
Vibrational Spectra of Zeolite Y as a Function of Ion Exchange


Abstract
1. Introduction
- sites I—located in the hexagonal prisms, which connect the so-called sodalite cages,
- sites I′—inside the sodalite cages facing site I,
- sites II—in front of the six rings inside the supercages and
- sites III and III′ or J—in the supercages near the 12-ring or four-ring windows.
2. Results and Discussion
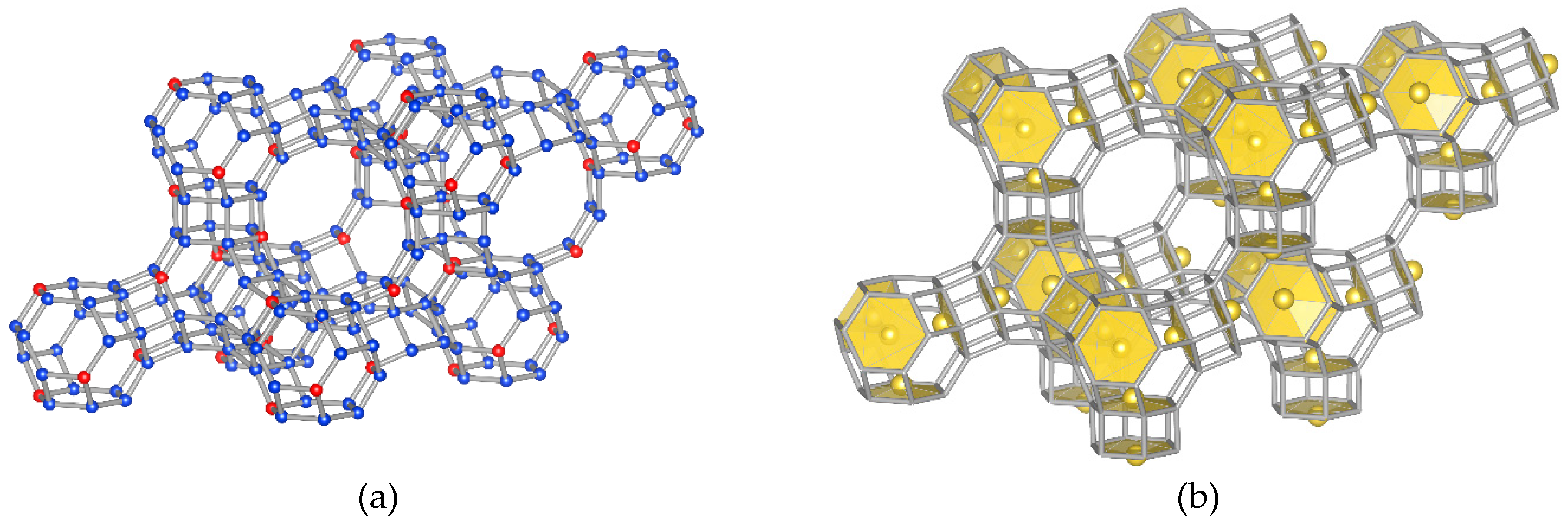
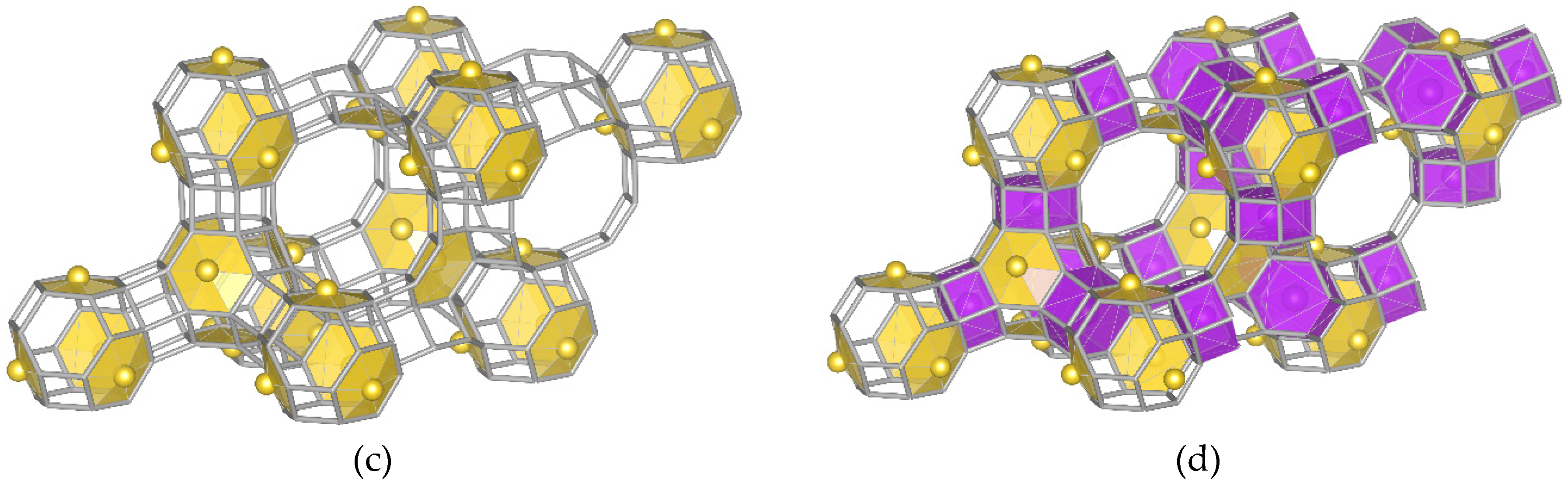
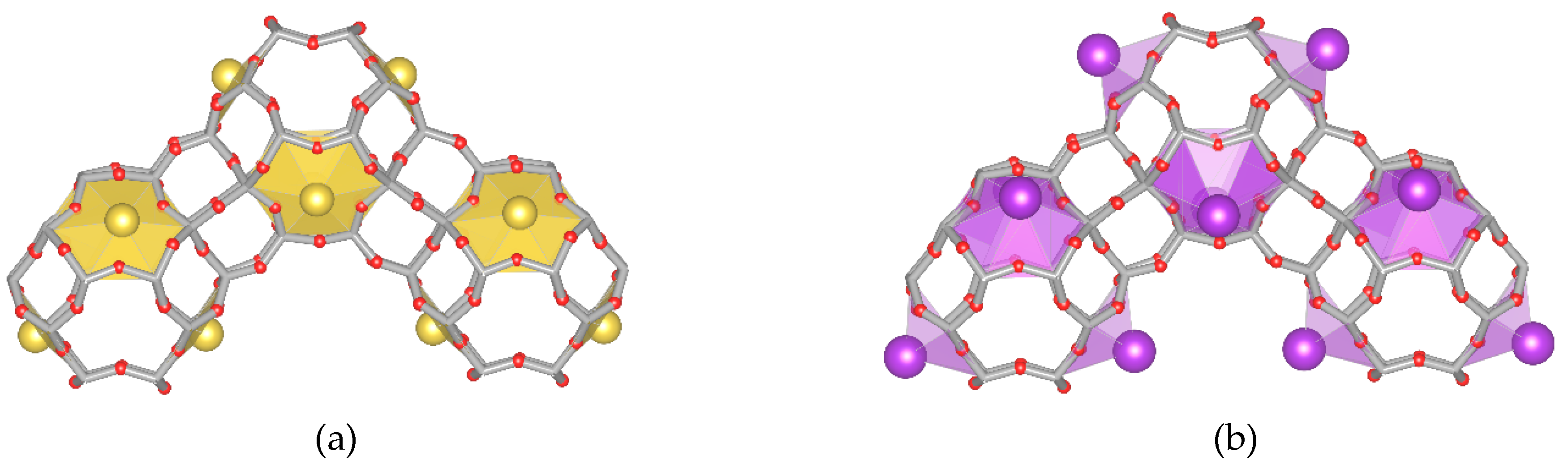
- Model “A”—32 Me+ (or 16 Me2+ cations with 0.5 occ. in each position) in SI′ sites (Wyckoff pos. 32e) in the sodalite cage in front of the six-ring window connected to the hexagonal prism (Figure 1b),
- Model “B”—32 Me+ (or 16 Me2+) cations in SII sites (Wyckoff pos. 32e) in the 12-ring windows of the supercages (Figure 1c) and
- Model “C”—16 Me+ (or 8 Me2+) cations in SI sites (Wyckoff pos. 16c) located in the hexagonal prism connecting two sodalite cages and 16 Me+ (or 8 Me2+) cations in SII sites (Figure 1d).
