
Synthesis, In Vitro, and In Silico Analysis of the Antioxidative Activity of Dapsone Imine Derivatives †

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Results
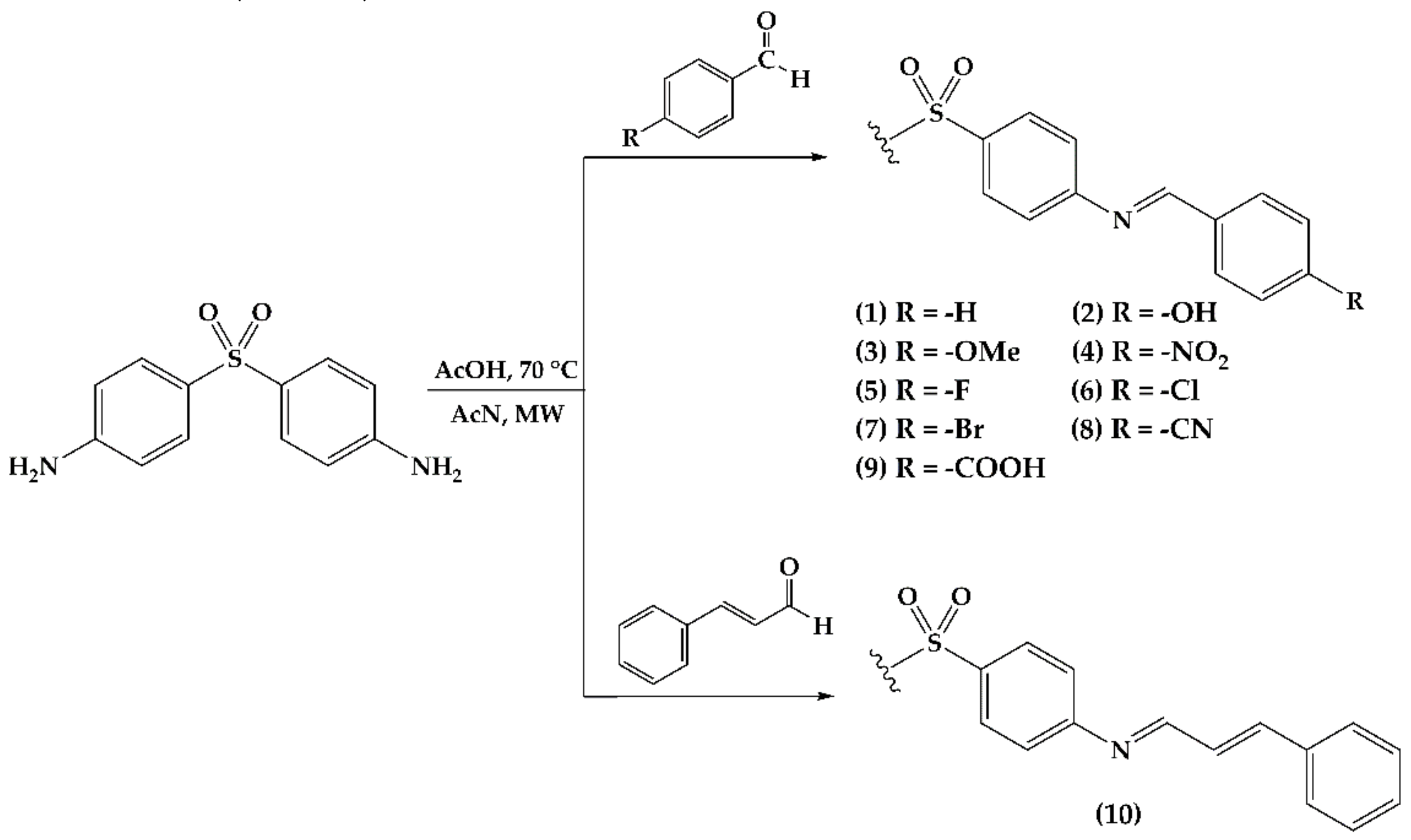
2.1. Imine Synthesis
2.2. Antioxidant Effects
2.2.1. DPPH Radical Scavenging Assay
2.2.2. Ferric Reducing/Antioxidant Power Assay (FRAP)
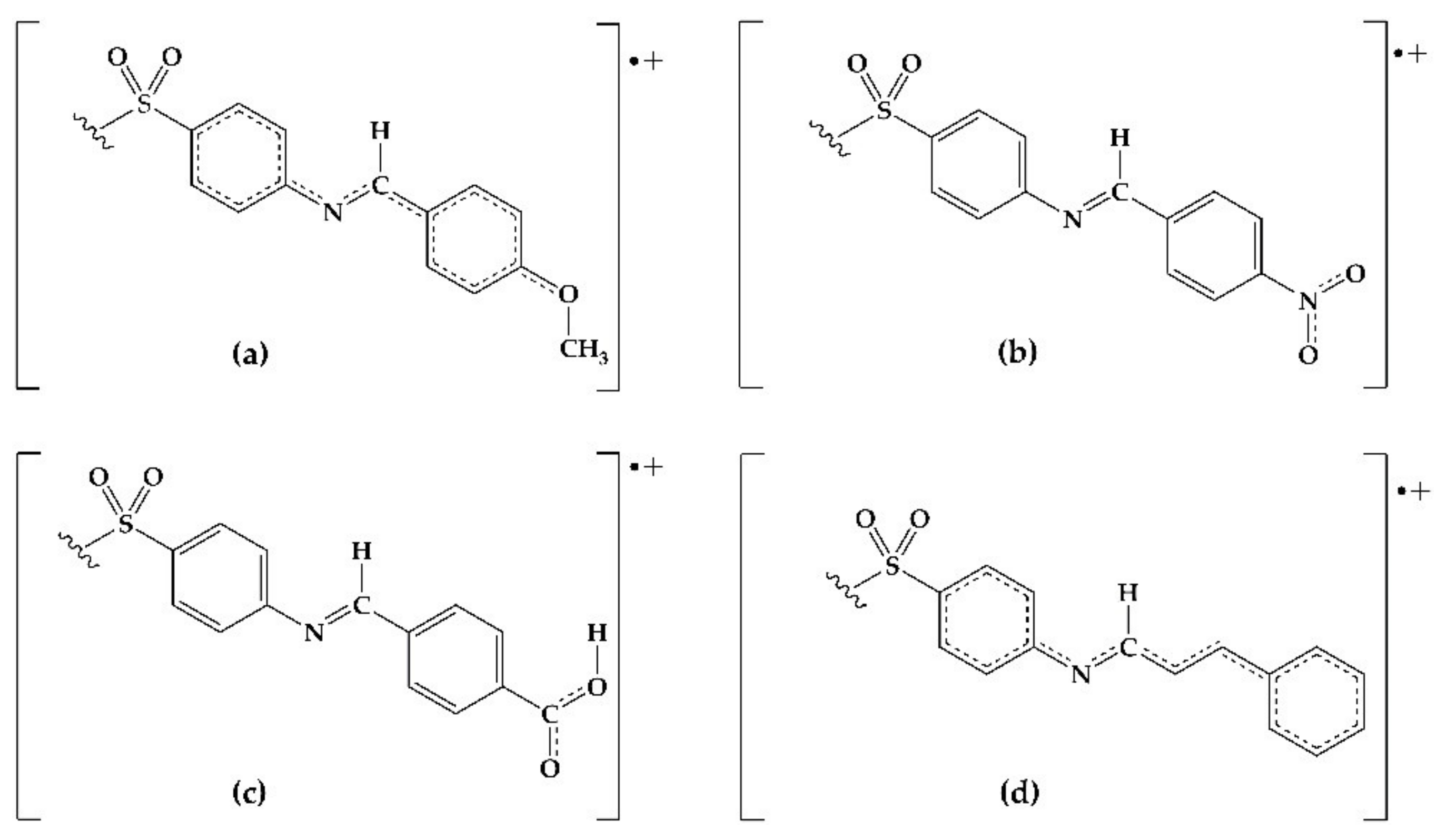
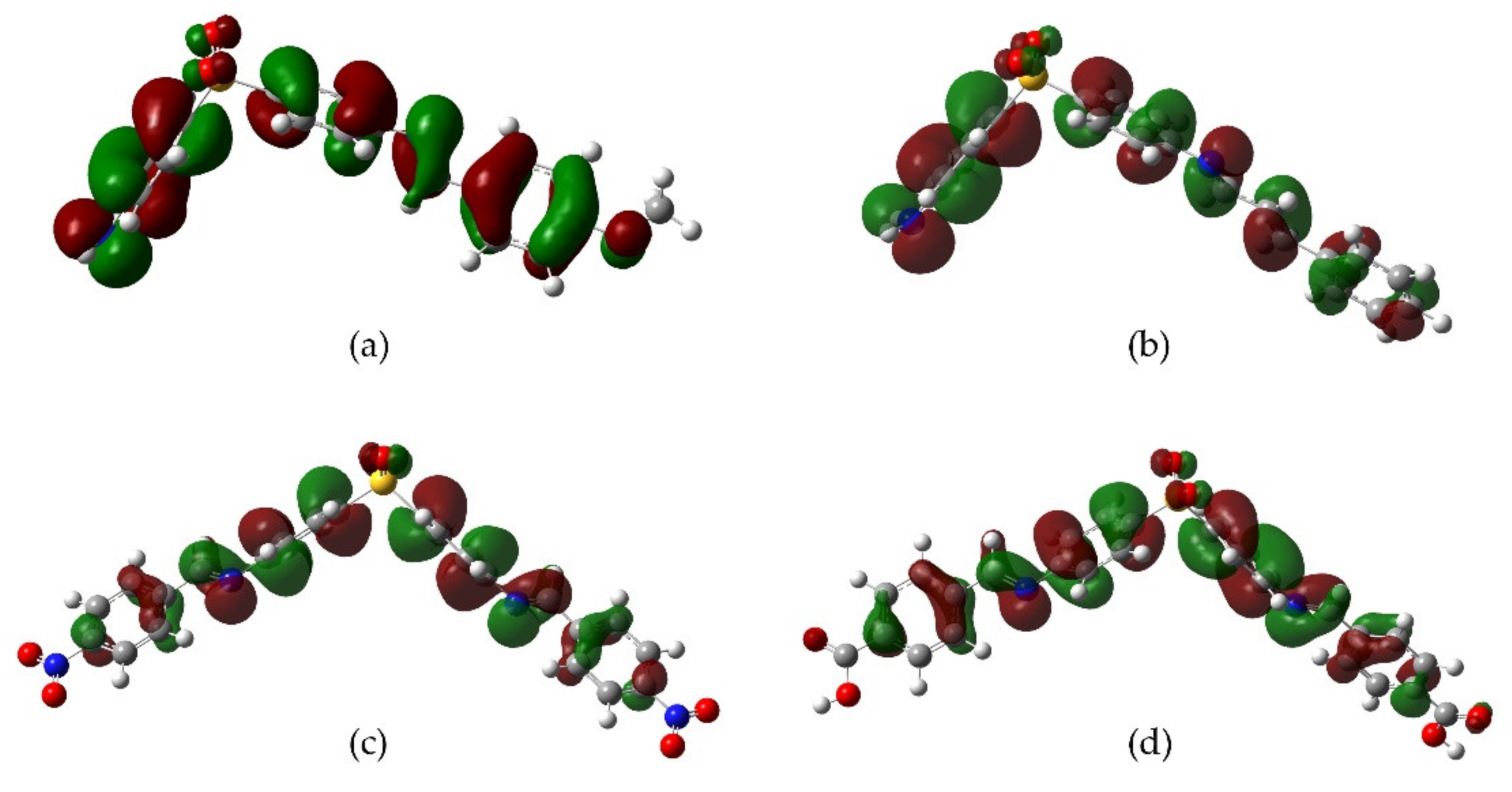
2.3. DFT Analysis
3. Discussion
4. Materials and Methods
4.1. Materials and Equipment
4.2. DDS Imine Derivative Synthesis
4.3. Antioxidant Activity
4.3.1. DPPH Radical Scavenging Assay
4.3.2. Ferric Reducing Antioxidant Power
4.3.3. Statistical Data Analysis
4.4. Computational Details
4.4.1. Electronic and Topological Parameters
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Leprosy. Available online: https://www.who.int/news-room/fact-sheets/detail/leprosy (accessed on 29 January 2020).
- Fischer, M. Leprosy–an overview of clinical features, diagnosis, and treatment. J. Dtsch. Dermatol. Ges. 2017, 15, 801–827. [Google Scholar] [CrossRef] [Green Version]
- Derouin, F.; Piketty, C.; Chastang, C.; Chau, F.; Rouveix, B.; Pocidalo, J.J. Anti-Toxoplasma effects of dapsone alone and combined with pyrimethamine. Antimicrob. Agents Chemother. 1991, 35, 252–255. [Google Scholar] [CrossRef] [Green Version]
- Girard, P.M.; Landman, R.; Gaudebout, C.; Olivares, R.; Saimot, A.G.; Jelazko, P.; Lecompte, T. Dapsone-pyrimethamine compared with aerosolized pentamidine as primary prophylaxis against Pneumocystis carinii pneumonia and toxoplasmosis in HIV infection. N. Engl. J. Med. 1993, 328, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Watkins, W.M.; Brandling-Bennett, A.D.; Nevill, C.G.; Carter, J.Y.; Boriga, D.A.; Howells, R.E.; Koech, D.K. Chlorproguanil/dapsone for the treatment of non-severe Plasmodium falciparum malaria in Kenya: A pilot study. Trans. R. Soc. Trop. Med. Hyg. 1988, 82, 398–403. [Google Scholar] [CrossRef]
