
Structural Basis for PPARs Activation by The Dual PPARα/γ Agonist Sanguinarine: A Unique Mode of Ligand Recognition


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
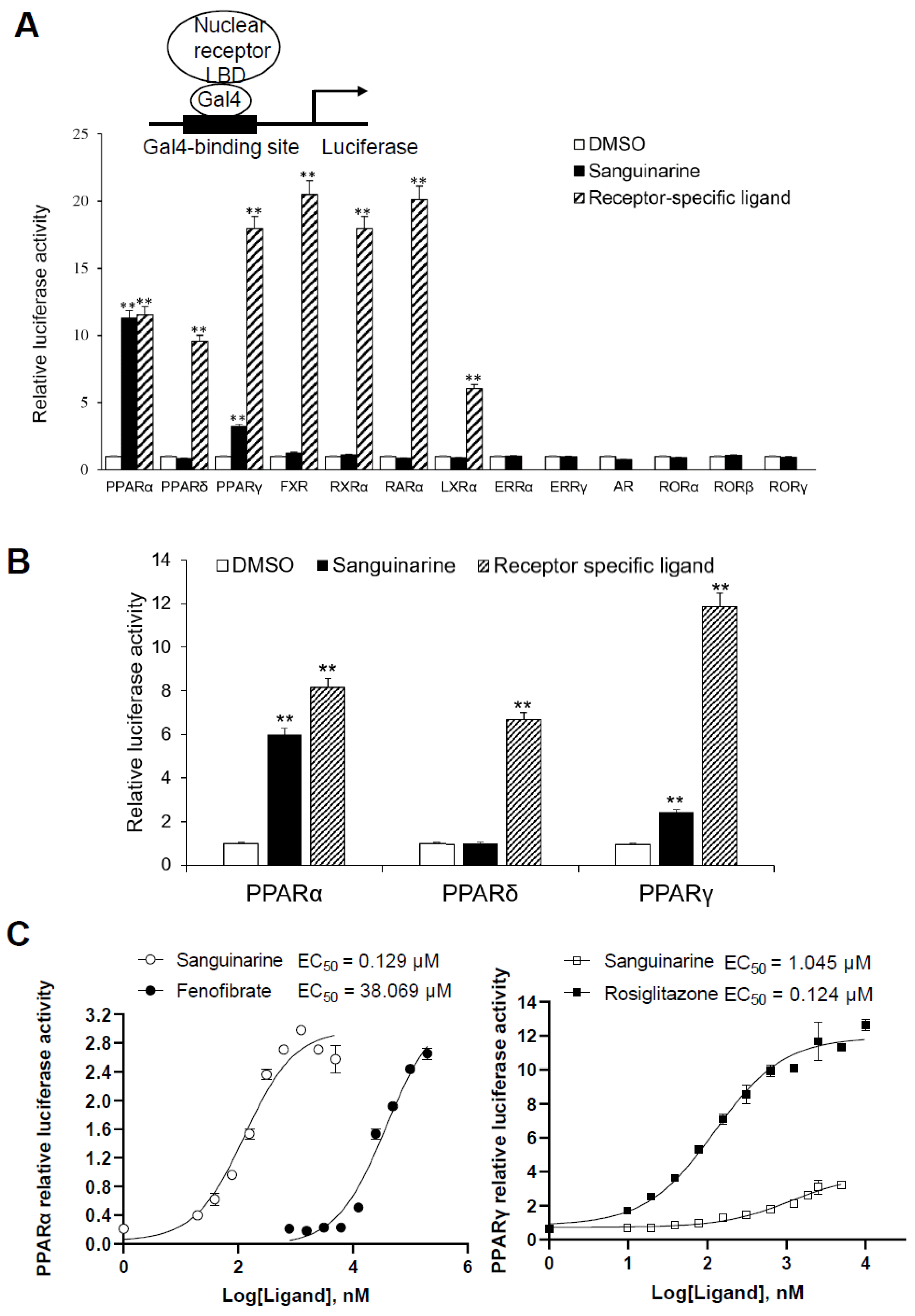
2.1. Identification of Sanguinarine as a Dual PPARα/γ Agonist
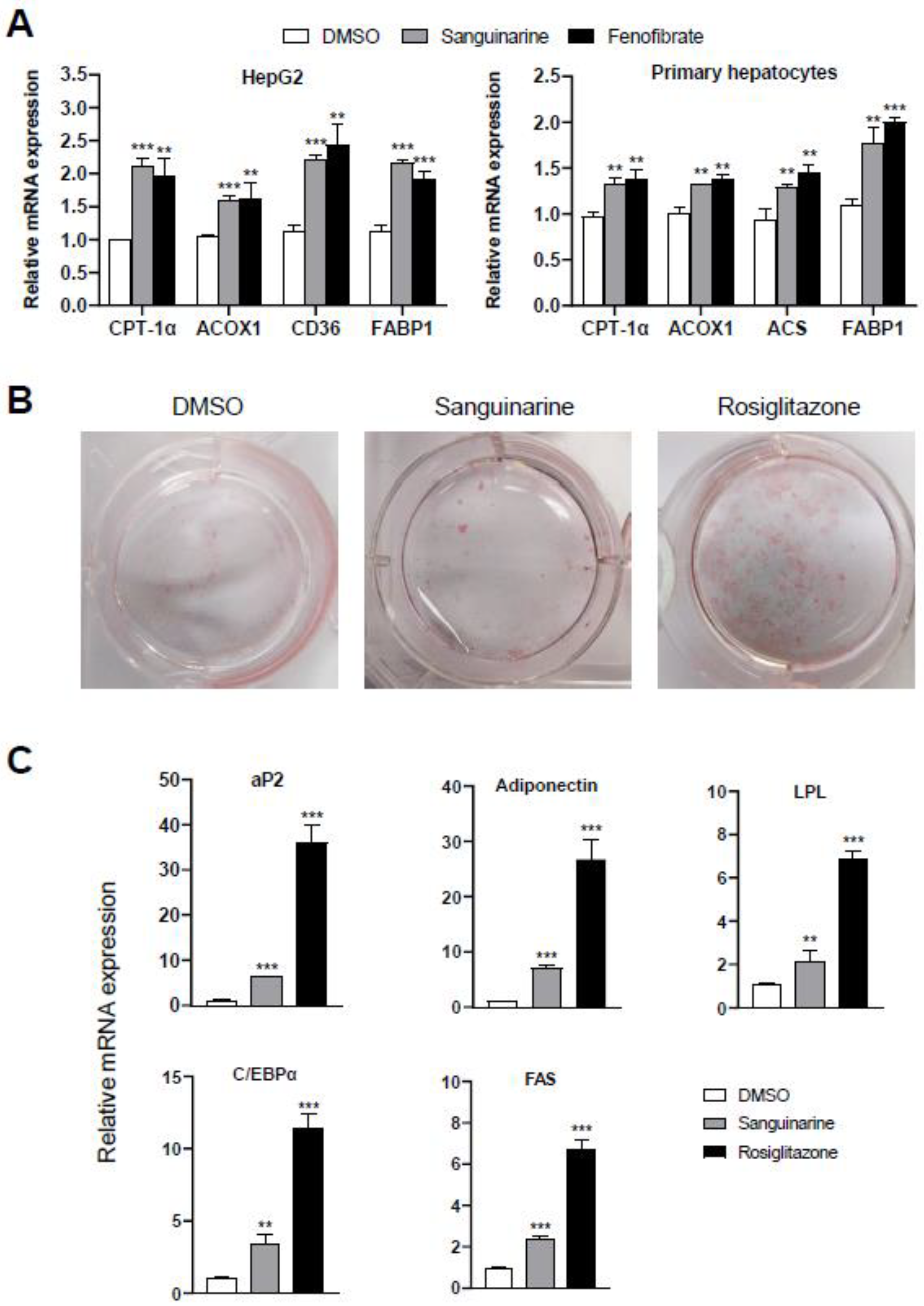
2.2. Sanguinarine Regulates PPARα-Target Genes in Hepatocytes and PPARγ-Target Genes in Adipocytes
2.3. Overall Structure of Sanguinarine and PPARα with Unique Binding Mode
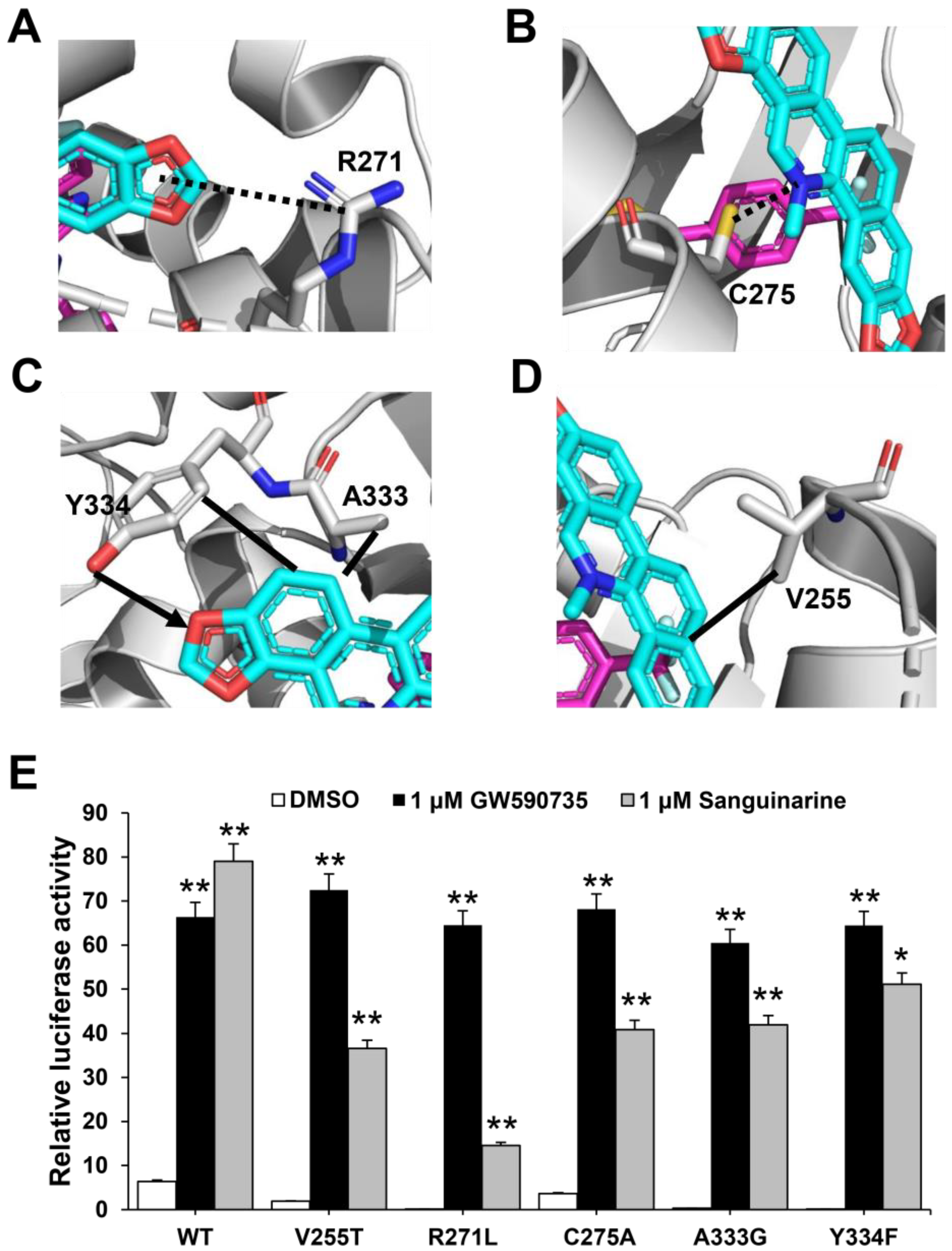
2.4. Functional Correlation of the Sanguinarine-PPARα Interactions
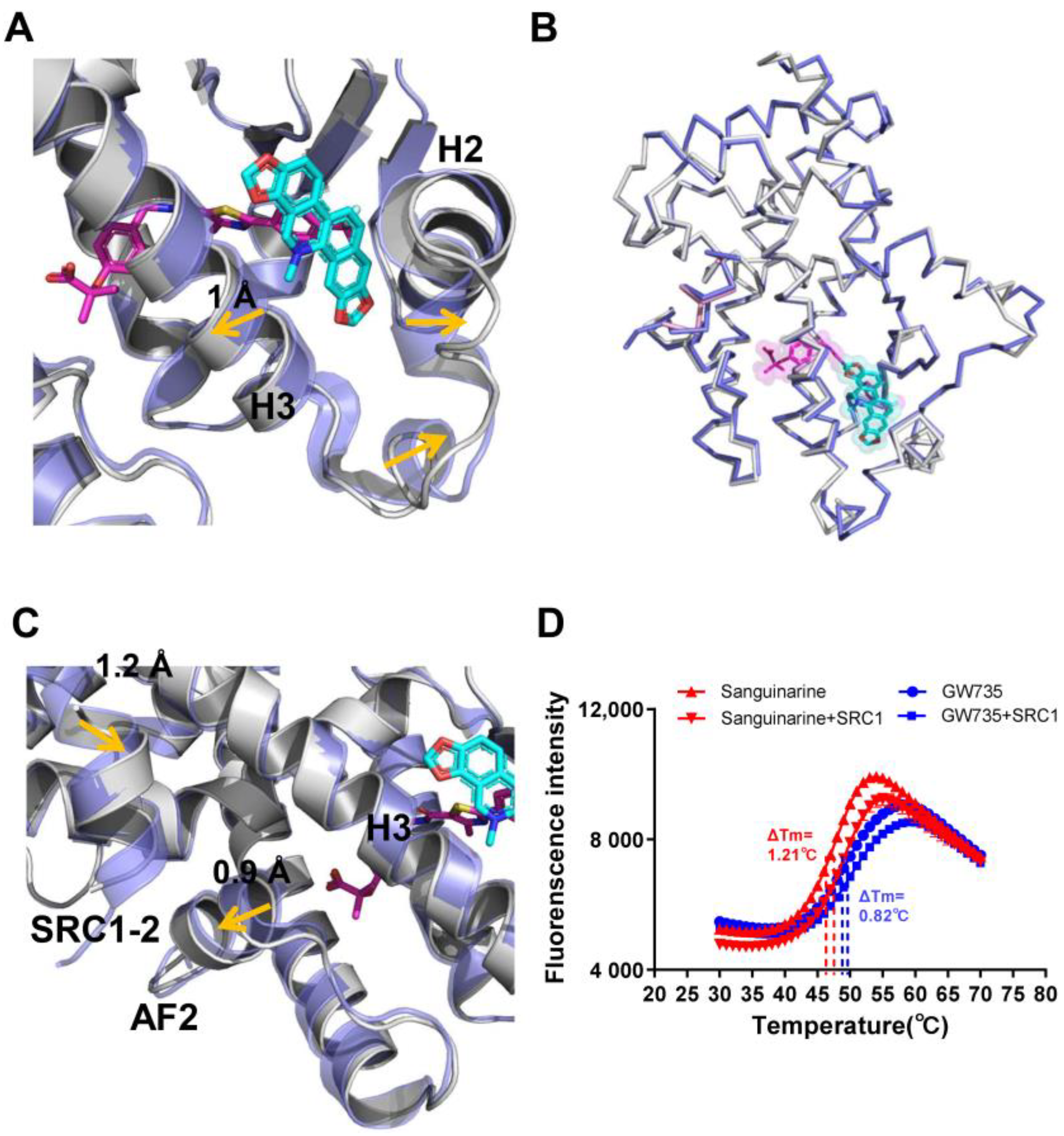
2.5. Conformational Changes of PPARα Induced by Sanguinarine Binding
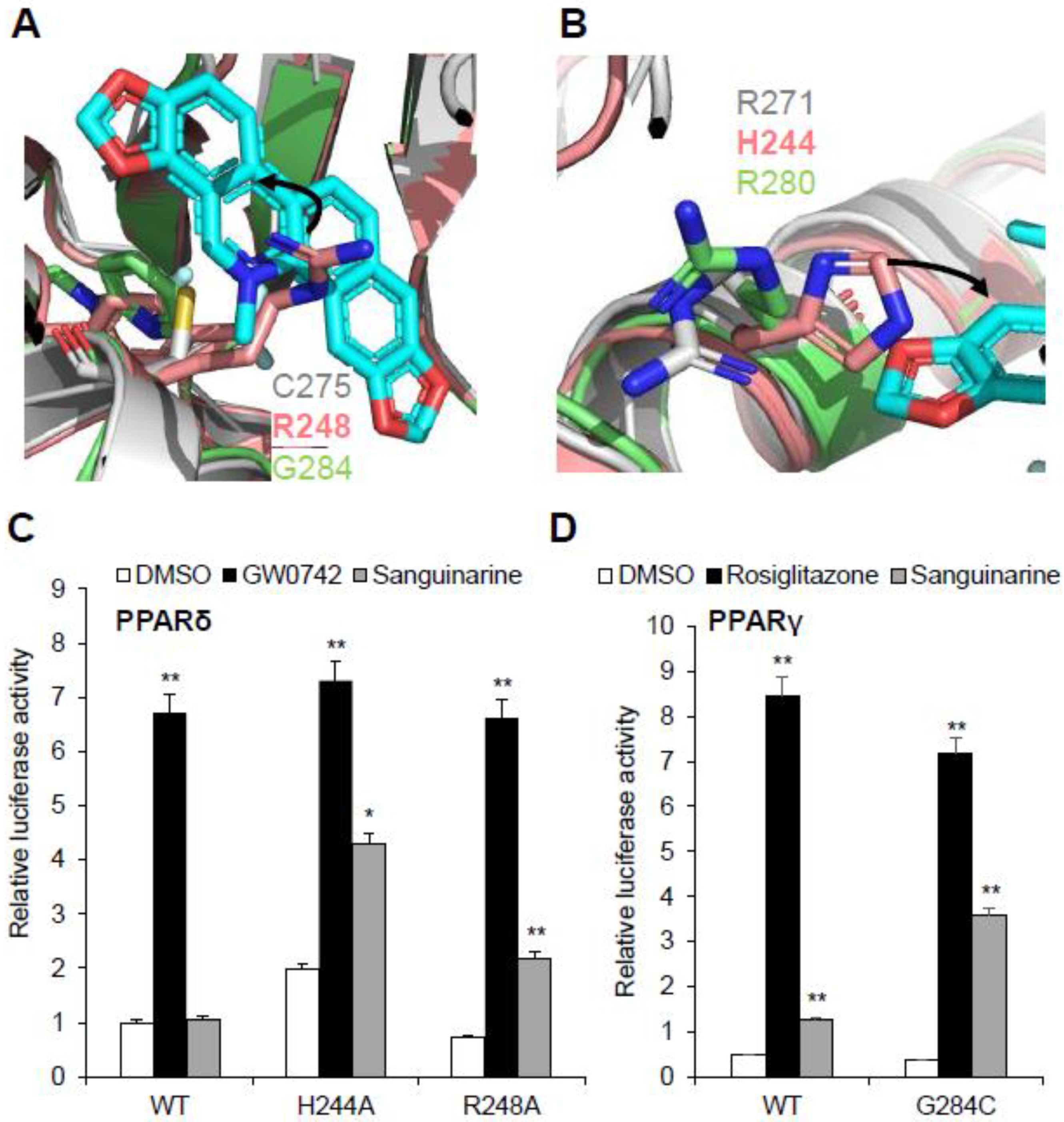
2.6. Molecular Determinants of PPAR Subtype Selectivity toward Sanguinarine
3. Discussion
4. Materials and Methods
4.1. Protein Preparation
4.2. Coactivator Binding Assays
4.3. Transient Transfection Assays
4.4. Thermal Shift Assay
4.5. Preparation of Mouse Primary Hepatocytes
4.6. 3T3-L1 Cells Differentiation
4.7. Quantitative Real-Time PCR (qPCR)
4.8. Crystallization and Structure Determination
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| PPAR | Peroxisome proliferator-activated receptors |
| AF-1 | activation function 1 |
| DBD | DNA-binding domain |
| LBD | ligand-binding domain |
| NASH | nonalcoholic steatohepatitis |
| TZDs | Thiazolidinediones |
| TSA | thermal shift assay |
References
