
Synthesis of the Hexahydropyrrolo-[3,2-c]-quinoline Core Structure and Strategies for Further Elaboration to Martinelline, Martinellic Acid, Incargranine B, and Seneciobipyrrolidine


Abstract
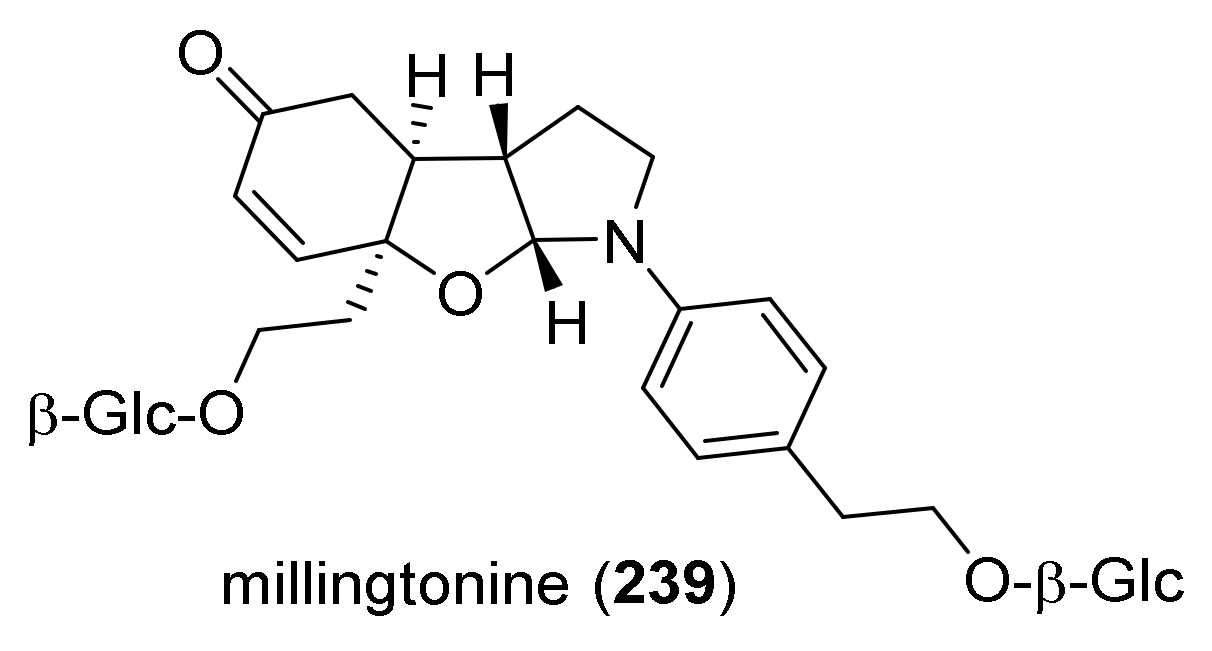
1. Introduction
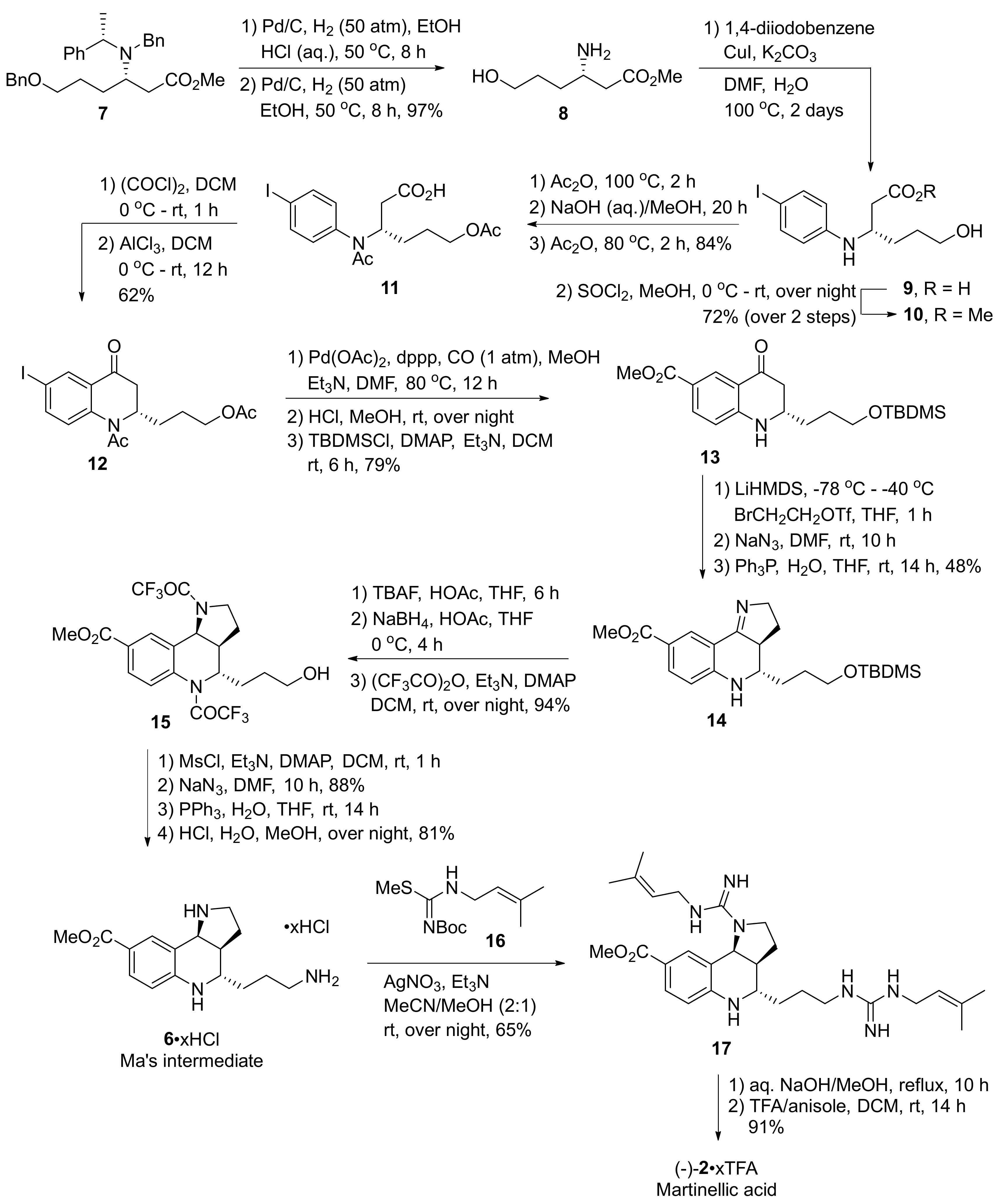
2. Total and Formal Synthesis of Martinellic Acid (2)
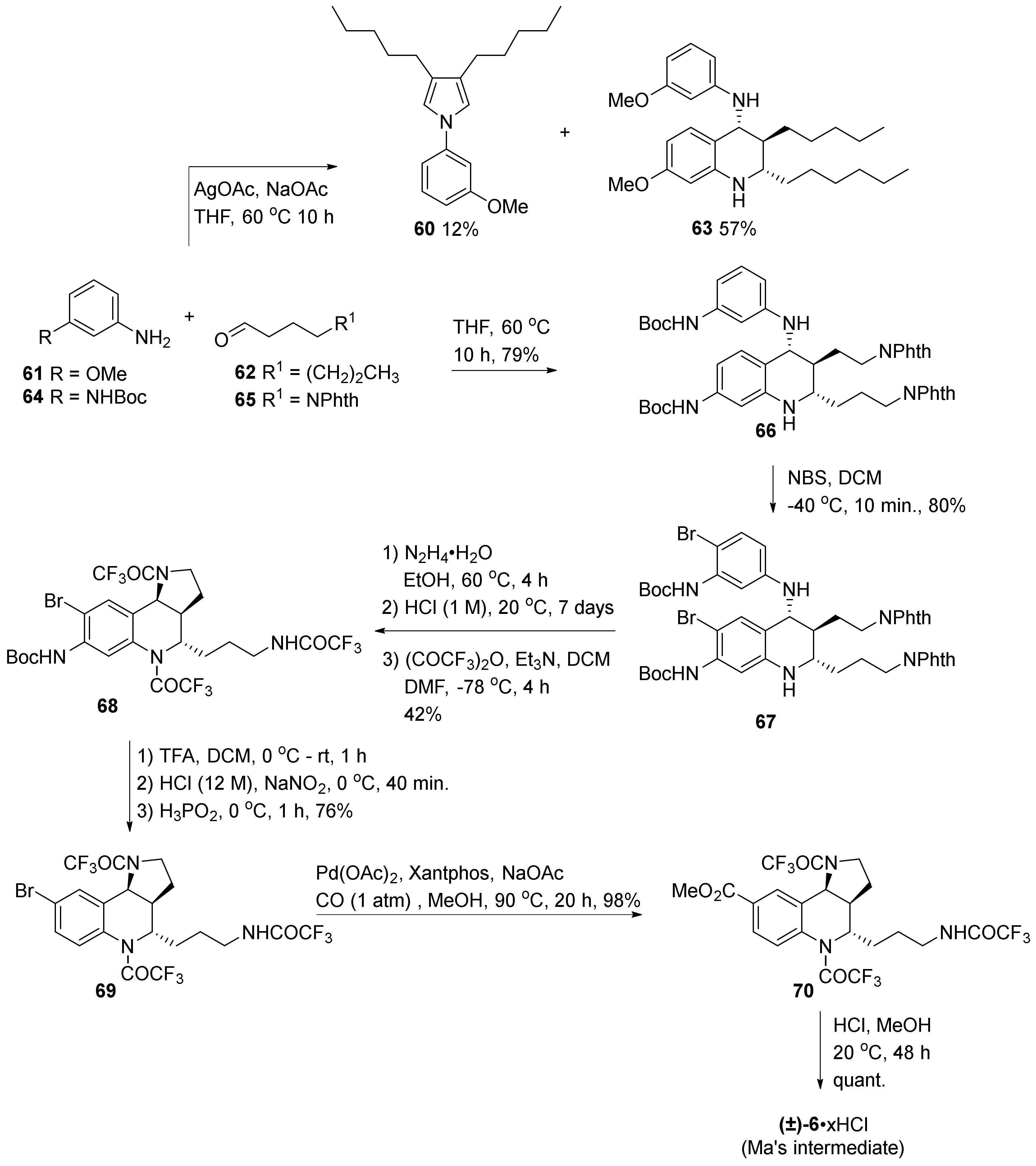
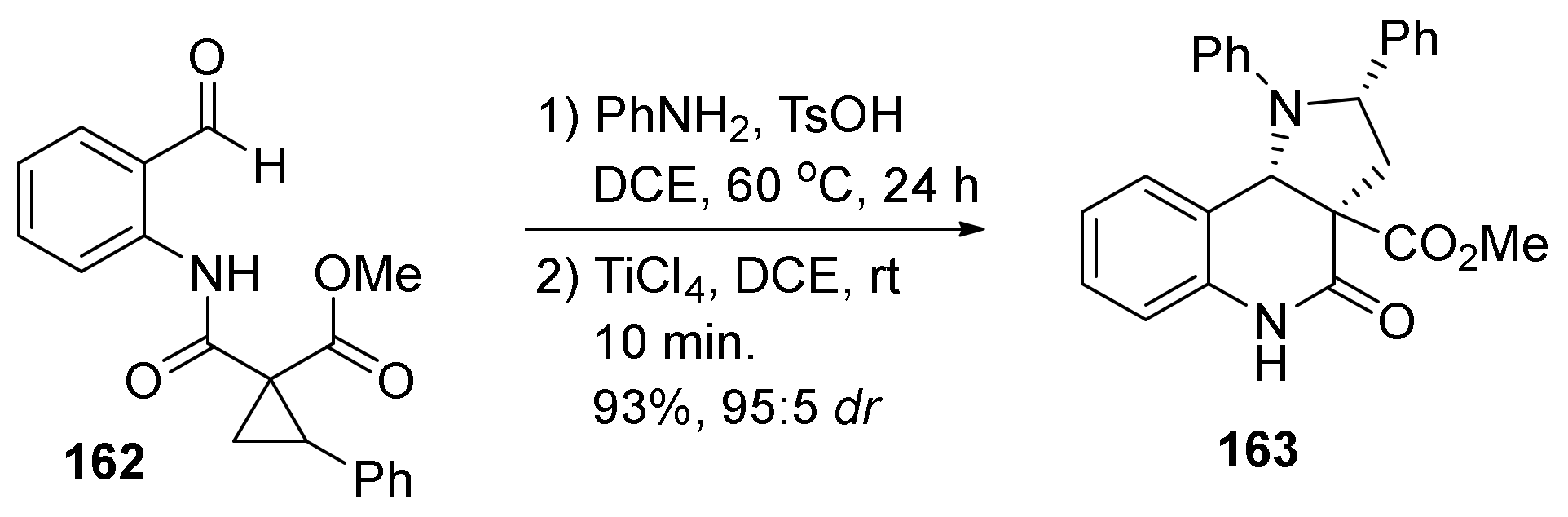
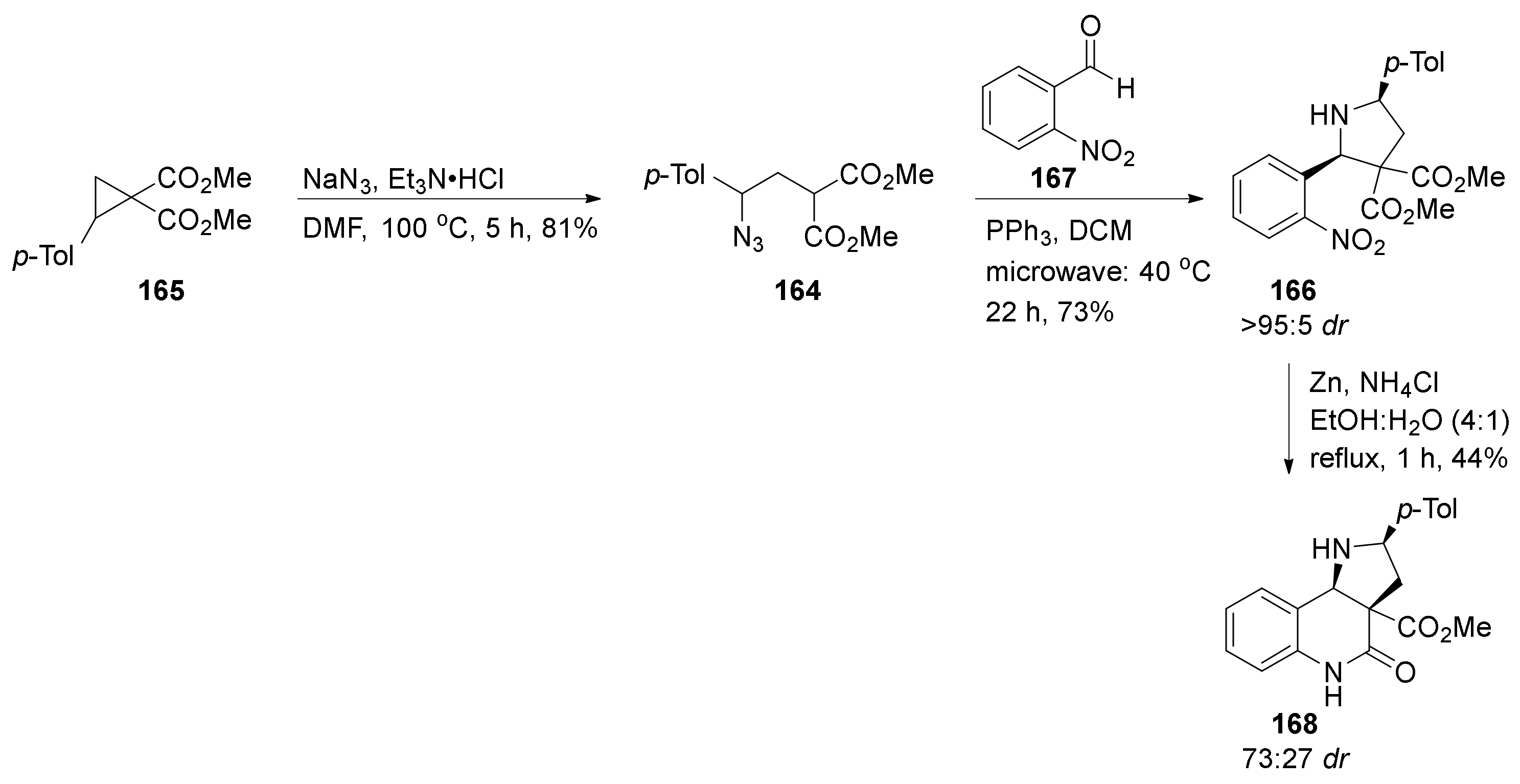
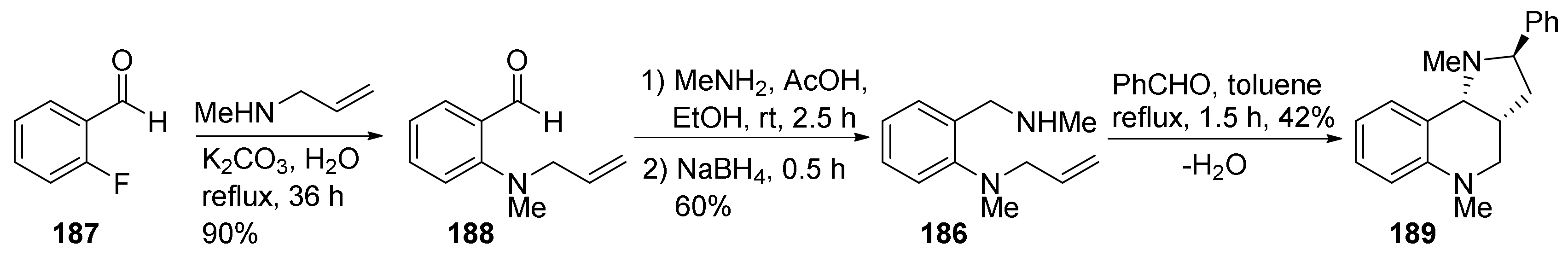
3. Tricyclic Core Scaffold Synthesis
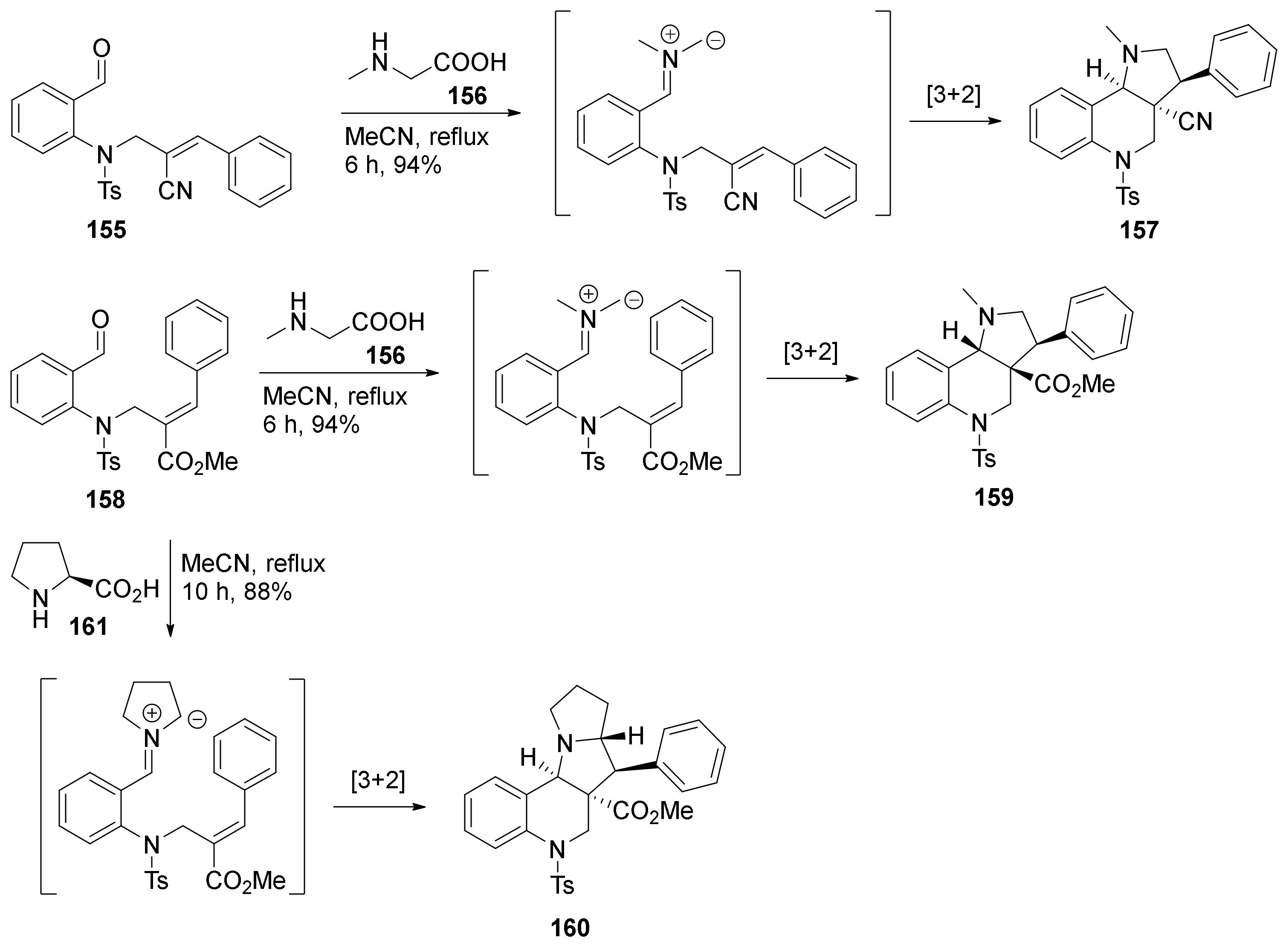
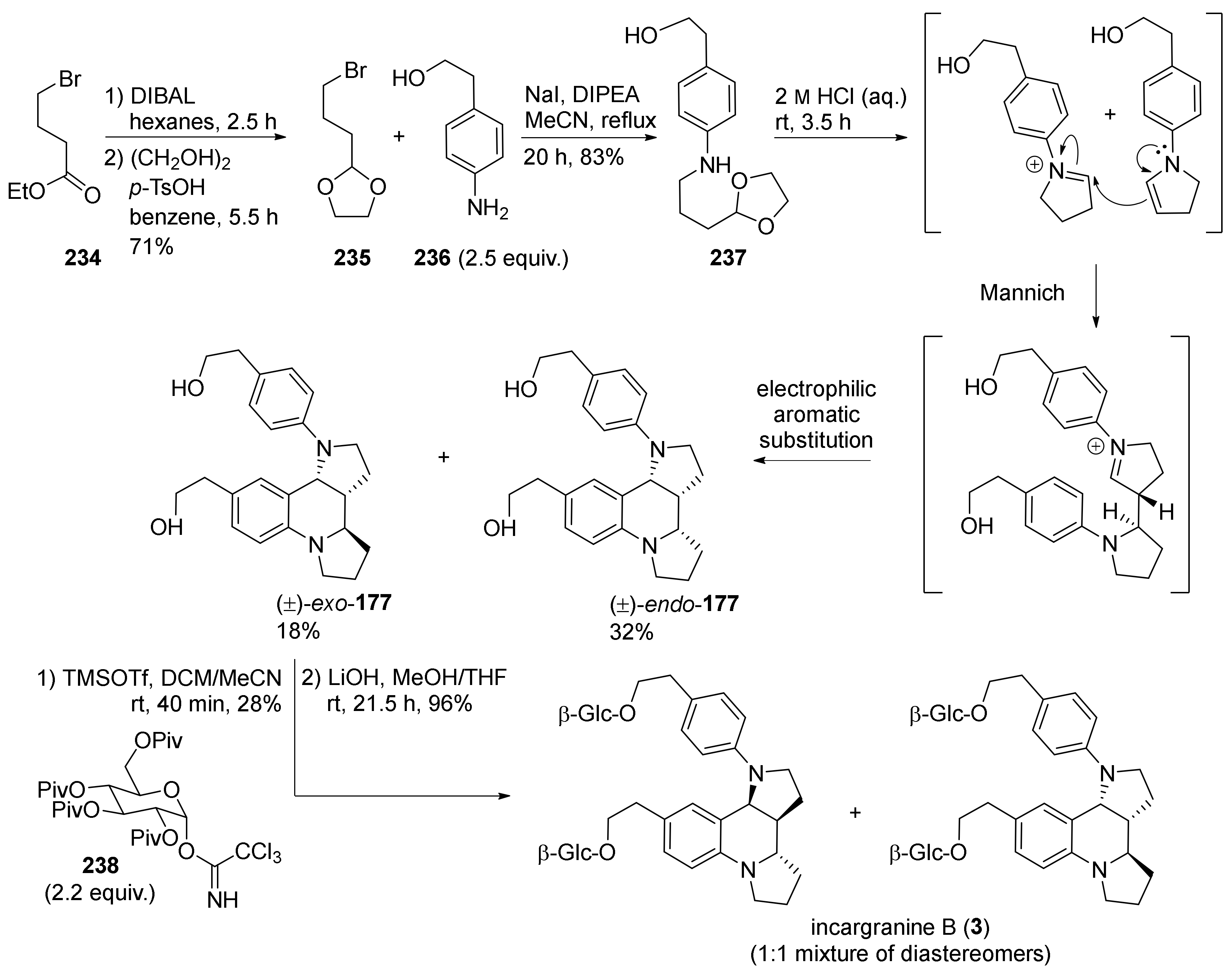
4. Synthesis of Dipyrroloquinolines Towards Natural Products Seneciobipyrrolidine (3) and Incargranine B (4)
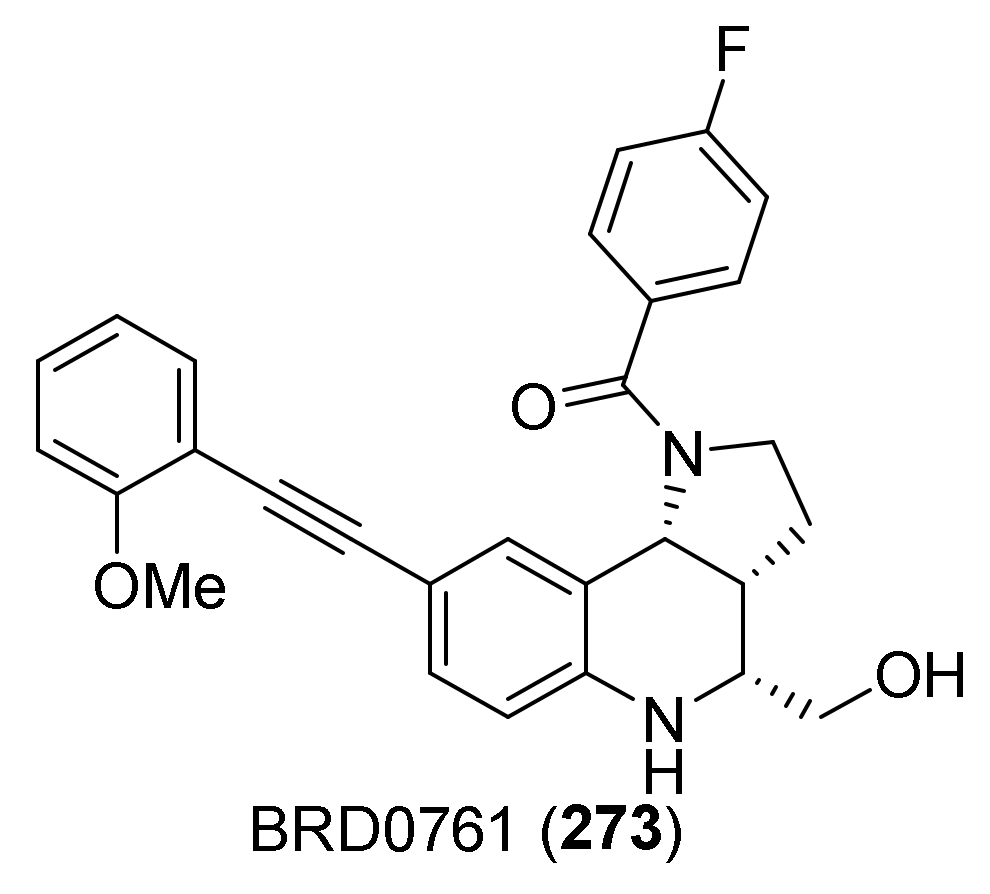
5. Elaborating Biological Properties
6. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, L.-D.; Chen, Y.; Xu, J. Total Synthesis of Daphniphyllum Alkaloids: From Bicycles to Diversified Caged Structures. Acc. Chem. Res. 2020, 53, 7390–7394. [Google Scholar] [CrossRef]
- Baudoin, O. Multiple Catalytic C–H Bond Functionalization for Natural Product Synthesis. Angew. Chem. Int. Ed. 2020, 59, 17798–17809. [Google Scholar] [CrossRef]
- Frontier, A.J.; Hernandez, J.J. New Twists in Nazarov Cyclization Chemistry. Acc. Chem. Res. 2020, 53, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Que, Y.; He, H. Advances in N-Heterocyclic Carbene Catalysis for Natural Product Synthesis. Eur. J. Org. Chem. 2020, 2020, 5917–5925. [Google Scholar] [CrossRef]
- Mandal, S.; Thirupathi, B. Strategies for the construction of γ-spirocyclic butenolides in natural product synthesis. Org. Biomol. Chem. 2020, 18, 5287–5314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, Y.-J.; Liang, X.-W. Total synthesis of natural products using photocycloaddition reactions of arenes. Org. Biomol. Chem. 2020, 18, 5558–5566. [Google Scholar] [CrossRef] [PubMed]
- Kalita, S.J.; Cheng, F.; Huang, Y.Y. Recent advances of applying boron-reagents in asymmetric total syntheses of natural products and bio-active molecules. Adv. Synth. Catal. 2020, 362, 2778–2800. [Google Scholar] [CrossRef]
- Fernandes, R.A.; Kumar, P.; Choudhary, P. Advances in catalytic and protecting-group-free total synthesis of natural products: A recent update. Chem. Commun. 2020, 56, 8569–8590. [Google Scholar] [CrossRef]
- Hu, Y.-J.; Li, L.-X.; Han, J.-C.; Min, L.; Li, C.-C. Recent Advances in the Total Synthesis of Natural Products Containing Eight-Membered Carbocycles (2009–2019). Chem. Rev. 2020, 120, 5910–5953. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J.; Snader, K.M. Natural Products in Drug Discovery and Development. J. Nat. Prod. 1997, 60, 52–60. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. The influence of natural products upon drug discovery. Nat. Prod. Rep. 2000, 17, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural product scaffolds as leads to drugs. Future Med. Chem. 2009, 1, 1415–1427. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Last 25 Years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Monteleone, S.; Fuchs, J.E.; Liedl, K.R. Molecular connectivity predefines polypharmacology: Aliphatic rings, chirality, and sp3 centers enhance target selectivity. Front. Pharmacol. 2017, 8, 552. [Google Scholar] [CrossRef]
- Witherup, K.M.; Ransom, R.W.; Graham, A.C.; Bernard, A.M.; Salvatore, M.J.; Lumma, W.C.; Anderson, P.S.; Pitzenberger, S.M.; Varga, S.L. Martinelline and martinellic acid, novel G-protein linked receptor antagonists from the tropical plant Martinella iquitosensis (Bignoniaceae). J. Am. Chem. Soc. 1995, 117, 6682–6685. [Google Scholar] [CrossRef]
- Elvis-Offiah, U.B.; Bafor, E.E.; Eze, G.I.; Igbinumwen, O.; Viegelmann, C.; Edrada-Ebel, R. In vivo investigation of female reproductive functions and parameters in nonpregnant mice models and mass spectrometric analysis of the methanol leaf extract of Emilia Coccinea (Sims) G Dons. Physiol. Rep. 2016, 4, e13047. [Google Scholar] [CrossRef][Green Version]