2.1. Na-Y Structure Optimization
2.2. Model Selection for Various Cationic Forms
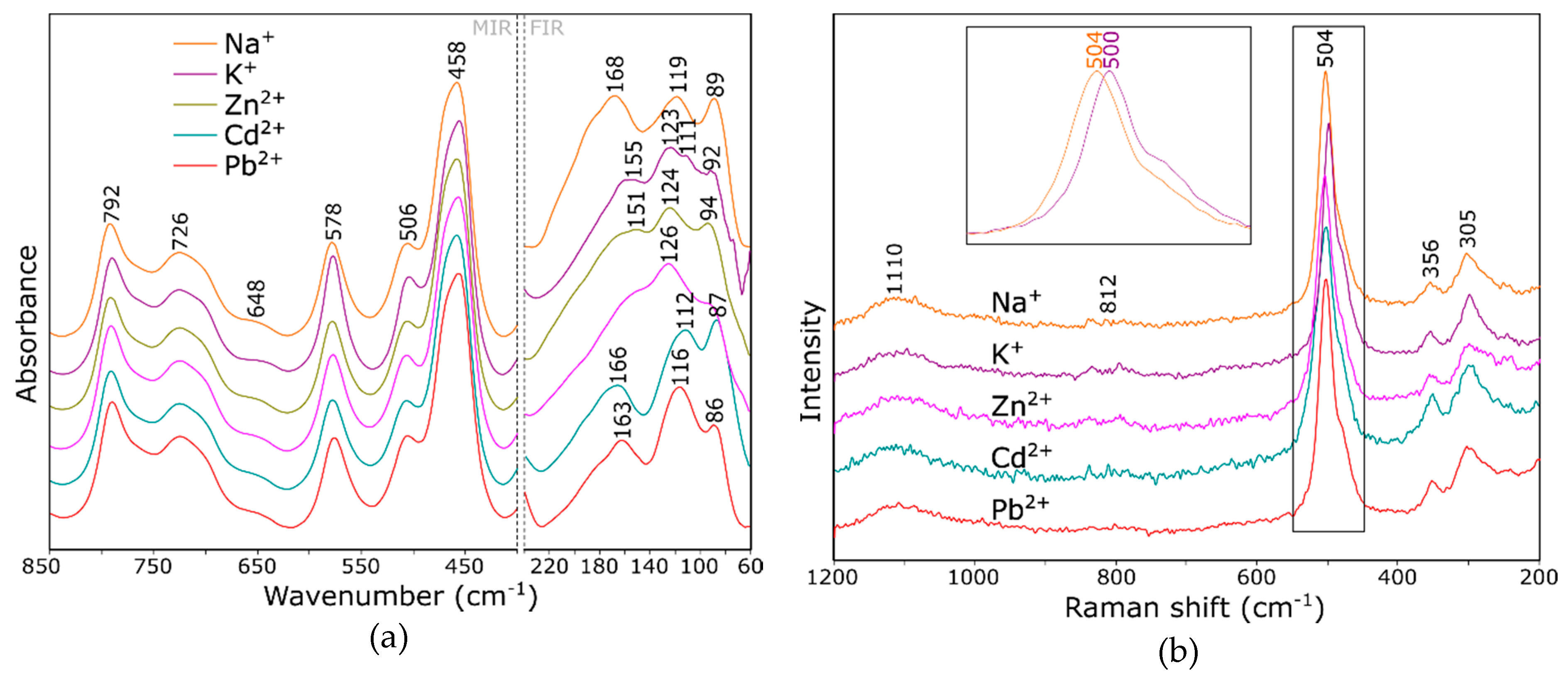
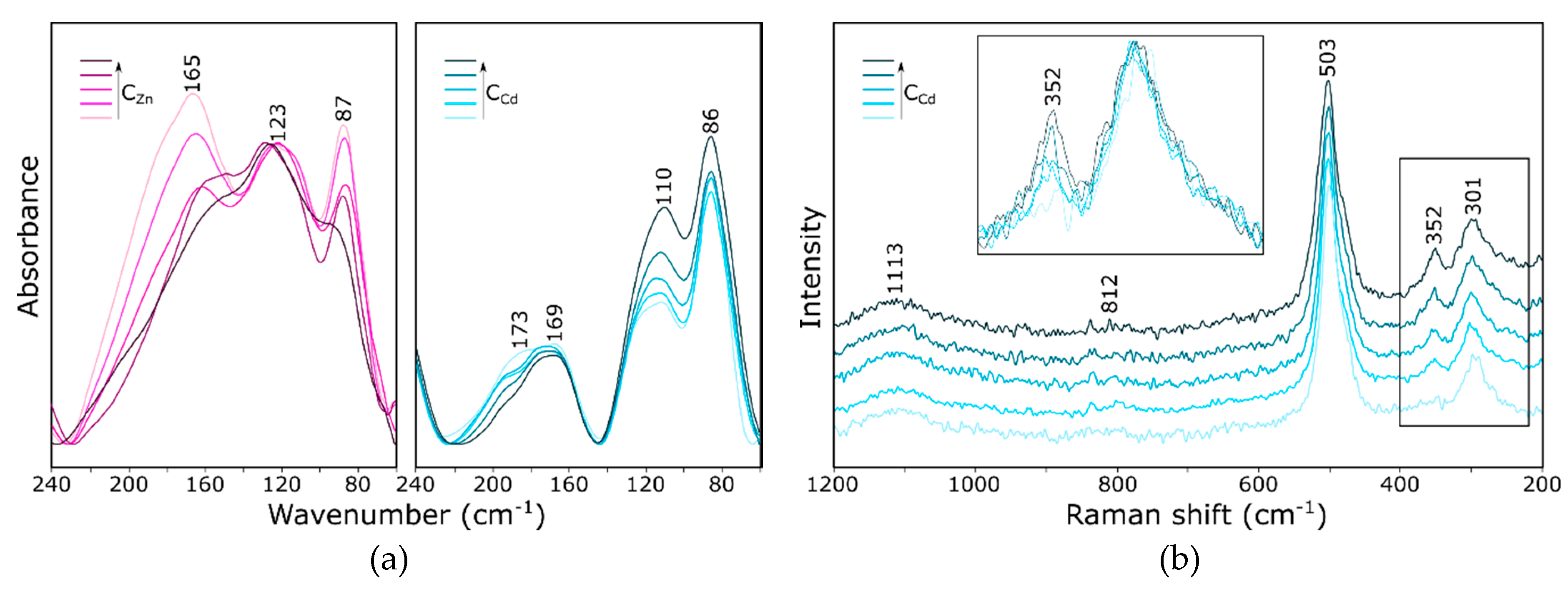
2.3. Influence of the Cation Guest on the Vibrational Spectra of Zeolite Y
3. Materials, Models and Methods
3.1. Computational Details
3.2. Experimental
4. Conclusions
- Theoretical spectra are a useful tool for the interpretation of experimental ones, allowing the assignment of bands to individual normal modes of vibrations.
