
Nitrogen-Based Linkers with a Mesitylene Core: Synthesis and Characterization

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
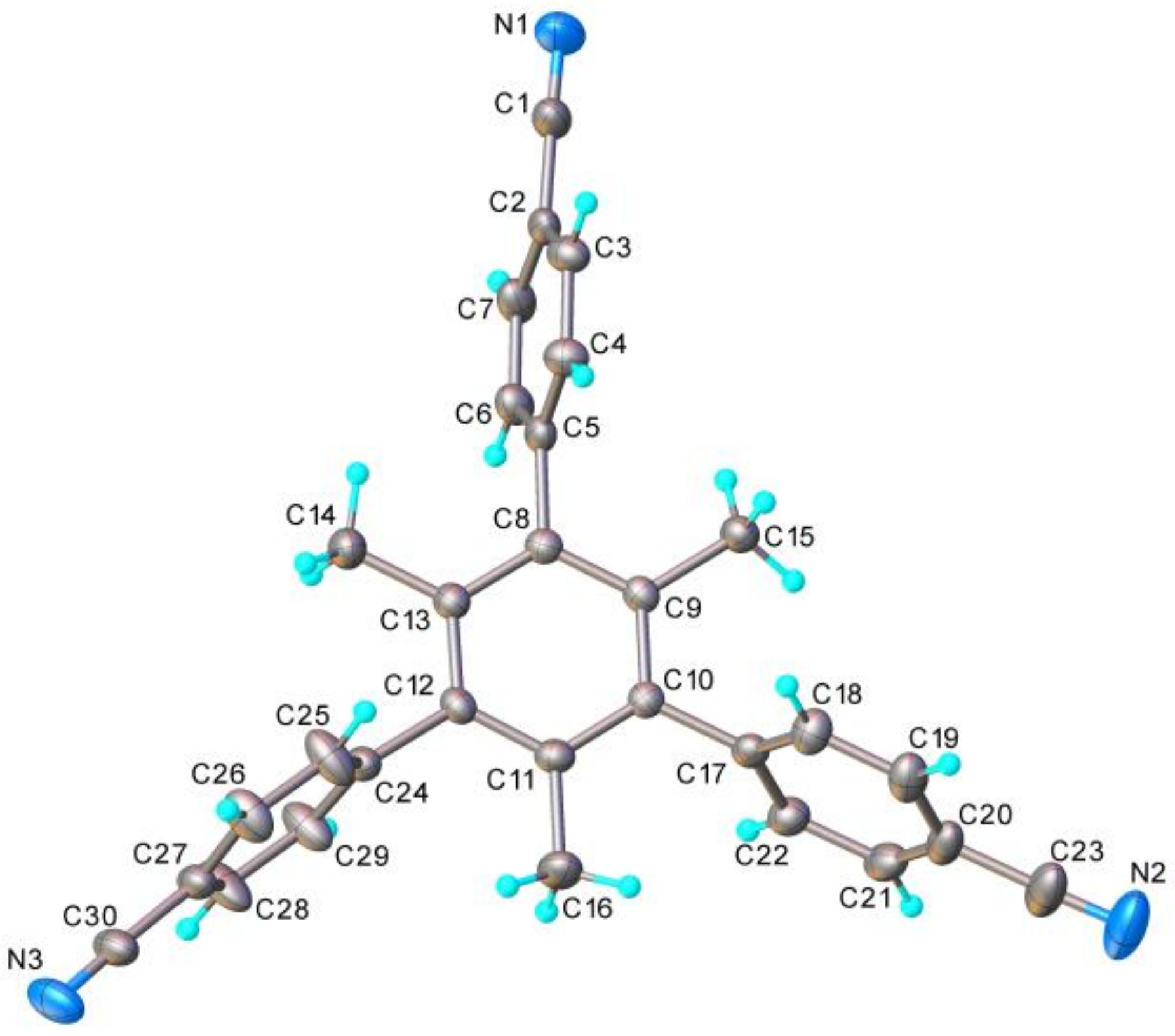
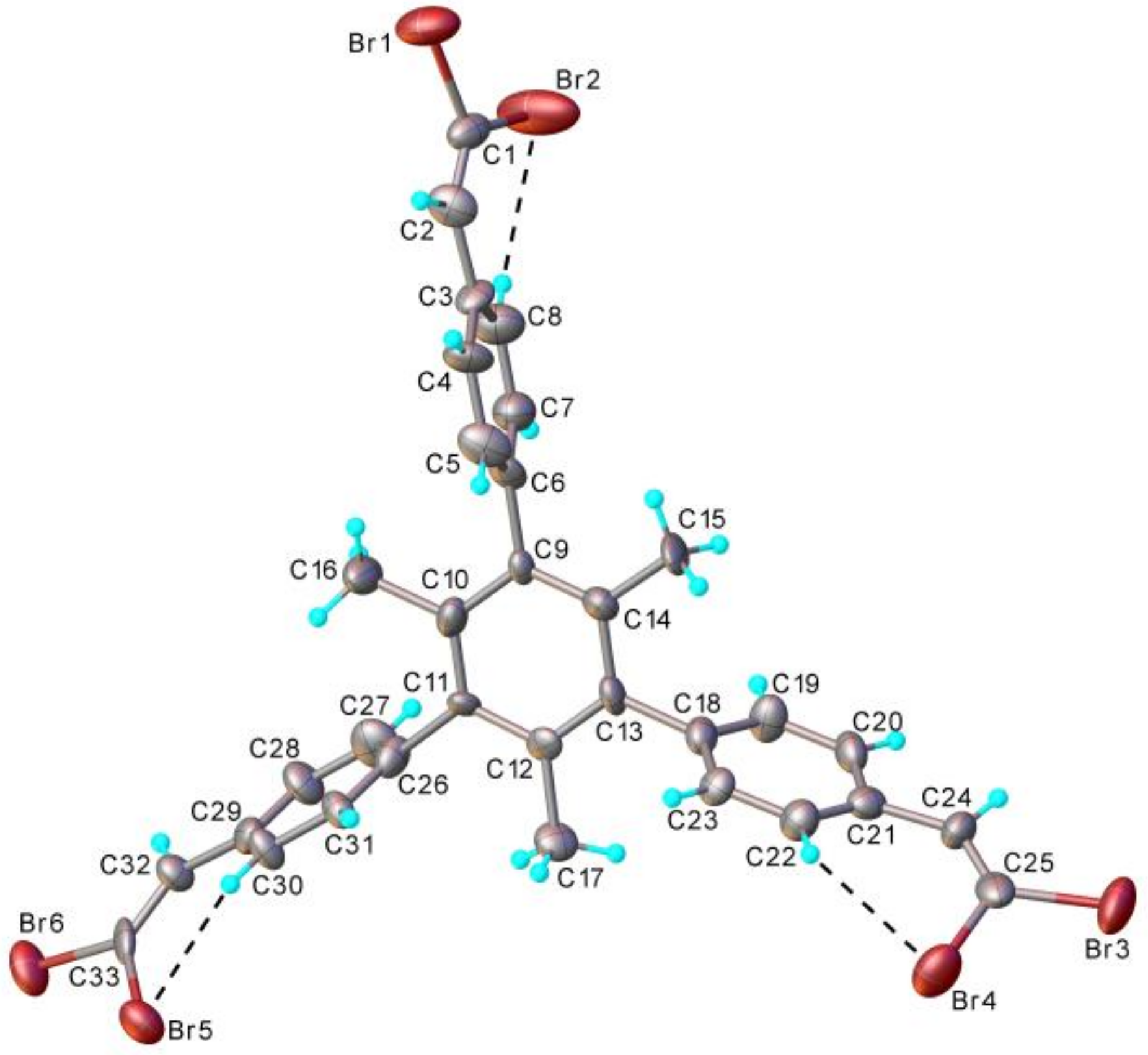
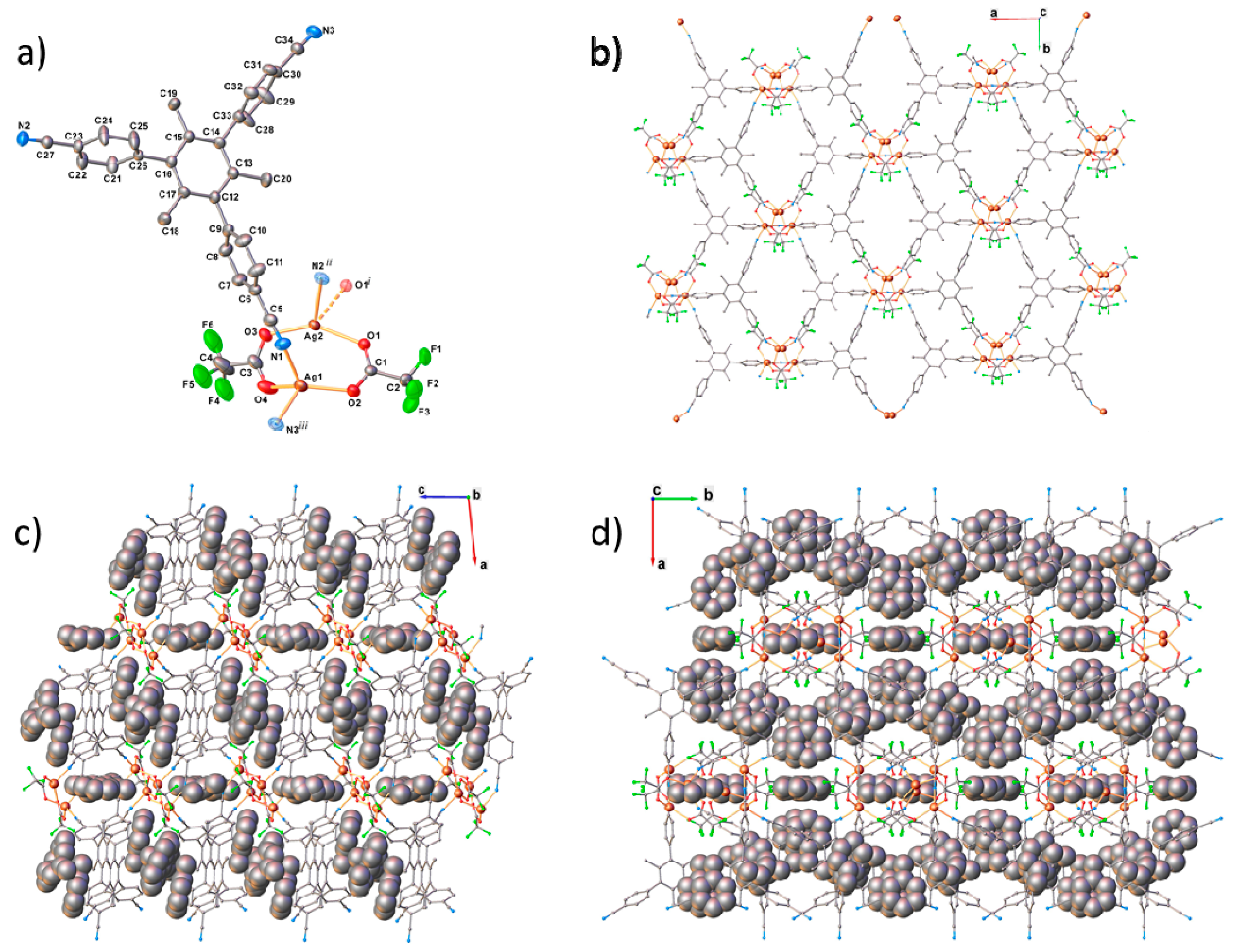
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of 2
3.1.2. Synthesis of 4
3.1.3. Synthesis of 6
3.1.4. Synthesis of 3
3.1.5. Synthesis of 5
3.1.6. Synthesis of 7
3.1.7. Synthesis of 9
3.1.8. Synthesis of 10
3.1.9. Synthesis of 11
3.1.10. Synthesis of 12
3.2. X-ray Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Szabo, B. Imidazoline antihypertensive drugs: A critical review on their mechanism of action. Pharmacol. Ther. 2002, 93, 1–35. [Google Scholar] [CrossRef]
- Peng, X.M.; Cai, G.X.; Zhou, C.H. Recent developments in azole compounds as antibacterial and antifungal agents. Curr. Top. Med. Chem. 2013, 13, 1963–2010. [Google Scholar] [CrossRef]
- Sabatini, J.J.; Moretti, J.D. High-nitrogen-based pyrotechnics: Perchlorate-free red-and green-light illuminants based on 5-aminotetrazole. Chem. Eur. J. 2013, 19, 12839–12845. [Google Scholar] [CrossRef]
- Biot, C.; Bauer, H.; Schirmer, R.H.; Davioud-Charvet, E. 5-Substituted tetrazoles as bioisosteres of carboxylic acids. Bioisosterism and mechanistic studies on glutathione reductase inhibitors as antimalarials. J. Med. Chem. 2004, 47, 5972–5983. [Google Scholar] [CrossRef]
- Janiak, C.; Vieth, J.K. MOFs, MILs and more: Concepts, properties and applications for porous coordination networks (PCNs). New J. Chem. 2010, 34, 2366–2388. [Google Scholar] [CrossRef]
- Kalaj, M.; Bentz, K.C.; Ayala, S., Jr.; Palomba, J.M.; Barcus, K.S.; Katayama, Y.; Cohen, S.M. MOF-polymer hybrid materials: From simple composites to tailored architectures. Chem. Rev. 2020, 120, 8267–8302. [Google Scholar] [CrossRef]
