 mL). Combined organic layer was washed with brine (1 mL × 15 mL) and was dried over Na2SO4. The solvent was evaporated in vacuo, and the residue was purified by CC (hexane–AcOEt = 91:9) to give 21 as a yellow oil (360 mg, 47%). IR (ATR) νmax cm−1: 3200–2800 (OH), 1655 (C=O). 1H-NMR (400 MHz, CDCl3) δ: 0.08 (6H, s, Si(CH3)2), 0.89 (9H, s, SiC(CH3)3), 1.95 (1H, dddd, J = 14.2, 8.2, 5.0, 5.0 Hz, one of C1′-H2), 2.10 (1H, dddd, J = 14.2, 8.2, 5.0, 5.0 Hz, one of C1′-H2), 3.01–3.12 (2H, m, C4-H2), 3.61 (3H, s, OCH3), 3.82 (1H, ddd, J = 10.5, 10.5, 5.3 Hz, one of C2′-H2), 3.90 (1H, ddd, 10.5, 8.2, 4.6 Hz, one of C2′-H2), 4.78 (1H, dddd, J = 9.2, 8.0, 4.6, 4.6 Hz, C3-H), 5.38 (2H, s, OCH2O), 7.01 (1H, s, C5-H), 7.10 (1H, dd, J = 7.8, 0.9 Hz, C8-H), 7.33 (1H, dd, J = 7.8, 0.9 Hz, C6-H), 7.49 (1H, dd, J = 7.8, 7.8 Hz, C7-H). 13C-NMR (100 MHz, CDCl3) δ: −5.48, −5.47, 18.2, 25.8, 33.5, 37.7, 56.4, 58.4, 76.6, 95.8, 102.4, 111.7, 116.0, 116.1, 121.3, 130.5, 133.3, 139.8, 156.3, 163.8, 171.0. [α + 12.7 (c 1.1, CHCl3). HREIMS m/z 432.1973 (calcd for C23H32O6Si: 432.1968).
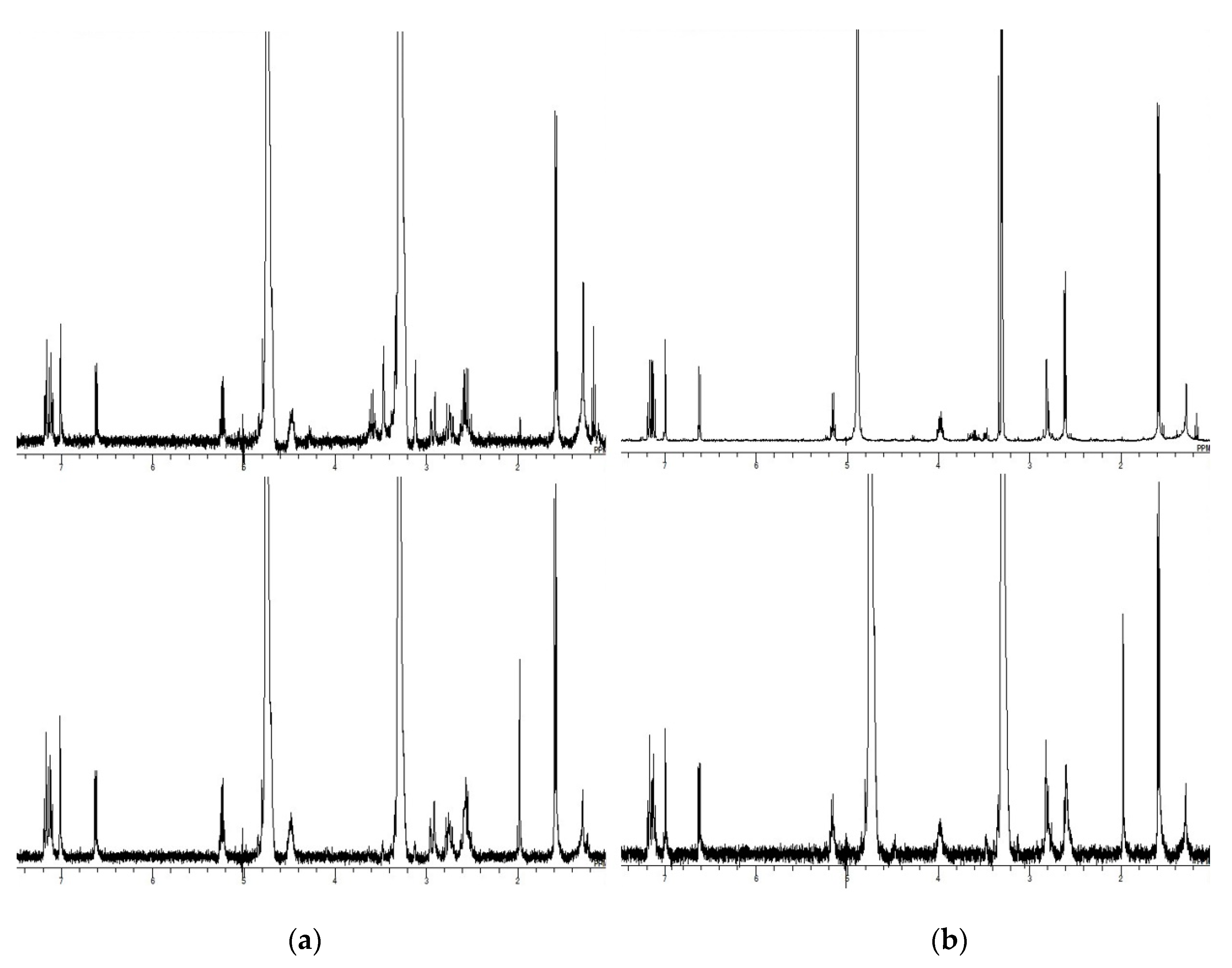
(1S,3S)-2-(3,4-Dihydro-10-hydroxy-9-methoxymethoxy-1-methyl-1H-naphtho[2,3-c]pyran-3-yl)ethanol (23) and (S)-3,4-Dihydro-9-hydroxy-3-(2-hydroxyethyl)-1-methyl-10H-naphtho[2,3-c]pyran-10-one (25). To a solution of 21 (102 mg, 0.24 mmol) in THF (1.6 mL), CH3Li (1.10 M in Et2O, 0.64 mL, 0.70 mmol) was added at 0 °C within 5 min. The whole was stirred at rt for 1 h. Saturated aqueous NH4Cl (3 mL) was added at 0 °C, and the whole was partitioned with CH2Cl2 (6 mL) and H2O (2 mL). Aqueous layer was extracted with CH2Cl2 (3 mL × 6 mL). Combined organic layer was washed with H2O (1 mL × 6 mL) and brine (1 mL × 6 mL) and dried over Na2SO4. Solvent was evaporated in vacuo to give 22 as a yellow oil (110.2 mg, 105%), which was used to next step without further purification. To a solution of crude 22 in CH2Cl2 (1 mL), TFA (0.054 mL, 0.71 mmol) and Et3SiH (0.11 mL, 0.69 mmol) were added successively at −78 °C. The mixture was warmed to −35 °C within 1 h and was stirred at rt for 1 h. Saturated aqueous NaHCO3 (5 mL) was added, and the whole was partitioned with CHCl3 (10 mL) and H2O (3 mL). The aqueous layer was extracted