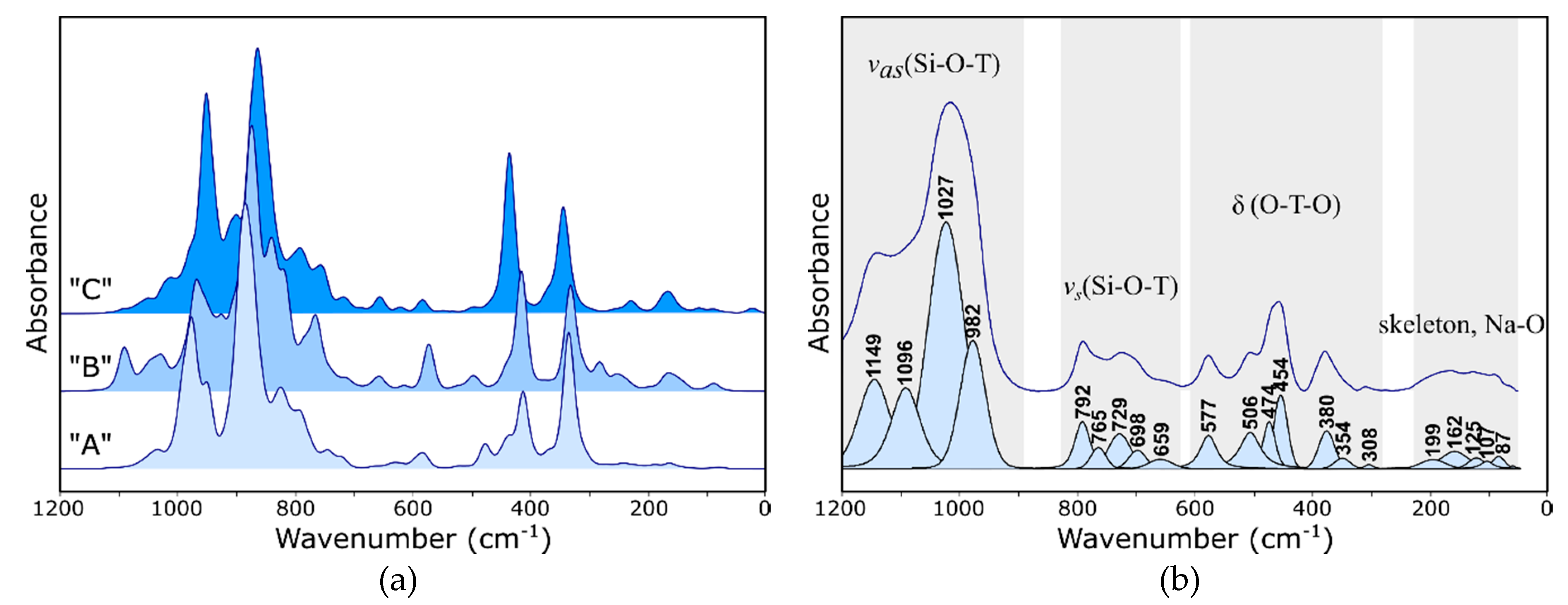
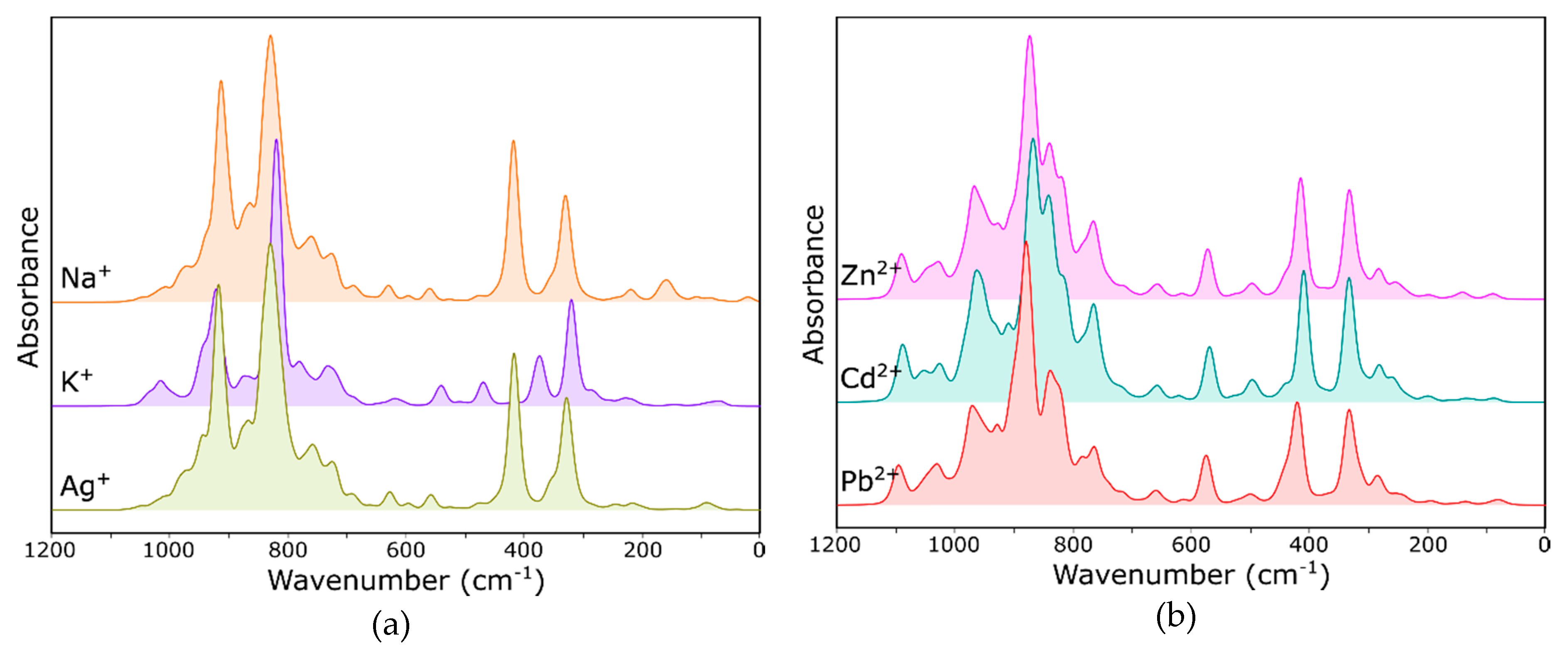
- Bands changing both the intensity and position, depending on the type of extra-framework cation in the structure, were identified both in the IR and Raman spectra of faujasite. Based on the literature data, these bands were assigned to fully symmetrical vibrations of six-membered rings—the so-called ring opening (RO) vibrations.
- In far IR, one can observe significant differences between the theoretical spectra, which results from different characters of respective interactions between extra-framework cations and the host aluminosilicate framework.
- MIR range spectra for the Y zeolite measured after the sorption process shows visible changes in the pseudo-lattice vibrations caused by the ion exchange process.
