
Spectroelectrochemical Properties and Catalytic Activity in Cyclohexane Oxidation of the Hybrid Zr/Hf-Phthalocyaninate-Capped Nickel(II) and Iron(II) tris-Pyridineoximates and Their Precursors

 , , , , , ,
, , , , , ,  ,
,  and
and 
Abstract
1. Introduction
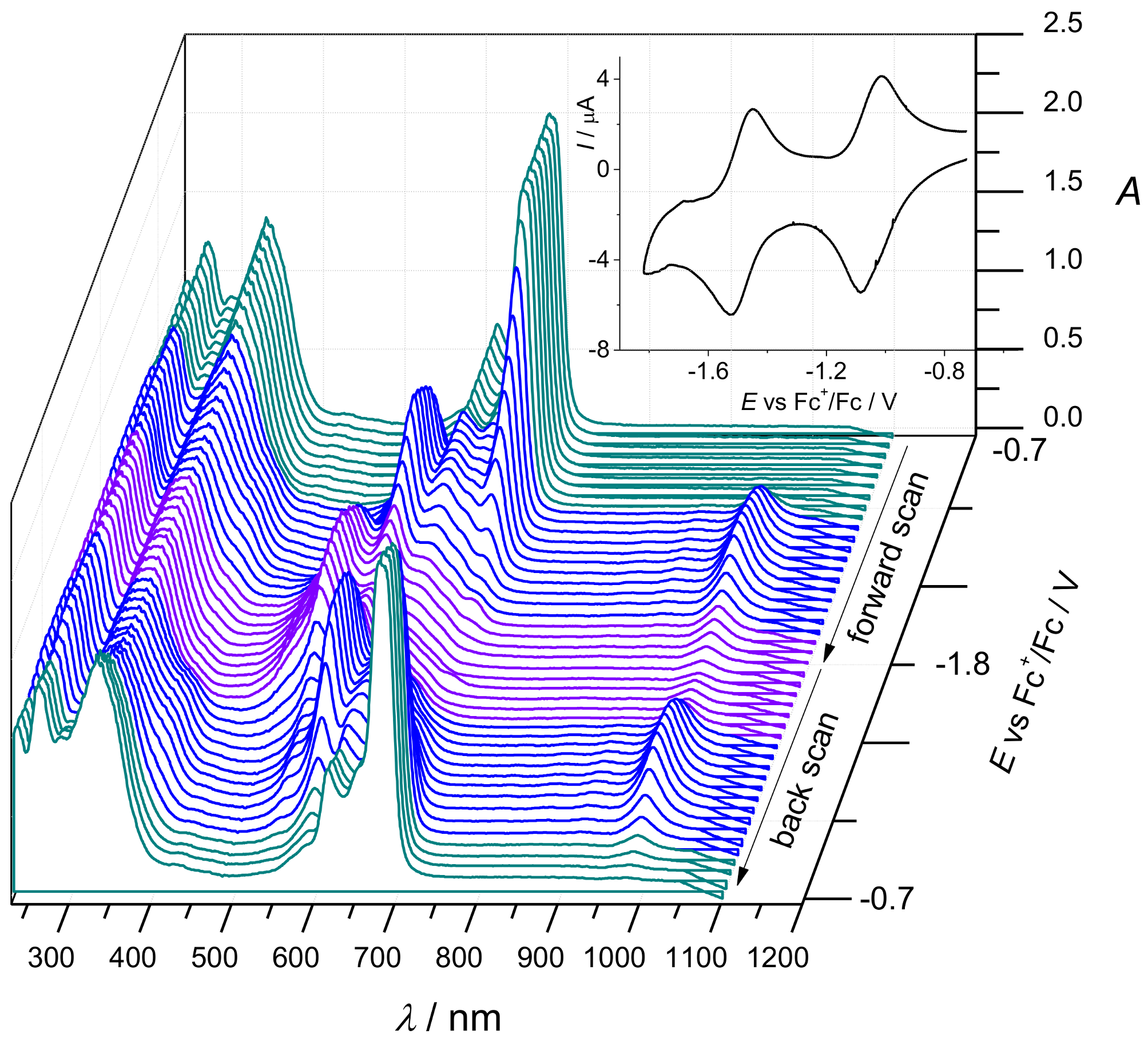
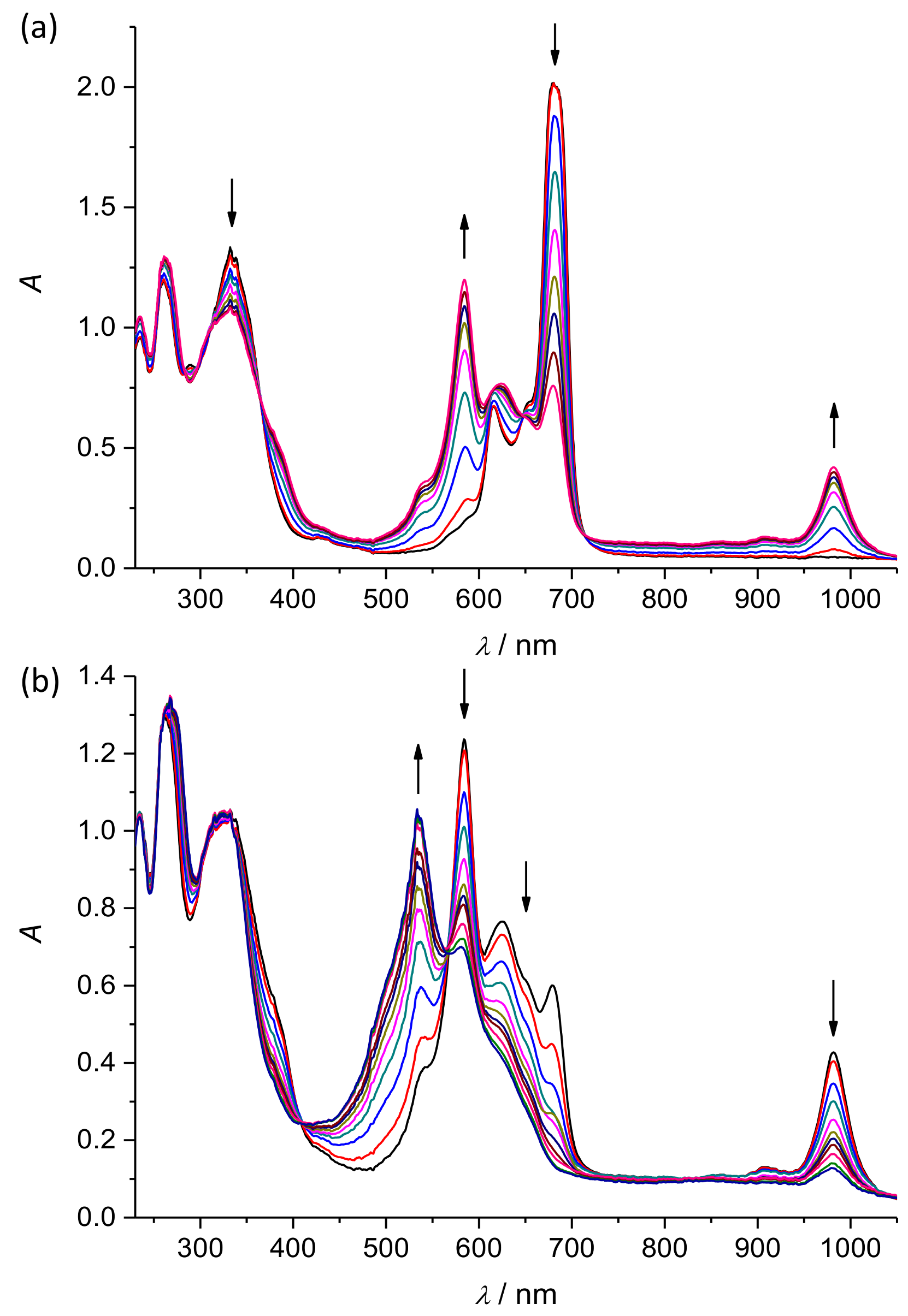
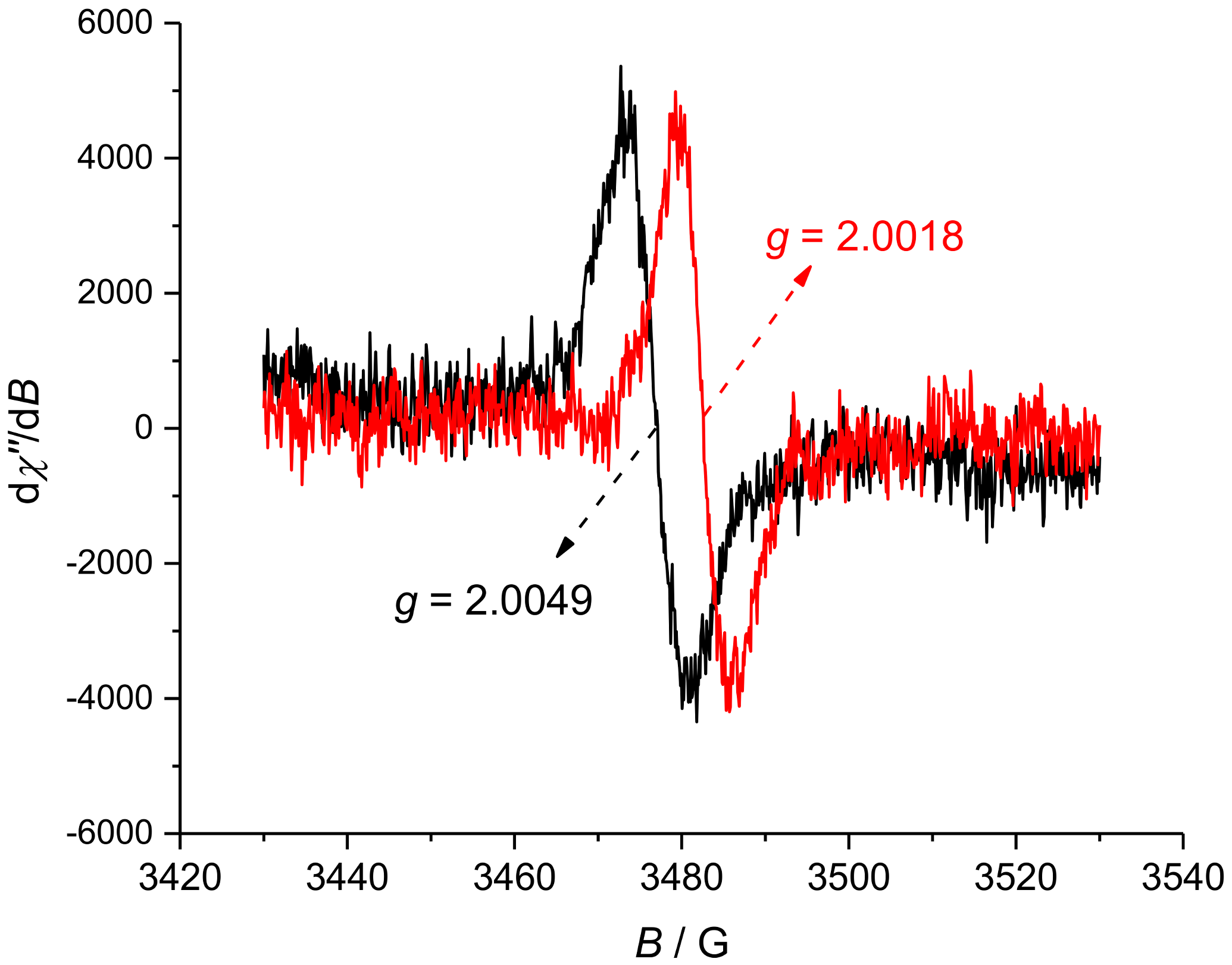
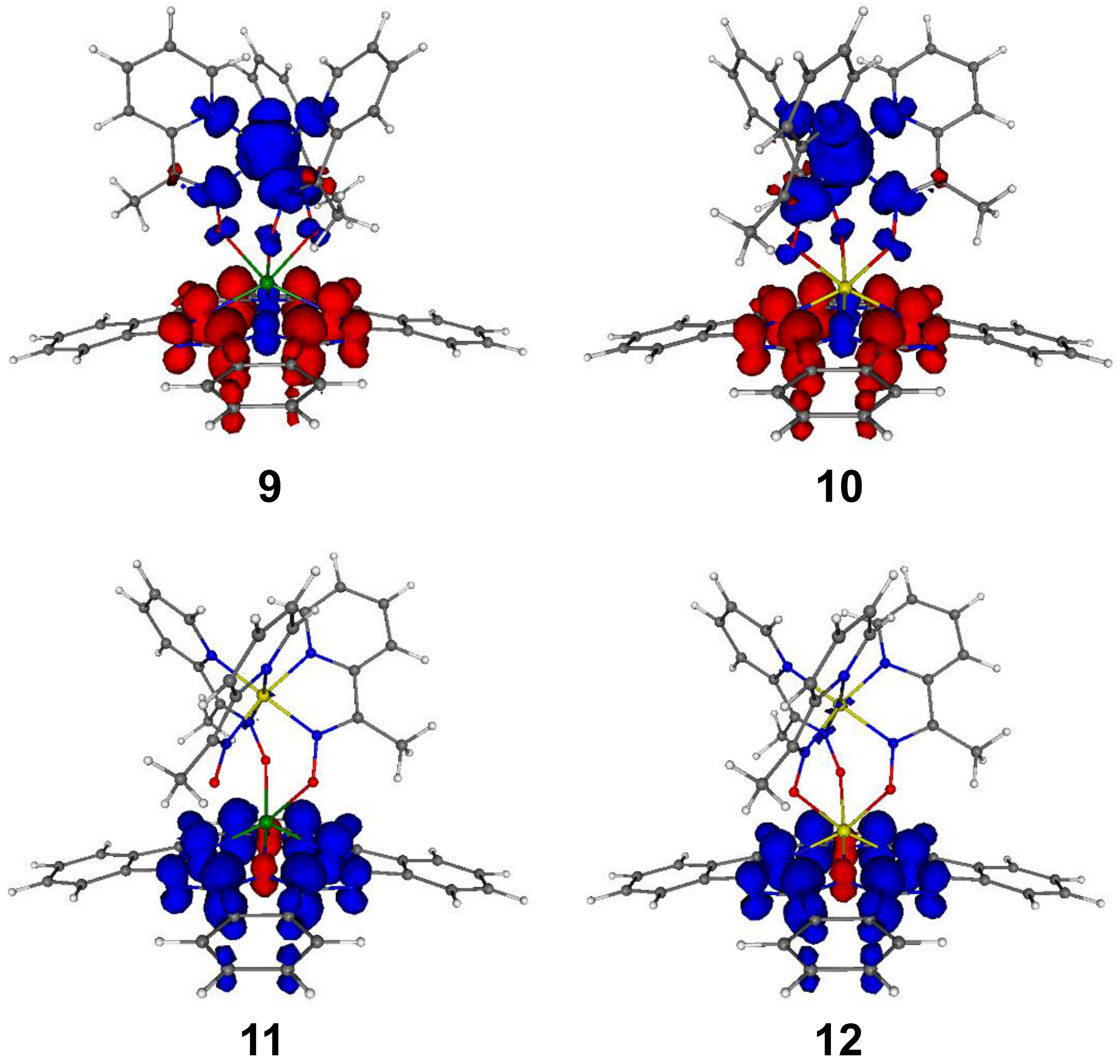
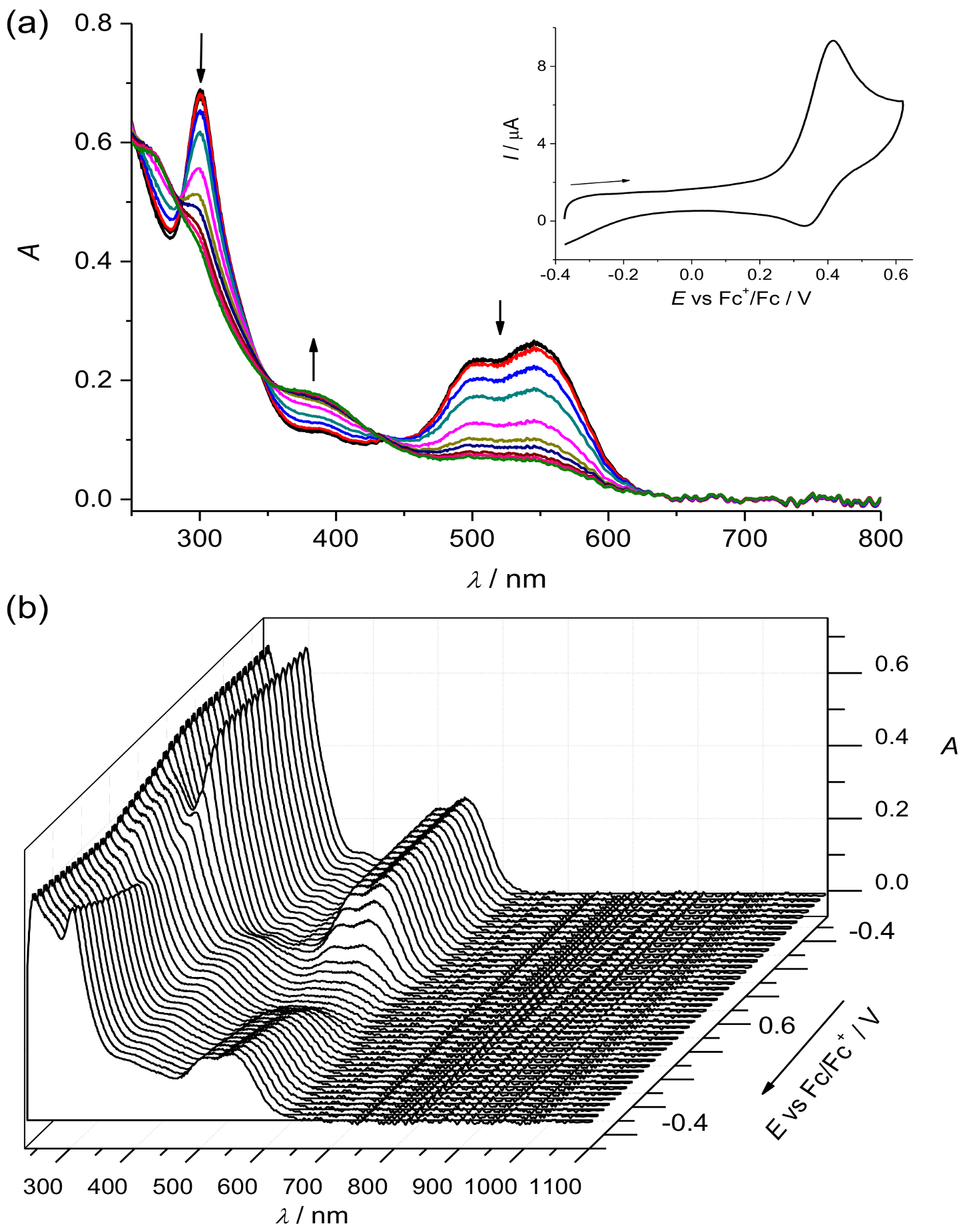
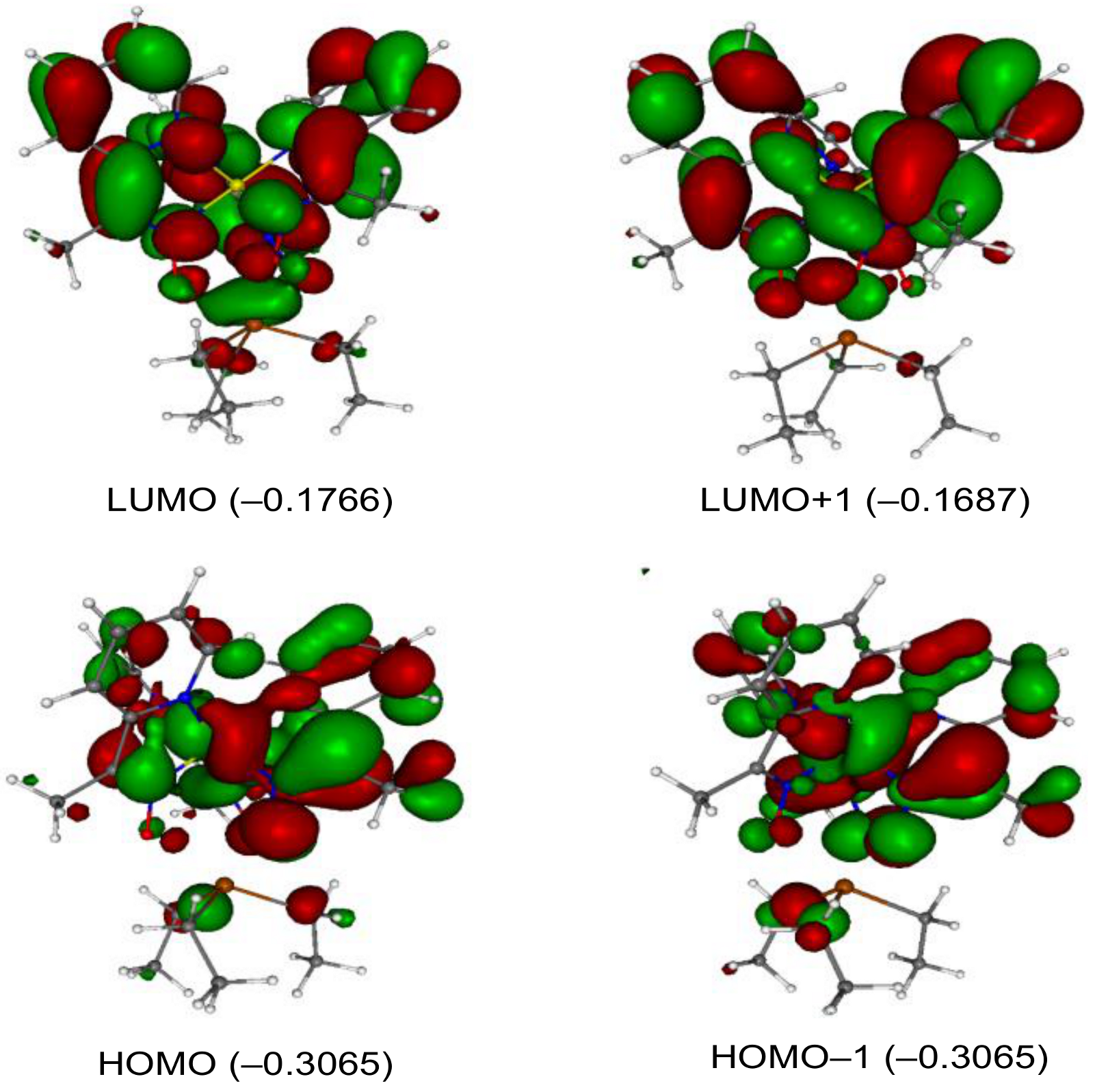
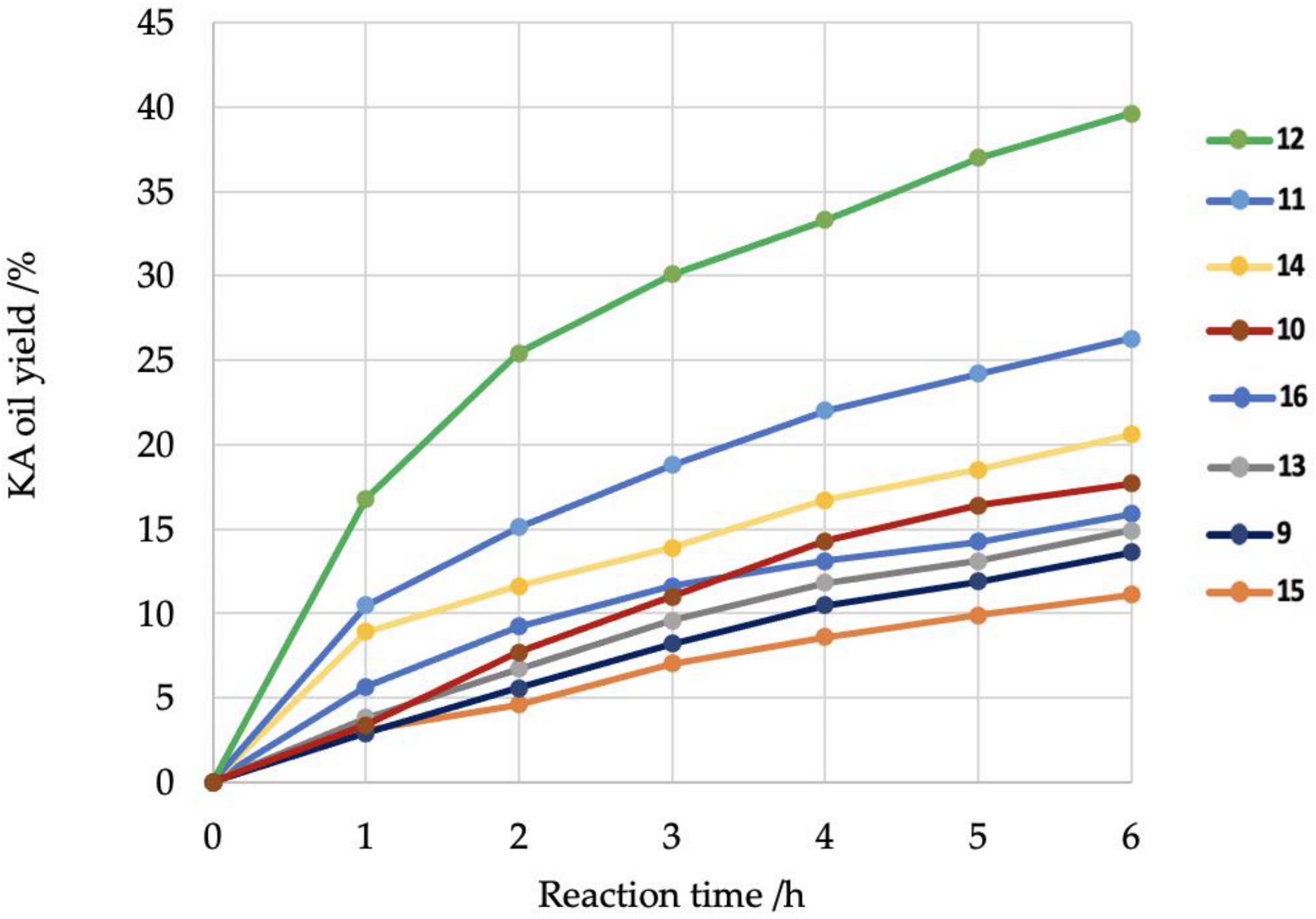
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Heath, J.R.; Ratner, M.A. Molecular Electronics. Physics Today 2003, 56, 43–49. [Google Scholar] [CrossRef]
- Nalwa, H.S. (Ed.) Encyclopedia of Nanoscience and Nanotechnology®; American Scientific Publ.: Stevenson Ranch, CA, USA, 2004. [Google Scholar]
- Weiss, J. Supramolecular Approaches to Nano and Molecular Electronics. Coord. Chem. Rev. 2010, 254, 2247–2248. [Google Scholar] [CrossRef]
- Lyshevski, S.E. (Ed.) Nano-and microscience, engineering, technology, and medicine series. In Nano and Molecular Electronics Handbook; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Bayley, H. Holes with an Edge. Nature 2010, 467, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Giacalone, F.; Martín, N. New Concepts and Applications in the Macromolecular Chemistry of Fullerenes. Adv. Mater. 2010, 22, 4220–4248. [Google Scholar] [CrossRef] [PubMed]
- Jurow, M.; Schuckman, A.E.; Batteas, J.D.; Drain, C.M. Porphyrins as Molecular Electronic Components of Functional Devices. Coord. Chem. Rev. 2010, 254, 2297–2310. [Google Scholar] [CrossRef]
- Chung, A.; Deen, J.; Lee, J.-S.; Meyyappan, M. Nanoscale Memory Devices. Nanotechnology 2010, 21, 412001. [Google Scholar] [CrossRef]
- Belosludov, R.V.; Farajian, A.A.; Kikuchi, Y.; Mizuseki, H.; Kawazoe, Y. Realization of Molecular Interconnection for Molecular Electronics: Theoretical Aspects. Comput. Mater. Sci. 2006, 36, 130–134. [Google Scholar] [CrossRef]
- Belosludov, R.V.; Farajian, A.A.; Baba, H.; Mizuseki, H.; Kawazoe, Y. Electronic and Transport Properties of Doped Organic Molecules for Molecular Wire Applications. Jpn. J. Appl. Phys. 2005, 44, 2823–2825. [Google Scholar] [CrossRef]
- Lee, S.U.; Belosludov, R.V.; Mizuseki, H.; Kawazoe, Y. Control of Electron Transport by Manipulating the Conjugated Framework. J. Phys. Chem. C 2007, 111, 15397–15403. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular Chemistry: Concepts and Perspectives: A Personal Account. Built upon the George Fisher Baker Lectures in Chemistry at Cornell University [and] Lezioni Lincee, Accademia Nazionale Dei Lincei, Roma; VCH: Weinheim, Germany; New York, NY, USA, 1995. [Google Scholar]