with CHCl3 (3 mL × 5 mL). Combined organic layer was washed with H2O (1 mL × 5 mL) and brine (1 mL × 5 mL) and was dried over Na2SO4. Solvent was evaporated in vacuo. The residue (a brown oil, 136 mg) was dissolved in THF (2.5 mL) and 10% HCl (0.5 mL) was added at rt. The mixture was stirred at rt for 14 h. The whole was diluted with H2O (3 mL) and was extracted with AcOEt (3 mL × 6 mL). Combined organic layer was washed with brine (1 mL × 6 mL) and was dried over Na2SO4. Solvent was evaporated in vacuo, and the residue was purified by CC (hexane–AcOEt = 55:45–34:66) to give 23 as a brown oil (30 mg, 40%) and 25 as dark green powder (8 mg, 12%). 23: IR (ATR) νmax cm−1: 3383 (OH). 1H-NMR (400 MHz, CDCl3) δ: 1.65 (3H, d, J = 6.4 Hz, C1-CH3), 1.88–2.00 (2H, m, C1′-H2), 2.73 (1H, dd, J = 15.6, 1.8 Hz, one of C4-H2), 2.96 (1H, dd, J = 15.6, 11.0 Hz, one of C4-H2), 3.15 (1H, s, C2′-OH), 3.59 (3H, s, OCH3), 3.83–3.91 (1H, m, C3-H), 3.90 (2H, t, J = 5.5 Hz, C2′-H), 5.26 (1H, q, J = 6.4 Hz, C1-H), 5.43 (1H, d, J = 9.2 Hz, one of OCH2O), 5.44 (1H, d, J = 9.2Hz, one of OCH2O), 6.98 (1H, dd, J = 7.8, 0.9 Hz, C8-H), 7.06 (1H, s, C5-H), 7.24 (1H, dd, J = 8.2, 7.8 Hz, C7-H), 7.36 (1H, d, J = 7.8 Hz, C6-H), 9.61 (1H, s, C10-OH). 13C-NMR (100 MHz, CDCl3) δ: 21.8, 36.0, 37.5, 56.9, 61.6, 71.3, 74.3, 95.8, 107.1, 113.9, 117.5, 121.1, 122.1, 125.5, 134.96, 135.02, 150.0, 153.6. HRESIMS m/z 341.1396 (calcd for C18H22NaO5: 341.1365). [α–133.7 (c 0.5, CHCl3). 25: mp 114.0–116.3 °C. IR (ATR) νmax cm−1: 3290 (O-H), 1635 (C=O). 1H-NMR (400 MHz, CDCl3) δ: 1.97 (1H, dddd, J = 14.6, 7.5, 5.7, 4.8 Hz, one of C1′-H2), 2.09 (1H, dddd, J = 14.6, 7.8, 5.0, 5.0 Hz, one of C1′-H2), 2.65 (3H, s, C1-CH3), 2.74 (1H, ddd, J = 16.0, 10.8, 1.6 Hz, one of C4-H2), 2.85 (1H, ddd, J = 16.0, 3.4, 0.9 Hz, one of C4-H2), 3.86–3.95 (2H, m, C2′-H), 4.50–4.57 (1H, m, C3-H), 6.26 (1H, s, C5-H), 6.74 (1H, dd, J = 7.8, 0.9 Hz, C8-H), 6.78 (1H, dd, J = 8.0, 0.9 Hz, C6-H), 7.39 (1H, dd, J = 8.0, 8.0 Hz, C7-H). 13C-NMR (125 MHz, CDCl3) δ: 23.4, 33.7, 36.7, 58.7, 111.3, 114.3, 115.7, 116.2, 116.9, 129.1, 135.1, 138.4, 163.5, 177.3, 188.8 (1C: missing, overlapped with a signal of CDCl3). HRESIMS m/z 273.1140 (calcd for C16H17O4: 273.1127). [α + 116.8 (c 0.3, CHCl3).