- Giampietro, L.; Laghezza, A.; Cerchia, C.; Florio, R.; Recinella, L.; Capone, F.; Ammazzalorso, A.; Bruno, I.; De Filippis, B.; Fantacuzzi, M.; et al. Novel Phenyldiazenyl Fibrate Analogues as PPAR α/γ/δ Pan-Agonists for the Amelioration of Metabolic Syndrome. ACS Med. Chem. Lett. 2019, 10, 545–551. [Google Scholar] [CrossRef]
- Bain, D.L.; Heneghan, A.F.; Connaghan-Jones, K.D.; Miura, M.T. Nuclear receptor structure: Implications for function. Annu. Rev. Physiol. 2007, 69, 201–220. [Google Scholar] [CrossRef]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.; Oplinger, J.A.; Kliewer, S.A.; Gampe, R.T., Jr.; McKee, D.D.; et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 13919–13924. [Google Scholar] [CrossRef] [Green Version]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef]
- Yu, S.; Reddy, J.K. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim. Biophys. Acta 2007, 1771, 936–951. [Google Scholar] [CrossRef]
- Kuwabara, N.; Oyama, T.; Tomioka, D.; Ohashi, M.; Yanagisawa, J.; Shimizu, T.; Miyachi, H. Peroxisome proliferator-activated receptors (PPARs) have multiple binding points that accommodate ligands in various conformations: Phenylpropanoic acid-type PPAR ligands bind to PPAR in different conformations, depending on the subtype. J. Med. Chem. 2012, 55, 893–902. [Google Scholar] [CrossRef]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Forder, P.; Pillai, A.; Davis, T.