- Zuidema, J.; Hilbers-Modderman, E.S.M.; Merkus, F.W.H.M. Clinical pharmacokinetics of dapsone. Clin. Pharmacokinet. 1986, 11, 299–315. [Google Scholar] [CrossRef]
- Reilly, T.P.; Woster, P.M.; Svensson, C.K. Methemoglobin formation by hydroxylamine metabolites of sulfamethoxazole and dapsone: Implications for differences in adverse drug reactions. J. Pharmacol. Exp. Ther. 1999, 288, 951–959. [Google Scholar]
- Tingle, M.D.; Mahmud, R.; Maggs, J.L.; Pirmohamed, M.; Park, B.K. Comparison of the metabolism and toxicity of dapsone in rat, mouse and man. J. Pharmacol. Exp. Ther. 1997, 283, 817–823. [Google Scholar] [PubMed]
- Coleman, M.D. Dapsone: Modes of action, toxicity and possible strategies for increasing patient tolerance. Brit. J. Dermatol. 1993, 129, 507–513. [Google Scholar] [CrossRef]
- Wozel, G.; Blasum, C. Dapsone in dermatology and beyond. Arch. Dermatol. Res. 2014, 306, 103–124. [Google Scholar] [CrossRef] [Green Version]
- Niwa, Y.; Sakane, T.; Miyachi, Y. Dissociation of the inhibitory effect of dapsone on the generation of oxygen intermediates—in comparision with that of colchicine and various scavengers. Biochem. Pharmacol. 1984, 33, 2355–2360. [Google Scholar] [CrossRef]
- Diaz-Ruiz, A.; Zavala, C.; Montes, S.; Ortiz-Plata, A.; Salgado-Ceballos, H.; Orozco-Suarez, S.; Nava-Ruiz, C.; Pérez-Neri, I.; Perez-Severiano, F.; Ríos, C. Antioxidant, antiinflammatory and antiapoptotic effects of dapsone in a model of brain ischemia/reperfusion in rats. J. Neurosci. Res. 2008, 86, 3410–3419. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kumar Singh, A.; Quraishi, M.A. Dapsone: A novel corrosion inhibitor for mild steel in acid media. Open Electrochem. J. 2010, 2, 43–51. [Google Scholar]
- Singh, A.; Avyaya, J.N.; Ebenso, E.E.; Quraishi, M.A. Schiff’s base derived from the pharmaceutical drug Dapsone (DS) as a new and effective corrosion inhibitor for mild steel in hydrochloric acid. Res. Chem. Intermed. 2013, 39, 537–551. [Google Scholar] [CrossRef]
