
Structure and Cytotoxicity of Novel Lignans and Lignan Glycosides from the Aerial Parts of Larrea tridentata


Abstract
:1. Introduction
2. Results and Discussion
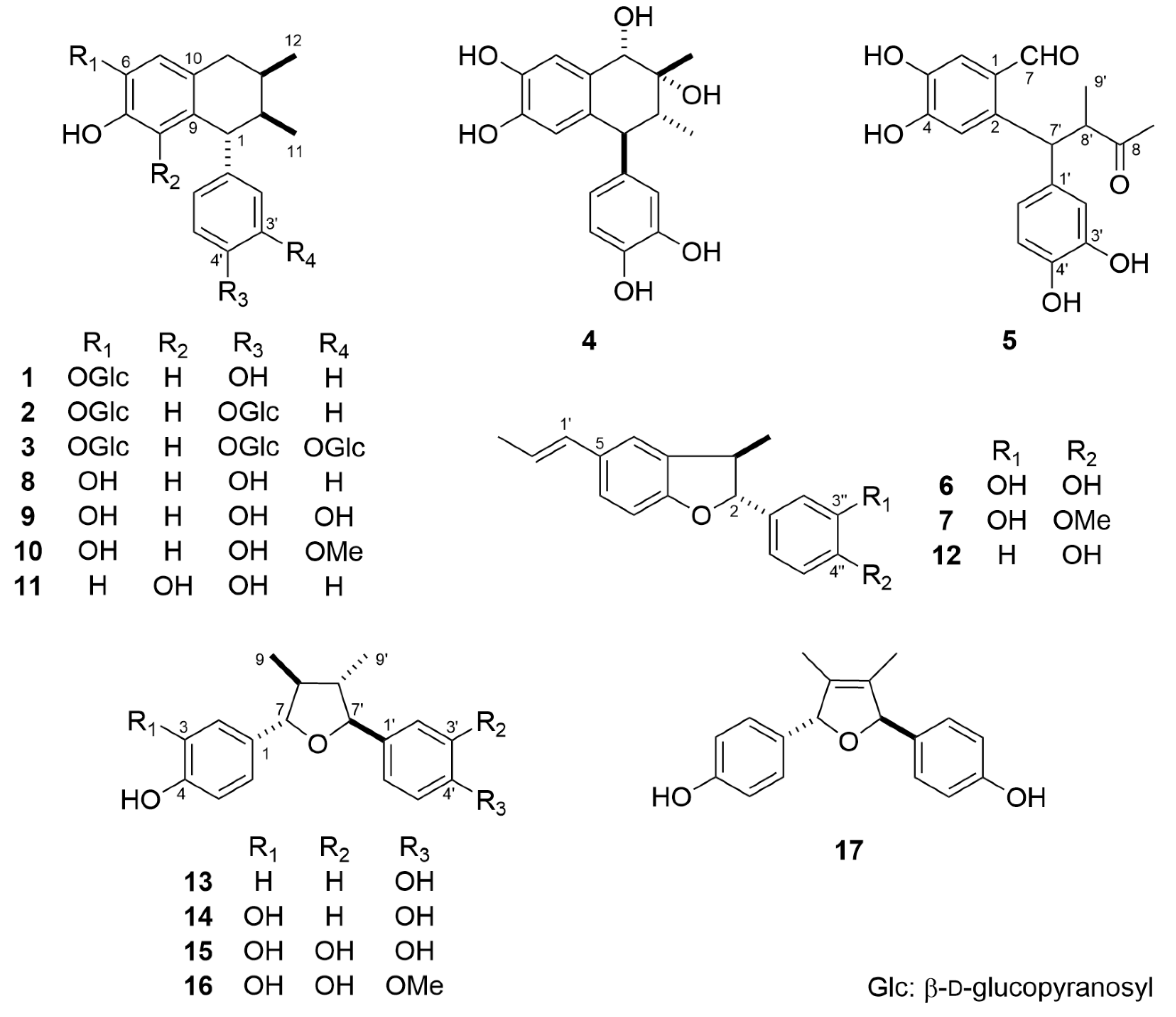
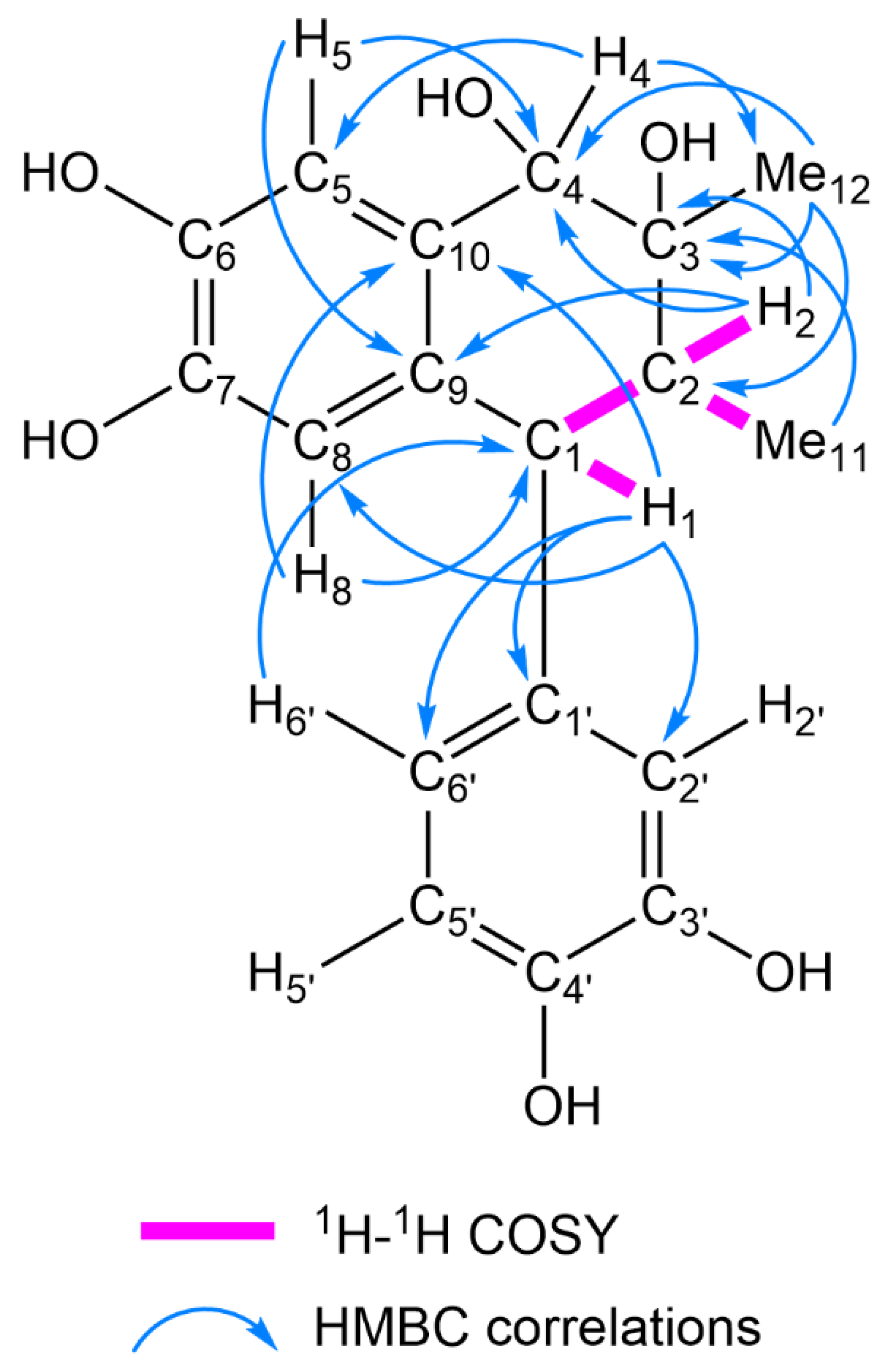
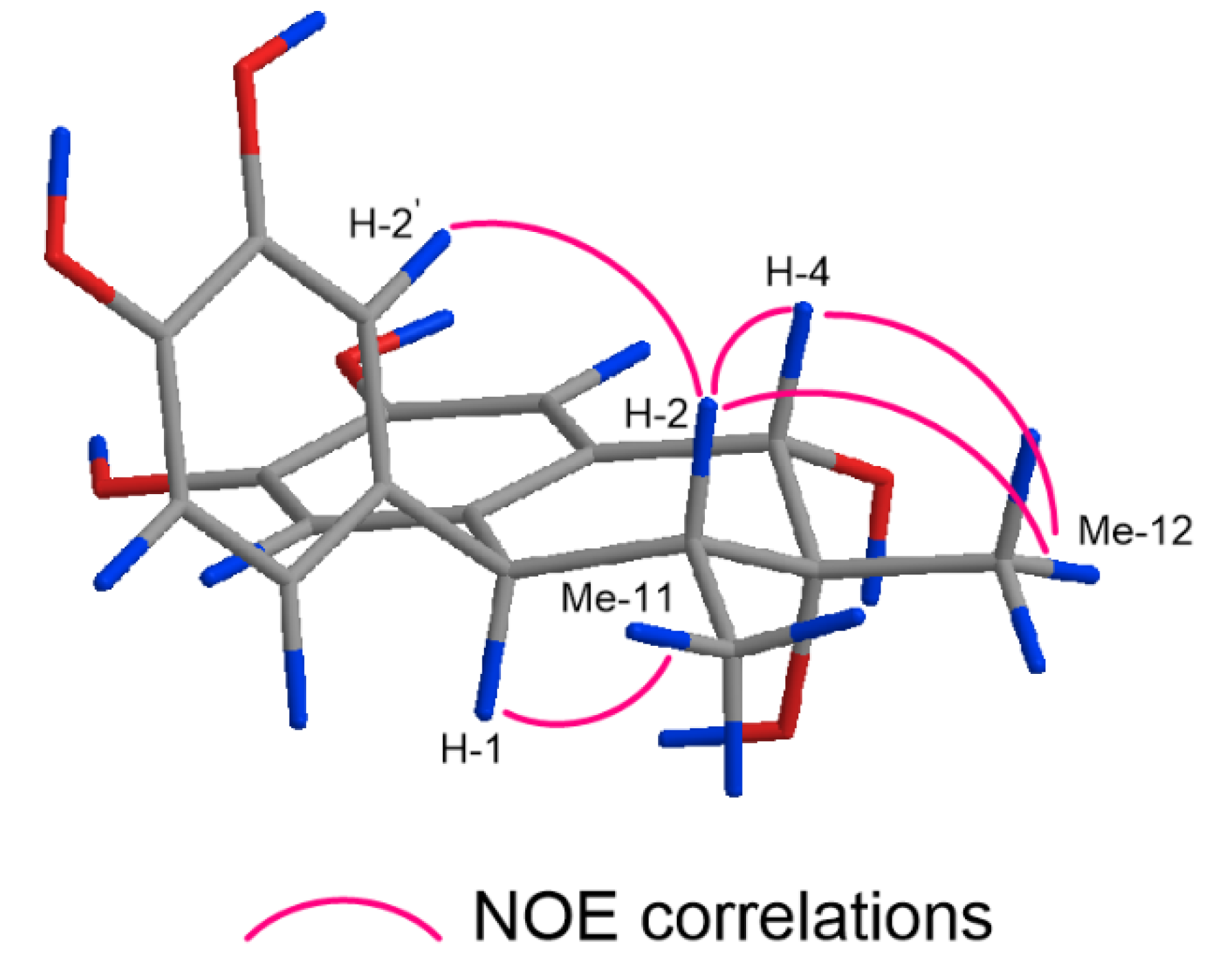
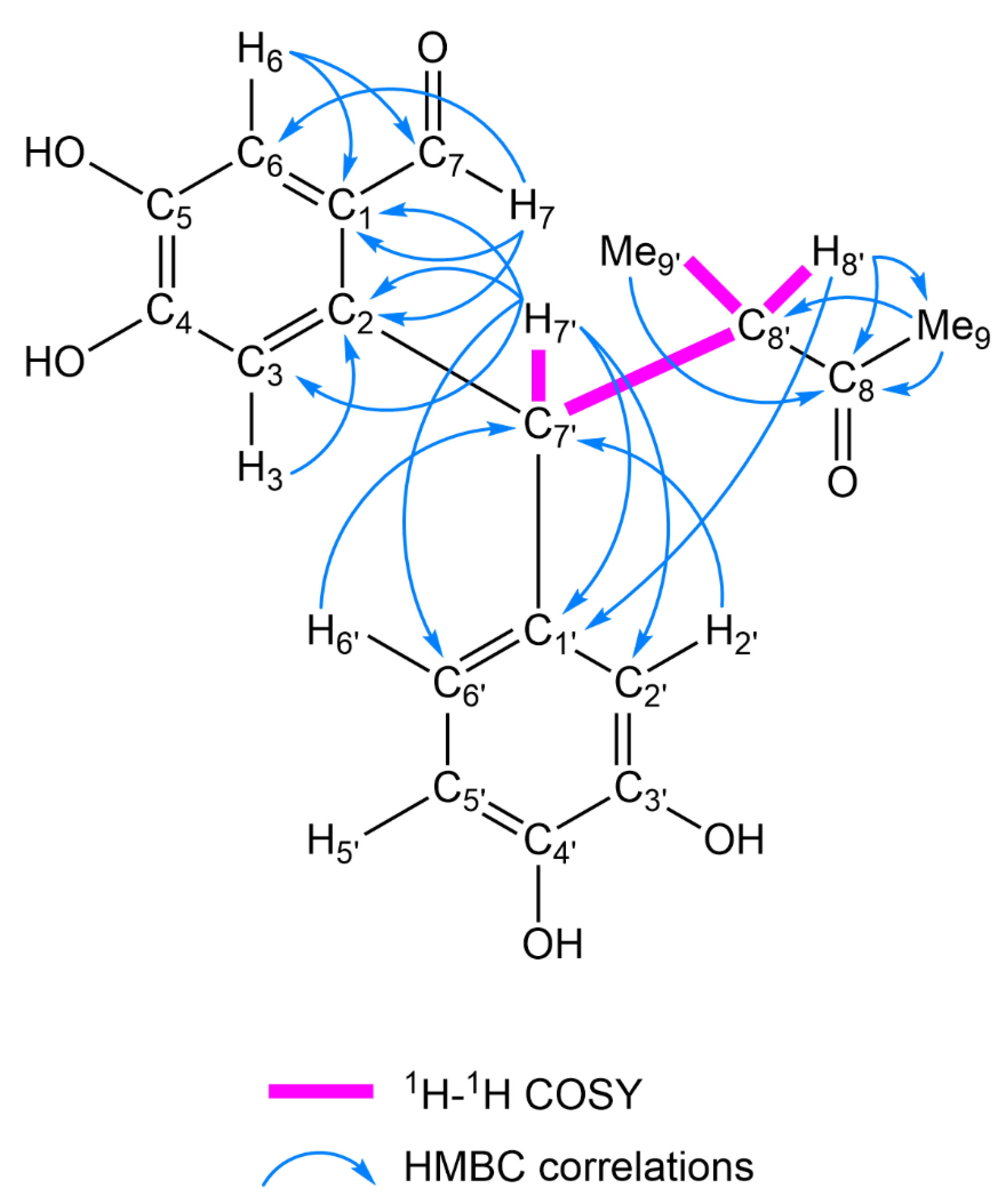
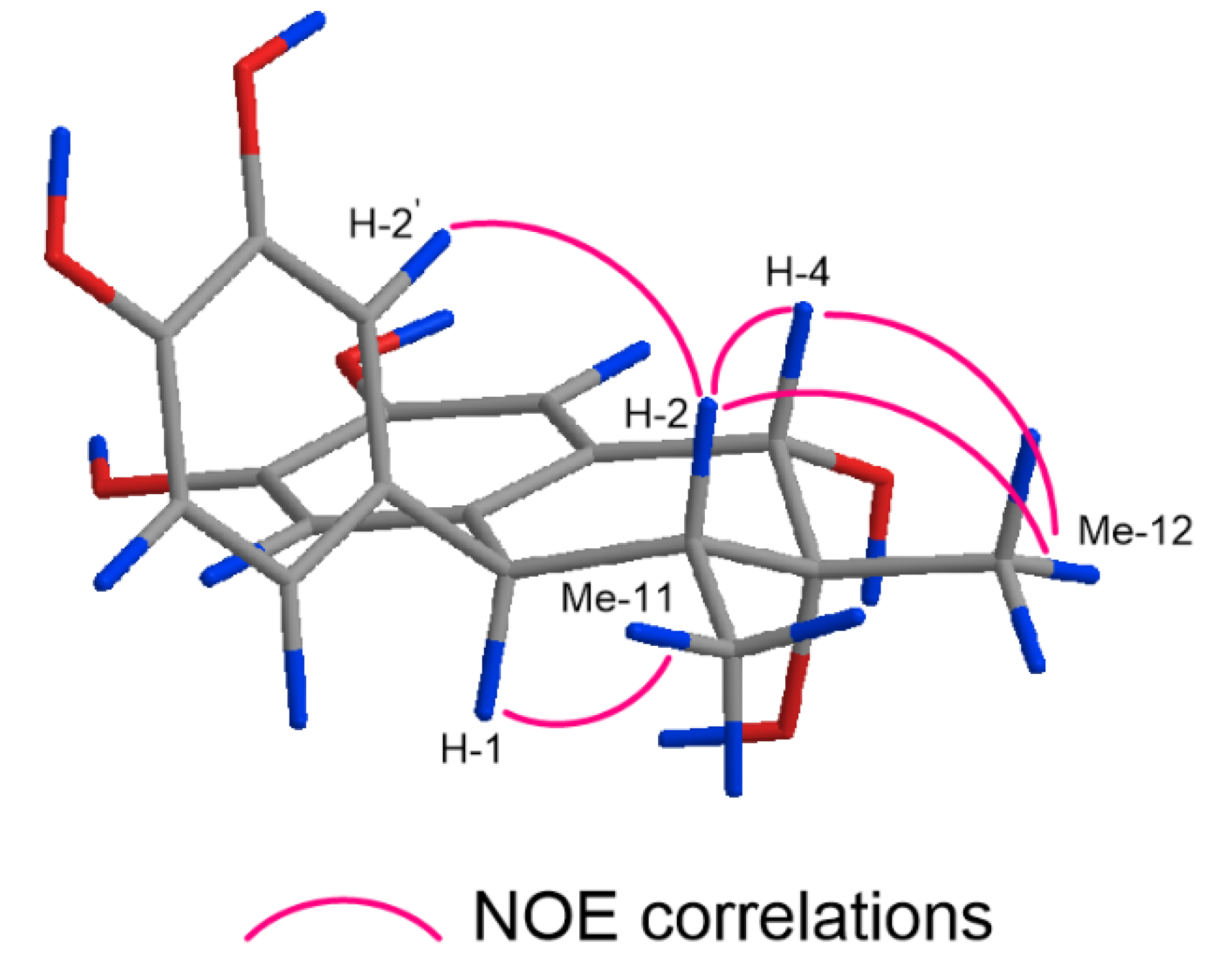
2.1. Structure Elucidation of 1–17
2.2. Cytotoxicity of 1–17
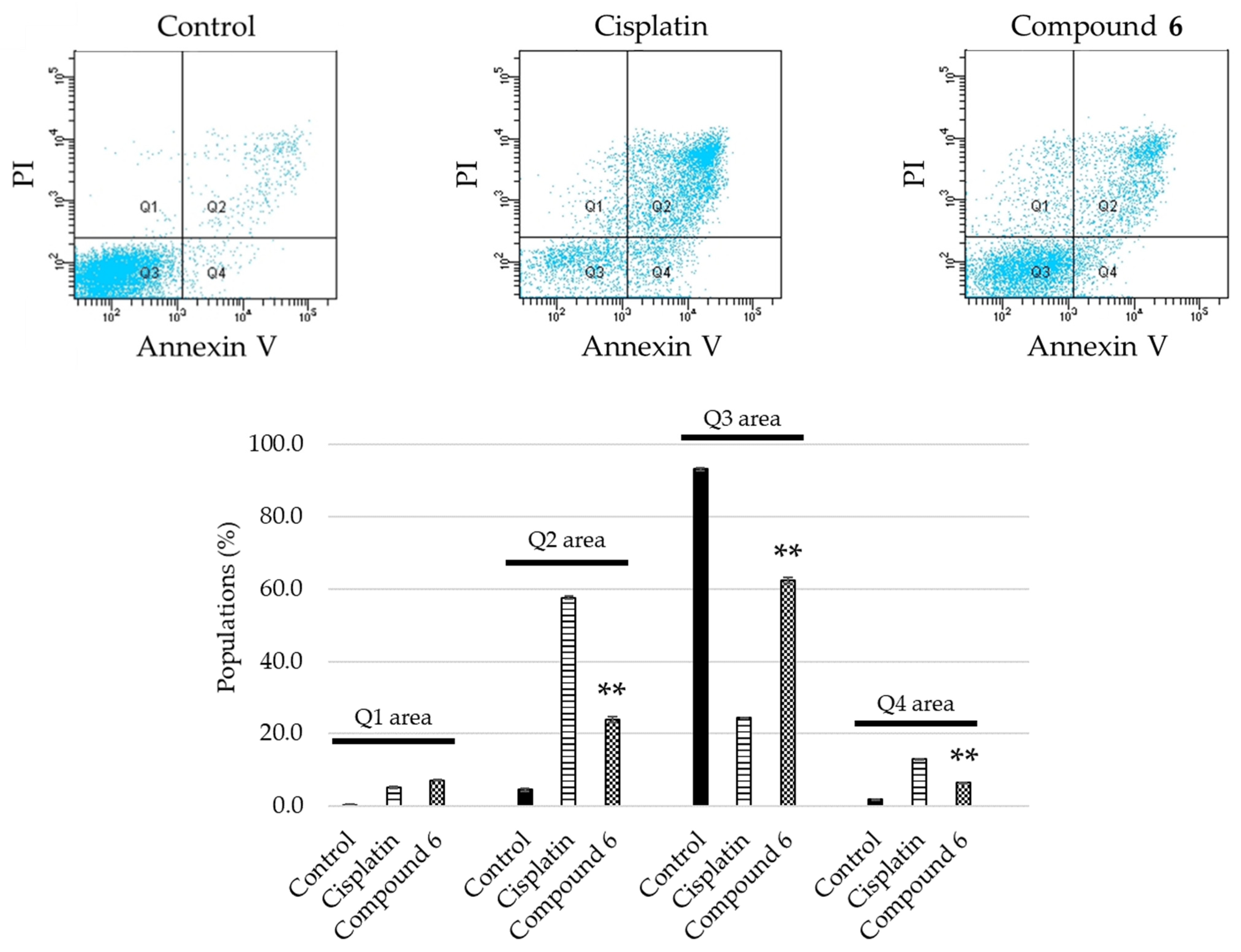
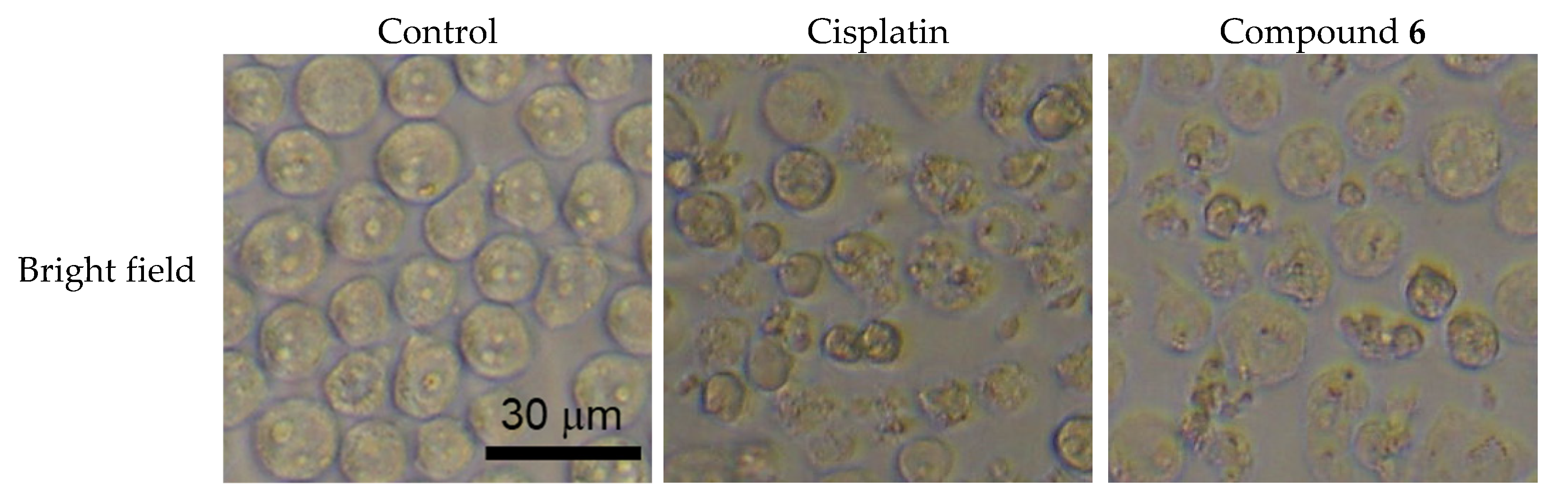
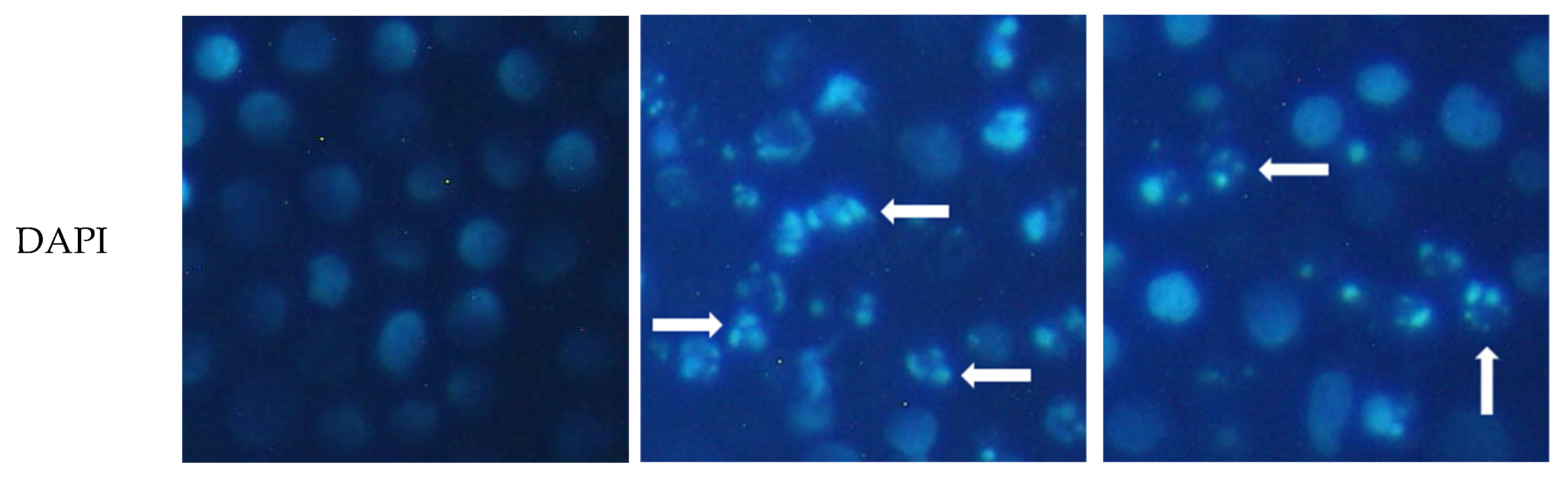
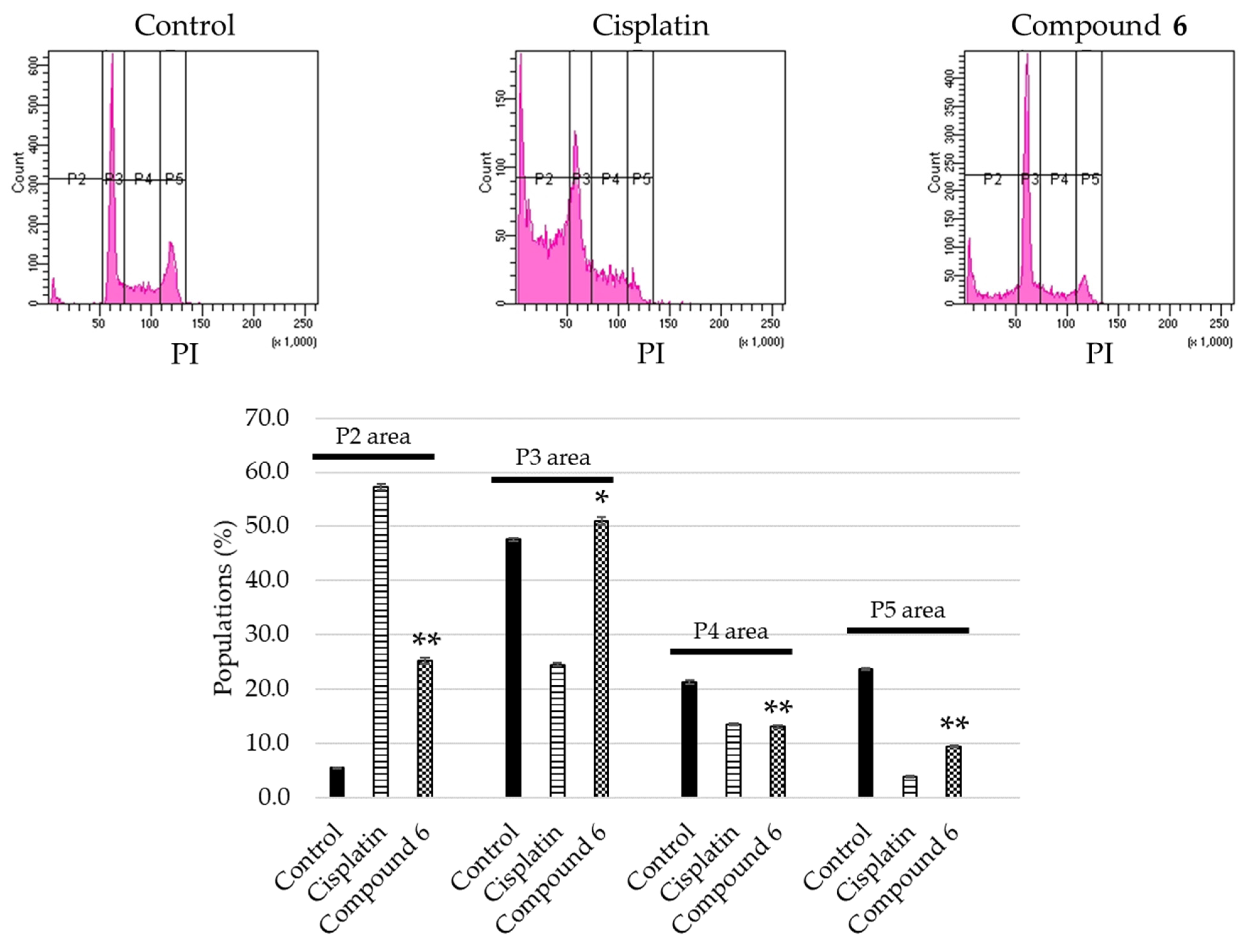
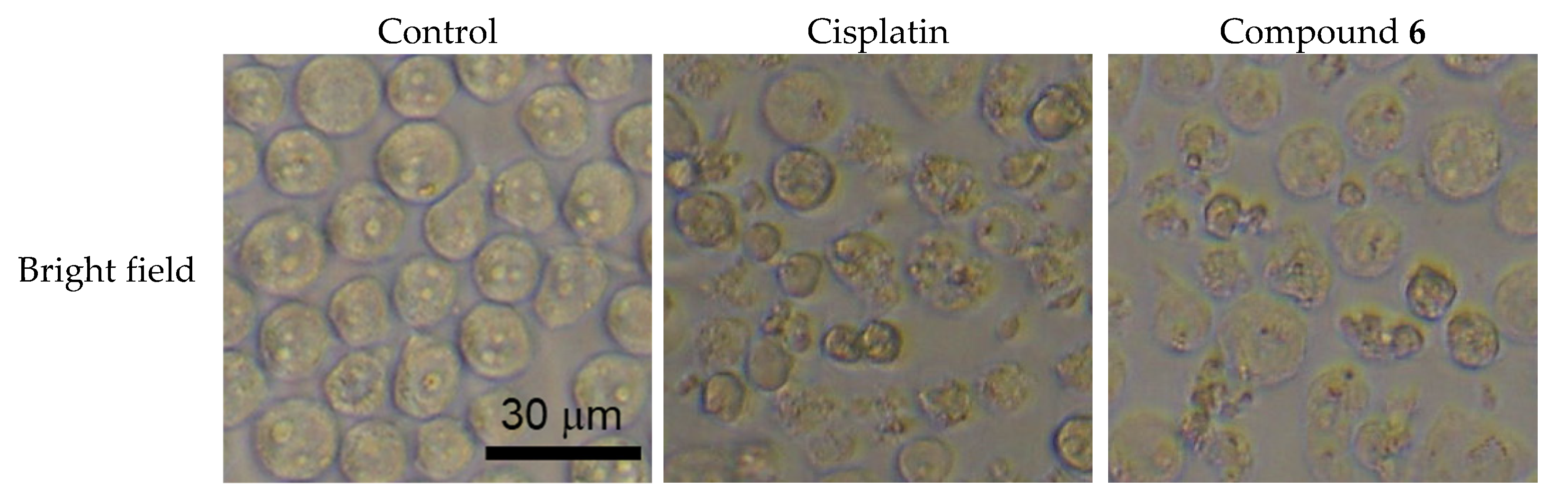