- Balzani, V.; Juris, A.; Venturi, M.; Campagna, S.; Serroni, S. Luminescent and Redox-Active Polynuclear Transition Metal Complexes. Chem. Rev. 1996, 96, 759–834. [Google Scholar] [CrossRef]
- Miller, J.S.; Epstein, A.J. Organische und metallorganische molekulare magnetische Materialien: Designer-Magnete. Angew. Chem. 1994, 106, 399–432. [Google Scholar] [CrossRef]
- Barlow, S.; O’Hare, D. Metal−Metal Interactions in Linked Metallocenes. Chem. Rev. 1997, 97, 637–670. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W.; Lahiri, G.K. Unconventional Mixed-Valent Complexes of Ruthenium and Osmium. Angew. Chem. Int. Ed. 2007, 46, 1778–1796. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W.; Sarkar, B. Mixed Valency in Ruthenium Complexes—Coordinative Aspects. Coord. Chem. Rev. 2007, 251, 584–594. [Google Scholar] [CrossRef]
- Venkatasubbaiah, K.; Doshi, A.; Nowik, I.; Herber, R.H.; Rheingold, A.L.; Jäkle, F. Examination of the Mixed-Valence State of the Doubly Boron-Bridged Diferrocene Cation [(FeCp)2{μ-C10H6(BPh)2}]+. Chem. Eur. J. 2008, 14, 444–458. [Google Scholar] [CrossRef] [PubMed]
- Hadt, R.G.; Nemykin, V.N. Exploring the Ground and Excited State Potential Energy Landscapes of the Mixed-Valence Biferrocenium Complex. Inorg. Chem. 2009, 48, 3982–3992. [Google Scholar] [CrossRef]
- Bucher, C.; Devillers, C.; Moutet, J.; Royal, G.; Saintaman, E. Ferrocene-Appended Porphyrins: Syntheses and Properties. Coord. Chem. Rev. 2009, 253, 21–36. [Google Scholar] [CrossRef]
- Burrell, A.K.; Campbell, W.M.; Jameson, G.B.; Officer, D.L.; Boyd, P.D.W.; Zhao, Z.; Cocks, P.A.; Gordon, K.C. Bis(Ferrocenyl)Porphyrins. Compounds with Strong Long-Range Metal–Metal Coupling. Chem. Commun. 1999, 637–638. [Google Scholar] [CrossRef]
- Shoji, O.; Tanaka, H.; Kawai, T.; Kobuke, Y. Single Molecule Visualization of Coordination-Assembled Porphyrin Macrocycles Reinforced with Covalent Linkings. J. Am. Chem. Soc. 2005, 127, 8598–8599. [Google Scholar] [CrossRef]
- Nemykin, V.N.; Rohde, G.T.; Barrett, C.D.; Hadt, R.G.; Bizzarri, C.; Galloni, P.; Floris, B.; Nowik, I.; Herber, R.H.; Marrani, A.G.; et al. Electron-Transfer Processes in Metal-Free Tetraferrocenylporphyrin. Understanding Internal Interactions To Access Mixed-Valence States Potentially Useful for Quantum Cellular Automata. J. Am. Chem. Soc. 2009, 131, 14969–14978. [Google Scholar] [CrossRef]
- Solomon, E.I.; Sarangi, R.; Woertink, J.S.; Augustine, A.J.; Yoon, J.; Ghosh, S. O 2 and N 2 O Activation by Bi-, Tri-, and Tetranuclear Cu Clusters in Biology. Acc. Chem. Res. 2007, 40, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Xi, B.; Liu, I.P.-C.; Xu, G.-L.; Choudhuri, M.M.R.; DeRosa, M.C.; Crutchley, R.J.; Ren, T. Modulation of Electronic Couplings within Ru 2 –Polyyne Frameworks. J. Am. Chem. Soc. 2011, 133, 15094–15104. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Kaizu, Y. Synthetic, Spectroscopic and Theoretical Study of Novel Supramolecular Structures Composed of Lanthanide Phthalocyanine Double-Decker Complexes. Coord. Chem. Rev. 2002, 226, 93–101. [Google Scholar] [CrossRef]