- Chakravarthy, M.P.; Mohana, K.N.; Kumar, C.P.; Badiea, A.M. Corrosion inhibition behaviour and adsorption characteristics of dapsone derivatives on mild steel in acid medium. Chem. Sci. Int. J. 2015, 8, 1–16. [Google Scholar] [CrossRef]
- Singh, P.; Chauhan, D.S.; Chauhan, S.S.; Singh, G.; Quraishi, M.A. Chemically modified expired dapsone drug as environmentally benign corrosion inhibitor for mild steel in sulphuric acid useful for industrial pickling process. J. Mol. Liq. 2019, 286, 110903. [Google Scholar] [CrossRef]
- Jornada, D.H.; dos Santos Fernandes, G.F.; Chiba, D.E.; De Melo, T.R.F.; Dos Santos, J.L.; Chung, M.C. The prodrug approach: A successful tool for improving drug solubility. Molecules 2016, 21, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, T.P.; Sil, D.; Shukla, N.M.; Anbanandam, A.; Day, V.W.; David, S.A. Imbuing aqueous solubility to amphotericin B and nystatin with a vitamin. Mol. Pharm. 2011, 8, 297–301. [Google Scholar] [CrossRef]
- Pochopin, N.L.; Charman, W.N.; Stella, V.J. Amino acid derivatives of dapsone as water-soluble prodrugs. Int. J. Pharm. 1995, 121, 157–167. [Google Scholar] [CrossRef]
- Wadher, S.J.; Puranik, M.P.; Karande, N.A.; Yeole, P.G. Synthesis and biological evaluation of Schiff base of dapsone and their derivative as antimicrobial agents. Int. J. PharmTech Res. 2009, 1, 22–33. [Google Scholar]
- Althagafi, I.I.; Gaffer, H.E. Synthesis, molecular modeling and antioxidant activity of new phenolic bis-azobenzene derivatives. J. Mol. Struct. 2019, 1182, 22–30. [Google Scholar] [CrossRef]
- Alaşalvar, C.; Soylu, M.S.; Güder, A.; Albayrak, Ç.; Apaydın, G.; Dilek, N. Crystal structure, DFT and HF calculations and radical scavenging activities of (E)-4, 6-dibromo-3-methoxy-2-[(3-methoxyphenylimino) methyl] phenol. Spectrochim. Acta A 2014, 125, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Horton, W.; Peerannawar, S.; Török, B.; Török, M. Theoretical and experimental analysis of the antioxidant features of substituted phenol and aniline model compounds. J. Struct. Chem. 2019, 30, 23–35. [Google Scholar] [CrossRef]