- Qian, Q.; Asinger, P.A.; Lee, M.J.; Han, G.; Rodriguez, K.M.; Lin, S.; Benedetti, F.M.; Wu, A.X.; Chi, W.S.; Smith, Z.P. MOF-based membranes for gas separations. Chem. Rev. 2020, 120, 8161–8266. [Google Scholar] [CrossRef] [PubMed]
- Bavykina, A.; Kolobov, N.; Khan, I.S.; Bau, J.A.; Ramirez, A.; Gascon, J. Metal–Organic Frameworks in heterogeneous catalysis: Recent progress, new trends, and future perspectives. Chem. Rev. 2020, 120, 8468–8535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alharbi, N.S.; Hu, B.; Hayat, T.; Rabah, S.O.; Alsaedi, A.; Zhuang, L.; Wang, X. Efficient elimination of environmental pollutants through sorption-reduction and photocatalytic degradation using nanomaterials. Front. Chem. Sci. Eng. 2020, 14, 1124–1135. [Google Scholar] [CrossRef]
- Yin, H.Q.; Yin, X.B. Metal–Organic Frameworks with multiple luminescence emissions: Designs and applications. Acc. Chem. Res. 2020, 53, 485–495. [Google Scholar] [CrossRef]
- Lu, Z.; Meng, F.; Du, L.; Jiang, W.; Cao, H.; Duan, J.; Huang, H.; He, H. A free tetrazolyl decorated Metal–Organic Framework exhibiting high and selective CO2 adsorption. Inorg. Chem. 2018, 57, 14018–14022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Meng, D.X.; Ge, F.Y.; Huang, J.H.; Wang, L.F.; Xv, Y.K.; Liu, X.G.; Meng, M.M.; Yan, H.; Lu, Z.Z.; et al. Tetrazole-based porous metal–organic frameworks for selective CO2 adsorption and isomerization studies. Dalton Trans. 2020, 49, 2145–2150. [Google Scholar] [CrossRef]
- Deng, S.Q.; Miao, Y.L.; Tan, Y.L.; Fang, H.N.; Li, Y.T.; Mo, X.J.; Cai, S.L.; Fan, J.; Zhang, W.G.; Zheng, S.R. An anionic nanotubular Metal–Organic Framework for high-capacity dye adsorption and dye degradation in darkness. Inorg. Chem. 2019, 58, 13979–13987. [Google Scholar] [CrossRef]