- Lent, C.S.; Isaksen, B.; Lieberman, M. Molecular Quantum-Dot Cellular Automata. J. Am. Chem. Soc. 2003, 125, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Steuerman, D.W.; Tseng, H.-R.; Peters, A.J.; Flood, A.H.; Jeppesen, J.O.; Nielsen, K.A.; Stoddart, J.F.; Heath, J.R. Molecular-Mechanical Switch-Based Solid-State Electrochromic Devices. Angew. Chem. Int. Ed. 2004, 43, 6486–6491. [Google Scholar] [CrossRef]
- Leigh, D.A.; Morales, M.Á.F.; Pérez, E.M.; Wong, J.K.Y.; Saiz, C.G.; Slawin, A.M.Z.; Carmichael, A.J.; Haddleton, D.M.; Brouwer, A.M.; Buma, W.J.; et al. Patterning through Controlled Submolecular Motion: Rotaxane-Based Switches and Logic Gates That Function in Solution and Polymer Films. Angew. Chem. Int. Ed. 2005, 44, 3062–3067. [Google Scholar] [CrossRef]
- Ohkubo, K.; Kotani, H.; Shao, J.; Ou, Z.; Kadish, K.M.; Li, G.; Pandey, R.K.; Fujitsuka, M.; Ito, O.; Imahori, H.; et al. Production of an Ultra-Long-Lived Charge-Separated State in a Zinc Chlorin–C60 Dyad by One-Step Photoinduced Electron Transfer. Angew. Chem. Int. Ed. 2004, 43, 853–856. [Google Scholar] [CrossRef]
- Sutton, L.R.; Scheloske, M.; Pirner, K.S.; Hirsch, A.; Guldi, D.M.; Gisselbrecht, J.-P. Unexpected Change in Charge Transfer Behavior in a Cobalt(II) Porphyrin−Fullerene Conjugate That Stabilizes Radical Ion Pair States. J. Am. Chem. Soc. 2004, 126, 10370–10381. [Google Scholar] [CrossRef]
- Baran, P.S.; Monaco, R.R.; Khan, A.U.; Schuster, D.I.; Wilson, S.R. Synthesis and Cation-Mediated Electronic Interactions of Two Novel Classes of Porphyrin−Fullerene Hybrids. J. Am. Chem. Soc. 1997, 119, 8363–8364. [Google Scholar] [CrossRef]
- Zhang, T.-G.; Zhao, Y.; Asselberghs, I.; Persoons, A.; Clays, K.; Therien, M.J. Design, Synthesis, Linear, and Nonlinear Optical Properties of Conjugated (Porphinato)Zinc(II)-Based Donor−Acceptor Chromophores Featuring Nitrothiophenyl and Nitrooligothiophenyl Electron-Accepting Moieties. J. Am. Chem. Soc. 2005, 127, 9710–9720. [Google Scholar] [CrossRef]
- Voloshin, Y.Z.; Varzatskii, O.A.; Tomilova, L.G.; Breusova, M.O.; Magdesieva, T.V.; Bubnov, Y.N.; Krämer, R. First Hybrid Oximehydrazonate Phthalocyaninoclathrochelates: The Synthesis and Properties of Lutetium Phthalocyanine-Capped Cage Iron(II) Complexes. Polyhedron 2007, 26, 2733–2740. [Google Scholar] [CrossRef]
