
Theoretical Evaluation of Novel Thermolysin Inhibitors from Bacillus thermoproteolyticus. Possible Antibacterial Agents


Abstract
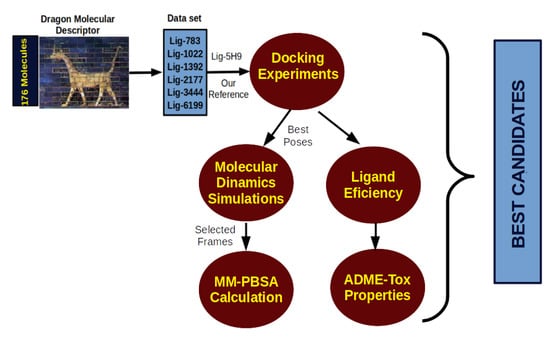
1. Introduction
2. Results and Discussion
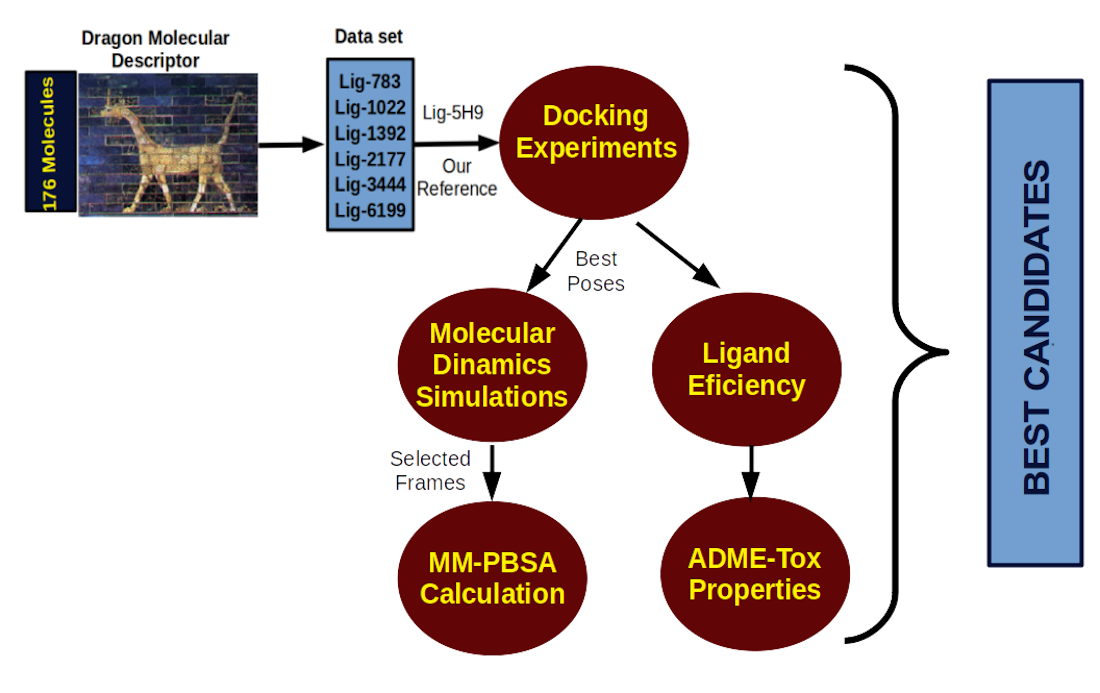
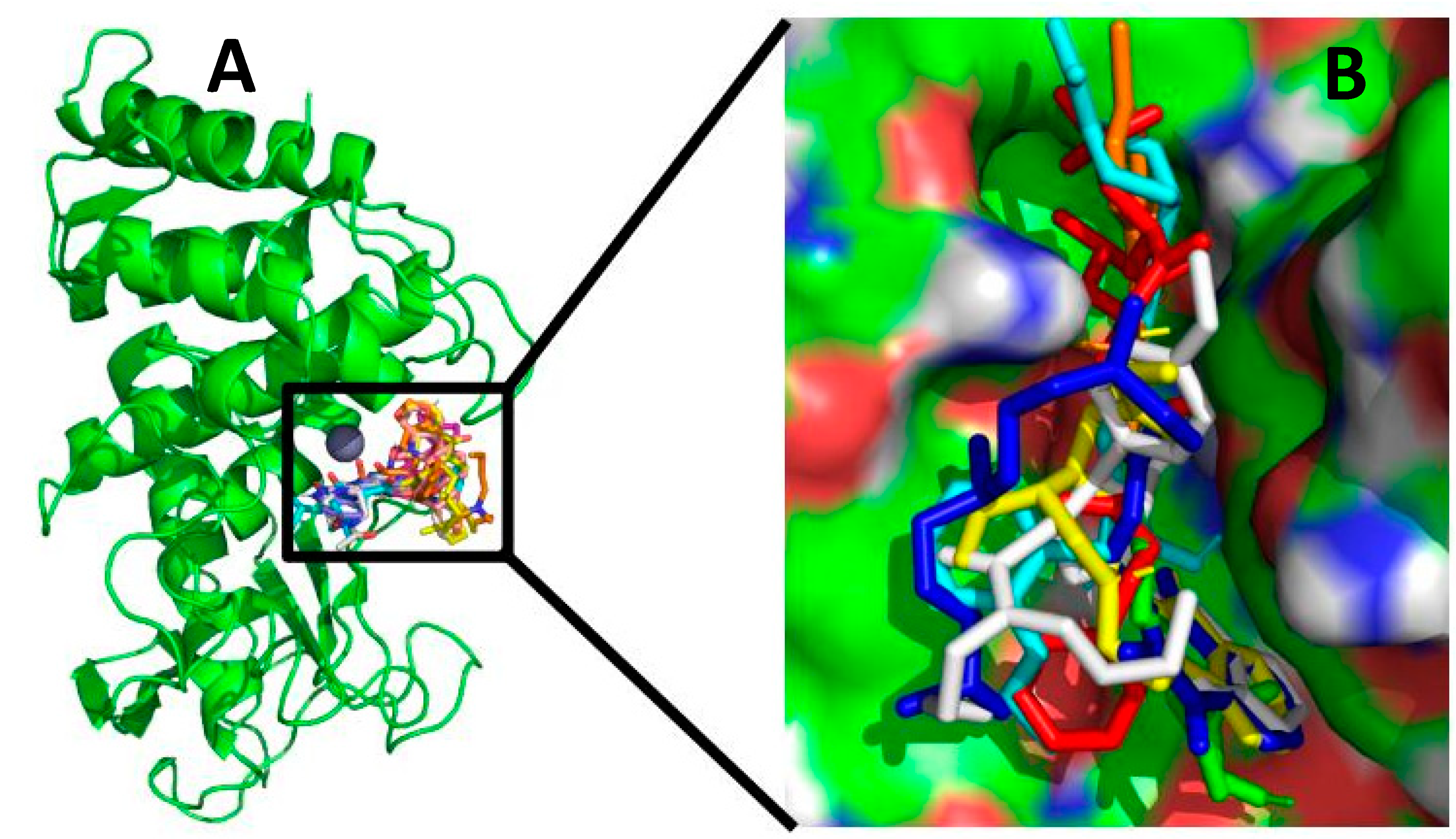
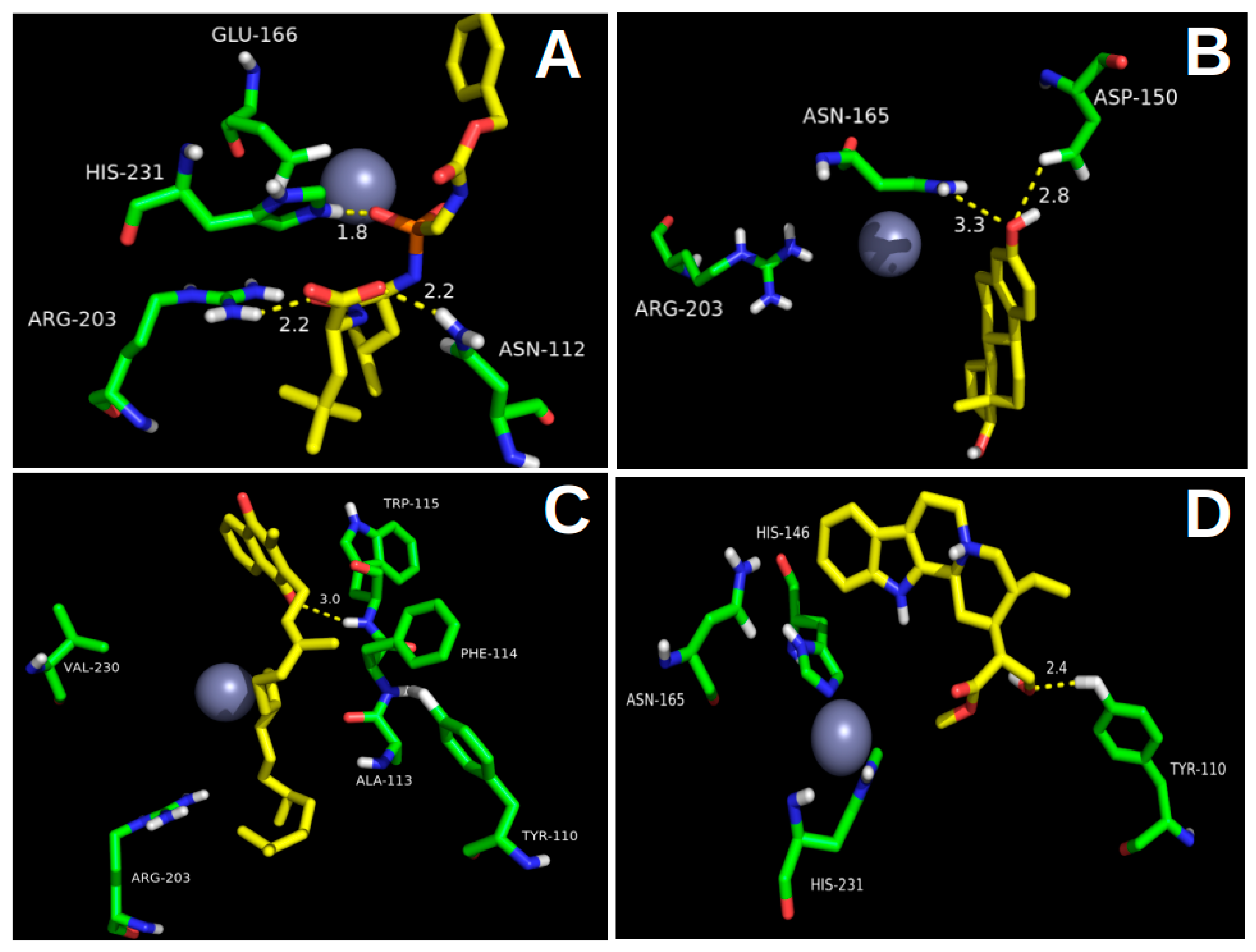
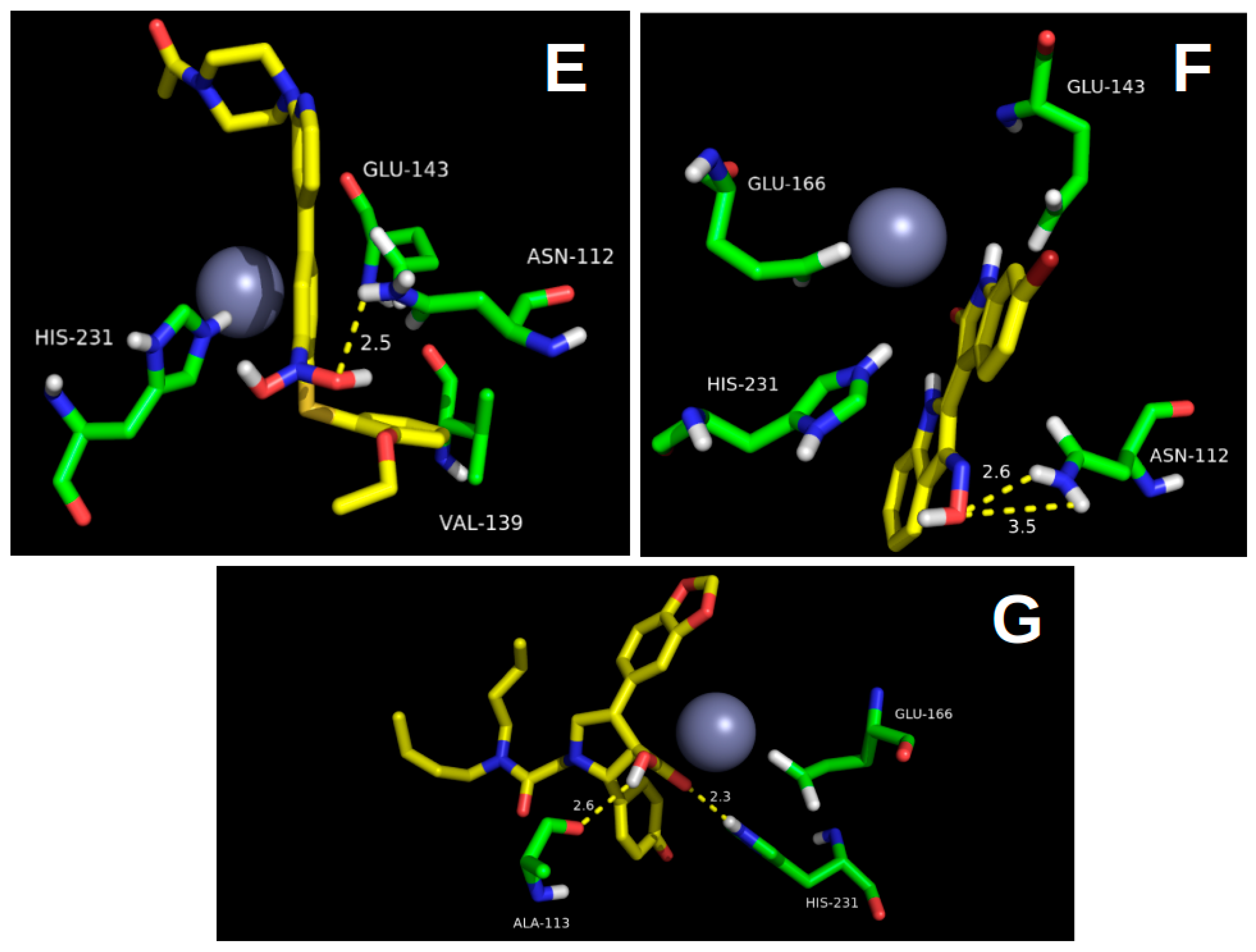
2.1. Molecular Docking
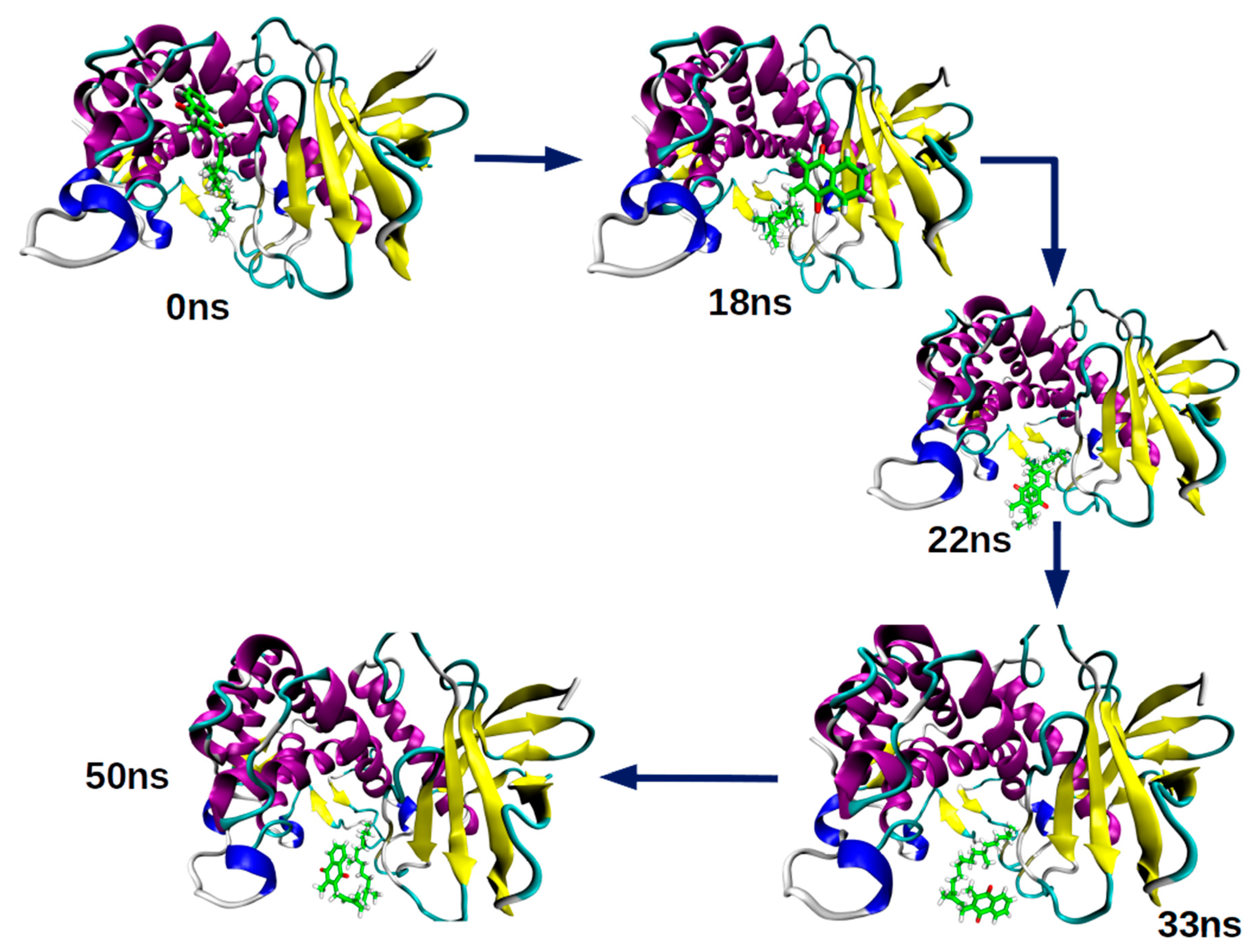
2.2. Molecular Dynamics Simulation
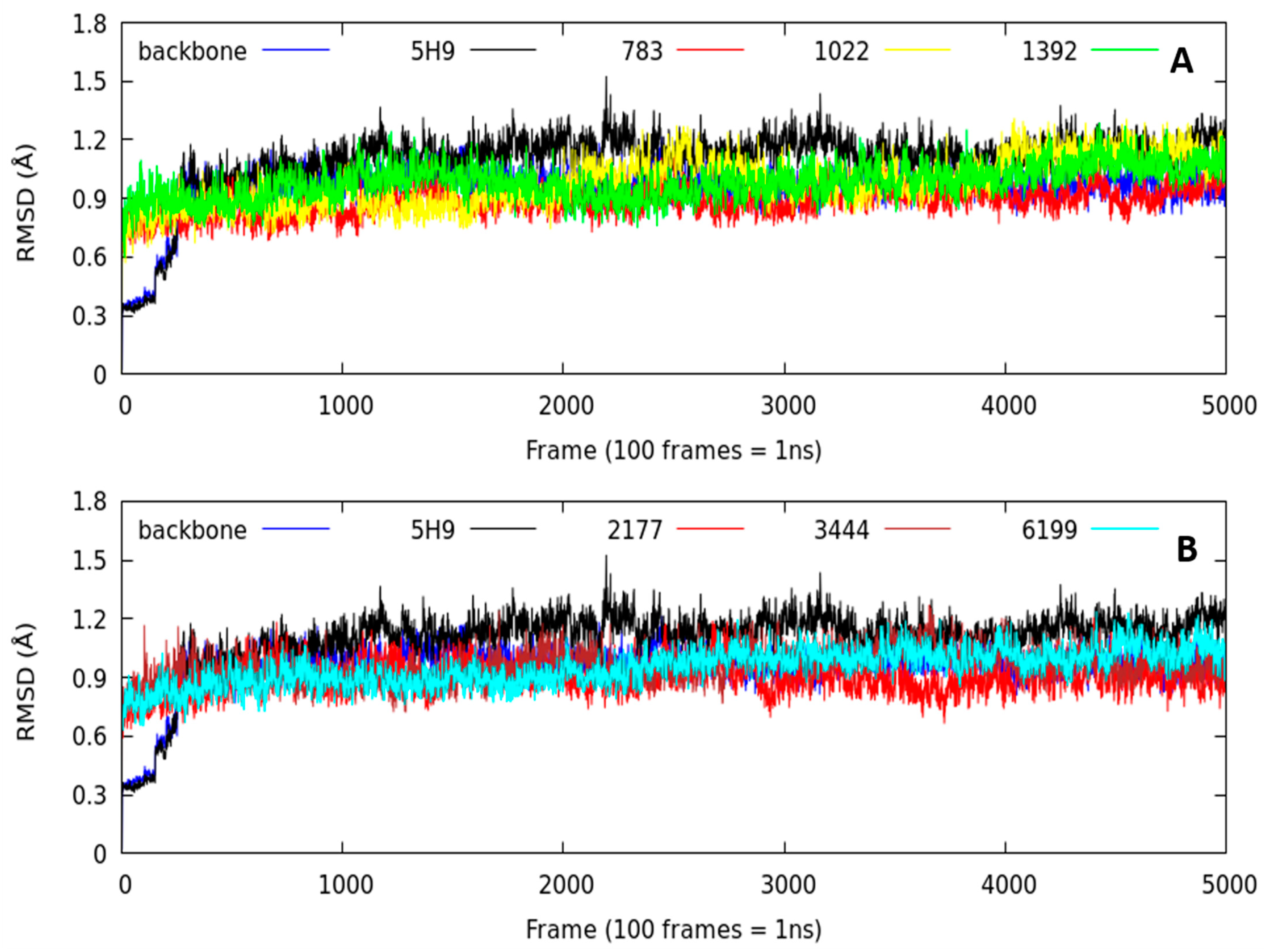
2.2.1. Root-Mean-Square Deviation (RMSD) Parameter
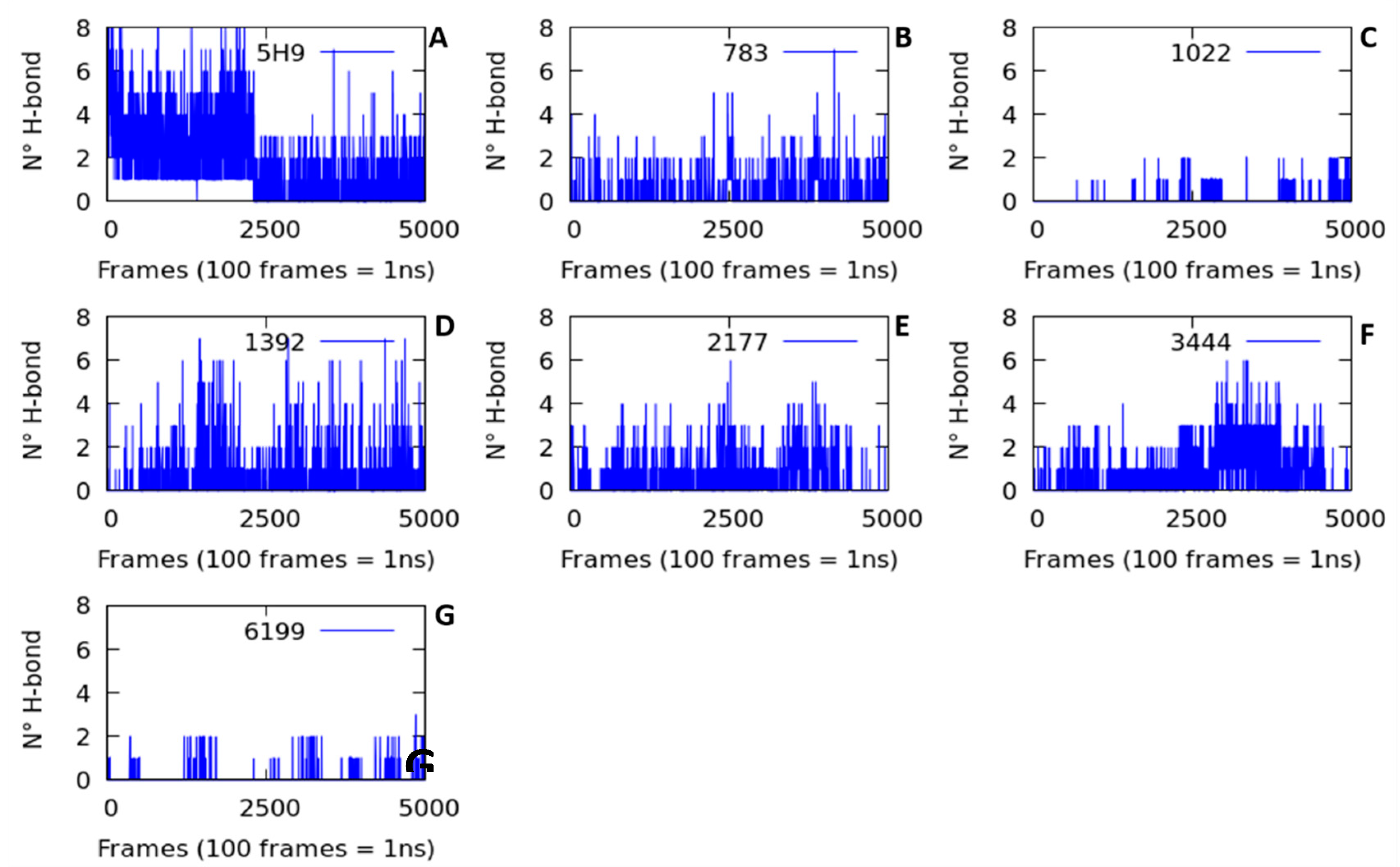
2.2.2. Hydrogen Bond Interactions (H-Bond)
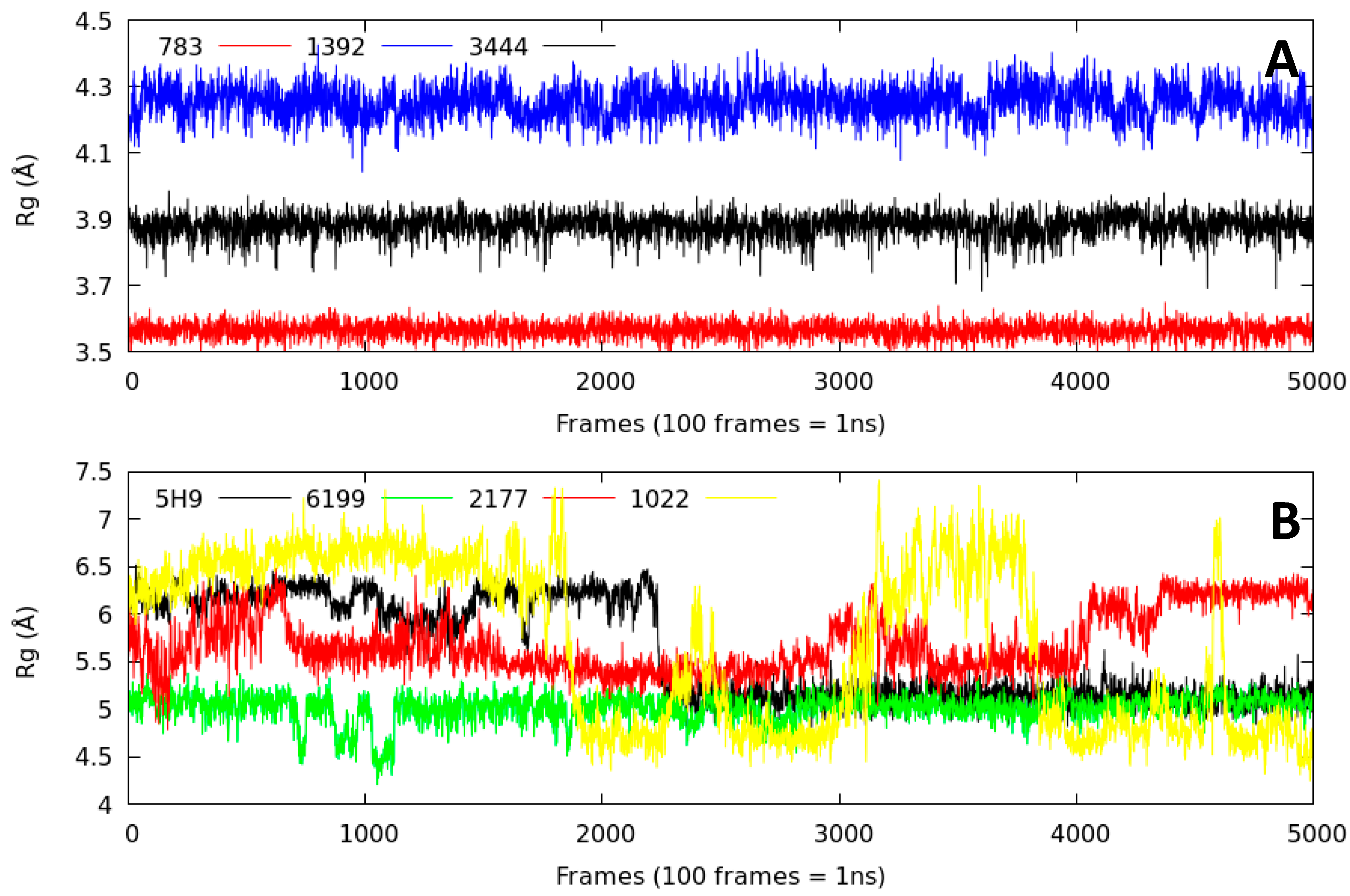
2.2.3. Radius of Gyration (Rg)
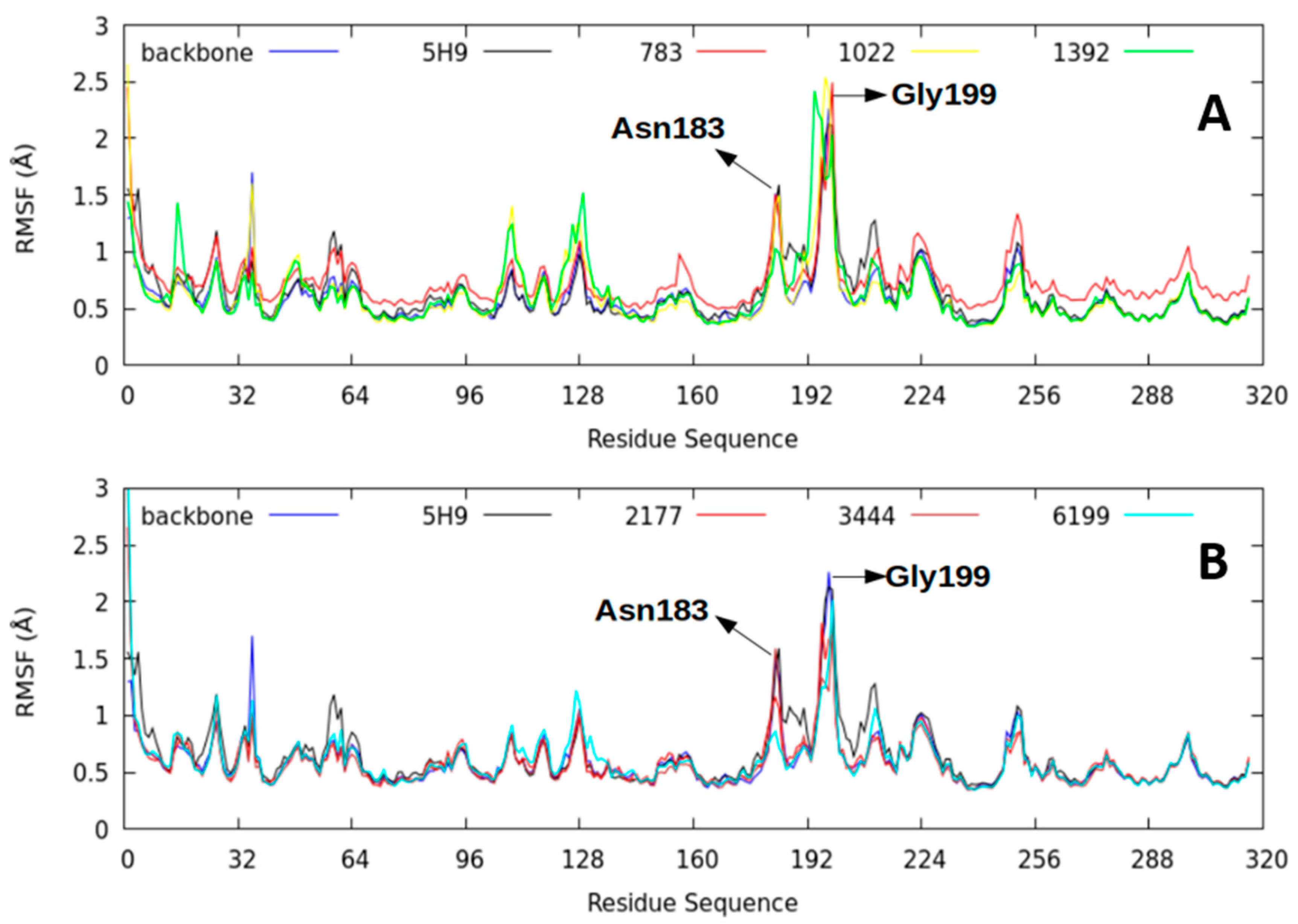
2.2.4. Root-Mean-Square Fluctuation (RMSF) Parameter
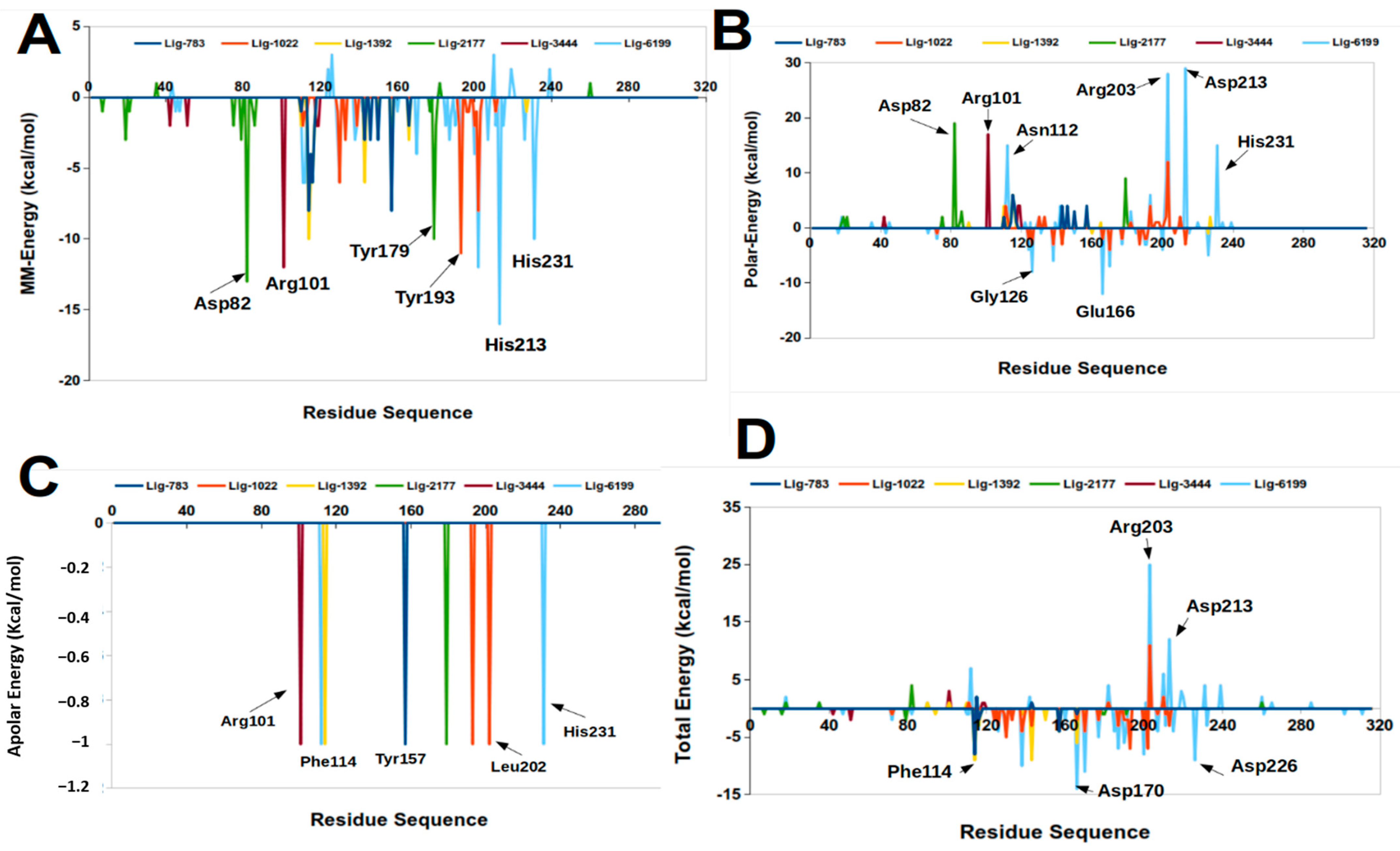
2.3. Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA)
2.4. Ligand Efficiency Calculation and Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADME-Tox) Properties
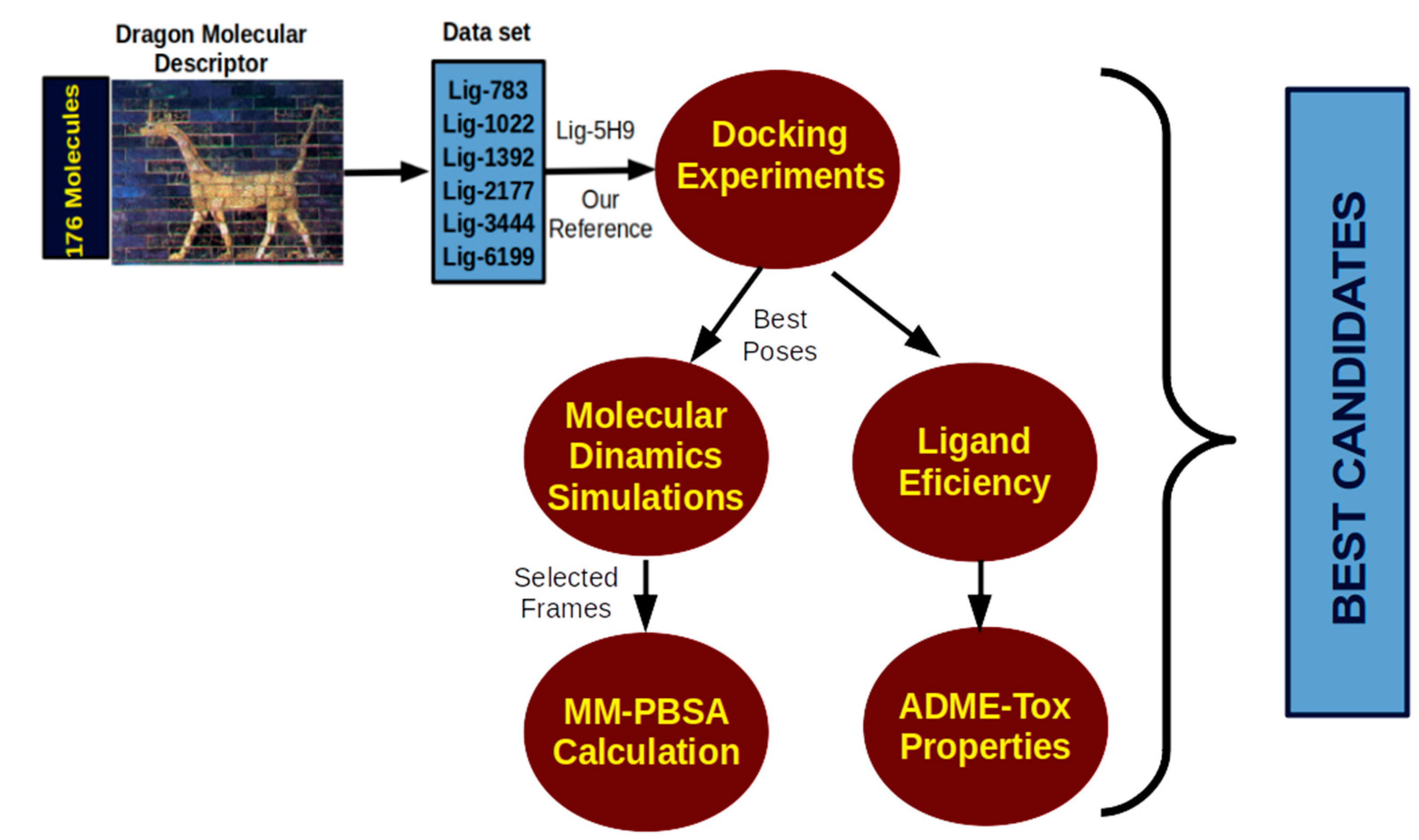
3. Computational Protocol
3.1. Docking Procedure
3.2. Molecular Dynamics Simulation
3.3. Free Energy Calculations
3.4. Ligand Efficiency Calculations
3.5. ADME-Tox Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Den Hoed, C.M.; Kuipers, E.J. 45-Helicobacter pylori Infection. In Hunter’s Tropical Medicine and Emerging Infectious Diseases, 10th ed.; Ryan, E.T., Hill, D.R., Solomon, T., Aronson, N.E., Endy, T.P., Eds.; Elsevier: London, UK, 2020; pp. 476–480. ISBN 978-0-323-55512-8. [Google Scholar]
- WHO. Antibacterial Agents in Clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Huang, J.; Zeng, B.; Liu, D.; Wu, R.; Zhang, J.; Liao, B.; He, H.; Bian, F. Classification and structural insight into vibriolysin-like proteases of Vibrio pathogenicity. Microb. Pathog. 2018, 117, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.; Dumitrescu, D.G.; Blankenship, L.R.; Herkert, D.; Hatzios, S.K. Functional characterisation of a subtilisin-like serine protease from Vibrio cholerae. J. Biol. Chem. 2019, 294, 9888–9900. [Google Scholar] [CrossRef] [PubMed]