- Mariyam, A.; Shahid, M.; Mantasha, I.; Khan, M.S.; Ahmad, M.S. Tetrazole based porous Metal Organic Framework (MOF): Topological analysis and dye adsorption properties. J. Inorg. Organomet. Polym. 2020, 30, 1935–1943. [Google Scholar] [CrossRef]
- Li, Y.; An, J.D.; Wang, T.T.; Shi, Y.F.; Huo, J.Z.; Wu, X.X.; Liu, Y.Y.; Ding, B. An ultra-stable cadmium(ii) coordination framework constructed from the new bi-functional ligand and application as fluorescent probe for acetylacetone and antibiotics. Dyes Pigment. 2021, 186, 109039. [Google Scholar] [CrossRef]
- Zhu, K.; Fan, R.; Wu, J.; Wang, B.; Lu, H.; Zheng, X.; Sun, T.; Gai, S.; Zhou, X.; Yang, Y. MOF-on-MOF membrane with cascading functionality for capturing dichromate ions and p-arsanilic acid turn-on sensing. ACS Appl. Mater. Interfaces 2020, 12, 58239–58251. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Xiong, X.; Xiong, J.; Krishna, R.; Li, L.; Fan, Y.; Luo, F.; Chen, B. A robust Th-azole framework for highly efficient purification of C2H4 from a C2H4/C2H2/C2H6 mixture. Nat. Commun. 2020, 11, 3163. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, S.; Deng, M.; Zhang, T.; Zhang, Q. Energetic Metal-Organic Frameworks incorporating NH3−OH+ for new high-energy-density materials. Inorg. Chem. 2019, 58, 12228–12233. [Google Scholar] [CrossRef]
- Lin, J.D.; Chen, F.; Xu, J.G.; Zheng, F.K.; Wen, N. Framework-interpenetrated nitrogen-rich Zn(II) Metal–Organic Frameworks for energetic materials. ACS Appl. Nano Mater. 2019, 2, 5116–5124. [Google Scholar] [CrossRef]
- Wang, Z.D.; Zang, Y.; Liu, Z.J.; Wang, P.P.R.; Zang, S.Q. Opening catalytic sites in the copper-triazoles framework via defect chemistry for switching on the proton reduction. Appl. Catal. B-Environ. 2021, 288, 119941. [Google Scholar] [CrossRef]
- Li, J.; He, Y.; Wang, L.; Pan, Q.; Song, Z.; Shi, X. Design and synthesis of photoluminescent active interpenetrating metal-organic frameworks using N-2-aryl-1,2,3-triazole ligands. Dalton Trans. 2020, 49, 5429–5433. [Google Scholar] [CrossRef]