- Voloshin, Y.Z.; Varzatskii, O.A.; Korobko, S.V.; Chernii, V.Y.; Volkov, S.V.; Tomachynski, L.A.; Pehn’o, V.I.; Antipin, M.Y.; Starikova, Z.A. Ditopic Macropolycyclic Complexes: Synthesis of Hybrid Phthalocyaninoclathrochelates. Inorg. Chem. 2005, 44, 822–824. [Google Scholar] [CrossRef] [PubMed]
- Sabin, J.R.; Varzatskii, O.A.; Voloshin, Y.Z.; Starikova, Z.A.; Novikov, V.V.; Nemykin, V.N. Insight into the Electronic Structure, Optical Properties, And Redox Behavior of the Hybrid Phthalocyaninoclathrochelates from Experimental and Density Functional Theory Approaches. Inorg. Chem. 2012, 51, 8362–8372. [Google Scholar] [CrossRef]
- Kadish, K.M.; Morrison, M.M. Solvent and Substituent Effects on the Redox Reactions of Para-Substituted Tetraphenylporphyrin. J. Am. Chem. Soc. 1976, 98, 3326–3328. [Google Scholar] [CrossRef] [PubMed]
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) The Porphyrin Handbook; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Voloshin, Y.Z.; Kostromina, N.A.; Krämer, R. (Eds.) Clathrochelates: Synthesis, Structure, and Properties, 1st ed.; Elsevier Science: Amsterdam, The Netherlands; Boston, MA, USA, 2002. [Google Scholar]
- Voloshin, Y.Z.; Dolganov, A.V.; Varzatskii, O.A.; Bubnov, Y.N. Efficient Electrocatalytic Hydrogen Production from H+ Ions Using Specially Designed Boron-Capped Cobalt Clathrochelates. Chem. Commun. 2011, 47, 7737–7739. [Google Scholar] [CrossRef]
- Ou, Z.; Zhan, R.; Tomachynski, L.A.; Chernii, V.Y.; Kadish, K.M. Electrochemistry and Spectroelectrochemistry of Zirconium(IV) and Hafnium(IV) Phthalocyanines with β-Diketone Axial Ligands in Nonaqueous Media. Macroheterocycles 2011, 5, 164–170. [Google Scholar] [CrossRef]
- Dudkin, S.V.; Erickson, N.R.; Vologzhanina, A.V.; Novikov, V.V.; Rhoda, H.M.; Holstrom, C.D.; Zatsikha, Y.V.; Yusubov, M.S.; Voloshin, Y.Z.; Nemykin, V.N. Preparation, X-Ray Structures, Spectroscopic, and Redox Properties of Di- and Trinuclear Iron–Zirconium and Iron–Hafnium Porphyrinoclathrochelates. Inorg. Chem. 2016, 55, 11867–11882. [Google Scholar] [CrossRef]
- D’Souza, F.; Ito, O. Photosensitized Electron Transfer Processes of Nanocarbons Applicable to Solar Cells. Chem. Soc. Rev. 2012, 41, 86–96. [Google Scholar] [CrossRef]
- Wróbel, D.; Graja, A. Photoinduced Electron Transfer Processes in Fullerene–Organic Chromophore Systems. Coord. Chem. Rev. 2011, 255, 2555–2577. [Google Scholar] [CrossRef]
- Zagal, J.H.; Griveau, S.; Silva, J.F.; Nyokong, T.; Bedioui, F. Metallophthalocyanine-Based Molecular Materials as Catalysts for Electrochemical Reactions. Coord. Chem. Rev. 2010, 254, 2755–2791. [Google Scholar] [CrossRef]
- Sorokin, A.B. Phthalocyanine Metal Complexes in Catalysis. Chem. Rev. 2013, 113, 8152–8191. [Google Scholar] [CrossRef] [PubMed]