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Barter, P.J.; Rye, K.A. Is there a role for fibrates in the management of dyslipidemia in the metabolic syndrome? Arterioscler. Thromb. Vasc. Biol. 2008, 28, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henke, B.R.; Blanchard, S.G.; Brackeen, M.F.; Brown, K.K.; Cobb, J.E.; Collins, J.L.; Harrington, W.W., Jr.; Hashim, M.A.; Hull-Ryde, E.A.; Kaldor, I.; et al. N-(2-Benzoylphenyl)-L-tyrosine PPARgamma agonists. 1. Discovery of a novel series of potent antihyperglycemic and antihyperlipidemic agents. J. Med. Chem. 1998, 41, 5020–5036. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Hao, C.; Cha, D.R.; Rao, R.; Lu, W.; Kohan, D.E.; Magnuson, M.A.; Redha, R.; Zhang, Y.; Breyer, M.D. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat. Med. 2005, 11, 861–866. [Google Scholar] [CrossRef]
- Wei, W.; Wang, X.; Yang, M.; Smith, L.C.; Dechow, P.C.; Sonoda, J.; Evans, R.M.; Wan, Y. PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone-induced bone loss. Cell Metab. 2010, 11, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Gaziano, R.; Moroni, G.; Buè, C.; Miele, M.T.; Sinibaldi-Vallebona, P.; Pica, F. Antitumor effects of the benzophenanthridine alkaloid sanguinarine: Evidence and perspectives. World J. Gastrointest. Oncol. 2016, 8, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Bessi, I.; Bazzicalupi, C.; Richter, C.; Jonker, H.R.; Saxena, K.; Sissi, C.; Chioccioli, M.; Bianco, S.; Bilia, A.R.; Schwalbe, H.; et al. Spectroscopic, molecular modeling, and