- Wang, X.M.; Wang, C.; Zhang, N.; Liu, D.Q.; Wang, Y.; Bai, F.Y. Multifunctional inorganic-organic U-MOF materials with nitrogen heterocyclic carboxylate: Synthesis, structure and properties. ChemistrySelect 2020, 5, 8625–8634. [Google Scholar] [CrossRef]
- Gontcharenko, V.E.; Lunev, A.M.; Taydakov, I.V.; Korshunov, V.M.; Drozdov, A.A.; Belousov, Y.A. Luminescent lanthanide-based sensor for H2O detection in aprotic solvents and D2O. IEEE Sens. J. 2019, 19, 7365–7372. [Google Scholar] [CrossRef]
- Wang, D.; Sun, L.; Hao, C.; Yan., Y.; Liang, Z. Lanthanide metal–organic frameworks based on a 1,2,3-triazole-containing tricarboxylic acid ligand for luminescence sensing of metal ions and nitroaromatic compounds. RSC Adv. 2016, 6, 57828–57834. [Google Scholar] [CrossRef]
- Yang, H.; Le, J.; Dinh, A.; Zhao, X.; Chen, X.; Peng, F.; Feng, F.; Bu, X. From MOF-74-Zn to triazolate-directed nonsymmetric assembly of chiral Zn6@Zn6 clusters. Chem. Eur. J. 2019, 25, 10590–10593. [Google Scholar] [CrossRef] [PubMed]
- Knippen, K.; Bredenkötter, B.; Kanschat, L.; Kraft, M.; Vermeyen, T.; Herrebout, W.; Sugimoto, K.; Bultinck, P.; Volkmer, D. CFA-18: A homochiral metal–organic framework (MOF) constructed from rigid enantiopure bistriazolate linker molecules. Dalton Trans. 2020, 49, 15758–15768. [Google Scholar] [CrossRef] [PubMed]
- Bahrin, L.G.; Sarbu, L.G.; Jones, P.G.; Birsa, L.M.; Hopf, H. [2.2]Paracyclophane-bis(triazole) systems: Synthesis and photochemical behavior. Chem. Eur. J. 2017, 23, 12338–12345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahrin, L.G.; Rosca, I.; Clima, L.; Shova, S.; Bejan, D.; Nicolescu, A.; Marangoci, N.L.; Sardaru, M.C.; Lozan, V.; Rotaru, A. Zinc(II) coordination polymer on the base of 3′-(1H-tetrazol-5-yl)-[1,1′-biphenyl]-4-carboxylic acid: Synthesis, crystal structure and antimicrobial properties. Inorg. Chem. Commun. 2018, 92, 60–63. [Google Scholar] [CrossRef]