- Losytskyy, M.; Chornenka, N.; Vakarov, S.; Meier-Menches, S.M.; Gerner, C.; Potocki, S.; Arion, V.B.; Gumienna-Kontecka, E.; Voloshin, Y.; Kovalska, V. Sensing of Proteins by ICD Response of Iron(II) Clathrochelates Functionalized by Carboxyalkylsulfide Groups. Biomolecules 2020, 10, 1602. [Google Scholar] [CrossRef] [PubMed]
- Voloshin, Y.; Belaya, I.; Krämer, R. Cage Metal. Complexes: Clathrochelates Revisited, 1st ed.; Springer International Publishing Imprint: Cham, Germany, 2017. [Google Scholar]
- Voloshin, Y.Z.; Buznik, V.M.; Dedov, A.G. New Types of the Hybrid Functional Materials Based on Cage Metal Complexes for (Electro) Catalytic Hydrogen Production. Pure Appl. Chem. 2020, 92, 1159–1174. [Google Scholar] [CrossRef]
- Harrowfield, J.M.; Koutsantonis, G.A.; Kraatz, H.-B.; Nealon, G.L.; Orlowski, G.A.; Skelton, B.W.; White, A.H. Cages on Surfaces: Thiol Functionalisation of CoIII Sarcophagine Complexes. Eur. J. Inorg. Chem. 2007, 263–278. [Google Scholar] [CrossRef]
- Antuch, M.; Millet, P.; Iwase, A.; Kudo, A.; Grigoriev, S.A.; Voloshin, Y.Z. Characterization of Rh:SrTiO3 Photoelectrodes Surface-Modified with a Cobalt Clathrochelate and Their Application to the Hydrogen Evolution Reaction. Electrochim. Acta 2017, 258, 255–265. [Google Scholar] [CrossRef]
- Ribeiro, S.; Cunha-Silva, L.; Balula, S.S.; Gago, S. Cobalt(III) Sepulchrate Complexes: Application as Sustainable Oxidative Catalysts. New J. Chem. 2014, 38, 2500–2507. [Google Scholar] [CrossRef]
- Chand, D.K.; Bharadwaj, P.K. A Cobalt(II) Cryptate of a Heteroditopic Cryptand L as an Efficient Oxygenation Catalyst of Organic Substrates Using Molecular Oxygen and 2-Methylpropanal. Inorg. Chem. 1997, 36, 5658–5660. [Google Scholar] [CrossRef]
- Hao, H.-G.; Zheng, X.-D.; Lu, T.-B. Photoinduced Catalytic Reaction by a Fluorescent Active Cryptand Containing an Anthracene Fragment. Angew. Chem. Int. Ed. 2010, 49, 8148–8151. [Google Scholar] [CrossRef]
- Tomyn, S.; Shylin, S.I.; Bykov, D.; Ksenofontov, V.; Gumienna-Kontecka, E.; Bon, V.; Fritsky, I.O. Indefinitely Stable Iron(IV) Cage Complexes Formed in Water by Air Oxidation. Nat. Commun. 2017, 8, 14099. [Google Scholar] [CrossRef]
- Shylin, S.I.; Pavliuk, M.V.; D’Amario, L.; Mamedov, F.; Sá, J.; Berggren, G.; Fritsky, I.O. Efficient Visible Light-Driven Water Oxidation Catalysed by an Iron(IV) Clathrochelate Complex. Chem. Commun. 2019, 55, 3335–3338. [Google Scholar] [CrossRef]
- Dudkin, S.V.; Belov, A.S.; Nelyubina, Y.V.; Savchuk, A.V.; Pavlov, A.A.; Novikov, V.V.; Voloshin, Y.Z. Synthesis, X-ray Structure and Electrochemical Properties of Hybrid Binuclear Metallophthalocyaninate-Capped Tris-Pyridineoximates. New J. Chem. 2017, 41, 3251–3259. [Google Scholar] [CrossRef]
- Nyokong, T.; Gasyna, Z.; Stillman, M.J. Phthalocyanine.Pi.-Cation-Radical Species: Photochemical and Electrochemical Preparation of [ZnPc(-1).+ in Solution. Inorg. Chem. 1987, 26, 548–553. [Google Scholar] [CrossRef]