- Zulfiker, A.H.M.; Sohrabi, M.; Qi, J.; Matthews, B.; Wei, M.Q.; Grice, I.D. Multi-constituent identification in Australian cane toad skin extracts using high-performance liquid chromatography high-resolution tandem mass spectrometry. J. Pharm. Biomed. Anal. 2016, 129, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Abhimannue, A.P.; Mohan, M.C.; Kumar, B.P. Inhibition of Tumor Necrosis Factor-α and Interleukin-1β Production in Lipopolysaccharide-Stimulated Monocytes by Methanolic Extract of Elephantopus scaber Linn and Identification of Bioactive Components. Appl. Biochem. Biotechnol. 2016, 179, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Gentry, A.H.; Cook, K. Martinella (Bignoniaceae): A widely used eye medicine of South America. J. Ethnopharmacol. 1984, 11, 337–343. [Google Scholar] [CrossRef]
- Ogunlesi, M.; Okiei, W.; Ademoye, M. Medicinal Plants Used in Treating Eye Infections in Nigeria. In A Textbook of Medicinal Plants from Nigeria; Odugbemi, T., Ed.; University of Lago Press: Lago, Nigeria, 2008; p. 305. [Google Scholar]
- Shen, Y.H.; Su, Y.Q.; Tian, J.M.; Lin, S.; Li, H.L.; Tang, J.; Zhang, W.D. A Unique Indolo-[1,7]naphthyridine Alkaloid from Incarvillea mairei var. grandiflora (Wehrh.) Grierson. Helv. Chim. Acta 2010, 93, 2393–2396. [Google Scholar] [CrossRef]
- Brown, P.D.; Willis, A.C.; Sherburn, M.S.; Lawrence, A.L. Total synthesis and structural revision of the alkaloid incargranine B. Angew. Chem. Int. Ed. 2013, 52, 13273–13275. [Google Scholar] [CrossRef]
- Tan, D.; Chou, G.; Wang, Z. Three New Alkaloids from Senecio scandens. Chem. Nat. Compd. 2014, 50, 329–332. [Google Scholar] [CrossRef]
- Wang, D.; Huang, L.; Chen, S. Senecio scandens Buch.-Ham.: A review on its ethnopharmacology, phytochemistry, pharmacology, and toxicity. J. Ethnopharmacol. 2013, 149, 1–23. [Google Scholar] [CrossRef]
- Ma, D.; Xia, C.; Jiang, J.; Zhang, J.; Tang, W. Aromatic nucleophilic substitution or CuI-catalyzed coupling route to martinellic acid. J. Org. Chem. 2003, 68, 442–451. [Google Scholar] [CrossRef]
- Ikeda, S.; Shibuya, M.; Iwabuchi, Y. Asymmetric total synthesis of martinelline and martinellic acid. Chem. Commun. 2007, 504–506. [Google Scholar] [CrossRef]
- Shirai, A.; Miyata, O.; Tohnai, N.; Miyata, M.; Procter, D.J.; Sucunza, D.; Naito, T. Total Synthesis of (−)-Martinellic Acid via Radical Addition–Cyclization–Elimination Reaction. J. Org. Chem. 2008, 73, 4464–4475. [Google Scholar] [CrossRef]
- Davies, S.G.; Fletcher, A.M.; Lee, J.A.; Lorkin, T.J.; Roberts, P.M.; Thomson, J.E. A diastereodivergent strategy for the asymmetric syntheses of (−)-martinellic acid and (−)-4-epi-martinellic acid. Tetrahedron 2013, 69, 9779–9803. [Google Scholar] [CrossRef]
- Nyerges, M. Construction of pyrrolo [3,2-c] quinolines: Recent advances in the synthesis of the martinelline alkaloids. Heterocycles 2004, 63, 1685–1712. [Google Scholar] [CrossRef]
- Lovely, C.J.; Bararinarayana, V. Synthetic studies toward the Martinella alkaloids. Curr. Org. Chem. 2008, 12, 1431–1453. [Google Scholar] [CrossRef]
- Eichholzer, J.V.; Lewis, I.A.; Macleod, J.K.; Oelrichs, P.B.; Vallely, P.J. Galegine and a new dihydroxyalkylacetamide from Verbesina enceloiodes. Phytochemistry 1982, 21, 97–99. [Google Scholar] [CrossRef]
- Reuter, G. Arginin als Vorstufe von Galegin in Galega officinalis L. Zur Biochemie und Physiologie von Galegin in Galega officinalis L., III. Mitt. Arch. Pharm. 1963, 296, 516–522. [Google Scholar] [CrossRef]
- Ma, D.; Xia, C.; Jiang, J.; Zhang, J. First total synthesis of martinellic acid, a naturally occurring bradykinin receptor antagonist. Org. Lett. 2001, 3, 2189–2191. [Google Scholar] [CrossRef]
- Powell, D.A.; Batey, R.A. Total synthesis of the alkaloids martinelline and martinellic acid via a hetero Diels–Alder multicomponent coupling reaction. Org. Lett. 2002, 4, 2913–2916. [Google Scholar] [CrossRef]
- Xia, C.; Heng, L.; Ma, D. Total synthesis of (±)-martinelline. Tetrahedron Lett. 2002, 43, 9405–9409. [Google Scholar] [CrossRef]
- Davies, S.G.; Fletcher, A.M.; Lee, J.A.; Lorkin, T.J.; Roberts, P.M.; Thomson, J.E. Asymmetric synthesis of (−)-martinellic acid. Org. Lett. 2013, 15, 2050–2053. [Google Scholar] [CrossRef]
- Rong, Z.; Li, Q.; Lin, W.; Jia, Y. Reagent-free synthesis of 2,3,4-polysubstituted tetrahydroquinolines: Application to the formal synthesis of (±)-martinellic acid and martinelline. Tetrahedron Lett. 2013, 54, 4432–4434. [Google Scholar] [CrossRef]