- Kavitt, R.T.; Lipowska, A.M.; Anyane-Yeboa, A.; Gralnek, I.M. Diagnosis and Treatment of Peptic Ulcer Disease. Am. J. Med. 2019, 132, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Vinasco, K.; Mitchell, H.M.; Kaakoush, N.O.; Castaño-Rodríguez, N. Microbial carcinogenesis: Lactic acid bacteria in gastric cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 188309. [Google Scholar] [CrossRef]
- Khan, M.T.H.; Fuskevåg, O.-M.; Sylte, I. Discovery of Potent Thermolysin Inhibitors Using Structure-Based Virtual Screening and Binding Assays. J. Med. Chem. 2009, 52, 48–61. [Google Scholar] [CrossRef]
- Goblirsch, B.R.; Wiener, M.C. Ste24: An Integral Membrane Protein Zinc Metalloprotease with Provocative Structure and Emergent Biology. J. Mol. Biol. 2020, 432, 5079–5090. [Google Scholar] [CrossRef]
- Ezawa, T.; Saito, R.; Suzuki, S.; Sugiyama, S.; Sylte, I.; Kurita, N. Protonation states of central amino acids in a zinc metalloprotease complexed with inhibitor: Molecular mechanics optimisations and ab initio molecular orbital calculations. Biophys. Chem. 2020, 261, 106368. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Gottwalt, S.; Beyer, P.; Butler, M.; Czaplewski, L.; Lienhardt, C.; Moja, L.; Paul, M.; Paulin, S.; Rex, J.H.; et al. Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect. Dis. 2019, 19, e40–e50. [Google Scholar] [CrossRef]
- Cañizares-Carmenate, Y.; Mena-Ulecia, K.; Perera-Sardiña, Y.; Torrens, F.; Castillo-Garit, J.A. An approach to identify new antihypertensive agents using thermolysin as model: In silico study based on QSARINS and docking. Arab. J. Chem. 2019, 12, 4861–4877. [Google Scholar] [CrossRef]
- MacLeod-Carey, D.; Solis-Céspedes, E.; Lamazares, E.; Mena-Ulecia, K. Evaluation of new antihypertensive drugs designed in silico using thermolysin as a target. Saudi Pharm. J. 2020, 28, 582–592. [Google Scholar] [CrossRef]
- Mena-Ulecia, K.; MacLeod-Carey, D. Interactions of 2-phenyl-benzotriazole xenobiotic compounds with human Cytochrome P450-CYP1A1 by means of docking, molecular dynamics simulations and MM-GBSA calculations. Comput. Biol. Chem. 2018, 74, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Quesada-Romero, L.; Mena-Ulecia, K.; Zuñiga, M.; De-la-Torre, P.; Rossi, D.; Tiznado, W.; Collina, S.; Caballero, J. Optimal graph-based and Simplified Molecular Input Line Entry System-based descriptors for quantitative structure-activity relationship analysis of arylalkylaminoalcohols, arylalkenylamines, and arylalkylamines as σ1 receptor ligands. J. Chemom. 2015, 29, 13–20. [Google Scholar] [CrossRef]