- Nyokong, T.; Gasyna, Z.; Stillman, M.J. Analysis of the Absorption and Magnetic Circular Dichroism Spectra of Zinc Phthalocyanine and the.Pi.-Cation-Radical Species [ZnPc(1-)].Cntdot.+. Inorg. Chem. 1987, 26, 1087–1095. [Google Scholar] [CrossRef]
- Nemykin, V.N.; Chernii, V.Y.; Volkov, S.V. Synthesis and Characterization of New Mixed-Ligand Lanthanide–Phthalocyanine Cation Radical Complexes. J. Chem. Soc., Dalton Trans. 1998, 2995–3000. [Google Scholar] [CrossRef]
- Hobbs, C.C. Hydrocarbon Oxidation. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons, Inc.: New York, NY, USA, 1995; Volume 13. [Google Scholar]
- Timokhin, I.; Pettinari, C.; Marchetti, F.; Pettinari, R.; Condello, F.; Galli, S.; Alegria, E.C.B.A.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Novel Coordination Polymers with (Pyrazolato)-Based Tectons: Catalytic Activity in the Peroxidative Oxidation of Alcohols and Cyclohexane. Cryst. Growth Des. 2015, 15, 2303–2317. [Google Scholar] [CrossRef]
- Sutradhar, M.; Martins, L.M.D.R.S.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Oxidovanadium Complexes with Tridentate Aroylhydrazone as Catalyst Precursors for Solvent-Free Microwave-Assisted Oxidation of Alcohols. Appl. Catal. A Gen. 2015, 493, 50–57. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Ribeiro, A.P.C.; Carabineiro, S.A.C.; Figueiredo, J.L.; Pombeiro, A.J.L. Highly Efficient and Reusable CNT Supported Iron(II) Catalyst for Microwave Assisted Alcohol Oxidation. Dalton Trans. 2016, 45, 6816–6819. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, G.R.; Lafferty, N.L.; Sumner, C.E. Catalyst Contg. Cobalt, Zirconium and Opt. Hafnium for Adipic Acid—Prepd. by Air Oxidn. of Cyclohexane in Presence of Acetic and per-Acetic Acids Affords High Yields and Selectivity. U.S. Patent US4902827-A, 1990. [Google Scholar]
- Feng, D.; Jiang, H.-L.; Chen, Y.-P.; Gu, Z.-Y.; Wei, Z.; Zhou, H.-C. Metal–Organic Frameworks Based on Previously Unknown Zr8/Hf8 Cubic Clusters. Inorg. Chem. 2013, 52, 12661–12667. [Google Scholar] [CrossRef]
- Khare, S.; Shrivastava, P.; Chokhare, R.; Kirar, J.S.; Parashar, S. Catalytic Liquid Phase Oxidation of Cyclohexane with Tert-Butylhydroperoxide over Transition Metal Exchanged Alpha-Zirconium Phosphate. Ind. J. Chem., Sect. A 2018, 57A, 424–434. [Google Scholar]
- Khosravi, H.B.; Rahimi, R.; Rabbani, M.; Maleki, A.; Mollahosseini, A. Design, Facile Synthesis and Characterization of Porphyrin-Zirconium-Ferrite@SiO2 Core-Shell and Catalytic Application in Cyclohexane Oxidation. Silicon 2020. [Google Scholar] [CrossRef]
- Ullmann’s Encyclopedia of Industrial Chemistry, 6th ed.; Wiley-VCH: Weinheim, Germany, 2016.
