
Structural Investigations on the SH3b Domains of Clostridium perfringens Autolysin through NMR Spectroscopy and Structure Simulation Enlighten the Cell Wall Binding Function

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
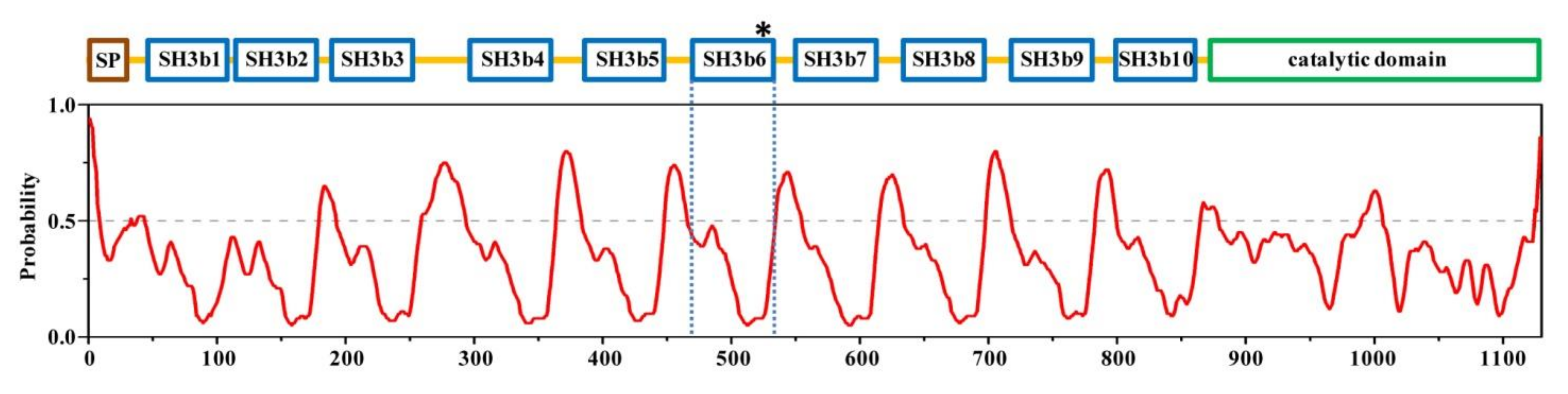
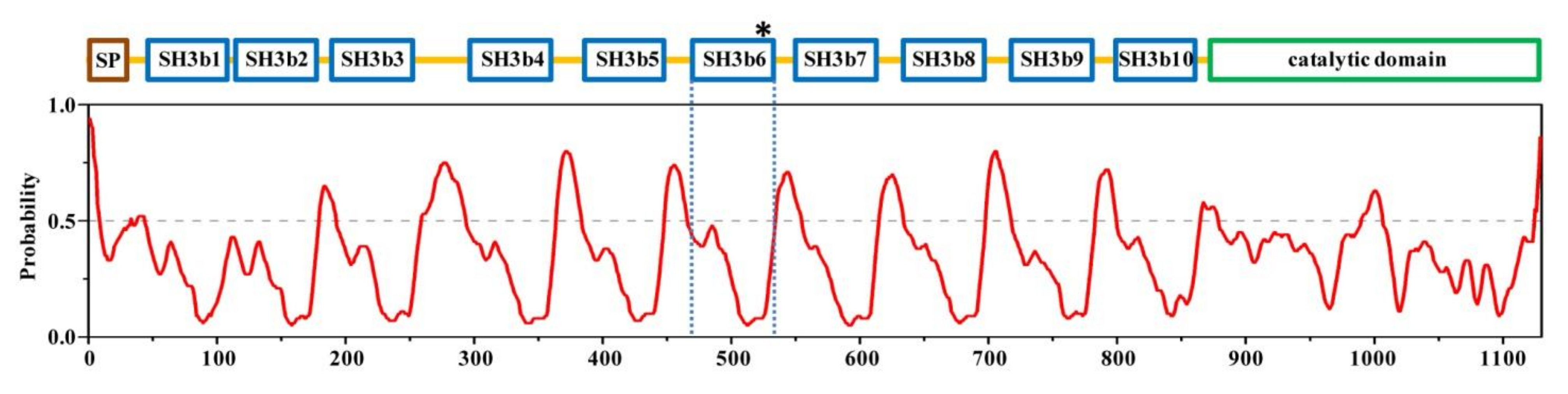