- Salsbury, F.R. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr. Opin. Pharmacol. 2010, 10, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Rivail, L.; Chipot, C.; Maigret, B.; Bestel, I.; Sicsic, S.; Tarek, M. Large-scale molecular dynamics of a G protein-coupled receptor, the human 5-HT4 serotonin receptor, in a lipid bilayer. J. Mol. Struct. Theochem 2007, 817, 19–26. [Google Scholar] [CrossRef]
- Bocharov, D.; Krack, M.; Rafalskij, Y.; Kuzmin, A.; Purans, J. Ab initio molecular dynamics simulations of negative thermal expansion in ScF3: The effect of the supercell size. Comput. Mater. Sci. 2020, 171, 109198. [Google Scholar] [CrossRef]
- Fegan, S.K.; Thachuk, M. A charge moving algorithm for molecular dynamics simulations of gas-phase proteins. J. Chem. Theory Comput. 2013, 9, 2531–2539. [Google Scholar] [CrossRef]
- Velázquez-Libera, J.L.; Durán-Verdugo, F.; Valdés-Jiménez, A.; Núñez-Vivanco, G.; Caballero, J. LigRMSD: A web server for automatic structure matching and RMSD calculations among identical and similar compounds in protein-ligand docking. Bioinformatics 2020, 36, 2912–2914. [Google Scholar] [CrossRef]
- Bell, E.W.; Zhang, Y. DockRMSD: An open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J. Cheminform. 2019, 11, 40. [Google Scholar] [CrossRef]
- Mena-Ulecia, K.; Vergara-Jaque, A.; Poblete, H.; Tiznado, W.; Caballero, J. Study of the Affinity between the Protein Kinase PKA and Peptide Substrates Derived from Kemptide Using Molecular Dynamics Simulations and MM/GBSA. PLoS ONE 2014, 9, e109639. [Google Scholar] [CrossRef]
- Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Study of the Differential Activity of Thrombin Inhibitors Using Docking, QSAR, Molecular Dynamics, and MM-GBSA. PLoS ONE 2015, 10, e0142774. [Google Scholar] [CrossRef]