NMR-spectroscopic investigation of the binding mode of the natural alkaloids berberine and sanguinarine to human telomeric G-quadruplex DNA. ACS Chem. Biol. 2012, 7, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Kumar, G.S. Sanguinarine and Its Role in Chronic Diseases. Adv. Exp. Med. Biol. 2016, 928, 155–172. [Google Scholar] [PubMed]
- Fu, C.; Guan, G.; Wang, H. The Anticancer Effect of Sanguinarine: A Review. Curr. Pharm. Des. 2018, 24, 2760–2764. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Molecular targets and anticancer potential of sanguinarine-a benzophenanthridine alkaloid. Phytomedicine 2017, 34, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Mackraj, I.; Govender, T.; Gathiram, P. Sanguinarine. Cardiovasc. Ther. 2008, 26, 75–83. [Google Scholar] [CrossRef]
- Singh, R.; Mackraj, I.; Naidoo, R.; Gathiram, P. Sanguinarine downregulates AT1a gene expression in a hypertensive rat model. J. Cardiovasc. Pharmacol. 2006, 48, 14–21. [Google Scholar] [CrossRef]
- Sierra, M.L.; Beneton, V.; Boullay, A.B.; Boyer, T.; Brewster, A.G.; Donche, F.; Forest, M.C.; Fouchet, M.H.; Gellibert, F.J.; Grillot, D.A.; et al. Substituted 2-[(4-aminomethyl)phenoxy]-2-methylpropionic acid PPARalpha agonists. 1. Discovery of a novel series of potent HDLc raising agents. J. Med. Chem. 2007, 50, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Kamata, S.; Oyama, T.; Saito, K.; Honda, A.; Yamamoto, Y.; Suda, K.; Ishikawa, R.; Itoh, T.; Watanabe, Y.; Shibata, T.; et al. PPARα Ligand-Binding Domain Structures with Endogenous Fatty Acids and Fibrates. iScience 2020, 23, 101727. [Google Scholar] [CrossRef]