- Mendes, A.P.; Schalcher, T.R.; Barros, T.G.; Almeida, E.D.; Maia, C.S.; Barros, C.A.; Monteiro, M.C.; Borges, R.S. A geometric and electronic study of dapsone. J. Comput. Theor. Nanosci. 2011, 8, 1428–1431. [Google Scholar] [CrossRef]
- Borges, R.S.; Vale, J.K.; Schalcher, T.R.; Almeida, E.D.; Maia, C.S.; Monteiro, M.C.; Orestes, E.; da Silva, A.B. A theoretical study of the dapsone derivatives on methemoglobin. J. Comput. Theor. Nanosci. 2013, 10, 2029–2033. [Google Scholar] [CrossRef]
- Martiínez, A.; Rodríguez-Gironés, M.A.; Barbosa, A.; Costas, M. Donator acceptor map for carotenoids, melatonin and vitamins. J. Phys. Chem. A 2008, 112, 9037–9042. [Google Scholar] [CrossRef]
- Mermer, A.; Demirbas, N.; Uslu, H.; Demirbas, A.; Ceylan, S.; Sirin, Y. Synthesis of novel Schiff bases using green chemistry techniques; antimicrobial, antioxidant, antiurease activity screening and molecular docking studies. J. Mol. Struct. 2019, 1181, 412–422. [Google Scholar] [CrossRef]
- Paşa, S.; Erdoğan, Ö.; Yenisey, Ç. Synthesis and structural identification of boron based Schiff compounds with Ishikawa endometrial cancer and antioxidant activity. J. Mol. Struct. 2019, 1186, 458–467. [Google Scholar] [CrossRef]
- Abdel-Wahab, B.F.; Awad, G.E.; Badria, F.A. Synthesis, antimicrobial, antioxidant, anti-hemolytic and cytotoxic evaluation of new imidazole-based heterocycles. Eur. J. Med. Chem. 2011, 46, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Berker, K.I.; Güçlü, K.; Tor, İ.; Apak, R. Comparative evaluation of Fe (III) reducing power-based antioxidant capacity assays in the presence of phenanthroline, batho-phenanthroline, tripyridyltriazine (FRAP), and ferricyanide reagents. Talanta 2007, 72, 1157–1165. [Google Scholar] [CrossRef]