- Kumar, K.M.; Anbarasu, A.; Ramaiah, S. Molecular docking and molecular dynamics studies on β-lactamases and penicillin-binding proteins. Mol. Biosyst. 2014, 10, 891–900. [Google Scholar] [CrossRef]
- Kumar, A.; Purohit, R. Use of Long Term Molecular Dynamics Simulation in Predicting Cancer-Associated SNPs. PLoS Comput. Biol. 2014, 10, e1003318. [Google Scholar] [CrossRef] [PubMed]
- Lavanya, P.; Ramaiah, S.; Anbarasu, A. A Molecular Docking and Dynamics Study to Screen Potent Anti-Staphylococcal Compounds Against Ceftaroline Resistant MRSA. J. Cell. Biochem. 2016, 117, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.; Marafie, S.K.; Alshawaf, E.; Abu-Farha, M.; Abubaker, J.; Al-Mulla, F. Structural analysis of ACE2 variant N720D demonstrates a higher binding affinity to TMPRSS2. Life Sci. 2020, 259, 118219. [Google Scholar] [CrossRef]
- Eisenhardt, M.; Schlupp, P.; Höfer, F.; Schmidts, T.; Hoffmann, D.; Czermak, P.; Pöppel, A.-K.; Vilcinskas, A.; Runkel, F. The therapeutic potential of the insect metalloproteinase inhibitor against infections caused by Pseudomonas aeruginosa. J. Pharm. Pharmacol. 2019, 71, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Murray, C.W.; Erlanson, D.A.; Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H.; Richmond, N.J. Validity of ligand efficiency metrics. ACS Med. Chem. Lett. 2014, 5, 616–618. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Abad-Zapatero, C. Ligand Efficiency Indices for Drug Discovery. Ligand Effic. Indices Drug Discov. 2013, 10, 469–488. [Google Scholar] [CrossRef]
- Kenny, P.W. The nature of ligand efficiency. J. Cheminform. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, X.; Chen, Y.; Chen, H.; Sun, H.; Li, W.; Xie, Q.; Yu, L.; Shao, L. Discovery of novel 20S proteasome inhibitors by rational topology-based scaffold hopping of Bortezomib. Bioorg. Med. Chem. Lett. 2018, 28, 2148–2152. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef]
- Kauthale, S.; Tekale, S.; Damale, M.; Sangshetti, J.; Pawar, R. Synthesis, biological evaluation, molecular docking, and ADMET studies of some isoxazole-based amides. Med. Chem. Res. 2018, 27, 429–441. [Google Scholar] [CrossRef]