- Li, Y.; Suino, K.; Daugherty, J.; Xu, H.E. Structural and biochemical mechanisms for the specificity of hormone binding and coactivator assembly by mineralocorticoid receptor. Mol. Cell 2005, 19, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, X.Y.; Wu, D.H.; Xu, Y. ROR nuclear receptors: Structures, related diseases, and drug discovery. Acta Pharmacol. Sin. 2015, 36, 71–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.W.; Tang, C.L.; Zheng, H.; Wu, J.X.; Wu, F.; Mo, Y.Y.; Liu, X.; Zhu, H.J.; Yin, C.L.; Cheng, B.; et al. Investigation of the hepatoprotective effect of Corydalis saxicola Bunting on carbon tetrachloride-induced liver fibrosis in rats by (1)H-NMR-based metabonomics and network pharmacology approaches. J. Pharm. Biomed. Anal. 2018, 159, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, K.A.; Reinking, J.L. Use of Differential Scanning Fluorimetry to Identify Nuclear Receptor Ligands. Methods Mol. Biol. 2016, 1443, 21–30. [Google Scholar]
- Rogue, A.; Renaud, M.P.; Claude, N.; Guillouzo, A.; Spire, C. Comparative gene expression profiles induced by PPARγ and PPARα/γ agonists in rat hepatocytes. Toxicol. Appl. Pharmacol. 2011, 254, 18–31. [Google Scholar] [CrossRef]