- In the IR spectrum, the band located at approx. 578 cm−1 can be assigned to bending the vibrations of Si–O–Al bridges; the position and intensity of this band changes significantly with a type of nontetrahedral cation and its position relative to the six-membered ring.
- The Raman spectrum, as more sensitive to changes in the symmetry of the system, is a good indicator of the sorption process. The bands at 503 and 352 cm−1 undergo changes in position and integral intensity, respectively.
- Similar observations can be made also in the case of spectra obtained for all analyzed “A”, “B” and “C” model structures. Since the spectra calculated using all three models are very similar to each other and to the experimental ones, one can safely assume that, in real structures, nontetrahedral cations will be distributed statistically (but with different probabilities) among all three positions (II, I′ and I).
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Gläser, R.; Weitkamp, J. The application of zeolites in catalysis. In Basic Principles in Applied Catalysis; Baerns, M., Ed.; Springer Series in Chemical Physics; Springer: Berlin/Heidelberg, Germany, 2004; Volume 75, pp. 159–212. [Google Scholar]
- Yang, S.; Navrotsky, A. Energetics of formation and hydration of ion-exchanged zeolite Y. Microporous Mesoporous Mater. 2000, 37, 175–186. [Google Scholar] [CrossRef]
- Flanigen, E.M.; Khatami, H.; Szymanski, H.A. Infrared structural studies of zeolite frameworks. Adv. Chem. Ser. 1971, 101, 201–229. [Google Scholar]
- Król, M.; Koleżyński, A.; Mikuła, A.; Mozgawa, W. Vibrational spectroscopy of zeolites. In Molecular Spectroscopy—Experiment and Theory. From Molecules to Functional Materials; Koleżyński, A., Król, M., Eds.; Challenges and Advances in Computational Chemistry and Physics; Springer Nature Switzerland AG: Cham, Switzerland, 2019; Volume 26, pp. 301–332. [Google Scholar]
- Gaigeot, M.P.; Martinez, M.; Vuilleumier, R. Infrared spectroscopy in the gas and liquid phase from first principle molecular dynamics simulations: Application to small peptides. Mol. Phys. 2007, 105, 2857–2878. [Google Scholar] [CrossRef]
- Chiariello, M.G.; Donati, G.; Rega, N. Time-Resolved vibrational analysis of excited state ab initio molecular dynamics to understand photorelaxation: The case of the pyranine photoacid in aqueous solution. J. Chem. Theory Comput. 2020, 16, 6007–6013. [Google Scholar] [CrossRef] [PubMed]
- Chiariello, M.G.; Raucci, U.; Coppola, F.; Rega, N. Unveiling anharmonic coupling by means of excited state ab initio dynamics: Application, to diarylethene photoreactivity. Phys. Chem. Chem. Phys. 2019, 3606–3614. [Google Scholar] [CrossRef]
- Rega, N. Vibrational analysis beyond the harmonic regime from ab-initio molecular dynamics. Theor. Chem. Acc. 2006, 116, 347–354. [Google Scholar] [CrossRef]
- Strachan, A. Normal modes and frequencies from covariances in molecular dynamics or Monte Carlo simulations. J. Chem. Phys. 2004, 120, 1–4. [Google Scholar] [CrossRef]
- Król, M.; Mozgawa, W.; Jastrzębski, W.; Barczyk, K. Application of IR spectra in the studies of zeolites from D4R and D6R structural groups. Microporous Mesoporous Mater. 2012, 156, 181–188. [Google Scholar] [CrossRef]
- Vitale, G.; Mellot, C.F.; Bull, L.M.; Cheetham, A.K. Neutron diffraction and computational study of zeolite NaX: Influence of SIII′ cations on its complex with benzene. J. Phys. Chem. B 1997, 101, 4559–4564. [Google Scholar] [CrossRef]
- Mortier, W.J. Compilation of Extra Framework Sites in Zeolites; Butterworth & Co: Leuven, Belgium, 1992. [Google Scholar]