- Kumar, N.; Goel, N.; Chand Yadav, T.; Pruthi, V. Quantum chemical, ADMET and molecular docking studies of ferulic acid amide derivatives with a novel anticancer drug target. Med. Chem. Res. 2017, 26, 1822–1834. [Google Scholar] [CrossRef]
- Tsaioun, K.; Blaauboer, B.J.; Hartung, T. Evidence-based absorption, distribution, metabolism, excretion (ADME) and its interplay with alternative toxicity methods. ALTEX 2016, 33, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Teotia, P.; Prakash Dw, S.; Dwivedi, N. In silico Molecular Docking and ADME/Tox Study on Benzoxazole Derivatives Against Inosine 5’-Monophosphate Dehydrogenase. Asian J. Biotechnol. 2018, 10, 1–10. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings1PII of original article: S0169-409X(96)00423-1. Adv. Drug Deliv. Rev. 1997, 23, 3–25, Erratum in 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualisation, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E.; Westbrook, Z.; Feng, G.; et al. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Krimmer, S.; Klebe, G. Thermodynamics of protein-ligand interactions as a reference for computational analysis: How to assess accuracy, reliability and relevance of experimental data. J. Comput. Aided Mol. Des. 2015, 29, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimisation, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Koebel, M.R.; Schmadeke, G.; Posner, R.G.; Sirimulla, S. AutoDock VinaXB: Implementation of XBSF, new empirical halogen bond scoring function, into AutoDock Vina. J. Cheminform. 2016, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Hendlich, M.; Klebe, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000, 295, 337–356. [Google Scholar] [CrossRef]
- Sahin, K. Investigation of novel indole-based HIV-1 protease inhibitors using virtual screening and text mining. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using PyMOL. J. Comput. Aided. Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, S.; Onck, P.R.; Giessen, E.; van der Giessen, E. CHARMM TIP3P Water Model Suppresses Peptide Folding by Solvating the Unfolded State. J. Phys. Chem. B 2016, 120, 3692–3698. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qiu, Y.; Baron, R.; Molinero, V. Coarse-Graining of TIP4P/2005, TIP4P-Ew, SPC/E, and TIP3P to Monatomic Anisotropic Water Models Using Relative Entropy Minimisation. J. Chem. Theory Comput. 