- Bejan, D.; Bahrin, L.G.; Shova, S.; Sardaru, M.; Clima, L.; Nicolescu, A.; Marangoci, N.; Lozan, V.; Janiak, C. Spontaneous resolution of non-centrosymmetric coordination polymers of zinc(II) with achiral imidazole-biphenyl-carboxylate ligands. Inorg. Chim. Acta 2018, 482, 275–283. [Google Scholar] [CrossRef]
- Bahrin, L.G.; Clima, L.; Shova, S.; Rosca, I.; Cojocaru, C.; Bejan, D.; Sardaru, M.C.; Marangoci, N.; Lozan, V.; Rotaru, A. Synthesis, structure, computational modeling, and biological activity of two novel bimesitylene derivatives. Res. Chem. Intermediat. 2019, 45, 453–469. [Google Scholar] [CrossRef]
- Bejan, D.; Bahrin, L.G.; Cojocaru, C.; Trandabat, A.F.; Marangoci, N.L.; Rotaru, A.; Shova, S. The use of C1 symmetry imidazole-carboxylate building block and auxiliary acetate co-ligand for assembly of a 2D wave-like zinc(II) coordination polymer: Experimental and theoretical study. J. Coord. Chem. 2020, 73, 2250–2264. [Google Scholar] [CrossRef]
- Bahrin, L.G.; Bejan, D.; Shova, S.; Gdaniec, M.; Fronc, M.; Lozan, V.; Janiak, C. Alkali- and alkaline-earth metal–organic networks based on a tetra(4-carboxyphenyl)bimesitylene-linker. Polyhedron 2019, 173, 114128. [Google Scholar] [CrossRef]
- Bejan, D.; Bahrin, L.G.; Shova, S.; Marangoci, N.L.; Kokcam-Demir, U.; Lozan, V.; Janiak, C. New microporous lanthanide organic frameworks. synthesis, structure, luminescence, sorption, and catalytic acylation of 2-naphthol. Molecules 2020, 25, 3055. [Google Scholar] [CrossRef] [PubMed]