- Allen, T.; Zhang, F.; Moodie, S.A.; Clemens, L.E.; Smith, A.; Gregoire, F.; Bell, A.; Muscat, G.E.; Gustafson, T.A. Halofenate is a selective peroxisome proliferator-activated receptor gamma modulator with antidiabetic activity. Diabetes 2006, 55, 2523–2533. [Google Scholar] [CrossRef] [Green Version]
- Madsen, L.; Petersen, R.K.; Sørensen, M.B.; Jørgensen, C.; Hallenborg, P.; Pridal, L.; Fleckner, J.; Amri, E.Z.; Krieg, P.; Furstenberger, G.; et al. Adipocyte differentiation of 3T3-L1 preadipocytes is dependent on lipoxygenase activity during the initial stages of the differentiation process. Biochem. J. 2003, 375 Pt 3, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Takada, I.; Makishima, M. Peroxisome proliferator-activated receptor agonists and antagonists: A patent review (2014-present). Expert Opin. Ther. Pat. 2020, 30, 1–13. [Google Scholar] [CrossRef]
- Lamers, C.; Schubert-Zsilavecz, M.; Merk, D. Therapeutic modulators of peroxisome proliferator-activated receptors (PPAR): A patent review (2008-present). Expert Opin. Ther. Pat. 2012, 22, 803–841. [Google Scholar] [CrossRef]
- Li, Y.; Lambert, M.H.; Xu, H.E. Activation of nuclear receptors: A perspective from structural genomics. Structure 2003, 11, 741–746. [Google Scholar] [CrossRef] [Green Version]
- Kalliora, C.; Drosatos, K. The Glitazars Paradox: Cardiotoxicity of the Metabolically Beneficial Dual PPARα and PPARγ Activation. J. Cardiovasc. Pharmacol. 2020, 76, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Sharma, B. Toxicological Effects of Berberine and Sanguinarine. Front. Mol. Biosci. 