(1S,3S)-2-{3,4-Dihydro-3-(2-hydroxyethyl)-9,10-bis(methoxymethoxy)-1-methyl-1H-naphtho[2,3-c]pyran-3-yl}ethanol (26). To a suspension of NaH (55%, 2.2 mg, 0.050 mmol) in THF (0.05 mL), a solution of 23 (14 mg, 0.042 mmol) in THF (0.2 mL) was added at 0 °C, and the whole was stirred at 0 °C for 10 min. A solution of MOMCl (0.0038 mL, 0.050 mmol) in THF (0.2 mL) was added at 0 °C, and the whole was stirred at rt for 2 h. Ice-H2O (1 mL) was added, and the whole was extracted with AcOEt (3 mL × 3 mL). Combined organic layer was washed with brine (1 mL × 1 mL) and was dried over Na2SO4. Solvent was evaporated in vacuo, and the residue was purified by CC (hexane–AcOEt = 57:43) to give 26 as a colorless oil (9 mg, 62%). IR (ATR) νmax cm−1: 3419 (O-H). 1H-NMR (400 MHz, CDCl3) δ: 1.71 (3H, d, J = 6.2 Hz, C1-CH3), 1.90–2.01 (2H, m, C1′-H2), 2.77 (1H, dd, J = 15.6, 2.1 Hz, one of C4-H2), 2.96 (1H, dd, J = 15.6, 11.0 Hz, one of C4-H2), 2.99 (1H, br, OH), 3.57 (3H, s, OCH3), 3.59 (3H, s, OCH3), 3.88–3.94 (3H, m, C3-H, C2′-H2), 4.96 (1H, d, J = 6.6 Hz, one of C10-OCH2O), 5.19 (1H, d, J = 6.6 Hz, one of C10-OCH2O), 5.28 (1H, d, J = 11.2 Hz, one of C9-OCH2O), 5.29 (1H, d, J = 11.2 Hz, one of C9-OCH2O), 5.39 (1H, q, J = 6.2 Hz, C1-H), 7.07 (1H, dd, J = 7.8, 0.9 Hz, C8-H), 7.30 (1H, dd, J = 8.2, 7.8 Hz, C7-H), 7.33 (1H, s, C5-H), 7.41 (1H, dd, J = 8.2, 0.9 Hz, C6-H). 13C-NMR (100 MHz, CDCl3) δ: 22.8, 35.8, 37.5, 56.3, 57.7, 61.5, 72.0, 74.1, 96.1, 101.5, 111.8, 118.9, 122.1, 123.2, 125.9, 130.3, 133.7, 135.9, 149.9, 152.5. HRESIMS m/z 363.1817 (calcd for C20H27O6: 363.1808). [α + 77.0 (c 0.5, CHCl3).
(1S,3S)-{9,10-Bis(methoxymethoxy)-3,4-dihydro-1-methyl-1H-naphtho[2,3-c]pyran-3-yl}acetic acid (27). To a solution of 26 (26 mg, 0.073 mmol) in CH2Cl2 (0.2 mL), PIDA (35 mg, 0.109 mmol) and TEMPO (1.2 mg, 0.0077 mmol) were added and the whole was stirred at rt for 12 h. 5% Aqueous Na2S2O3 (2 mL) was added and the whole was extracted with CHCl3 (4 mL × 5 mL). Combined organic layer was washed with brine (1 mL × 5 mL) and was dried over Na2SO4. Solvent was evaporated in vacuo. The residue (a yellow oil, 61 mg) was dissolved with acetone (0.43 mL), t-BuOH (0.86 mL), and H2O (0.21 mL). 2-Methyl-2-butene (0.21 mL, 1.98 mmol) and NaH2PO4·2H2O (39 mg, 0.25 mmol) were added. NaClO2 (16 mg, 0.18 mmol) was added at rt, and the whole was stirred at rt for 1 h; then, 5% HCl (0.5 mL) was added at rt and the whole was extracted with CHCl3 (4 mL × 5 mL). Combined organic layer was washed with brine (1 mL × 5 mL) and dried over Na2SO4. Solvent was evaporated in vacuo, and the residue was purified by CC (CHCl3–MeOH = 99:1–91:9) to give 27 as a yellow oil (24 mg, 86%). IR (ATR) νmax cm−1: 3500–2900 (O-H), 1711 (C=O). 1H-NMR (400 MHz, CDCl3) δ: 1.71 (3H, d, J = 6.2 Hz, C1-CH3), 2.71 (1H, dd, J = 15.8, 5.5 Hz, one of C4-H2), 2.79 (1H, dd, J = 15.8, 7.3 Hz, one of C4-H2), 2.87–2.97 (2H, m, C1′-H2), 3.57 (3H, s, OCH3), 3.59 (3H, s, OCH3), 4.08–4.14 (1H, m, C3-H), 4.96 (1H, d, J = 6.4 Hz, one of C10-OCH2O), 5.20 (1H, d, J = 6.4 Hz, one of C10-OCH2O), 5.28 (1H, d, J = 10.8 Hz, one of C9-OCH2O), 5.30 (1H, d, J = 10.8 Hz, one of C9-OCH2O), 5.42 (1H, q, J = 6.4 Hz, C1-H), 7.07 (1H, dd, J = 7.6, 0.9 Hz, C8-H), 7.30 (1H, dd, J = 8.2, 7.6 Hz, C7-H), 7.34 (1H, s, C5-H), 7.41 (1H, d, J = 7.6 Hz, C6-H). 13C-NMR (100 MHz, CDCl3) δ: 22.6, 35.3, 40.7, 56.5, 57.7, 69.8, 72.3, 96.1, 101.5, 111.1, 119.0, 122.1, 123.3, 125.9, 130.1, 133.0, 135.9, 149.9, 152.5, 175.3. HRESIMS m/z 377.1617 (calcd for C20H24O7: 377.1600). [α + 77.6 (c 1.2, CHCl3).