- Yehye, W.A.; Rahman, N.A.; Ariffin, A.; Abd Hamid, S.B.; Alhadi, A.A.; Kadir, F.A.; Yaeghoobi, M. Understanding the chemistry behind the antioxidant activities of butylated hydroxytoluene (BHT): A review. Eur. J. Med. Chem. 2015, 101, 295–312. [Google Scholar] [CrossRef]
- Nieva-Echevarría, B.; Manzanos, M.J.; Goicoechea, E.; Guillén, M.D. 2, 6-Di-tert-butyl-hydroxytoluene and its metabolites in foods. Compr. Rev. Food Sci. Food Saf. 2015, 14, 67–80. [Google Scholar] [CrossRef]
- Meščić Macan, A.; Gazivoda Kraljević, T.; Raić-Malić, S. Therapeutic perspective of vitamin C and its derivatives. Antioxidants 2019, 8, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaddouri, Y.; Abrigach, F.; Yousfi, E.B.; El Kodadi, M.; Touzani, R. New thiazole, pyridine and pyrazole derivatives as antioxidant candidates: Synthesis, DFT calculations and molecular docking study. Heliyon 2020, 6, e03185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mıhçıokur, Ö.; Özpozan, T. Molecular structure, vibrational spectroscopic analysis (IR & Raman), HOMO-LUMO and NBO analysis of anti-cancer drug sunitinib using DFT method. J. Mol. Struct. 2017, 1149, 27–41. [Google Scholar]
- Shankar, R.; Senthilkumar, K.; Kolandaivel, P. Calculation of ionization potential and chemical hardness: A comparative study of different methods. Int. J. Quantum Chem. 2009, 109, 764–771. [Google Scholar] [CrossRef]
- Simons, J.; Jordan, K.D. Ab initio electronic structure of anions. Chem. Rev. 1987, 87, 535–555. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Akhtari, K.; Hassanzadeh, K.; Fakhraei, B.; Fakhraei, N.; Hassanzadeh, H.; Zarei, S.A. A density functional theory study of the reactivity descriptors and antioxidant behavior of Crocin. Comput. Theor. Chem. 2013, 1013, 123–129. [Google Scholar] [CrossRef]
- Avelar, M.; Martínez, A. Do casiopeinas® prevent cancer disease by acting as antiradicals? A chemical reactivity study applying density functional theory. J. Mex. Chem. Soc. 2012, 56, 250–256. [Google Scholar]
- Gázquez, J.L. Perspectives on the density functional theory of chemical reactivity. J. Mex. Chem. Soc. 2008, 52, 3–10. [Google Scholar]