2014, 10, 4104–4120. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Aurélien, G.; Olivier, M. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Soteras, G.I.; Lin, F.-Y.; Vanommeslaeghe, K.; Lemkul, J.A.; Armacost, K.A.; Brooks, C.L.; MacKerell, A.D. Parametrisation of halogen bonds in the CHARMM general force field: Improved treatment of ligand-protein interactions. Bioorg. Med. Chem. 2016, 24, 4812–4825. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Yang, M.; MacKerell, A.D. Robustness in the fitting of molecular mechanics parameters. J. Comput. Chem. 2015, 36, 1083–1101. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.K. Molecular Dynamics with coupling to an external bath. J. Chem. Physic 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalised born model. Proteins Struct. Funct. Bioinform. 2004, 55, 383–394. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Dalke, W.; Humphrey, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Abad-Zapatero, C.; Perišić, O.; Wass, J.; Bento, A.P.; Overington, J.; Al-Lazikani, B.; Johnson, M.E. Ligand efficiency indices for an effective mapping of chemico-biological space: The concept of an atlas-like representation. Drug Discov. Today 2010, 15, 804–811. [Google Scholar] [CrossRef]
- Meneses, L.; Cuesta, S. Determinación Computacional de la Afinidad y Eficiencia de Enlace de Antinflamatorios No Esteroideos Inhibidores de la Ciclooxigenasa-2. Rev. Ecuat. Med. Cienc. Biol. 2017, 36, 17. [Google Scholar] [CrossRef][Green Version]
- Reynolds, C.H.; Tounge, B.A.; Bembenek, S.D. Ligand Binding Efficiency: Trends, Physical Basis, and Implications. J. Med. Chem. 2008, 51, 2432–2438. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.W.; Leitão, A.; Montanari, C.A. Ligand efficiency metrics considered harmful. J. Comput. Aided. Mol. Des. 2014, 28, 699–710. [Google Scholar] [CrossRef]
- Polanski, J.; Tkocz, A.; Kucia, U. Beware of ligand efficiency (LE): Understanding LE data in modeling structure-activity and structure-economy relationships. J. Cheminform. 2017, 9, 49. [Google Scholar] [CrossRef]