- Yoshitomi, Y.; Arai, H.; Makino, K.; Hamada, Y. Enantioselective synthesis of martinelline chiral core and its diastereomer using asymmetric tandem Michael–aldol reaction. Tetrahedron 2008, 64, 11568–11579. [Google Scholar] [CrossRef]
- He, Y.; Mahmud, H.; Moningka, R.; Lovely, C.J.; Dias, H.R. Cyclization reactions of N-acryloyl-2-aminobenzaldehyde derivatives: Formal total synthesis of martinellic acid. Tetrahedron 2006, 62, 8755–8769. [Google Scholar] [CrossRef]
- Hadden, M.; Nieuwenhuyzen, M.; Osborne, D.; Stevenson, P.J.; Thompson, N.; Walker, A.D. Synthesis of the heterocyclic core of martinelline and martinellic acid. Tetrahedron 2006, 62, 3977–3984. [Google Scholar] [CrossRef]
- Snider, B.B.; Ahn, Y.; O’Hare, S.M. Total synthesis of (±)-martinellic acid. Org. Lett. 2001, 3, 4217–4220. [Google Scholar] [CrossRef] [PubMed]
- Hadden, M.; Nieuwenhuyzen, M.; Osborne, D.; Stevenson, P.J.; Thompson, N. Synthesis of the heterocyclic core of the alkaloids martinelline and martinellic acid. Tetrahedron Lett. 2001, 42, 6417–6419. [Google Scholar] [CrossRef]
- Batey, R.A.; Powell, D.A. Multi-component coupling reactions: Synthesis of a guanidine containing analog of the hexahydropyrrolo [3,2-c] quinoline alkaloid martinelline. Chem. Commun. 2001, 22, 2362–2363. [Google Scholar] [CrossRef]
- Davies, S.G.; Ichihara, O.; Walters, I. Asymmetric-synthesis of syn-a-alkyl-b-amino acids. J. Chem. Soc. Perkin Trans. 1994, 9, 1141–1147. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Xie, H.; Zu, L.; Jiang, W.; Duesler, E.N.; Wang, W. Chiral Diphenylprolinol TES Ether Promoted Conjugate Addition–Aldol-Dehydration Reactions between α, β-Unsaturated Aldehydes and 2-N-Protected Amino Benzaldehydes. Org. Lett. 2007, 9, 965–968. [Google Scholar] [CrossRef]
- Hara, O.; Sugimoto, K.; Makino, K.; Hamada, Y. New synthesis of a pyrroloquinoline skeleton, the martinelline core, using a tandem Michael-aldol strategy. Synlett 2004, 2004, 1625–1627. [Google Scholar]
- Pappoppula, M.; Aponick, A. Enantioselective Total Synthesis of (−)-Martinellic Acid. Angew. Chem. Int. Ed. 2015, 54, 15827–15830. [Google Scholar] [CrossRef]
- Lackey, K.E. Compounds and Methods of Treatment. U.S. Patent 20080234267A1, 25 September 2008. [Google Scholar]
- Behenna, D.C.; Stoltz, B.M. The enantioselective Tsuji allylation. J. Am. Chem. Soc. 2004, 126, 15044–15045. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Kawai, S.; Hayashi, M.; Naito, T.; Miyata, O. Efficient entry into 2-substituted tetrahydroquinoline systems through alkylative ring expansion: Stereoselective formal synthesis of (±)-martinellic acid. J. Org. Chem. 2010, 75, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Batey, R.; Simoncic, P.; Smyj, R.; Lough, A. A three-component coupling protocol for the synthesis of substituted hexahydropyrrolo [3,2-c] quinolines. Chem. Commun. 1999, 7, 651–652. [Google Scholar] [CrossRef]
- Kouznetsov, V.V. Recent synthetic developments in a powerful imino Diels-Alder reaction (Povarov reaction): Application to the synthesis of N-polyheterocycles and related alkaloids. Tetrahedron 2009, 65, 2721–2750. [Google Scholar] [CrossRef]
- Fochi, M.; Caruana, L.; Bernardi, L. Catalytic asymmetric aza-Diels–Alder reactions: The Povarov cycloaddition reaction. Synthesis 2014, 46, 135–157. [Google Scholar] [CrossRef]
- Hadden, M.; Stevenson, P.J. Regioselective synthesis of pyrroloquinolines—Approaches to Martinelline. Tetrahedron Lett. 1999, 40, 1215–1218. [Google Scholar] [CrossRef]
- He, L.; Liu, H.-B.; Zhao, L.; Wang, D.-X.; Wang, M.-X. Lewis acid-catalyzed reaction between tertiary enamides and imines of salicylaldehydes: Expedient synthesis of novel 4-chromanamine derivatives. Tetrahedron 2015, 71, 523–531. [Google Scholar] [CrossRef]
- Yu, J.; Jiang, H.-J.; Zhou, Y.; Luo, S.-W.; Gong, L.-Z. Sodium Salts of Anionic Chiral Cobalt(III) Complexes as Catalysts of the Enantioselective Povarov Reaction. Angew. Chem. Int. Ed. 2015, 54, 11209–11213. [Google Scholar] [CrossRef]
- Liu, X.; Toy, P.H. Halogen Bond-Catalyzed Povarov Reactions. Adv. Synth. Catal. 2020, 362, 3437–3441. [Google Scholar] [CrossRef]