2.3. Apoptosis Inducing Activity of 6
3. Material and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation Procedures
3.4. Structural Elucidation
3.5. Assay for Cytotoxic Activity
3.6. Detection of Apoptosis
3.7. DAPI Staining
3.8. Cell Cycle Distribution Analysis
3.9. Statistical Anlysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- Arteaga, S.; Andrade-Cetto, A.; Cárdenas, R. Larrea tridentata (Creosote bush), an abundant plant of Mexico and US-American deserts and its metabolite nordihydroguaiaretic acid. J. Ethnopharmacol. 2005, 98, 231–239. [Google Scholar] [CrossRef]
- Núñez-Mojica, G.; Vázquez-Ramírez, A.L.; García, A.; Rivas-Galindo, V.M.; Garza-González, E.; González-Bravo, G.E.C.; Toscano, R.A.; Moo-Puc, R.E.; Villanueva-Toledo, J.R.; Marchand, P.; et al. New cyclolignans of Larrea tridentata and their antibacterial and cytotoxic activities. Phytochem. Lett. 2021, 43, 212–218. [Google Scholar] [CrossRef]
- Favela-Hernández, J.M.J.; García, A.; Garza-González, E.; Rivas-Galindo, V.M.; Camacho-Corona, M.R. Antibacterial and antimycrobacterial lignans and flavonoids from Larrea tridentata. Phytother. Res. 2012, 26, 1957–1960. [Google Scholar] [CrossRef]
- Lambert, J.D.; Sang, S.; Dougherty, A.; Galdwell, C.G.; Meyers, R.O.; Dorr, R.T.; Timmermann, B.N. Cytotoxic lignans from Larrea tridentata. Phytochemistry 2005, 66, 811–815. [Google Scholar] [CrossRef]