- Scott, J.S.; Waring, M.J. Practical application of ligand efficiency metrics in lead optimisation. Bioorganic Med. Chem. 2018, 26, 3006–3015. [Google Scholar] [CrossRef] [PubMed]
- Duchowics, P.R.; Talevi, A.; Bellera, C.; Bruno-Blanch, L.E.; Castro, E.A.; Duchowicz, P.R.; Talevi, A.; Bellera, C.; Bruno-Blanch, L.E.; Castro, E.A. Application of descriptors based on Lipinski’s rules in the QSPR study of aqueous solubilities. Bioorg. Med. Chem. 2007, 15, 3711–3719. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Docking Pose-1 | Docking Pose-2 | Docking Pose-3 | ||||
|---|---|---|---|---|---|---|
| ΔGbinding | RMSD | ΔGbinding | RMSD | ΔGbinding | RMSD | |
| Lig5H9-5DPF 1 | −8.2 | 0.94 | −7.7 | 1.80 | −7.5 | 2.36 |
| Lig783-5DPF | −8.0 | 1.07 | −8.0 | 1.16 | −7.8 | 1.80 |
| Lig1022-5DPF | −7.9 | 0.90 | −7.8 | 1.88 | −7.7 | 1.46 |
| Lig1392-5DPF | −7.3 | 3.02 | −7.2 | 4.02 | −6.8 | 5.06 |
| Lig2177-5DPF | −8.0 | 2.45 | −7.9 | 3.01 | −7.8 | 3.76 |
| Lig3444-5DPF | −8.1 | 1.16 | −8.0 | 4.18 | −7.9 | 4.57 |
| Lig6199-5DPF | −7.0 | 6.76 | −6.9 | 6.81 | −6.6 | 6.85 |
| Complexes | RMSD (Å) | Number of H-Bond | Rg (Å) |
|---|---|---|---|
| Lig5H9-5DPF 1 | 1.11 ± 0.13 | 1.48 ± 1.68 | 5.36 ± 0.52 |
| Lig783-5DPF | 0.93 ± 0.08 | 0.17 ± 0.54 | 3.56 ± 0.02 |
| Lig1022-5DPF | 1.02 ± 0.11 | 0.09 ± 0.32 | 5.88 ± 0.86 |
| Lig1392-5DPF | 1.10 ± 0.15 | 0.35 ± 0.82 | 4.25 ± 0.04 |
| Lig2177-5DPF | 0.90 ± 0.07 | 0.46 ± 0.73 | 5.78 ± 0.29 |
| Lig3444-5DPF | 0.98 ± 0.08 | 0.74 ± 1.26 | 3.87 ± 0.03 |
| Lig6199-5DPF | 0.97 ± 0.08 | 0.05 ± 0.26 | 4.99 ± 0.14 |
| Complexes | ∆Gbinding | ∆Eelect | ∆Evdw | ∆Gpolar | ∆GApolar |
|---|---|---|---|---|---|
| Lig5H9-5DPF 1 | −146.79 ± 8.30 | −150.45 ± 13.06 | −227.00 ± 7.61 | 254.72 ± 9.29 | −24.05 ± 0.74 |
| Lig783-5DPF | −60.84 ± 11.32 | −24.72 ± 14.56 | −81.13 ± 0.29 | 55.27 ± 17.78 | −10.26 ± 0,84 |
| Lig1022-5DPF | −114.11 ± 25.88 | −1.84 ± 5.19 | −120.97 ± 20.03 | 25.89 ± 25.87 | −17.12 ± 2.35 |
| Lig1392-5DPF | −75.79 ± 11.25 | −23.69 ± 6.66 | −87.35 ± 8.27 | 46.35 ± 12.71 | −11.09 ± 1.40 |
| Lig2177-5DPF | −37.06 ± 10.44 | −42.08 ± 10.52 | −63.36 ± 15.97 | 78.06 ± 17.56 | −9.07 ± 2.28 |
| Lig3444-5DPF | −27.31 ± 11.72 | −7.94 ± 6.26 | −49.88 ± 20.82 | 36.92 ± 21.27 | −6.41 ± 2.67 |