- Reyes-Gutiérrez, P.E.; Amatov, T.T.; Švec, P.; Císařová, I.; Šaman, D.; Pohl, R.; Teplý, F.; Pospíšil, L. Helquats as Promoters of the Povarov Reaction: Synthesis of 1,2,3,4-Tetrahydroquinoline Scaffolds Catalyzed by Helicene-Viologen Hybrids. ChemPlusChem 2020, 85, 2212–2218. [Google Scholar] [CrossRef]
- Škopić, M.K.; Götte, K.; Gramse, C.; Dieter, M.; Pospich, S.; Raunser, S.; Weberskirch, R.; Brunschweiger, A. Micellar Brønsted Acid Mediated Synthesis of DNA-Tagged Heterocycles. J. Am. Chem. Soc. 2019, 141, 10546–10555. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zuend, S.J.; Woll, M.G.; Tao, Y.; Jacobsen, E.N. Asymmetric Cooperative Catalysis of Strong Brønsted Acid–Promoted Reactions Using Chiral Ureas. Science 2010, 327, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Gerard, B.; O’Shea, M.W.; Donckele, E.; Kesavan, S.; Akella, L.B.; Xu, H.; Jacobsen, E.N.; Marcaurelle, L.A. Application of a catalytic asymmetric Povarov reaction using chiral ureas to the synthesis of a tetrahydroquinoline library. ACS Comb. Sci. 2012, 14, 621–630. [Google Scholar] [CrossRef]
- Kinzel, T.; Zhang, Y.; Buchwald, S.L. A new palladium precatalyst allows for the fast Suzuki–Miyaura coupling reactions of unstable polyfluorophenyl and 2-heteroaryl boronic acids. J. Am. Chem. Soc. 2010, 132, 14073–14075. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.-L.; Ding, C.-H.; Yang, X.-F.; Wan, X.-L.; Hou, X.-L. Kinetic resolution of 2, 3-dihydro-2-substituted 4-quinolones by palladium-catalyzed asymmetric allylic alkylation. J. Am. Chem. Soc. 2009, 131, 18250–18251. [Google Scholar] [CrossRef]
- Gigant, N.; Gillaizeau, I. Construction of nitrogen-fused tetrahydroquinolines via a domino reaction. Org. Lett. 2012, 14, 4622–4625. [Google Scholar] [CrossRef]
- Song, Z.; Zhao, Y.-M.; Zhai, H. One-Step Construction of Tetrahydro-5 H-indolo [3,2-c] quinolines from Benzyl Azides and Indoles via a Cascade Reaction Sequence. Org. Lett. 2011, 13, 6331–6333. [Google Scholar] [CrossRef]
- Desai, P.; Schildknegt, K.; Agrios, K.A.; Mossman, C.; Milligan, G.L.; Aube, J. Reactions of alkyl azides and ketones as mediated by Lewis acids: Schmidt and Mannich reactions using azide precursors. J. Am. Chem. Soc. 2000, 122, 7226–7232. [Google Scholar] [CrossRef]
- Dagousset, G.; Retailleau, P.; Masson, G.; Zhu, J. Chiral phosphoric acid-catalyzed enantioselective three-component Povarov reaction using cyclic enethioureas as dienophiles: Stereocontrolled access to enantioenriched hexahydropyrroloquinolines. Chem. Eur. J. 2012, 18, 5869–5873. [Google Scholar] [CrossRef]
- Roy, S.; Reiser, O. A Catalytic Multicomponent Approach for the Stereoselective Synthesis of cis-4,5-Disubstituted Pyrrolidinones and Tetrahydro-3H-pyrrolo[3,2-c]quinolines. Angew. Chem. Int. Ed. 2012, 51, 4722–4725. [Google Scholar] [CrossRef]
- Wood, J.; Bagi, C.M.; Akuche, C.; Bacchiocchi, A.; Baryza, J.; Blue, M.-L.; Brennan, C.; Campbell, A.-M.; Choi, S.; Cook, J.H.; et al. 4,5-Disubstituted cis-pyrrolidinones as inhibitors of type II 17β-hydroxysteroid dehydrogenase. Part 3. Identification of lead candidate. Bioorg. Med. Chem. Lett. 2006, 16, 4965–4968. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Hirao, H.; Li, Y.; Zhou, J. Palladium-Catalyzed Asymmetric Intermolecular Cyclization. Angew. Chem. Int. Ed. 2013, 52, 8676–8680. [Google Scholar] [CrossRef] [PubMed]
- Comesse, S.; Sanselme, M.; Daïch, A. New and expeditious tandem sequence aza-Michael/intramolecular nucleophilic substitution route to substituted γ-lactams: Synthesis of the tricyclic core of (±)-Martinellines. J. Org. Chem. 2008, 73, 5566–5569. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, R.; Lawson, A.M.; Daïch, A.; Comesse, S. Synthesis of highly functionalized pyrrolidines as tunable templates for the direct access to (±)-coerulescine and the tricyclic core of martinellines. Org. Biomol. Chem. 2013, 11, 1818–1821. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, T.; Bao, W. FeCl3-Catalyzed Cascade Cyclization in One Pot: Synthesis of Ring-Fused Tetrahydroquinoline Derivatives from Arylamines and N-Substituted Lactams. J. Org. Chem. 2013, 78, 8155–8160. [Google Scholar] [CrossRef]