- Frising, T.; Leflaive, P. Extraframework cation distributions in X and Y faujasite zeolites: A review. Microporous Mesoporous Mater. 2008, 114, 27–63. [Google Scholar] [CrossRef]
- Di Lella, A.; Desbiens, N.; Boutin, A.; Demachy, I.; Ungerer, P.; Bellatc, J.P.; Fuchs, A.H. Molecular simulation studies of water physisorption in zeolites. Phys. Chem. Chem. Phys. 2006, 8, 5396–5406. [Google Scholar] [CrossRef] [PubMed]
- Jeffroy, M.; Draghi, C.N.; Boutin, A. A new molecular simulation method to determine both aluminum and cation location in cationic zeolites. Chem. Mater. 2017, 29, 513–523. [Google Scholar] [CrossRef]
- Maurin, G.; Llewellyn, P.; Poyet, T.; Kuchta, B. Influence of extra-framework cations on the adsorption properties of X-faujasite systems: Microcalorimetry and molecular simulations. J. Phys. Chem. B 2005, 109, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Thang, H.V.; Grajciar, L.; Nachtigall, P.; Bludsky, O.; Arean, C.O.; Frydova, E.; Bulanek, R. Adsorption of CO2 in FAU zeolites: Effect of zeolite composition. Catal. Today 2014, 227, 50–56. [Google Scholar] [CrossRef]
- Abrioux, C.; Coasne, B.; Maurin, G.; Henn, F.; Jeffroy, M.; Boutin, A. Cation behavior in haujasite zeolites upon water adsorption: A combination of Monte Carlo and molecular dynamics simulations. J. Phys. Chem. C 2009, 113, 10696–10705. [Google Scholar] [CrossRef]
- Higgins, F.M.; de Leeuw, N.H.; Parker, S.C. Modelling the effect of water on cation exchange in zeolite A. J. Mater. Chem. 2002, 12, 124–131. [Google Scholar]
- Gale, J.D. GULP: A computer program for the symmetry-adapted simulation of solids. JCS Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The general utility lattice program (GULP). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Baerlocher, C.; McCusker, L.B. Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases/ (accessed on 7 December 2020).
- Bornhauser, P.; Calzaferri, G. Ring-Opening vibrations of spherosiloxanes. J. Phys. Chem. 1996, 100, 2035–2044. [Google Scholar] [CrossRef]
- Mozgawa, W.; Jastrzȩbski, W.; Handke, M. Vibrational spectra of D4R and D6R structural units. J. Mol. Struct. 2005, 744–747, 663–670. [Google Scholar] [CrossRef]
- Mikosch, H.; Uzunova, E.L.; Nikolov, G.S. Vibrational modes of double six-member rings of oxygen-bridged silicon and aluminum atoms in zeolites: A DFT study. J. Phys. Chem. B 2004, 108, 13200–13207. [Google Scholar] [CrossRef]
- Blackwell, C.S. Investigation of zeolite frameworks by vIbrational properties. 2. The double-six-ring in group 4 zeolites. J. Phys. Chem. 1979, 83, 3257–3261. [Google Scholar] [CrossRef]
- Baker, M.D.; Godber, J.; Ozin, G.A. Frequency and intensity considerations in the Far-IR spectroscopy of faujasite zeolites: Experiment and theory. Metal cation vibrational assignments, site locations, and populations. J. Am. Chem. Soc. 1985, 107, 3033–3043. [Google Scholar] [CrossRef]
- Kaduk, J. Extraframework species in zeolite Y at nonambient conditions. Crystallogr. Rev. 2005, 11, 1–19. [Google Scholar] [CrossRef]
- Mikuła, A.; Król, M.; Koleżyński, A. Periodic model of an LTA framework. J. Mol. Model. 2015, 21, 275. [Google Scholar] [CrossRef] [PubMed]
- Król, M.; Mozgawa, W.; Barczyk, K.; Bajda, T.; Kozanecki, M. Changes in the vibrational spectra of zeolites due to sorption of heavy metal cations. J. Appl. Spectrosc. 2013, 80, 662–668. [Google Scholar]
- Ferwerda, R.; Maas, J.H. The influence of adsorbed molecules on the framework vibrations of Na-Faujasites studied with FT Raman spectroscopy. Spectrochim. Acta A 1995, 51, 2147–2159. [Google Scholar] [CrossRef]