2018, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Ulrichová, J.; Dvorák, Z.; Vicar, J.; Lata, J.; Smrzová, J.; Sedo, A.; Simánek, V. Cytotoxicity of natural compounds in hepatocyte cell culture models. The case of quaternary benzo[c]phenanthridine alkaloids. Toxicol. Lett. 2001, 125, 125–132. [Google Scholar] [CrossRef]
- Ullman, E.F.; Kirakossian, H.; Singh, S.; Wu, Z.P.; Irvin, B.R.; Pease, J.S.; Switchenko, A.C.; Irvine, J.D.; Dafforn, A.; Skold, C.N.; et al. Luminescent oxygen channeling immunoassay: Measurement of particle binding kinetics by chemiluminescence. Proc. Natl. Acad. Sci. USA 1994, 91, 5426–5430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadowski, I.; Ma, J.; Triezenberg, S.; Ptashne, M. GAL4-VP16 is an unusually potent transcriptional activator. Nature 1988, 335, 563–564. [Google Scholar] [CrossRef] [PubMed]
- Fields, S.; Song, O. A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef]
- Kim, Y.I.; Hirai, S.; Takahashi, H.; Goto, T.; Ohyane, C.; Tsugane, T.; Konishi, C.; Fujii, T.; Inai, S.; Iijima, Y.; et al. 9-oxo-10(E),12(E)-Octadecadienoic acid derived from tomato is a potent PPAR α agonist to decrease triglyceride accumulation in mouse primary hepatocytes. Mol. Nutr. Food Res. 2011, 55, 585–593. [Google Scholar] [CrossRef]
- Yoon, S.; Kim, J.; Lee, H.; Lee, H.; Lim, J.; Yang, H.; Shin, S.S.; Yoon, M. The effects of herbal composition Gambigyeongsinhwan (4) on hepatic steatosis and inflammation in Otsuka Long-Evans Tokushima fatty rats and HepG2 cells. J. Ethnopharmacol. 2017, 195, 204–213. [Google Scholar] [CrossRef]