- Yang, M.; Su, B.; Wang, Y.; Chen, K.; Jiang, X.; Zhang, Y.-F.; Zhang, X.-S.; Chen, G.; Cheng, Y.; Cao, Z.; et al. Silver-catalysed direct amination of unactivated C–H bonds of functionalized molecules. Nat. Commun. 2014, 5, 4707. [Google Scholar] [CrossRef]
- Yang, Y.-J.; Zhang, H.-R.; Zhu, S.-Y.; Zhu, P.; Hui, X.-P. Highly stereoselective synthesis of functionalized pyrrolo [3, 2-c] quinolines via N-heterocyclic carbene catalyzed cascade sequence. Org. Lett. 2014, 16, 5048–5051. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Sun, J.; Wang, H.; Jin, Z.; Chi, Y.R. Carbene-Catalyzed Enantioselective Addition of Benzylic Carbon to Unsaturated Acyl Azolium for Rapid Synthesis of Pyrrolo[3,2-c]quinolines. ACS Catal. 2018, 8, 9859–9864. [Google Scholar] [CrossRef]
- Boomhoff, M.; Yadav, A.K.; Appun, J.; Schneider, C. Modular, flexible, and stereoselective synthesis of pyrroloquinolines: Rapid assembly of complex heterocyclic scaffolds. Org. Lett. 2014, 16, 6236–6239. [Google Scholar] [CrossRef]
- Appun, J.; Stolz, F.; Naumov, S.; Abel, B.; Schneider, C. Modular Synthesis of Dipyrroloquinolines: A Combined Synthetic and Mechanistic Study. J. Org. Chem. 2018, 83, 1737–1744. [Google Scholar] [CrossRef]
- Bakthadoss, M.; Kannan, D.; Srinivasan, J.; Vinayagam, V. Highly regio-and diastereo-selective synthesis of novel tri-and tetra-cyclic perhydroquinoline architectures via an intramolecular [3+2] cycloaddition reaction. Org. Biomol. Chem. 2015, 13, 2870–2874. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.-A.; Li, J.; Xia, P.-J.; Zhou, Z.-F.; Deng, Z.-X.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Diastereoselective Intramolecular [3+2]-Annulation of Donor–Acceptor Cyclopropane with Imine-Assembling Hexahydropyrrolo [3,2-c] quinolinone Scaffolds. J. Org. Chem. 2016, 81, 11185–11194. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, A.S.; Ivanova, O.A.; Chagarovskiy, A.O.; Stebunov, N.S.; Orlov, N.V.; Shumsky, A.N.; Budynina, E.M.; Rybakov, V.B.; Trushkov, I.V. Domino Staudinger/aza-Wittig/Mannich Reaction: An Approach to Diversity of Di- and Tetrahydropyrrole Scaffolds. Chem. Eur. J. 2016, 22, 17967–17971. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, K.L.; Villemson, E.V.; Budynina, E.M.; Ivanova, O.A.; Trushkov, I.V.; Melnikov, M.Y. Ring Opening of Donor–Acceptor Cyclopropanes with the Azide Ion: A Tool for Construction of N-Heterocycles. Chem. Eur. J. 2015, 21, 4975–4987. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, C.; Huang, K.; Liu, L.; Chang, W.; Li, J. Copper-catalyzed cascade reaction via intramolecular hydroamination cyclization of homopropargylic amines and intermolecular Povarov reaction with imines. Org. Lett. 2016, 18, 2367–2370. [Google Scholar] [CrossRef]
- Liu, L.; Wang, C.; Liu, Q.; Kong, Y.; Chang, W.; Li, J. Copper (II) Trifluoromethanesulfonate Catalyzed Hydroamination Cyclization–Dimerization Cascade Reaction of Homopropargylic Amines for the Construction of Complex Fused Nitrogen-Containing Tetracycles. Eur. J. Org. Chem. 2016, 2016, 3684–3690. [Google Scholar] [CrossRef]
- Li, J.; Lin, N.; Yu, L.; Zhang, Y. Synthesis of β-prolinols via [3+2] cycloaddition and one-pot programmed reduction: Valuable building blocks for polyheterocycles. Tetrahedron Lett. 2016, 57, 5777–5780. [Google Scholar] [CrossRef]
- Xie, H.; Gong, B.; Zhong, X.; Cui, H.; Xiang, J. Intramolecular Cycloaddition of Azomethine Ylides Activated by Aromatic Rings: Scope and Limitations. Chem. HeterocycIic Comp. 2016, 52, 484–492. [Google Scholar] [CrossRef]
- Cai, J.; Li, F.; Deng, G.-J.; Ji, X.; Huang, H. The cyclopropylimine rearrangement/Povarov reaction cascade for the assembly of pyrrolo [3, 2-c] quinoline derivatives. Green Chem. 2016, 18, 3503–3506. [Google Scholar] [CrossRef]
- Kondo, Y.; Nishikimi, Y. Process for Production of Optically Active Hexahydropyrroloquinoline and Intermediate for the Process. U.S. Patents WO 2010038434, 8 April 2010. [Google Scholar]
- Tatsuta, K.; Kondo, Y. Process for Preparation of Hexahydropyrroloquinoline. U.S. Patents JP 2011256110A, 30 September 2011. [Google Scholar]
- Yamada, M.; Usutani, H.; Ito, T.; Yamano, M. Construction of a (3aR, 4R, 9bR)-Hexahydropyrrolo- quinoline by Stereoselective Hydrogen-Mediated Domino Cyclization. Org. Process Res. Dev. 2019, 23, 535–547. [Google Scholar] [CrossRef]