- Rahman, S.; Ansari, R.A.; Rehman, H.; Parvez, S.; Raisuddin, S. Nordihydroguaiaretic acid from Creosote Bush (Larrea tridentata) mitigates 12-O-tetradecanoylphorbol-13-acetate-induced inflammatory and oxidative stress response of tumor promotion cascade in mouse skin. Evid.-Based Complement. Altern. Med. 2009, 2011, 734785. [Google Scholar] [CrossRef] [Green Version]
- Camacho-Corona, M.d.R.; Ramírez-Cabrera, M.A.; González-Santiago, O.; Garza-González, E.; Palacios, I.d.P.; Luna-Herrera, J. Activity against drug resistant-tuberculosis strains of plants used in Mexican traditional medicine to treat tuberculosis and other respiratory diseases. Phytother. Res. 2008, 22, 82–85. [Google Scholar] [CrossRef]
- Vargas-Arispuro, I.; Contreras-Valenzuela, A.; Martínez-Téllez, M.A. Lignans from Larrea tridentata (creosote bush) as fungal β-1,3-glucanase inhibitors. Pest. Biochem. Physiol. 2009, 94, 60–63. [Google Scholar] [CrossRef]
- Bashyal, B.; Li, L.; Bains, T.; Debnath, A.; LaBarbera, D.V. Larrea tridentata: A novel source for antiparasitic agents active against Entamoeba histolytica, Giardia lamblia and Naegleria fowleri. PLoS Neglect. Trop. Dis. 2017, 11, e0005832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jitsuno, M.; Mimaki, Y. Triterpene glycosides from the aerial parts of Larrea tridentata. Phytochemistry 2010, 71, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, A.; Matsuo, Y.; Jitsuno, M.; Adachi, K.; Mimaki, Y. Larrealignans A and B, novel lignan glycosides from the aerial parts of Larrea tridentata. Chem. Pharm. Bull. 2011, 59, 1467–1470. [Google Scholar] [CrossRef] [Green Version]
- Cancer Statistics. Cancer Information Service, National Cancer Center, Japan (National Cancer Registry, Ministry of Health, Labour and Welfare). Available online: https://ganjoho.jp/reg_stat/statistics/data/dl/index.html (accessed on 12 October 2021).