- Martinez, A. Donator acceptor map of psittacofulvins and anthocyanins: Are they good antioxidant substances? J. Phys. Chem. B 2009, 113, 4915–4921. [Google Scholar] [CrossRef] [PubMed]
- Laguninl, A.A.; Dubovskaja, V.I.; Rudik, A.V.; Druzhilovskiy, D.S.; Gloriozova, T.A.; Filimonov, D.A.; Sastry, N.D.; Poroikov, V.V. CLC-Pred: A freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. PLoS ONE 2018, 13, e0191838. [Google Scholar] [CrossRef] [Green Version]
- García-Jacas, C.R.; Marrero-Ponce, Y.; Cortés-Guzmán, F.; Suárez-Lezcano, J.; Martinez-Rios, F.O.; García-González, L.A.; PupoMeriño, M.; Martinez-Mayorga, K. Enhancing acute oral toxicity predictions by using consensus modeling and algebraic form-based 0D-to-2D molecular encodes. Chem. Res. Toxicol. 2019, 32, 1178–1192. [Google Scholar] [CrossRef]
- Vogel, A.I.; Furniss, B.S.; Hannaford, A.J.; Smith, P.W.; Tatchell, A.R. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; John Wiley & Sons: New York, NY, USA, 1989; pp. 395–469. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Cortes, E.; Mora, J.; Márquez, E. Modelling the anti-methicillin-resistant Staphylococcus aureus (MRSA) activity of cannabinoids: A QSAR and Docking Study. Crystals 2020, 10, 692. [Google Scholar] [CrossRef]
- Márquez, E.; Mora, J.R.; Flores-Morales, V.; Insuasty, D.; Calle, L. Modeling the antileukemia activity of ellipticine-related compounds: QSAR and Molecular Docking Study. Molecules 2020, 25, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Derivative | Substituent (R) | Time (h) | %Yield | Melting Point °C |
|---|---|---|---|---|
| 1 | Hydrogen | 2 | 90 | 213.0–214.6 |
| 2 | 4-hydroxyl | 3 | 70 | 129.8–130.9 |
| 3 | 4-methoxy | 3 | 90 | 227.0–228.2 |
| 4 | 4-nitro | 3 | 81 | 234.0–236.0 |
| 5 | 4-fluoro | 5 | 81 | 163.1–164.3 |
| 6 | 4-chloro | 3 | 90 | 204.5–205.3 |
| 7 | 4-bromo | 7 | 75 | 191.7–194.1 |
| 8 | 4-cyano | 3 | 75 | 123.9–124.7 |