- Krause, K.; Geidel, E.; Kindler, J.; Fijrster, H.; Smimov, K.S. A comparative computer modelling study of spectral characteristics of cation exchanged zeolite Y. Vib. Spectrosc. 1996, 12, 45–52. [Google Scholar] [CrossRef]
- Bougeard, D.; Brémard, C.; Dumont, D.; Maire, M. Spectroscopic investigations and molecular dynamics studies of cationic exchanged EMT zeolites. J. Phys. Chem. B 1998, 102, 10805–10816. [Google Scholar] [CrossRef]
- Koleżyński, A.; Mikuła, A.; Król, M. Periodic model of an LTA framework containing various non-tetrahedral cations. Spectrochim. Acta A 2016, 157, 17–25. [Google Scholar] [CrossRef]
- Mikuła, A.; Król, M.; Koleżyński, A. Experimental and theoretical spectroscopic studies of Ag-, Cd- and Pb-sodalite. J. Mol. Struct. 2016, 1126, 110–116. [Google Scholar] [CrossRef]
- Dick, B.G.; Overhauser, A.W. Theory of the dielectric constants of alkali halide crystals. Phys. Rev. 1958, 112, 90–103. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Structure | Total Energy (eV) |
|---|---|
| 0.5 × I′ + 0.5 × II | −5868.37 |
| 0.666 × I + 0.666 × II | −5869.32 |
| 0.666 × I′ + 0.333 × II | −5869.84 |
| 0.333 × I′ + 0.666 × II | −5870.07 |
| I + 0.5 × II | −5873.83 |
| Total Energy (eV) | |||
|---|---|---|---|
| Cation | Model “A” | Model “B” | Model “C” |
| Na+ | −5920.48 | −5921.07 | −5937.11 |
| K+ | −5897.30 | −5906.96 | −5916.51 |
| Ag+ | −5921.83 | −5922.54 | −5940.92 |
| Ni2+ | −5926.12 | −5926.55 | −5915.52 |
| Cu2+ | −5927.76 | −5928.13 | −5918.72 |
| Zn2+ | −5919.86 | −5920.30 | −5910.80 |
| Cd2+ | −5915.26 | −5916.41 | −5908.17 |
| Pb2+ | −5928.04 | −5928.41 | −5918.63 |
| Interaction | A (eV) | ρ (Å) | C (eVÅ6) | rmin (Å) | rmax (Å) |
|---|---|---|---|---|---|
| OS-OS | 22,764.3 | 0.149 | 27.879 | 0 | 12 |
| SiC-OS | 1283.907 | 0.32052 | 10.66158 | 0 | 10 |
| AlC-OS | 1460.3 | 0.29912 | 10 | 0 | 10 |
| NaC-OS | 280.1 | 0.35771 | 10 | 0 | 10 |
| KC-OS | 1000.3 | 0.36198 | 10.569 | 0 | 10 |
| AgC-OS | 1417.03 | 0.2851 | 10 | 0 | 10 |
| AgC-AgC | 1335.89 | 0.1424 | 10 | 0 | 10 |
| NiS-OS | 1582.5 | 0.2882 | 10 | 0 | 10 |
| CuS-OS | 5950.2 | 0.2427 | 10 | 0 | 12 |
| ZnC-OS | 539.7 | 0.3581 | 10 | 0 | 10 |
| CdC-OS | 868.3 | 0.35 | 10 | 0 | 10 |
| PbC-OS | 2950.4 | 0.261 | 10 | 0 | 10 |
| Interaction | k3b (eV rad−2) | θ0 (deg) | Cutoffs (Å) | ||
|---|---|---|---|---|---|
| OS-SiC-OS | 2.09724 | 109.47 | 1.9 | 1.9 | 3.5 |
| OS-AlC-OS | 2.09724 | 109.47 | 1.9 | 1.9 | 3.5 |
| Species | Core Charge | Shell Charge | kCS (eVÅ−2) | kCS4 (eVÅ−4) |
|---|---|---|---|---|
| Al | 3 | - | - | - |
| Si | 4 | - | - | - |
| O | 0.86902 | −2.86902 | 74.92 | 0 |
| Na | 1 | - | - | - |
| K | 1 | - | - | - |
| Ag | 1 | - | - | - |
| Cu | 1 | 1 | 100 | 0 |
| Ni | −1.344 | 3.344 | 50 | 0 |
| Zn | 2 | - | - | - |
| Cd | 2 | - | - | - |
| Pb | 2 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Król, M.; Koleżyński, A.; Mozgawa, W. Vibrational Spectra of Zeolite Y as a Function of Ion Exchange. Molecules 2021, 26, 342. https://doi.org/10.3390/molecules26020342
Król M, Koleżyński A, Mozgawa W. Vibrational Spectra of Zeolite Y as a Function of Ion Exchange. Molecules. 2021; 26(2):342. https://doi.org/10.3390/molecules26020342
Chicago/Turabian StyleKról, Magdalena, Andrzej Koleżyński, and Włodzimierz Mozgawa. 2021. "Vibrational Spectra of Zeolite Y as a Function of Ion Exchange" Molecules 26, no. 2: 342. https://doi.org/10.3390/molecules26020342
APA StyleKról, M., Koleżyński, A., & Mozgawa, W. (2021). Vibrational Spectra of Zeolite Y as a Function of Ion Exchange. Molecules, 26(2), 342. https://doi.org/10.3390/molecules26020342