- Lindbäck, E.; Sydnes, M.O. Catalytic Enantioselective Synthesis of the Partially Reduced Tricyclic Pyrrolo [3,2-c] quinoline Core Structure of the Martinella Alkaloids. ChemistrySelect 2016, 1, 1837–1840. [Google Scholar] [CrossRef]
- Kolb, H.C.; Andersson, P.G.; Sharpless, K.B. Toward an understanding of the high enantioselectivity in the osmium-catalyzed asymmetric dihydroxylation (AD). 1. Kinetics. J. Am. Chem. Soc. 1994, 116, 1278–1291. [Google Scholar] [CrossRef]
- Reddy, J.S.; Rao, B.V. A short, efficient, and stereoselective total synthesis of a pyrrolidine alkaloid: (−)-codonopsinine. J. Org. Chem. 2007, 72, 2224–2227. [Google Scholar] [CrossRef] [PubMed]
- Haarr, M.B.; Sydnes, M.O. Synthetic approach towards the martinella alkaloids. Unpublished work. 2020. [Google Scholar]
- Wittig, G.; Sommer, H. Zum Verhalten ungesättigter Ammoniumsalze gegenüber Protonenacceptoren. Justus Liebigs Ann. Chem. 1955, 594, 1–14. [Google Scholar] [CrossRef]
- Swan, G.A.; Wilcock, J.D. Reduction of some N-alkyl- and N-aryl-pyrrolidin-2-ones and-piperidin-2-ones by lithium aluminium hydride. J. Chem. Soc. Perkin Trans. 1974, 885–891. [Google Scholar] [CrossRef]
- Kerr, G.H.; Meth-Cohn, O.; Mullock, E.B.; Suschitzky, H. Reactions of NN-dialkylanilines with diethyl azodicarboxylate and with ozone. J. Chem. Soc. Perkin Trans. 1974, 1614–1619. [Google Scholar] [CrossRef]
- Khandelwal, G.D.; Swan, G.A.; Roy, R.B. Dehydrogenation of some aromatic tertiary amines by gamma radiation and by peroxides. J. Chem. Soc. Perkin Trans. 1974, 891–896. [Google Scholar] [CrossRef]
- Rao, G.A.; Periasamy, M. Cycloaddition of enamine and iminium ion intermediates formed in the reaction of N-arylpyrrolidines with T-HYDRO. Synlett 2015, 26, 2231–2236. [Google Scholar] [CrossRef]
- Minakata, S.; Ohshima, Y.; Takemiya, A.; Ryu, I.; Komatsu, M.; Ohshiro, Y. Catalytic oxidation of amines utilizing binuclear copper (II) complex of 7-azaindole. Chem. Lett. 1997, 26, 311–312. [Google Scholar] [CrossRef]
- Buswell, M.; Fleming, I. The reaction of phenyldimethylsilyllithium with N-phenylpyrrolidone. Chem. Commun. 2003, 202–203. [Google Scholar] [CrossRef] [PubMed]
- Min, C.; Sanchawala, A.; Seidel, D. Dual C–H Functionalization of N-Aryl Amines: Synthesis of Polycyclic Amines via an Oxidative Povarov Approach. Org. Lett. 2014, 16, 2756–2759. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yao, X.; Zhang, L.; Ni, P.; Cheng, R.; Ye, J. Direct Arylation of α-Amino C(sp3)-H Bonds by Convergent Paired Electrolysis. Angew. Chem. Int. Ed. 2019, 58, 16548–16552. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Yang, Q.; Zhang, L.; Luo, S. Photoredox Mediated Acceptorless Dehydrogenative Coupling of Saturated N-Heterocycles. ACS Catal. 2019, 9, 3589–3594. [Google Scholar] [CrossRef]
- Wang, L.; Liu, L.; Chang, W.; Li, J. The Divergent Cascade Reactions of Arylalkynols with Homopropargylic Amines or Electron-Deficient Olefins: Access to the Spiro-Isobenzofuran-b-pyrrolo- quinolines or Bridged-Isobenzofuran Polycycles. J. Org. Chem. 2018, 83, 7799–7813. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.-Q.; Xu, J.-T.; Feng, Z.-T.; Liang, H.; Wang, Z.-Y.; Qin, Y.; Xu, P.-F. Dual C(sp3)–H Bond Functionalization of N-Heterocycles through Sequential Visible-Light Photocatalyzed Dehydrogenation/ [2+2] Cycloaddition Reactions. Angew. Chem. Int. Ed. 2018, 57, 5110–5114. [Google Scholar] [CrossRef]
- Kong, Y.; Liu, Y.; Wang, B.; Li, S.; Liu, L.; Chang, W.; Li, J. The Catalyst-Controlled Divergent Cascade Reactions of Homo-Propargylic Amines and Nitrones: Synthesis of Pyrrolo-Isoxazolidines and γ-Lactams. Adv. Synth. Catal. 2018, 360, 1240–1252. [Google Scholar] [CrossRef]
- Wang, F.; He, Y.; Tian, M.; Zhang, X.; Fan, X. Synthesis of α-Formylated N-Heterocycles and Their 1,1-Diacetates from Inactivated Cyclic Amines Involving an Oxidative Ring Contraction. Org. Lett. 2018, 20, 864–867. [Google Scholar] [CrossRef]
- Snider, B.B.; Ahn, Y.; Foxman, B.M. Synthesis of the tricyclic triamine core of martinelline and martinellic acid. Tetrahedron Lett. 1999, 40, 3339–3342. [Google Scholar] [CrossRef]