| 9 | 4-carboxylate | 3 | 77 | 388 (decomp.) |
| 10 | 2-phenylethylen | 3 | 71 | 212.4–213.5 |
| Derivative | Substituent | %Capture | Ratio Derivative/DSS | Ratio Derivative/BHT |
|---|---|---|---|---|
| 1 | Hydrogen | 1.5 ± 1.2 ns | 0.60 | 0.017 |
| 2 | 4-hydroxyl | 5.8 ± 0.6 ** | 1.92 | 0.064 |
| 3 | 4-methoxy | 3.3 ± 1.9 ns | 1.32 | 0.036 |
| 4 | 4-nitro | 0.0 ± 1.8 ns | 0.00 | 0.000 |
| 5 | 4-fluoro | 1.7 ± 0.4 ns | 0.68 | 0.019 |
| 6 | 4-chloro | 0.0 ± 1.2 * | 0.00 | 0.000 |
| 7 | 4-bromo | 0.1 ± 0.5 * | 0.04 | 0.001 |
| 8 | 4-cyano | 1.6 ± 1.6 ns | 0.64 | 0.018 |
| 9 | 4-carboxyl | 66.2 ± 0.5 *** | 26.48 | 0.730 |
| 10 | 2-phenylethylen | 4.0 ± 0.8 ns | 1.60 | 0.044 |
| DDS | 2.5 ± 0.3 | 1.00 | 0.028 | |
| BHT | 90.7 ± 0.3 | 36.28 | 1.00 |
| Compound | Substituent | %Reduction | Ratio Derivative/DDS | Ratio Derivative/AA |
|---|---|---|---|---|
| 1 | Hydrogen | 15.0 ± 0.7 ** | 0.9 | 0.15 |
| 2 | 4-hydroxyl | 16.9 ± 1.3 ns | 1.0 | 0.17 |
| 3 | 4-methoxy | 28.4 ± 0.8 *** | 1.6 | 0.28 |
| 4 | 4-nitro | 40.7 ± 0.2 *** | 2.4 | 0.41 |
| 5 | 4-fluoro | 13.2 ± 0.3 *** | 0.8 | 0.13 |
| 6 | 4-chloro | 14.5 ± 0.4 *** | 0.8 | 0.15 |
| 7 | 4-bromo | 19.3 ± 1.4 ** | 1.1 | 0.19 |
| 8 | 4-cyano | 20.0 ± 0.8 *** | 1.2 | 0.20 |
| 9 | 4-carboxyl | 44.8 ± 0.7 *** | 2.6 | 0.45 |
| 10 | 2-phenylethylen | 39.6 ± 0.1 *** | 2.3 | 0.40 |
| DDS | 17.3 ± 0.6 | 1.0 | 0.17 | |
| AA | 100.0 ± 0.3 | 5.8 | 1.00 |
| Derivative | HOMO (eV) | LUMO (eV) | GapHOMO-LUMO (eV) |
|---|---|---|---|
| 1 | −7.82 | −0.99 | 6.83 |
| 2 | −7.42 | −0.69 | 6.72 |
| 3 | −7.37 | −1.93 | 6.70 |
| 4 | −8.33 | −2.03 | 6.31 |
| 5 | −7.52 | −0.89 | 6.63 |
| 6 | −7.96 | −1.23 | 6.73 |
| 7 | −7.54 | −1.05 | 6.49 |
| 8 | −7.66 | −1.57 | 6.09 |
| 9 | −8.05 | −1.50 | 6.55 |
| 10 | −7.43 | −1.15 | 6.28 |
| DDS | −7.25 | −0.30 | 6.95 |
| BHT | −7.17 | 1.23 | 8.40 |
| AA | −8.22 | 0.42 | 8.64 |
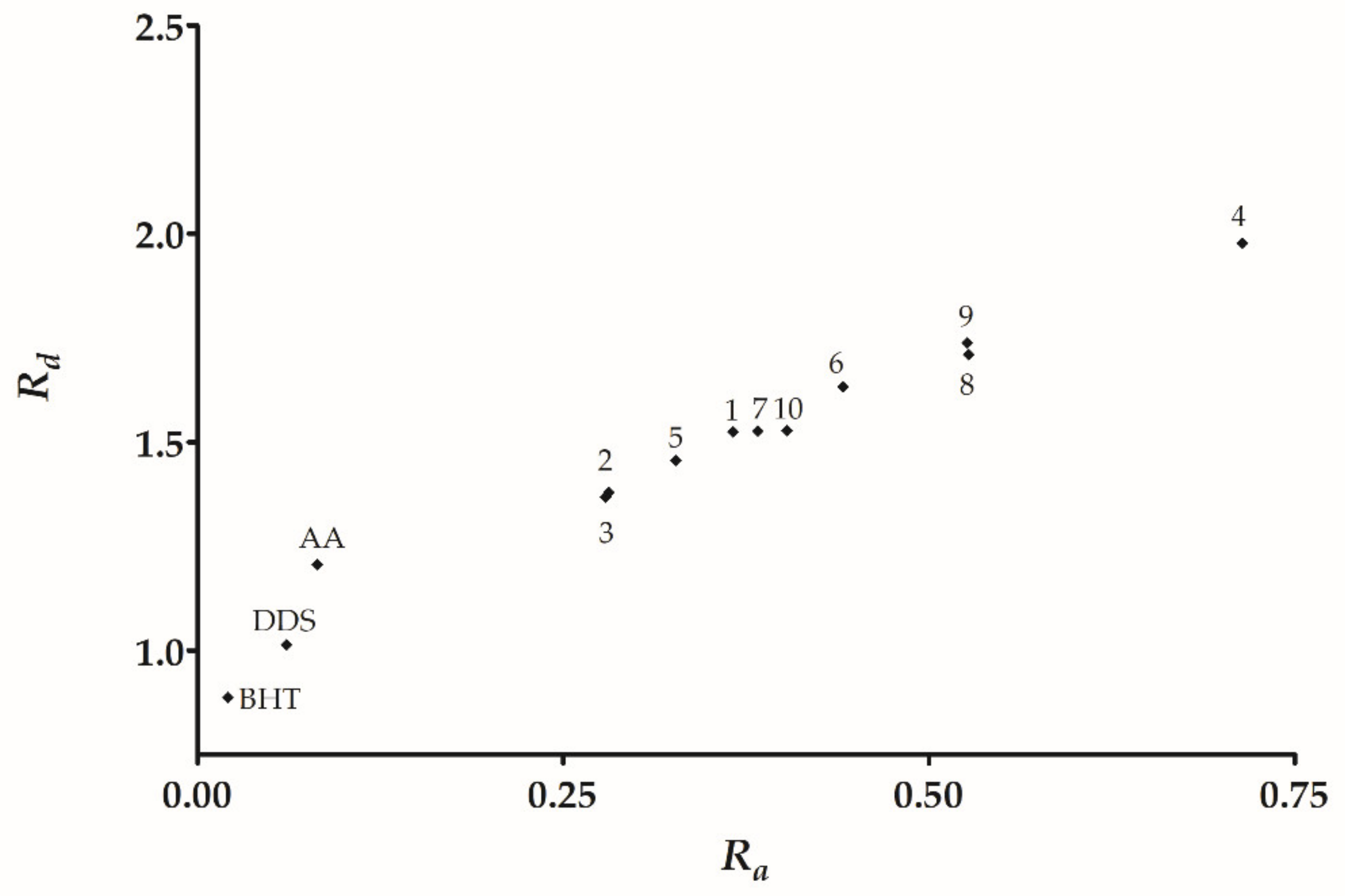
| Compound | I | A | ω− | ω+ | Ra | Rd |
|---|---|---|---|---|---|---|