- Shul’pin, G. Hydrocarbon Oxygenations with Peroxides Catalyzed by Metal Compounds. MROC 2009, 6, 95–104. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Methods of Electronic Structure Theory, 1977th ed.; Schaefer, H.F., Ed.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Fox. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Flükiger, P.; Lüthi, H.P.; Portmann, S.; Weber, J. MOLEKEL 4.3; Swiss Center for Scientific Computing: Manno, Switzerland, 2000. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
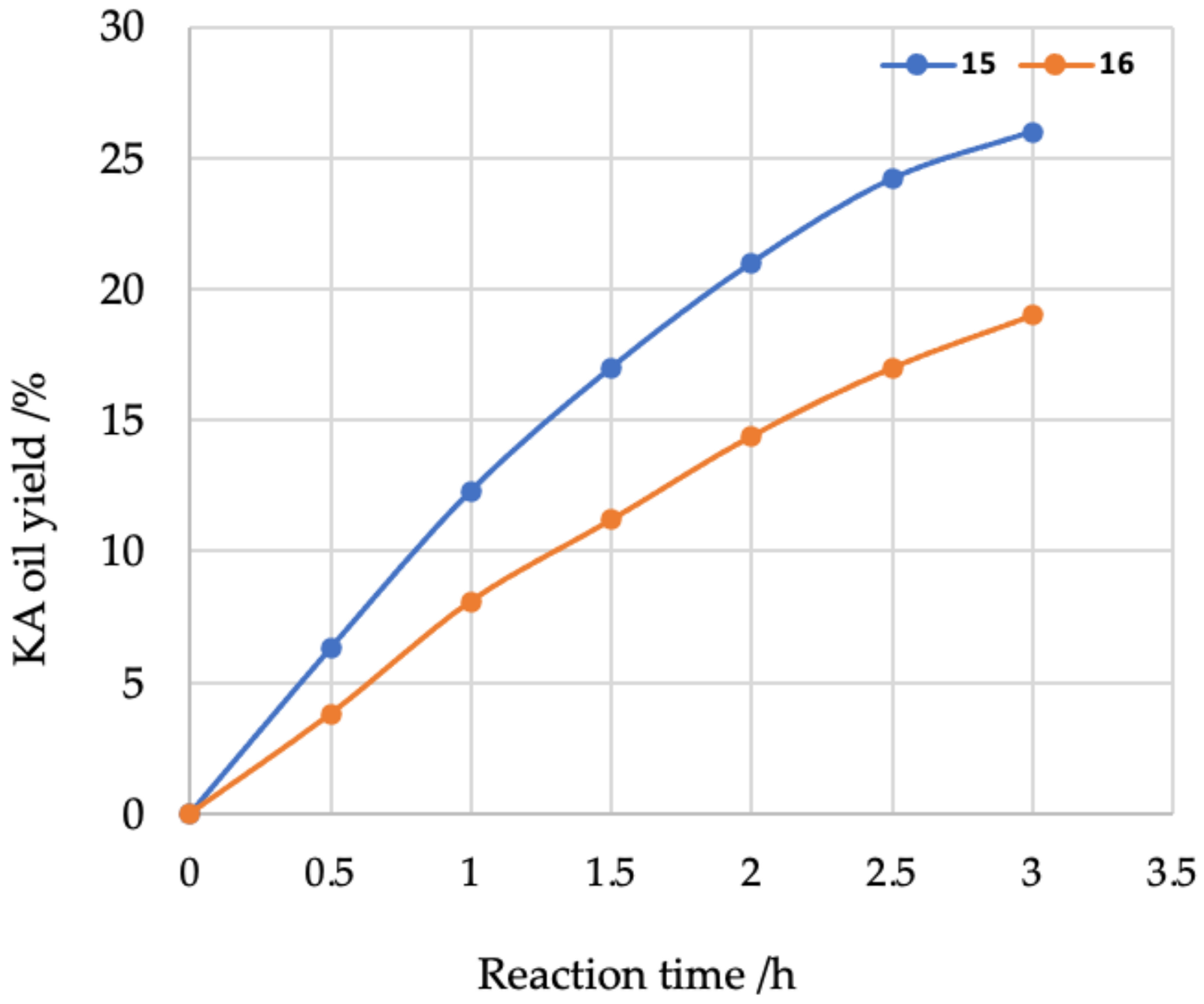{kind=link}
{kind=link}
{kind=link}
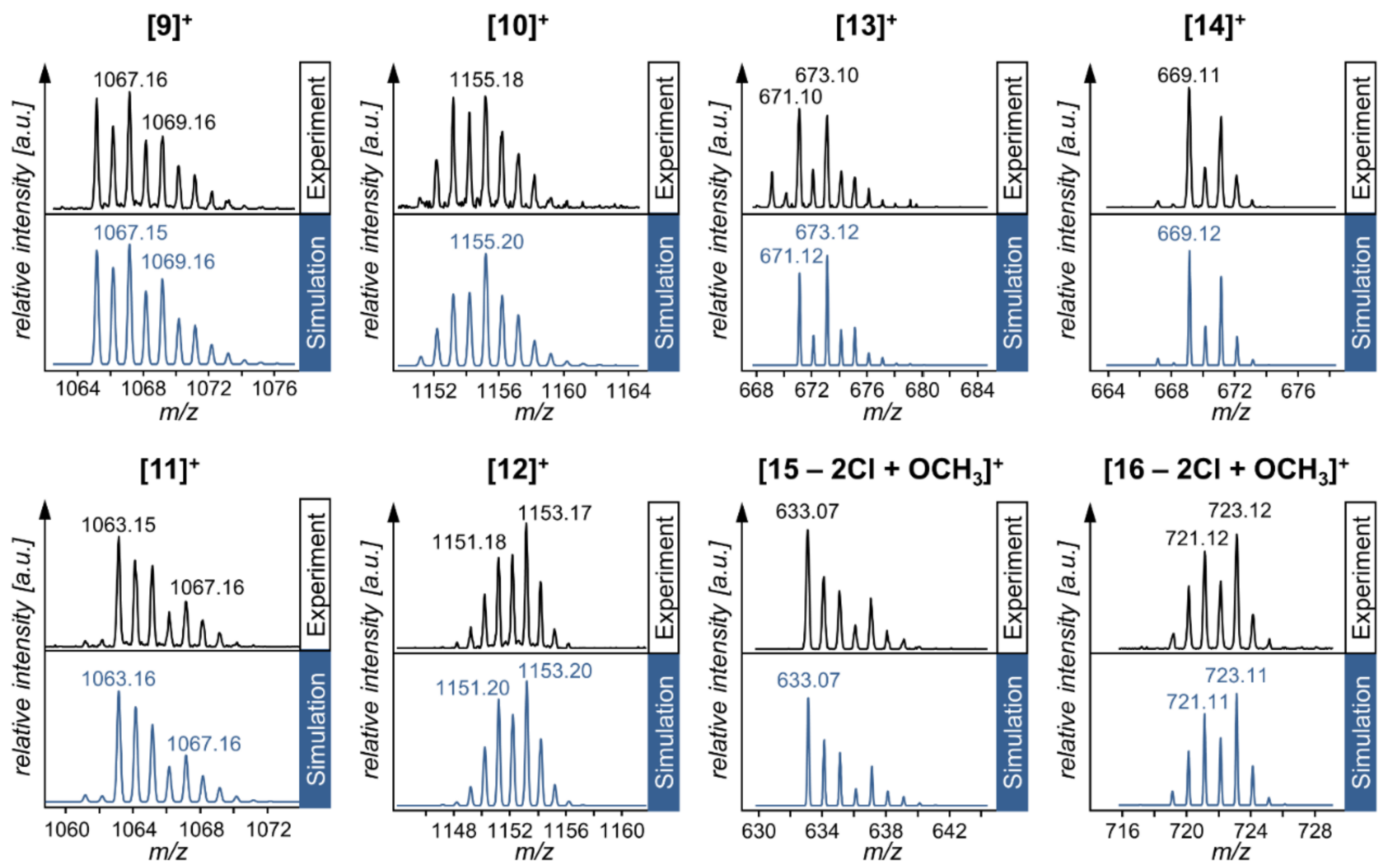
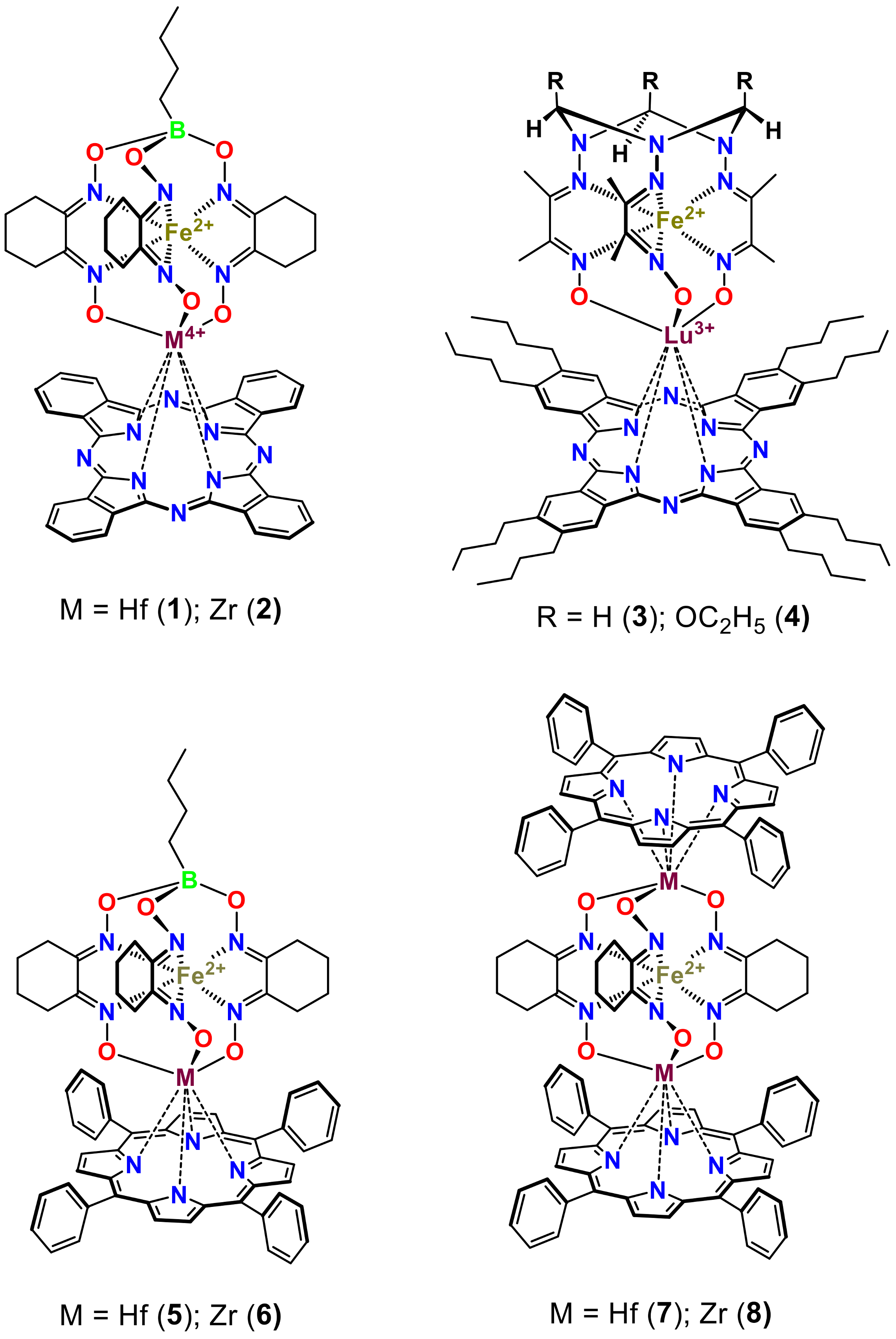
| Compound | Species Observed | mexp | mtheor | |
|---|---|---|---|---|
| Hetero-Bimetallic Coordination Complexes | 9 | [M]+ | 1067.16 | 1067.15 |
| 10 | [M]+ | 1155.18 | 1155.20 | |
| 11 | [M]+ | 1063.15 | 1063.16 | |
| 12 | [M]+ | 1153.17 | 1153.20 | |
| Synthetic Precursors | 13 | [M]+ | 673.10 | 673.12 |
| 14 | [M]+ | 669.11 | 669.12 | |
| 15 | [M − 2Cl + OCH3]+ | 633.07 | 633.07 | |
| 16 | [M − 2Cl + OCH3]+ | 723.12 | 723.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voloshin, Y.Z.; Dudkin, S.V.; Belova, S.A.; Gherca, D.; Samohvalov, D.; Manta, C.-M.; Lungan, M.-A.; Meier-Menches, S.M.; Rapta, P.; Darvasiová, D.; et al. Spectroelectrochemical Properties and Catalytic Activity in Cyclohexane Oxidation of the Hybrid Zr/Hf-Phthalocyaninate-Capped Nickel(II) and Iron(II) tris-Pyridineoximates and Their Precursors. Molecules 2021, 26, 336. https://doi.org/10.3390/molecules26020336