- Dascalu, I.A.; Mikhalyova, E.A.; Shova, S.; Bratanovici, B.I.; Ardeleanu, R.; Marangoci, N.; Lozan, V.; Roman, G. Synthesis, crystal structure and luminescent properties of isoreticular lanthanide–organic frameworks based on a tetramethyl-substituted terphenyldicarboxylic acid. Polyhedron 2021, 194, 114929. [Google Scholar] [CrossRef]
- Jiao, T.; Chen, L.; Yang, D.; Li, X.; Wu, G.; Zeng, P.; Zhou, A.; Yin, Q.; Pan, Y.; Wu, B.; et al. Trapping white phosphorus within a purely organic molecular container produced by imine condensation. Angew. Chem. Int. Ed. 2017, 56, 14545–14550. [Google Scholar] [CrossRef] [PubMed]
- Bates, C.G.; Saejueng, P.; Murphy, J.M.; Venkataraman, D. Synthesis of 2-arylbenzo[b]furans via copper(i)-catalyzed coupling of o-iodophenols and aryl acetylenes. Org. Lett. 2002, 26, 4727–4729. [Google Scholar] [CrossRef]
- CrysAlis RED. Version 1.171.36.32; Oxford Diffraction Ltd.: Abingdon, UK, 2003. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahrin, L.G.; Nicolescu, A.; Shova, S.; Marangoci, N.L.; Birsa, L.M.; Sarbu, L.G. Nitrogen-Based Linkers with a Mesitylene Core: Synthesis and Characterization. Molecules 2021, 26, 5952. https://doi.org/10.3390/molecules26195952
Bahrin LG, Nicolescu A, Shova S, Marangoci NL, Birsa LM, Sarbu LG. Nitrogen-Based Linkers with a Mesitylene Core: Synthesis and Characterization. Molecules. 2021; 26(19):5952. https://doi.org/10.3390/molecules26195952
Chicago/Turabian StyleBahrin, Lucian Gabriel, Alina Nicolescu, Sergiu Shova, Narcisa Laura Marangoci, Lucian Mihail Birsa, and Laura Gabriela Sarbu. 2021. "Nitrogen-Based Linkers with a Mesitylene Core: Synthesis and Characterization" Molecules 26, no. 19: 5952. https://doi.org/10.3390/molecules26195952
APA StyleBahrin, L. G., Nicolescu, A., Shova, S., Marangoci, N. L., Birsa, L. M., & Sarbu, L. G. (2021). Nitrogen-Based Linkers with a Mesitylene Core: Synthesis and Characterization. Molecules, 26(19), 5952. https://doi.org/10.3390/molecules26195952