(1S,3S)-(3,4-Dihydro-10-hydroxy-9-methoxymethoxy-1-methyl-1H-naphtho[2,3-c]pyran-3-yl)acetic acid (28) and (1S,3S)-(3,4-dihydro-9,10-dihydroxy-1-methyl-1H-naphtho[2,3-c]pyran-3-yl)acetic acid (7). To a solution of 27 (20 mg, 0.053 mmol) in THF (0.6 mL), 10% HCl (0.1 mL) was added at rt, and the whole was stirred at rt for 30 min and at 50 °C for 2 h. Solvent was evaporated in vacuo, and the residue was partitioned with AcOEt (8 mL) and H2O (2 mL). The aqueous layer was extracted with AcOEt (2 mL × 5 mL). Combined organic layer was washed with brine (1 mL × 5 mL) and dried over Na2SO4. Solvent was evaporated in vacuo and the residue was purified by CC (CHCl3–MeOH = 91:9–82:18) to give 28 as a yellow oil (6.5 mg, 37%) and 7 as a colorless oil (2.2 mg, 14%). 28: IR (ATR) νmax cm−1: 3384 (O-H), 1711 (C=O). 1H-NMR (400 MHz, CDCl3) δ: 1.66 (3H, d, J = 6.4 Hz, C1-CH3), 2.72 (1H, dd, J = 16.0, 5.0 Hz, one of C4 or C1′-H2), 2.78 (1H, dd, J = 16.0, 7.3 Hz, one of C4 or C1′-H2), 2.87 (1H, d, J = 15.1 Hz, one of C4 or C1′-H2), 2.92 (1H, dd, J = 15.1, 11.0 Hz, one of C4 or C1′-H2), 3.58 (3H, s, OCH3), 4.02–4.13 (1H, m, C3-H), 5.31 (1H, q, J = 6.4 Hz, C1-H), 5.42 (1H, d, J = 8.7 Hz, one of C9-OCH2O), 5.44 (1H, d, J = 8.7 Hz, one of C9-OCH2O), 6.98 (1H, d, J = 7.3 Hz, C8-H), 7.06 (1H, s, C5-H), 7.24 (1H, dd, J = 8.2, 7.8 Hz, C7-H), 7.35 (1H, d, J = 8.2 Hz, C6-H), 9.63 (1H, s, OH). 13C-NMR (100 MHz, CDCl3) δ: 21.7, 35.4, 40.6, 56.9, 70.0, 71.8, 95.9, 107.3, 114.0, 117.6, 120.2, 122.1, 125.7, 133.8, 135.0, 150.0, 153.6, 172.6. HRESIMS m/z 333.1333 (calcd for C18H21O6: 333.1338). [α–104.1 (c 0.2, CHCl3). 7: IR (ATR) νmax cm−1: 3206 (O-H), 1704 (C=O). 1H-NMR (400 MHz, CD3OD) δ: 1.59 (3H, d, J = 6.2 Hz, C1-CH3), 2.61 (2H, d, J = 6.4 Hz, C4 or C1′-H2), 2.79 (1H, dd, J = 15.3, 9.4 Hz, one of C4 or C1′-H2), 2.83 (1H, d, J = 15.3 Hz, one of C4 or C1′-H2), 3.98 (1H, dddd, J = 9.4, 6.4, 6.4, 4.1 Hz, C3-H), 5.16 (1H, q, J = 6.2 Hz, C1-H), 6.63 (1H, dd, J = 7.3, 1.4 Hz, C8-H), 7.00 (1H, s, C5-H), 7.13 (1H, dd, J = 8.2, 7.3 Hz, C7-H), 7.18 (1H, dd, J = 8.2, 1.4 Hz, C6-H). 13C-NMR (100 MHz, CD3OD) δ: 22.0, 36.7, 41.9, 71.8, 72.7, 108.2, 118.1, 120.5, 120.7, 126.9, 135.6, 136.8, 144.1, 152.0, 154.6, 175.1. HRESIMS m/z 289.1087 (calcd for C16H17O5: 289.1076. [α − 104.6 (c 0.1, CHCl3).
(S)-3,4-Dihydro-3-(2-hydroxyethyl)-10-hydroxy-9-(methoxymethoxy)-1H-naphtho[2,3-c]pyran-1-one (30). To a solution of 21 (84 mg, 0.19 mmol) in DMF (0.5 mL), NaH (55%, 11 mg, 0.25 mmol) was added at 0 °C. After 15 min, a solution of Ph2HSiCl (0.041 mL, 0.21 mmol) in DMF (0.5 mL) was added, and the whole was stirred at rt for 1.5 h. Ice-H2O (2 mL) was added, and the whole was extracted with AcOEt (3 mL × 15 mL). Combined organic layer was washed with brine (1 mL × 15 mL) and was dried over Na2SO4. Solvent was evaporated in vacuo, and the residue was purified by CC (hexane–AcOEt = 36:64–15:85) to give 30 as colorless solids (50 mg, 82%) and 31 as a colorless oil (61 mg, quant). 30: mp 100.3–102.4 °C. IR (ATR) νmax cm−1: 3519 (O-H), 1635 (C=O). 1H-NMR (400 MHz, CDCl3) δ: 1.95 (1H, dddd, J = 14.7, 8.2, 5.5, 5.0 Hz, one of C1′-H2), 2.10 (1H, dddd, J = 14.7, 8.2, 5.0, 5.0 Hz, one of C1′-H2), 3.06 (2H, d, J = 7.3 Hz, C4-H2), 3.60 (3H, s, OCH3), 3.89 (1H, ddd, J = 11.0, 5.5, 5.0 Hz, one of C2′-H2), 3.90 (1H, ddd, J = 11.0, 8.2, 5.0 Hz, one of C2′-H2), 4.82 (1H, dtd, J = 8.2, 7.3, 4.6 Hz, C3-H), 5.37 (2H, s, OCH2O), 6.99 (1H, s, C5-H), 7.09 (1H, dd, J = 8.2, 0.9 Hz, C8-H), 7.32 (1H, d, J = 7.8 Hz, C6-H), 7.48 (1H, dd, J = 8.2, 7.8 Hz, C7-H). 13C-NMR (100 MHz, CDCl3) δ: 33.5, 37.3, 56.48, 56.51, 58.4, 95.8, 102.3, 111.7, 116.1, 116.2, 121.4, 130.7, 133.1, 139.8, 156.2, 163.9, 170.9. HRESIMS m/z 319.1182 (calcd for C17H19O6: 319.1182). [α+16.7 (c 1.0, CHCl3). 31: 1H-NMR (400 MHz, CDCl3) δ: 0.06 (6H, s, Si(CH3)2), 0.88 (9H, s, SiC(CH3)3), 7.32–7.74 (6H, m, Ar-H), 7.59 (4H, dd, J = 7.8, 1.8 Hz, Ar-H).