- Waki, H.; Park, K.W.; Mitro, N.; Pei, L.; Damoiseaux, R.; Wilpitz, D.C.; Reue, K.; Saez, E.; Tontonoz, P. The small molecule harmine is an antidiabetic cell-type-specific regulator of PPARgamma expression. Cell Metab. 2007, 5, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 4, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, S.; Wang, R.; Chen, S.; He, J.; Zheng, W.; Li, Y. Structural Basis for PPARs Activation by The Dual PPARα/γ Agonist Sanguinarine: A Unique Mode of Ligand Recognition. Molecules 2021, 26, 6012. https://doi.org/10.3390/molecules26196012
Tian S, Wang R, Chen S, He J, Zheng W, Li Y. Structural Basis for PPARs Activation by The Dual PPARα/γ Agonist Sanguinarine: A Unique Mode of Ligand Recognition. Molecules. 2021; 26(19):6012. https://doi.org/10.3390/molecules26196012
Chicago/Turabian StyleTian, Siyu, Rui Wang, Shuming Chen, Jialing He, Weili Zheng, and Yong Li. 2021. "Structural Basis for PPARs Activation by The Dual PPARα/γ Agonist Sanguinarine: A Unique Mode of Ligand Recognition" Molecules 26, no. 19: 6012. https://doi.org/10.3390/molecules26196012
APA StyleTian, S., Wang, R., Chen, S., He, J., Zheng, W., & Li, Y. (2021). Structural Basis for PPARs Activation by The Dual PPARα/γ Agonist Sanguinarine: A Unique Mode of Ligand Recognition. Molecules, 26(19), 6012. https://doi.org/10.3390/molecules26196012