| Lig6199-5DPF | −88.56 ± 19.45 | −74.95 ± 15.19 | −159.04 ± 16.52 | 165.67 ± 33.93 | −20.23 ± 1.22 |
| Ligands | MW (kDa) | Kd | clogP | LE | BEI | LLE | HBA | HBD | TPSA (Å2) | RB |
|---|---|---|---|---|---|---|---|---|---|---|
| Lig5H9 1 | 0.4855 | 9.78 × 10−7 | 2.71 | 0.248 | 12.37 | 3.29 | 8 | 5 | 163.87 | 16 |
| Lig783 | 0.2723 | 1.37 × 10−6 | 2.60 | 0.400 | 21.52 | 3.26 | 2 | 2 | 40.46 | 0 |
| Lig1022 | 0.4507 | 1.62 × 10−6 | 5.86 | 0.239 | 12.84 | −0.07 | 2 | 0 | 34.14 | 14 |
| Lig1392 | 0.3574 | 4.46 × 10−6 | 2.81 | 0.280 | 14.96 | 2.54 | 3 | 3 | 74.35 | 5 |
| Lig2177 | 0.4805 | 1.37 × 10−6 | 3.45 | 0.235 | 12.19 | 2.41 | 5 | 2 | 114.67 | 0 |
| Lig3444 | 0.3561 | 1.15 × 10−6 | 2.31 | 0.368 | 16.66 | 3.62 | 3 | 3 | 74.72 | 0 |
| Lig6199 | 0.5106 | 7.64 × 10−6 | 4.06 | 0.189 | 10.04 | 1.07 | 7 | 1 | 88.56 | 13 |
| Properties | Oral Availability | Toxicity | |
|---|---|---|---|
| Lipinski Rules | Veber Rules | Pfizer 3/75 Rules | |
| MW | ≤500 | - | - |
| cLogP | ≤5 | - | ≤3 |
| HBA | ≤10 | - | - |
| HBD | ≤5 | - | - |
| TPSA | - | ≤140 | ≤75 |
| RB | - | ≤10 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamazares, E.; MacLeod-Carey, D.; Miranda, F.P.; Mena-Ulecia, K. Theoretical Evaluation of Novel Thermolysin Inhibitors from Bacillus thermoproteolyticus. Possible Antibacterial Agents. Molecules 2021, 26, 386. https://doi.org/10.3390/molecules26020386
Lamazares E, MacLeod-Carey D, Miranda FP, Mena-Ulecia K. Theoretical Evaluation of Novel Thermolysin Inhibitors from Bacillus thermoproteolyticus. Possible Antibacterial Agents. Molecules. 2021; 26(2):386. https://doi.org/10.3390/molecules26020386
Chicago/Turabian StyleLamazares, Emilio, Desmond MacLeod-Carey, Fernando P. Miranda, and Karel Mena-Ulecia. 2021. "Theoretical Evaluation of Novel Thermolysin Inhibitors from Bacillus thermoproteolyticus. Possible Antibacterial Agents" Molecules 26, no. 2: 386. https://doi.org/10.3390/molecules26020386
APA StyleLamazares, E., MacLeod-Carey, D., Miranda, F. P., & Mena-Ulecia, K. (2021). Theoretical Evaluation of Novel Thermolysin Inhibitors from Bacillus thermoproteolyticus. Possible Antibacterial Agents. Molecules, 26(2), 386. https://doi.org/10.3390/molecules26020386