(3S)-3-(2-{[Bis(2-propyl)silyl]oxy}ethyl)-3,4-dihydro-10-hydroxy-9-(methoxymethoxy)-1H-naphtho[2,3-c]pyran-1-one (32). To a solution of 30 (40 mg, 0.13 mmol) and imidazole (17 mg, 0.25 mmol) in THF (1.6 mL), a solution of chlorodiisopropylsilane (0.027 mL, 0.16 mmol) in THF (0.1 mL) was added, and the whole was stirred at rt for 1 h. H2O (2 mL) was added at 0 °C and the whole was extracted with Et2O (3 mL × 5 mL). Combined organic layer was washed with H2O (1 mL × 5 mL) and brine (1 mL × 5 mL) and was dried over Na2SO4. Solvent was evaporated in vacuo, and the residue was purified by CC (neutral SiO2, hexane–AcOEt = 9:1) to give 32 as a colorless oil (36 mg, 65%). IR (ATR) νmax cm−1: 3400–2500 (O-H), 2089 (Si-H), 1655 (C=O). 1H-NMR (400 MHz, CDCl3) δ: 0.08 (14H, m, Si(i-Pr)2), 1.98 (1H, dddd, J = 13.3, 7.8, 5.5, 5.5 Hz, one of C1′-H2), 2.13 (1H, dddd, J = 13.3, 7.8, 5.0, 5.0 Hz, one of C1′-H2), 3.05 (1H, ddd, J = 16.0, 9.2, 0.9 Hz, one of C4-H2), 3.09 (1H, dd, J = 16.0, 4.1 Hz, one of C4-H2), 3.61 (3H, s, OCH3), 3.91 (1H, ddd, J = 10.1, 5.0, 5.0 Hz, one of C2′-H2), 3.99 (1H, ddd, J = 10.1, 8.2, 5.0 Hz, one of C2′-H2), 4.81 (1H, dddd, J = 9.6, 8.2, 4.6, 4.6 Hz, C3-H), 5.37 (2H, s, OCH2O), 7.01 (1H, s, C5-H), 7.10 (1H, dd, J = 7.8, 0.9 Hz, C8-H), 7.33 (1H, dd, J = 8.2, 0.9 Hz, C6-H), 7.49 (1H, dd, J = 8.2, 7.8 Hz, C7-H), 13.07 (1H, s, OH). 13C-NMR (100 MHz, CDCl3) δ: 12.25, 12.29, 17.3, 17.4, 33.5, 37.7, 56.50, 56.54, 60.9, 95.8, 102.5, 111.7, 116.2 (overlapped), 121.4, 130.7, 133.3, 139.9, 156.3, 163.9, 171.0. HRESIMS m/z 433.2067 (calcd for C23H33O6Si: 433.2046). [α + 14.9 (c 1.2, CHCl3).
(1R,5S)-11-Methoxymethoxy-1-methyl-3,4,5,6-tetrahydro-1,5-epoxy-1H-naphtho[2,3-c]oxocin-12-ol (33). To a solution of 32 (33 mg, 0.079 mmol) in THF (0.5 mL), CH3Li (1.10 M in Et2O, 0.28 mL, 0.31 mmol) was added at 0 °C, and the whole was stirred at rt for 30 min. Saturated aqueous NH4Cl (3 mL) was added at 0 °C, and the whole was extracted with CH2Cl2 (4 mL × 6 mL). The combined organic layer was washed with H2O (1 mL × 6 mL) and brine (1 mL × 6 mL) and was dried over Na2SO4. Solvent was evaporated in vacuo, and the residue (55 mg, 161%) was used for the next step without purification. To a solution of crude hemiacetal (55 mg) in CH2Cl2 (0.4 mL), TFA (0.02 mL, 0.23 mmol) in CH2Cl2 (0.1 mL) was added at −78 °C, and the whole was stirred at −40 °C for 20 min and at −20 °C for 15 min. Saturated aqueous NaHCO3 (3 mL) was added and the whole was partitioned with CHCl3 (6 mL) and H2O (1.5 mL). Aqueous layer was extracted with CHCl3 (3 mL × 5 mL). Combined organic layer was washed with H2O (1 mL × 5 mL) and brine (1 mL × 5 mL) and dried over Na2SO4. The solvent was evaporated in vacuo, and the residue was purified by CC (hexane–AcOEt = 79:21–59:41) to give 33 as a colorless oil (9 mg, 34%). IR (neat) νmax cm−1: 3385 (O-H). 1H-NMR (400 MHz, CDCl3) δ: 1.40 (1H, dif.dd, J = 13.6, 3.2 Hz, one of C6-H2), 2.07 (3H, s, C1-CH3), 2.51 (1H, m, one of C6-H2), 2.79 (1H, d, J = 17.4 Hz, one of C4-H2), 3.56–3.64 (2H, m, one of C4-H2 and C7-H2), 3.59 (3H, s, OCH3), 3.79 (1H, dd, J = 11.9, 6.9 Hz, one of C7-H2), 4.54 (1H, dd, J = 6.9, 6.9 Hz, C5-H), 5.42 (1H, d, J = 15.1 Hz, one of C11-OCH2O), 5.44 (1H, d, J = 15.1 Hz, one of C11-OCH2O), 7.02 (1H, dd, J = 6.9, 0.9 Hz, C10-H), 7.11 (1H, s, C7-H), 7.28 (1H, t, J = 7.8 Hz, C9-H), 7.36 (1H, d, J = 8.2 Hz, C8-H), 10.00 (1H, s, OH). 13C-NMR (125 MHz, CDCl3) δ: 27.1, 29.3, 30.4, 57.1, 59.5, 66.2, 95.9, 96.1, 107.3, 114.4, 117.0, 117.9, 121.9, 126.5, 135.9, 136.1, 151.7, 154.4. HRESIMS m/z 317.1384 (calcd for C18H21O5: 317.1389). [α − 71.7 (c 0.3, CHCl3).