- Cancer Statistics. Cancer Information Service, National Cancer Center, Japan (Vital Statistics of Japan, Ministry of Health, Labour and Welfare). Available online: https://ganjoho.jp/reg_stat/statistics/data/dl/index.html (accessed on 12 October 2021).
- Konno, C.; Xue, H.Z.; Lu, Z.Z.; Ma, B.X.; Erdelmeier, C.A.J.; Che, C.T.; Cordell, G.A.; Soejarto, D.D.; Waller, D.P.; Fong, H.H.S. 1-aryl tetralin lignans from Larrea tridentata. J. Nat. Prod. 1989, 52, 1113–1117. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Rzeppa, S.; Kaiser, M.; Brun, R. Larrea tridentata-Absolute configuration of its epoxylignans and investigations on its antiprotozoal activity. Phytochem. Lett. 2012, 5, 632–638. [Google Scholar] [CrossRef]
- Liu, J.S.; Huang, M.F.; Gao, Y.L. The structure of chicanine, a new lignan from Schisandra sp. Can. J. Chem. 1981, 59, 1680–1684. [Google Scholar] [CrossRef]
- Ganbre, J.; Huang, R.C.C.; Bates, R.B.; Burns, J.J.; Caldera, S.; Malcomson, M.E.; McClure, K.J. Characterization of anti-HIV lignans from Larrea tridentata. Tetrahedron 1995, 51, 12203–12210. [Google Scholar]
- Achenbach, H.; Grob, J.; Dominguez, X.A.; Cano, G.; Star, J.V.; Brussolo, L.D.C.; Muñoz, G.; Salgado, F.; López, L. Lignans, neolignans and norneolignans from Krameria cystisoides. Phytochemistry 1987, 26, 1159–1166. [Google Scholar] [CrossRef]
- Konno, C.; Lu, Z.Z.; Xue, H.Z.; Erdelmeier, C.A.J.; Meksuriyen, D.; Che, C.T.; Cordell, G.A.; Soejarto, D.D.; Waller, D.P.; Fong, H.H.S. Furanoid lignans from Larrea tridentata. J. Nat. Prod. 1990, 53, 396–406. [Google Scholar] [CrossRef]
- Abou-Gazar, H.; Bedir, E.; Takamatsu, S.; Ferreira, D.; Khan, I.A. Antioxidant lignans from Larrea tridentata. Phytochemistry 2004, 65, 2499–2505. [Google Scholar] [CrossRef]
- Hulbert, P.B.; Klyne, W.; Scopes, P.M. Chiroptical studies. Part 100: Lignans. J. Chem. Res. (S). 1981, 27. [Google Scholar]
- Iguchi, T.; Kuroda, M.; Naito, R.; Watanabe, T.; Matsuo, Y.; Yokosuka, A.; Mimaki, Y. Cholestane glycosides from Ornithogalum sundersiae bulbs and the induction of apoptosis in HL-60 cells by OSW-1 through a mitochondrial-independent signaling pathway. J. Nat. Med. 2019, 73, 131–145. [Google Scholar] [CrossRef]
- Iguchi, T.; Kuroda, M.; Naito, R.; Watanabe, T.; Matsuo, Y.; Yokosuka, A.; Mimaki, Y. Structural characterization of cholestane rhamnosides from Ornithogalum saundersiae bulbs and their cytotoxic activity against cultured tumor celds. Molecules 2017, 22, e1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokosuka, A.; Iguchi, T.; Kawahata, R.; Mimaki, Y. Cytotoxic bufadienolides from the whole plants of Helleborus foetidus. Phytochem. Lett. 2018, 23, 94–99. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (μM) | Compounds | IC50 (μM) | ||||
|---|---|---|---|---|---|---|---|
| 1 | >20 | 10 | 4.2 | ± | 0.25 | ||
| 2 | >20 | 11 | 2.7 | ± | 0.23 | ||
| 3 | >20 | 12 | >20 | ||||
| 4 | 16 | ± | 0.95 | 13 | >20 | ||
| 5 | 17 | ± | 0.83 | 14 | 5.1 | ± | 0.14 |
| 6 | 4.1 | ± | 0.52 | 15 | 16 | ± | 1.5 |
| 7 | 15 | ± | 1.9 | 16 | 9.3 | ± | 0.45 |
| 8 | 9.2 | ± | 0.44 | 17 | >20 | ||
| 9 | 11 | ± | 0.17 | Cisplatin | 1.1 | ± | 0.19 |
| 1 | 2 | 3 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Positions | δH | J (Hz) | Positions | δH | J (Hz) | Positions | δH | J (Hz) | ||||||
| Glc 1″ | 4.78 | d | 8.0 | Glc 1″ | 4.78 | d | 7.9 | Glc 1″ | 5.53 | d | 8.0 | |||
| 2″ | 3.52 | dd | 9.0, 8.0 | 2″ | 3.52 | m | 2″ | 4.34 | m | |||||
| 3″ | 3.51 | dd | 9.0, 8.5 | 3″ | 3.50 | m | 3″ | 4.28 | m | |||||
| 4″ | 3.45 | dd | 9.5, 8.5 | 4″ | 3.42 | m | 4″ | 4.36 | m | |||||