- Fustero, S.; Bello, P.; Miró, J.; Sánchez-Roselló, M.; Maestro, M.A.; Gonzalez, J.; del Pozo, C. Gold catalyzed stereoselective tandem hydroamination–formal aza-Diels–Alder reaction of propargylic amino esters. Chem. Commun. 2013, 49, 1336–1338. [Google Scholar] [CrossRef]
- Ma, C.-L.; Li, X.-H.; Yu, X.-L.; Zhu, X.-L.; Hu, Y.-Z.; Dong, X.-W.; Tan, B.; Liu, X.-Y. Gold-catalyzed tandem synthesis of bioactive spiro-dipyrroloquinolines and its application in the one-step synthesis of incargranine B aglycone and seneciobipyrrolidine (I). Org. Chem. Front. 2016, 3, 324–329. [Google Scholar] [CrossRef]
- Yu, X.-L.; Kuang, L.; Chen, S.; Zhu, X.-L.; Li, Z.-L.; Tan, B.; Liu, X.-Y. Counteranion-controlled unprecedented diastereo-and enantioselective tandem formal Povarov reaction for construction of bioactive octahydro-dipyrroloquinolines. ACS Catal. 2016, 6, 6182–6190. [Google Scholar] [CrossRef]
- Li, S.-S.; Zhou, L.; Wang, L.; Zhao, H.; Yu, L.; Xiao, J. Organocatalytic C (sp3)–H functionalization via carbocation-initiated cascade [1,5]-hydride transfer/cyclization: Synthesis of dihydrodibenzo [b,e] azepines. Org. Lett. 2018, 20, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wang, M.; Pan, L.; Li, Y.; Liu, Q. Csp 3–H bond functionalization of amines via tunable iminium ions: Divergent synthesis of trifluoromethylated arylamines. Chem. Commun. 2018, 54, 8721–8724. [Google Scholar] [CrossRef] [PubMed]
- Kajino, M.; Hird, N.W.; Tarui, N.; Banno, H.; Kawano, Y.; Inatomi, N. Preparation of Fused Quinoline Derivatives as NK2 Receptor Antagonists for Functional Gastrointestinal Diseases. U.S. Patent WO 2005105802, 10 November 2005. [Google Scholar]
- Tanaka, T.; Matsumoto-Okano, S.; Inatomi, N.; Fujioka, Y.; Kamiguchi, H.; Yamaguchi, M.; Imanishi, A.; Kawamoto, M.; Miura, K.; Nishikawa, Y. Establishment and validation of a rabbit model for in vivo pharmacodynamic screening of tachykinin NK2 antagonists. J. Pharmacol. Sci. 2012, 118, 487–495. [Google Scholar] [CrossRef]
- Tanaka, T.; Tanaka, A.; Nakamura, A.; Matsushita, K.; Imanishi, A.; Matsumoto-Okano, S.; Inatomi, N.; Miura, K.; Toyoda, M.; Mizojiri, G. Effects of TAK-480, a Novel Tachykinin NK2–Receptor Antagonist, on Visceral Hypersensitivity in Rabbits and Ricinoleic Acid–Induced Defecation in Guinea Pigs. J. Pharmacol. Sci. 2012, 120, 15–25. [Google Scholar] [CrossRef]
- Kajiwara, T.; Konishi, T.; Yamano, M. Asymmetric catalytic hydrogenation for large scale preparation of optically active 2-(N-benzoylamino) cyclohexanecarboxylic acid derivatives. Catal. Sci. Technol. 2012, 2, 2146–2152. [Google Scholar] [CrossRef]
- Furuya, K.; Yamamoto, N.; Ohyabu, Y.; Makino, A.; Morikyu, T.; Ishige, H.; Kuzutani, K.; Endo, Y. The Novel Non-steroidal Selective Androgen Receptor Modulator S-101479 Has Additive Effects with Bisphosphonate, Selective Estrogen Receptor Modulator, and Parathyroid Hormone on the Bones of Osteoporotic Female Rats. Biol. Pharm. Bull. 2012, 35, 1096–1104. [Google Scholar] [CrossRef]
- Furuya, K.; Yamamoto, N.; Ohyabu, Y.; Morikyu, T.; Ishige, H.; Albers, M.; Endo, Y. Mechanism of the tissue-specific action of the selective androgen receptor modulator S-101479. Biol. Pharm. Bull. 2013, 36, 442–451. [Google Scholar] [CrossRef]
- Duvall, J.R.; Bedard, L.; Naylor-Olsen, A.M.; Manson, A.L.; Bittker, J.A.; Sun, W.; Fitzgerald, M.E.; He, Z.; Lee, M.D., IV; Marie, J.-C.; et al. Identification of highly specific diversity-oriented synthesis-derived inhibitors of Clostridium difficile. ACS Infect. Dis. 2017, 3, 349–359. [Google Scholar] [CrossRef]
- Prosser, G.A.; Rodenburg, A.; Khoury, H.; de Chiara, C.; Howell, S.; Snijders, A.P.; de Carvalho, L.P.S. Glutamate racemase is the primary target of β-chloro-D-alanine in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2016, 60, 6091–6099. [Google Scholar] [CrossRef] [PubMed]
- Geng, B.; Basarab, G.; Comita-Prevoir, J.; Gowravaram, M.; Hill, P.; Kiely, A.; Loch, J.; MacPherson, L.; Morningstar, M.; Mullen, G. Potent and selective inhibitors of Helicobacter pylori glutamate racemase (MurI): Pyridodiazepine amines. Bioorg. Med. Chem. Lett. 2009, 19, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, T.; Fisher, S.L.; Kern, G.; Folmer, R.H.; Xue, Y.; Newton, D.T.; Keating, T.A.; Alm, R.A.; de Jonge, B.L. Exploitation of structural and regulatory diversity in glutamate racemases. Nature 2007, 447, 817–822. [Google Scholar] [CrossRef]