Synthesis of 25. To a solution of 21 (274 mg, 0.63 mmol) in THF (3.5 mL), CH3Li (1.10 M in Et2O, 1.7 mL, 1.87 mmol) was added at 0 °C, and the whole was stirred at rt for 1 h. Saturated aqueous NH4Cl (6 mL) was added at 0 °C, and the whole was extracted with CH2Cl2 (3 mL × 10mL). Combined organic layer was washed with H2O (1 mL × 5 mL) and brine (1 mL × 5 mL) and was dried over Na2SO4. Solvent was evaporated in vacuo to give crude 22 as a yellow oil (285 mg), which was used to next reaction without purification. To a solution of crude 22 in THF (2.5 mL), 10% HCl (0.5 mL) was added at rt, and the whole was stirred at rt for 30 min. H2O (9 mL) was added at rt and the whole was extracted with AcOEt (3 mL × 15mL). Combined organic layer was washed with brine (1 mL × 10 mL) and was dried over Na2SO4. The solvent was evaporated in vacuo, and the residue was washed with a mixed solvent of hexane and AcOEt (1:1) to give 25 as yellow powder (132 mg, 77%). The physical properties of 25 obtained here were identical with those prepared as described above.
(1R,5S)-11,12-Bis(methoxymethoxy)-1-methyl-3,4,5,6-tetrahydro-1H-1,5-epoxynaphtho[2,3-c]oxocine(38). To a suspension of NaH (60%, 75 mg, 1.87 mmol) in THF (1 mL), 25 (206 mg, 0.76 mmol) in THF (6 mL) was added, and the whole was stirred at 0 °C for 20 min. MOMCl (0.13 mL, 1.71 mmol) was added, and the whole was stirred at rt for 1 h. NaH (60%, 30 mg, 0.75 mmol) and MOMCl (0.06 mL, 0.79 mmol) was added at 0 °C, and the whole was stirred at rt for 30 min. Ice-H2O (5 mL) was added, and the whole was extracted with AcOEt (1 mL × 15 mL, 2 mL × 10 mL). Combined organic layer was washed with brine (1 mL × 10 mL) and was dried over Na2SO4. Solvent was evaporated in vacuo, and the residue was purified by CC (hexane–AcOEt = 76:24–54:45) to give 38 as a yellow oil (128 mg, 47%) and 33 (a yellow oil, 13 mg, 5%). 38: IR (ATR) νmax cm−1: no characteristic absorption. 1H-NMR (400 MHz, CDCl3) δ: 1.43 (1H, dd, J = 13.7, 3.2 Hz, one of C6-H2), 2.11 (3H, s, C1-CH3), 2.51 (1H, m, one of C6-H2), 2.82 (1H, d, J = 16.9 Hz, one of C4-H2), 3.56–3.67 (2H, m, one of C3- and C4-H2), 3.58 (3H, s, OCH3), 3.67 (3H, s, OCH3), 3.75 (1H, dd, J = 11.9, 6.9 Hz, one of C3-H2), 4.55 (1H, dd, J = 6.9, 6.9 Hz, C5-H), 4.87 (1H, d, J = 4.1 Hz, one of C11-OCH2O), 5.10 (1H, d, J = 4.1 Hz, one of C11-OCH2O), 5.30 (1H, d, J = 6.5 Hz, one of C12-OCH2O), 5.33 (1H, d, J = 6.5 Hz, one of C12-OCH2O), 7.10 (1H, dd, J = 7.8, 0.9 Hz, C10-H), 7.34 (1H, dd, J = 8.2, 7.8 Hz, C9-H), 7.37 (1H, s, C7-H), 7.40 (1H, dd, J = 8.2, 1.3 Hz, C8-H). 13C-NMR (100 MHz, CDCl3) δ: 27.4, 29.3, 33.0, 53.4, 56.5, 58.9, 65.7, 95.9, 96.3, 100.7, 111.6, 120.0, 121.7, 122.5, 126.7, 127.5, 134.7, 136.5, 149.7, 153.4. HRESIMS m/z 361.1658 (calcd for C20H25O6: 361.1651). [α − 216.1 (c 1.5, CHCl3).
(1R,3S)-2-[3,4-Dihydro-9,10-bis(methoxymethoxy)-1-methyl-1H-naphtho[2,3-c]pyran-3-yl]ethanol (39). (Table 1run 7). To a suspension of LiAlH
4 (17 mg, 0.43 mmol) in Et
2O (1.0 mL), a suspension of AlCl
3 (18 mg, 0.14 mmol) in Et
2O (1 mL) was added at 0 °C, and the whole was stirred at 0 °C for 30 min. A solution of
38 (49 mg, 0.14 mmol) in Et
2O (1 mL) was added at −60 °C, and the whole was stirred at −60 °C for 21 h. Saturated aqueous NH
4Cl (5 mL) was added. The whole was filtered through a pad of Celite, and the precipitate was washed with AcOEt (5 mL × 1 mL). The filtrate was extracted with AcOEt (3 mL × 10 mL). Combined organic layer was washed with H
2O (1 mL × 10 mL) and brine (1 mL × 10 mL) and was dried over Na
2SO
4. Solvent was evaporated in vacuo, and the residue was purified by CC (hexane–AcOEt = 72:28–51:49) to give
39 as a yellow oil (25 mg, 50%) and
26 as a yellow oil (5 mg, 11%).
39: IR (ATR) ν
max cm
−1: 3413 (O-H).