| 5″ | 3.46 | m | 5″ | 3.47 | m | 5″ | 4.10 | m | ||||||
| 6″ | a | 3.96 | dd | 12.0, 2.0 | 6″ | a | 3.95 | dd | 12.1, 1.8 | 6″ | a | 4.57 | br d | 11.0 |
| b | 3.76 | dd | 12.0, 5.0 | b | 3.76 | dd | 12.1, 5.4 | b | 4.42 | br d | 11.0 | |||
| Glc 1″′ | 4.92 | d | 7.9 | Glc 1″′ | 5.62 | d | 7.5 | |||||||
| 2″′ | 3.50 | m | 2″′ | 4.28 | m | |||||||||
| 3″′ | 3.48 | m | 3″′ | 4.30 | m | |||||||||
| 4″′ | 3.42 | m | 4″′ | 4.32 | m | |||||||||
| 5″′ | 3.45 | m | 5″′ | 3.93 | m | |||||||||
| 6″′ | a | 3.92 | br d | 12.2 | 6″′ | a | 4.44 | br d | 11.3 | |||||
| b | 3.72 | dd | 12.2, 4.6 | b | 4.38 | br d | 11.3 | |||||||
| Glc 1″″ | 5.56 | d | 7.4 | |||||||||||
| 2″″ | 4.32 | m | ||||||||||||
| 3″″ | 4.32 | m | ||||||||||||
| 4″″ | 4.32 | m | ||||||||||||
| 5″″ | 4.01 | m | ||||||||||||
| 6″″ | a | 4.50 | br d | 10.7 | ||||||||||
| b | 4.39 | br d | 10.7 | |||||||||||
| Positions | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 1 | 49.4 | 49.5 | 50.9 | 48.3 | 125.2 | - | - |
| 2 | 40.2 | 40.2 | 40.8 | 43.6 | 140.3 | 92.4 | 92.1 |
| 3 | 28.7 | 28.7 | 28.8 | 72.3 | 113.4 | 44.6 | 44.7 |
| 4 | 34.2 | 34.2 | 34.6 | 73.5 | 151.1 | 120.0 | 120.0 |
| 5 | 116.9 | 117.0 | 119.6 | 112.5 | 143.1 | 130.8 | 130.8 |
| 6 | 143.3 | 143.4 | 145.4 | 142.7 | 116.4 | 125.4 | 125.4 |
| 7 | 144.2 | 144.3 | 147.4 | 143.2 | 190.4 | 107.9 | 108.0 |
| 8 | 116.7 | 116.7 | 118.9 | 115.0 | 213.2 | 157.8 | 157.7 |
| 9 | 133.2 | 132.8 | 133.6 | 131.5 | 27.1 | 131.9 | 131.8 |
| 10 | 127.1 | 127.0 | 127.5 | 128.7 | 16.6 | 16.6 | |
| 11 | 14.3 | 14.2 | 15.4 | 11.0 | |||
| 12 | 14.1 | 14.1 | 16.4 | 21.9 | |||
| 1′ | 137.4 | 140.5 | 143.0 | 137.6 | 133.3 | 130.3 | 130.3 |
| 2′ | 129.1 | 129.1 | 120.8 | 115.5 | 114.6 | 121.5 | 121.5 |
| 3′ | 113.9 | 115.5 | 148.3 | 144.4 | 144.5 | 16.5 | 16.6 |
| 4′ | 154.5 | 155.4 | 147.2 | 142.7 | 143.1 | ||
| 5′ | 113.9 | 115.5 | 118.9 | 114.1 | 114.4 | ||
| 6′ | 129.1 | 129.1 | 124.1 | 120.3 | 118.9 | ||
| 7′ | 44.7 | ||||||
| 8′ | 51.1 | ||||||
| 9′ | 15.1 | ||||||
| 1″ | 102.7 | 102.7 | 105.1 | 131.8 | 133.3 | ||
| 2″ | 73.1 | 73.1 | 75.2 | 112.3 | 112.1 | ||
| 3″ | 75.8 | 76.1 | 78.3 | 144.7 | 145.9 | ||
| 4″ | 69.5 | 69.5 | 71.2 | 144.7 | 147.2 | ||
| 5″ | 76.4 | 76.2 | 79.0 | 114.3 | 110.8 | ||
| 6″ | 60.6 | 60.6 | 62.3 | 117.1 | 116.8 | ||
| OMe | 54.6 | ||||||
| 1″′ | 100.5 | 103.8 | |||||
| 2″′ | 73.1 | 75.0 | |||||
| 3″′ | 75.8 | 78.3 | |||||
| 4″′ | 69.5 | 71.2 | |||||
| 5″′ | 76.4 | 78.5 | |||||
| 6″′ | 60.6 | 62.2 | |||||
| 1″″ | 104.1 | ||||||
| 2″″ | 75.1 | ||||||
| 3″″ | 78.1 | ||||||
| 4″″ | 71.2 | ||||||
| 5″″ | 78.8 | ||||||
| 6″″ | 62.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yokosuka, A.; Iguchi, T.; Jitsuno, M.; Mimaki, Y. Structure and Cytotoxicity of Novel Lignans and Lignan Glycosides from the Aerial Parts of Larrea tridentata. Molecules 2021, 26, 6186. https://doi.org/10.3390/molecules26206186
Yokosuka A, Iguchi T, Jitsuno M, Mimaki Y. Structure and Cytotoxicity of Novel Lignans and Lignan Glycosides from the Aerial Parts of Larrea tridentata. Molecules. 2021; 26(20):6186. https://doi.org/10.3390/molecules26206186
Chicago/Turabian StyleYokosuka, Akihito, Tomoki Iguchi, Maki Jitsuno, and Yoshihiro Mimaki. 2021. "Structure and Cytotoxicity of Novel Lignans and Lignan Glycosides from the Aerial Parts of Larrea tridentata" Molecules 26, no. 20: 6186. https://doi.org/10.3390/molecules26206186
APA StyleYokosuka, A., Iguchi, T., Jitsuno, M., & Mimaki, Y. (2021). Structure and Cytotoxicity of Novel Lignans and Lignan Glycosides from the Aerial Parts of Larrea tridentata. Molecules, 26(20), 6186. https://doi.org/10.3390/molecules26206186