- Park, S.W.; Casalena, D.E.; Wilson, D.J.; Dai, R.; Nag, P.P.; Liu, F.; Boyce, J.P.; Bittker, J.A.; Schreiber, S.L.; Finzel, B.C. Target-based identification of whole-cell active inhibitors of biotin biosynthesis in Mycobacterium tuberculosis. Chem. Biol. 2015, 22, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Maji, B.; Gangopadhyay, S.A.; Lee, M.; Shi, M.; Wu, P.; Heler, R.; Mok, B.; Lim, D.; Siriwardena, S.U.; Paul, B. A high-throughput platform to identify small-molecule inhibitors of CRISPR-Cas9. Cell 2019, 177, 1067–1079. [Google Scholar] [CrossRef]
- Choudhary, A.; Fox, K.; Subramanian, H.; Franco, E. Compositions and Methods for Regulating Proteins and Nucleic Acids Activities. U.S. Patent 20200239879A1, 30 July 2020. [Google Scholar]
- Choudhary, A.; Maji, B.; Gangopadhyay, S.A.; Lee, M.; Shi, M. Inhibitors of RNA-Guided Nuclease Target Binding and Uses Thereof. U.S. Patent WO 2020068304, 2 April 2020. [Google Scholar]























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Optical Rotation [αD] | Concentration (mg/ 10 cm3) | Reference |
|---|---|---|---|
| 1 (isolated) | +9.4 | 0.02 | [19] |
| (−)-1 | −108.0 | 0.09 | [30] |
| (+)-1 | +98.6 | 0.02 | [30] |
| 2 (isolated) | −8.5 | 0.01 | [19] |
| (−)-2 | −122.7 | 0.37 | [29] |
| (−)-2 | −118 | 0.3 | [32] |
| (−)-2 | −164.3 | 0.14 | [30] |
| (+)-2 | +165.5 | 0.11 | [30] |
| (−)-2 | −164.8 | 0.33 | [31] |
| 3 (isolated) | −12 | 0.275 | [25] |
| (±)-3 | −16.7 | 0.275 | [26] |
| 4 (isolated) | −72.9 | 0.10 | [27] |
| Entry | Reaction Conditions | R1 | R2 | Enamide | Yield | Endo:Exo | Reference |
|---|---|---|---|---|---|---|---|
| 1 | InCl3 (2 equiv.), MeCN, rt, 30 min | H | H | 73b | 41% | 1:1 | [58] |
| 2 | H | 2-NO2 | 73b | 50% | 2:1 | ||
| 3 | Zn(OTf)2 (10 mol%), DCM, rt | H | 2-OH | 73c | 42% | >20:1 | [59] |
| 4 | 76 (10 mol%), 5 Å MS, n-hexane −40 °C, 72 h | H | H | 73a | 94% | >20:1 | [60] |
| 5 | 77 (10 mol%), MeCN, rt, 0.5–1 h | H | H | 73a | 95% | 43:57 | [61] |
| 6 | 78 (10 mol%), MeCN, rt, 27 h | 4-OMe | H | 73d | 86% | 53:47 | [62] |
| 7 | Micellar-SO3H, H2O, 25 °C, 18 h | H | 4-O-DNA | 73d | >90% | NR | [63] |
| Entry | Starting Material | Conditions | Yield a | Endo:Exo | Reference |
|---|---|---|---|---|---|
| 1 | 240 | LiAlH4, ether, rt, 3.5 h | 27% | 38:36 | [100] |
| 2 | 242 | O3, n-hexane, 0 °C | 41% | NR | [101] |
| 3 | 242 | 1) DEAD, cyclohexane, reflux, 2 h, 80% 2) xylene, reflux, 15 h | 50% | 28:22 | [101] |
| 4 | 242 | γ-irradiation, 17 d | 9% | ~1:1 | [102] |
| 5 | 242 | di-t-butylperoxide, 140 °C, 44 h | NR | ~1:1 | [102] |
| 6 | 242 | Dibenzoyl peroxide, MeCN, 0 °C, 9 h | 28% | 1:0 | [102] |
| 7 | 242 | t-BuOOH, NaOAc•3H2O, cyclohexane 70 °C, 24 h | 72% | 1:0 | [103] |
| 8 | 242 | Cu(OAc)2, O2, Et3N | 26% | 11:15 | [104] |
| 9 | 240 | PhMe2SiLi, −78 to −20 °C | 47% | 31:16 | [105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haarr, M.B.; Sydnes, M.O. Synthesis of the Hexahydropyrrolo-[3,2-c]-quinoline Core Structure and Strategies for Further Elaboration to Martinelline, Martinellic Acid, Incargranine B, and Seneciobipyrrolidine. Molecules 2021, 26, 341. https://doi.org/10.3390/molecules26020341
Haarr MB, Sydnes MO. Synthesis of the Hexahydropyrrolo-[3,2-c]-quinoline Core Structure and Strategies for Further Elaboration to Martinelline, Martinellic Acid, Incargranine B, and Seneciobipyrrolidine. Molecules. 2021; 26(2):341. https://doi.org/10.3390/molecules26020341
Chicago/Turabian StyleHaarr, Marianne B., and Magne O. Sydnes. 2021. "Synthesis of the Hexahydropyrrolo-[3,2-c]-quinoline Core Structure and Strategies for Further Elaboration to Martinelline, Martinellic Acid, Incargranine B, and Seneciobipyrrolidine" Molecules 26, no. 2: 341. https://doi.org/10.3390/molecules26020341
APA StyleHaarr, M. B., & Sydnes, M. O. (2021). Synthesis of the Hexahydropyrrolo-[3,2-c]-quinoline Core Structure and Strategies for Further Elaboration to Martinelline, Martinellic Acid, Incargranine B, and Seneciobipyrrolidine. Molecules, 26(2), 341. https://doi.org/10.3390/molecules26020341