1H-NMR (400 MHz, CDCl
3) δ: 1.69 (3H, d,
J = 6.9 Hz, C1-C
H3), 1.92 (2H, dt,
J = 6.4, 5.5 Hz, C1′-
H2), 2.86 (1H, ddd,
J = 16.5, 4.1, 0.9 Hz, one of C4-
H2), 2.93 (1H, ddd,
J = 16.5, 10.5, 1.4 Hz, one of C4-
H2), 3.57 (3H, s, OC
H3), 3.60 (3H, s, OC
H3), 3.90 (2H, t,
J = 5.5 Hz, C2′-
H), 4.27 (1H, dtd,
J = 10.5, 6.4, 4.1 Hz, C3-
H), 5.01 (1H, d,
J = 6.4 Hz, one of C10-OC
H2O), 5.23 (1H, d,
J = 6.4 Hz, one of C10-OC
H2O), 5.28 (1H, d,
J = 12.4 Hz, one of C9-OC
H2O), 5.30 (1H, d,
J = 12.4 Hz, one of C9-OC
H2O), 5.39 (1H, q,
J = 6.9 Hz, C1-
H), 7.06 (1H, dd,
J = 7.8, 0.9 Hz, C8-
H), 7.30 (1H, dd,
J = 8.2, 7.8 Hz, C7-
H), 7.35 (1H, s, C5-
H), 7.40 (1H, dd,
J = 8.2, 0.9 Hz, C6-
H).
13C-NMR (100 MHz, CDCl
3) δ: 20.4, 34.4, 37.7, 56.5, 57.6, 61.4, 67.1, 69.4, 96.1, 101.7, 110.8, 118.8, 122.13, 123.7, 125.7, 130.3, 132.2, 136.0, 148.8, 152.5. HRESIMS
m/z 363.1801 (calcd for C
20H
27O
6: 363.1808). [α
-196 (
c 0.5, CHCl
3).
(1R,3S)-{9,10-Bis(methoxymethoxy)-3,4-dihydro-1-methyl-1H-naphtho[2,3-c]pyran-3-yl}acetic acid (40). To a solution of 39 (16 mg, 0.043 mmol) in CH2Cl2 (0.2 mL), PIDA (21 mg, 0.064 mmol) was added at rt. After 5 min, TEMPO (1 mg, 0.0064 mmol) was added at rt, and the whole was stirred at rt for 14 h. Then, 5% aqueous Na2S2O3 (1.5 mL) was added, and the whole was extracted with CHCl3 (4 mL × 5 mL). Combined organic layer was washed with brine (1 mL × 5 mL) and dried over Na2SO4. Solvent was evaporated in vacuo, to give a yellow oil (19 mg). The residue was dissolved in acetone (0.25 mL), t-BuOH (0.51 mL), and H2O (0.12 mL). 2-Methyl-2-butene (0.13 mL, 1.23 mmol), NaH2PO4·2H2O (29 mg, 0.19 mmol), and NaClO2 (10 mg, 0.11 mmol) was successively added in each 10 min, and the whole was stirred at rt for 1 h. 5% HCl (0.3 mL) was added, and the whole was extracted with CHCl3 (4 mL × 5 mL). Combined organic layer was washed with brine (1 mL × 5 mL) and dried over Na2SO4. Solvent was evaporated in vacuo, and the residue was purified by CC (CHCl3–MeOH = 60:40) to give 40 as a yellow oil (11 mg, 65%). IR (ATR) νmax cm−1: 3500–2800 (O-H), 1711 (C=O). 1H-NMR (400 MHz, CDCl3) δ: 1.68 (3H, d, J = 6.9 Hz, C1-CH3), 2.70 (2H, brs, C4-H2), 2.85–2.98 (2H, m, C1′-H), 3.56 (3H, s, OCH3), 3.59 (3H, s, OCH3), 4.48 (1H, brs, C3-H), 4.99 (1H, d, J = 6.4 Hz, one of C10-OCH2O), 5.21 (1H, d, J = 6.4 Hz, one of C10-OCH2O), 5.27 (1H, d, J = 11.4 Hz, one of C9-OCH2O), 5.29 (1H, d, J = 11.4 Hz, one of C9-OCH2O), 5.59 (1H, q, J = 6.4 Hz, C1-H), 7.05 (1H, d, J = 7.8 Hz, C8-H), 7.28 (1H, dd, J = 7.8, 7.8 Hz, C7-H), 7.33 (1H, s, C5-H), 7.37 (1H, d, J = 7.8 Hz, C6-H). 13C-NMR (100 MHz, CDCl3) δ: 20.1, 33.7, 41.0, 56.5, 57.5, 63.8, 69.8, 96.0, 101.7, 110.8, 118.9, 122.2, 123.8, 125.8, 130.0, 131.4, 136.0, 148.8, 152.5, 175.3. HRESIMS m/z 377.1616 (calcd for C20H25O7: 377.1600). [α − 144.0 (c 1.1, CHCl3).