| 1 | 8.14 | 0.64 | 5.24 | 0.84 | 0.37 | 1.53 |
| 2 | 7.84 | 0.33 | 4.73 | 0.65 | 0.28 | 1.38 |
| 3 | 7.77 | 0.33 | 4.69 | 0.64 | 0.28 | 1.37 |
| 4 | 8.63 | 1.65 | 6.79 | 1.65 | 0.71 | 1.98 |
| 5 | 7.98 | 0.51 | 5.00 | 0.75 | 0.33 | 1.46 |
| 6 | 8.27 | 0.90 | 5.60 | 1.02 | 0.44 | 1.63 |
| 7 | 8.00 | 0.71 | 5.24 | 0.88 | 0.38 | 1.53 |
| 8 | 8.14 | 1.17 | 5.87 | 1.22 | 0.53 | 1.71 |
| 9 | 8.35 | 1.16 | 5.97 | 1.21 | 0.53 | 1.74 |
| 10 | 7.83 | 0.80 | 5.24 | 0.93 | 0.40 | 1.53 |
| DDS | 7.78 | −1.10 | 3.48 | 0.14 | 0.06 | 1.01 |
| BHT | 7.63 | −1.64 | 3.04 | 0.05 | 0.02 | 0.89 |
| AA | 9.06 | −1.16 | 4.14 | 0.19 | 0.08 | 1.21 |
| F | 21.26 | 1.84 | 13.86 | 2.31 | 1.00 | 4.04 |
| Na | 5.38 | 0.40 | 3.43 | 0.54 | 0.23 | 1.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzmán-Ávila, R.; Avelar, M.; Márquez, E.A.; Rivera-Leyva, J.C.; Mora, J.R.; Flores-Morales, V.; Rivera-Islas, J. Synthesis, In Vitro, and In Silico Analysis of the Antioxidative Activity of Dapsone Imine Derivatives. Molecules 2021, 26, 5747. https://doi.org/10.3390/molecules26195747
Guzmán-Ávila R, Avelar M, Márquez EA, Rivera-Leyva JC, Mora JR, Flores-Morales V, Rivera-Islas J. Synthesis, In Vitro, and In Silico Analysis of the Antioxidative Activity of Dapsone Imine Derivatives. Molecules. 2021; 26(19):5747. https://doi.org/10.3390/molecules26195747
Chicago/Turabian StyleGuzmán-Ávila, Ricardo, Mayra Avelar, Edgar A. Márquez, Julio C. Rivera-Leyva, José R. Mora, Virginia Flores-Morales, and Jesús Rivera-Islas. 2021. "Synthesis, In Vitro, and In Silico Analysis of the Antioxidative Activity of Dapsone Imine Derivatives" Molecules 26, no. 19: 5747. https://doi.org/10.3390/molecules26195747
APA StyleGuzmán-Ávila, R., Avelar, M., Márquez, E. A., Rivera-Leyva, J. C., Mora, J. R., Flores-Morales, V., & Rivera-Islas, J. (2021). Synthesis, In Vitro, and In Silico Analysis of the Antioxidative Activity of Dapsone Imine Derivatives. Molecules, 26(19), 5747. https://doi.org/10.3390/molecules26195747