Voloshin YZ, Dudkin SV, Belova SA, Gherca D, Samohvalov D, Manta C-M, Lungan M-A, Meier-Menches SM, Rapta P, Darvasiová D, et al. Spectroelectrochemical Properties and Catalytic Activity in Cyclohexane Oxidation of the Hybrid Zr/Hf-Phthalocyaninate-Capped Nickel(II) and Iron(II) tris-Pyridineoximates and Their Precursors. Molecules. 2021; 26(2):336. https://doi.org/10.3390/molecules26020336
Chicago/Turabian StyleVoloshin, Yan Z., Semyon V. Dudkin, Svetlana A. Belova, Daniel Gherca, Dumitru Samohvalov, Corina-Mihaela Manta, Maria-Andreea Lungan, Samuel M. Meier-Menches, Peter Rapta, Denisa Darvasiová, and et al. 2021. "Spectroelectrochemical Properties and Catalytic Activity in Cyclohexane Oxidation of the Hybrid Zr/Hf-Phthalocyaninate-Capped Nickel(II) and Iron(II) tris-Pyridineoximates and Their Precursors" Molecules 26, no. 2: 336. https://doi.org/10.3390/molecules26020336
APA StyleVoloshin, Y. Z., Dudkin, S. V., Belova, S. A., Gherca, D., Samohvalov, D., Manta, C.-M., Lungan, M.-A., Meier-Menches, S. M., Rapta, P., Darvasiová, D., Malček, M., Pombeiro, A. J. L., Martins, L. M. D. R. S., & Arion, V. B. (2021). Spectroelectrochemical Properties and Catalytic Activity in Cyclohexane Oxidation of the Hybrid Zr/Hf-Phthalocyaninate-Capped Nickel(II) and Iron(II) tris-Pyridineoximates and Their Precursors. Molecules, 26(2), 336. https://doi.org/10.3390/molecules26020336