(1R,3S)-(3,4-Dihydro-10-hydroxy-9-methoxymethoxy-1-methyl-1H-naphtho[2,3-c]pyran-3-yl)acetic acid (41) and (1R,3S)-(3,4-Dihydro-9,10-dihydroxy-1-methyl-1H-naphtho[2,3-c]pyran-3-yl)acetic acid (DDHK, 3). To a solution of 40 (23 mg, 0.061 mmol) in THF (1 mL), 6 M HCl (0.1 mL) was added, and the whole was stirred at 50 °C for 20 min. After cooling to rt, CH2Cl2 (3 mL) was added, and the whole was washed with H2O (1 mL × 2 mL). Aqueous layer was extracted with CH2Cl2 (1 mL × 3 mL). The combined organic layer was washed with brine (1 mL × 2 mL) and dried over Na2SO4. Solvent was evaporated in vacuo, and the residue was purified by CC (CHCl3–MeOH = 97:3–90:10) to give 41 as a brown oil (14 mg. 71%) and 3 as a brown oil (5 mg, 27%). 41: IR (ATR) νmax cm−1: 3390 (O-H), 1709 (C=O). 1H-NMR (400 MHz, CDCl3) δ: 1.65 (3H, d, J = 6.6 Hz, C1-CH3), 2.73 (2H, d, J = 6.2 Hz, two of C4 or C1′-H2), 2.87 (1H, dd, J = 15.8, 10.5 Hz, one of C4 or C1′-H2), 2.96 (1H, dd, J = 16.4, 3.2 Hz, one of C4 or C1′-H2), 3.59 (3H, s, OCH3), 4.47–4.54 (1H, m, C3-H), 5.43 (1H, q, J = 6.4 Hz, C1-H), 5.44 (2H, s, C9-OCH2O), 6.98 (1H, d, J = 7.3 Hz, C8-H), 7.09 (1H, s, C5-H), 7.24 (1H, dd, J = 8.2, 7.8 Hz, C7-H), 7.36 (1H, d, J = 8.2 Hz, C6-H), 9.56 (1H, s, OH). 13C-NMR (100 MHz, CDCl3) δ: 18.9, 33.9, 40.5, 56.9, 63.9, 69.5, 95.8, 107.2, 113.7, 117.8, 120.6, 122.1, 125.6, 132.0, 135.1, 149.1, 153.6, 173.1. HRESIMS m/z 333.1335 (calcd. for C18H21O6: 333.1338). [α + 10.6 (c 0.3, CHCl3). 3: IR (ATR) νmax cm−1: 2924 (O-H), 1712 (C=O). 1H-NMR (400 MHz, CD3OD) δ: 1.49 (3H, d, J = 6.4 Hz, C1-CH3), 2.45 (1H, dd, J = 15.3, 8.2 Hz, one of C4-H2), 2.52 (1H, dd, J = 15.3, 4.8 Hz, C4-H2), 2.65 (1H, dd, J = 15.8, 10.1 Hz, one of C1′-H2), 2.84 (1H, dd, J = 15.8, 3.2 Hz, one of C1′-H2), 4.34–4.43 (1H, m, C3-H), 5.16 (1H, q, J = 6.6 Hz, C1-H), 6.53 (1H, dd, J = 7.6, 0.9 Hz, C8-H), 6.92 (1H, s, C5-H), 7.03 (1H, dd, J = 8.2, 7.6 Hz, C7-H), 7.09 (1H, d, J = 8.2 Hz, C6-H). 13C-NMR (125 MHz, CD3OD) δ: 19.4, 35.1, 42.0, 65.7, 70.2, 108.2, 114.6, 120.6, 120.8, 126.8, 133.7, 136.9, 151.0, 154.6, 174.9. HRESIMS m/z 289.1082 (calcd for C16H17O5: 289.1076). [α + 30.9 (c 0.2, MeOH).
Transformation of MOM ether 41 to DDHK (3). To a solution of 41 (5 mg, 0.016 mmol) in CH2Cl2 (0.4 mL), BCl3 (1.0 M in CH2Cl2, 0.05 mL, 0.05 mmol) was added at −78 °C, and the mixture was stirred at −78 °C for 45 min. H2O (1.3 mL) was added, and the whole was extracted with AcOEt (1 mL × 10 mL, 3 mL × 5 mL). The combined organic layer was washed with brine (1 mL × 5 mL) and was dried over Na2SO4. The solvent was evaporated in vacuo, and the residue was purified by CC (CHCl3–MeOH = 9:1) to give 3 as a yellow oil (3 mg, 69%). The physical properties of 3 obtained here were identical with those prepared as described above.
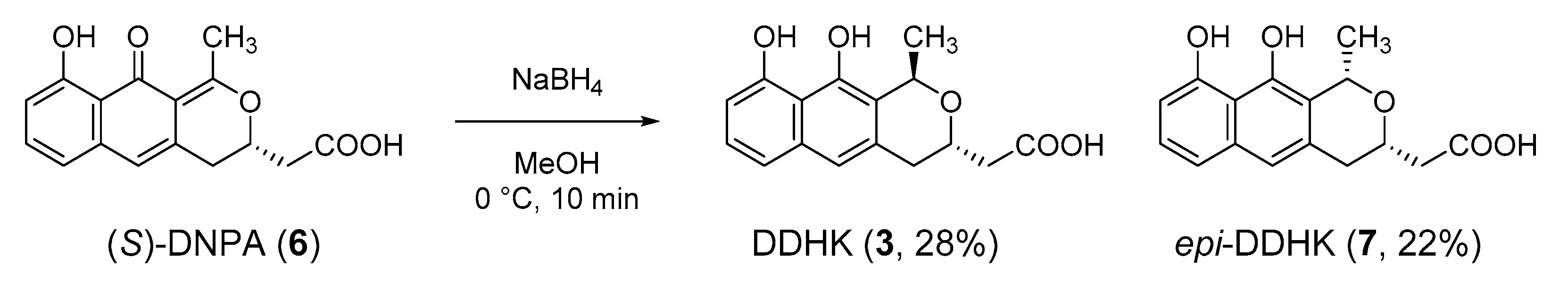
Semisynthesis of DDHK (3) and epi-DDHK (7) from (S)-DNPA (6). To a solution of (S)-DNPA (6) (2 mg, 6.3 x 10–3 mmol) in MeOH (0.2 mL), NaBH4 (2.5 mg, 6.8 × 10−2 mmol) was added at 0 °C, and the mixture was stirred at 0 °C for 10 min. The solvent was evaporated in vacuo, and the residue was partitioned with AcOEt (1 mL) and 10% HCl (0.5 mL). Aqueous layer was extracted with AcOEt (2 mL × 1 mL). Combined organic layer was washed with brine (1 mL × 1 mL) and dried over Na2SO4. The solvent was evaporated in vacuo. The residue was purified by RP HPLC (Cosmosil AR-II, 26 mm × 250 mm, CH3OH-H2O 2:1, 2 mL/min) to give DDHK (3) (0.5 mg, 28%) and epi-DDHK (7) (0.4 mg, 22%).


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