2.1. Structural Architecture of CpAcp
2.2. NMR Structure of CpAcp SH3b6
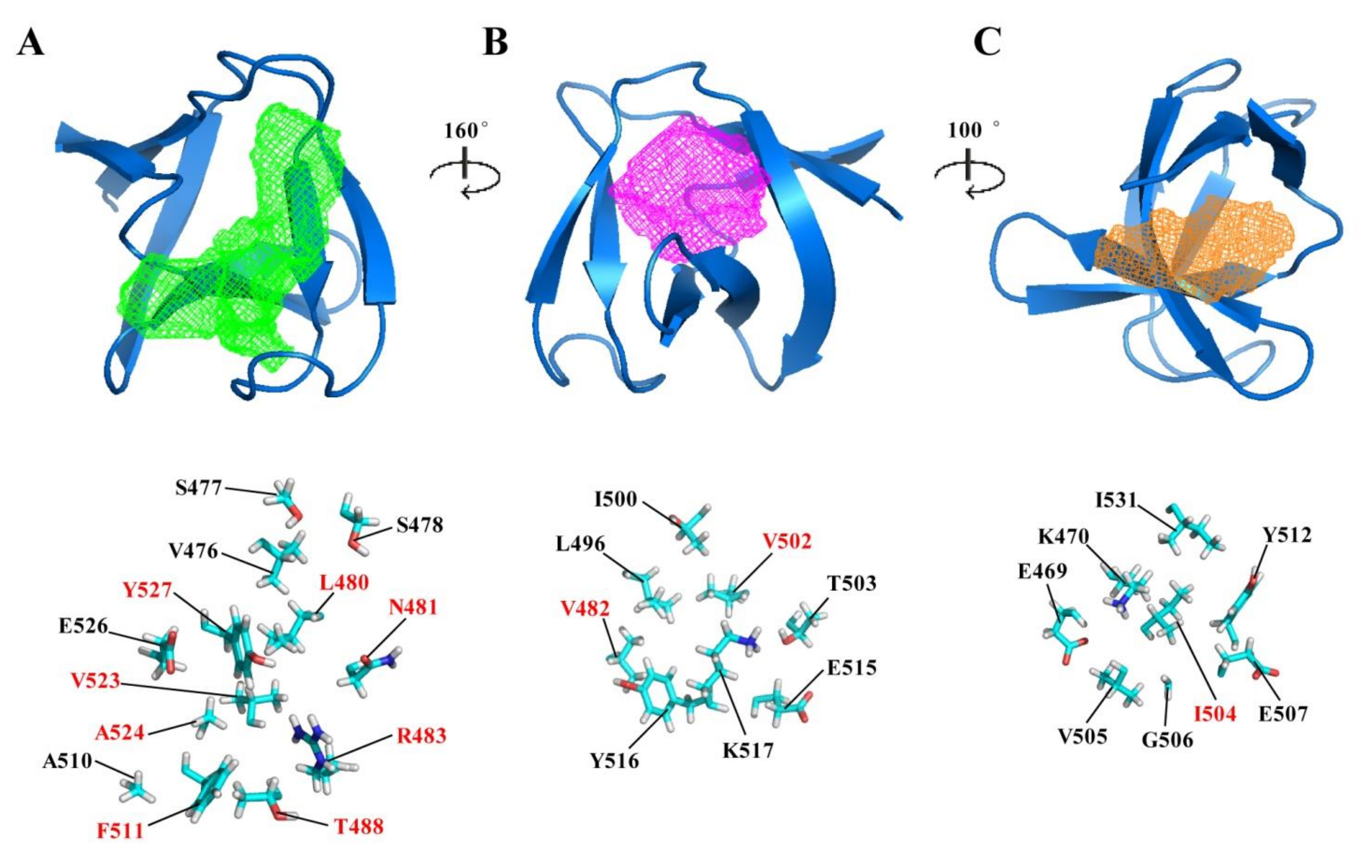
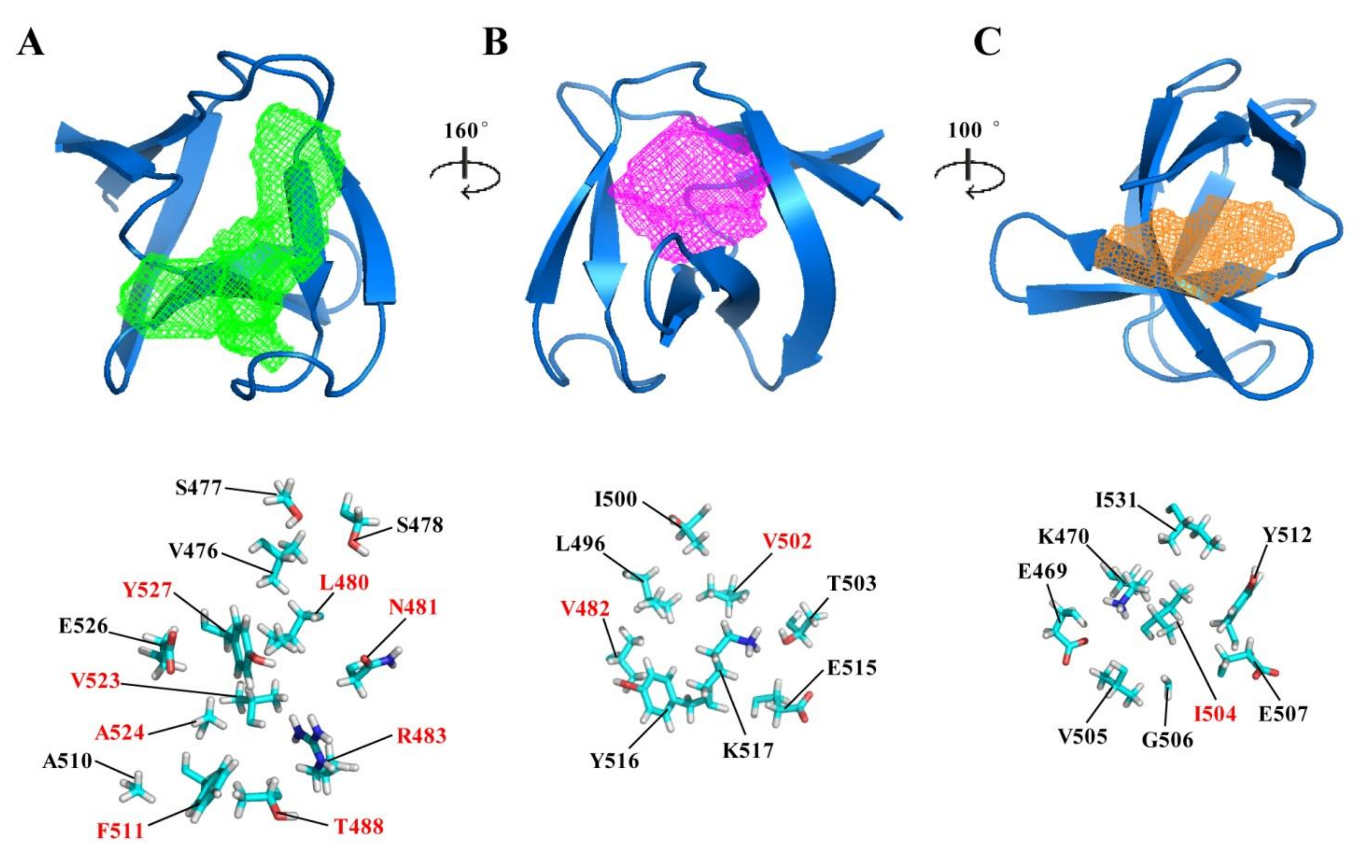
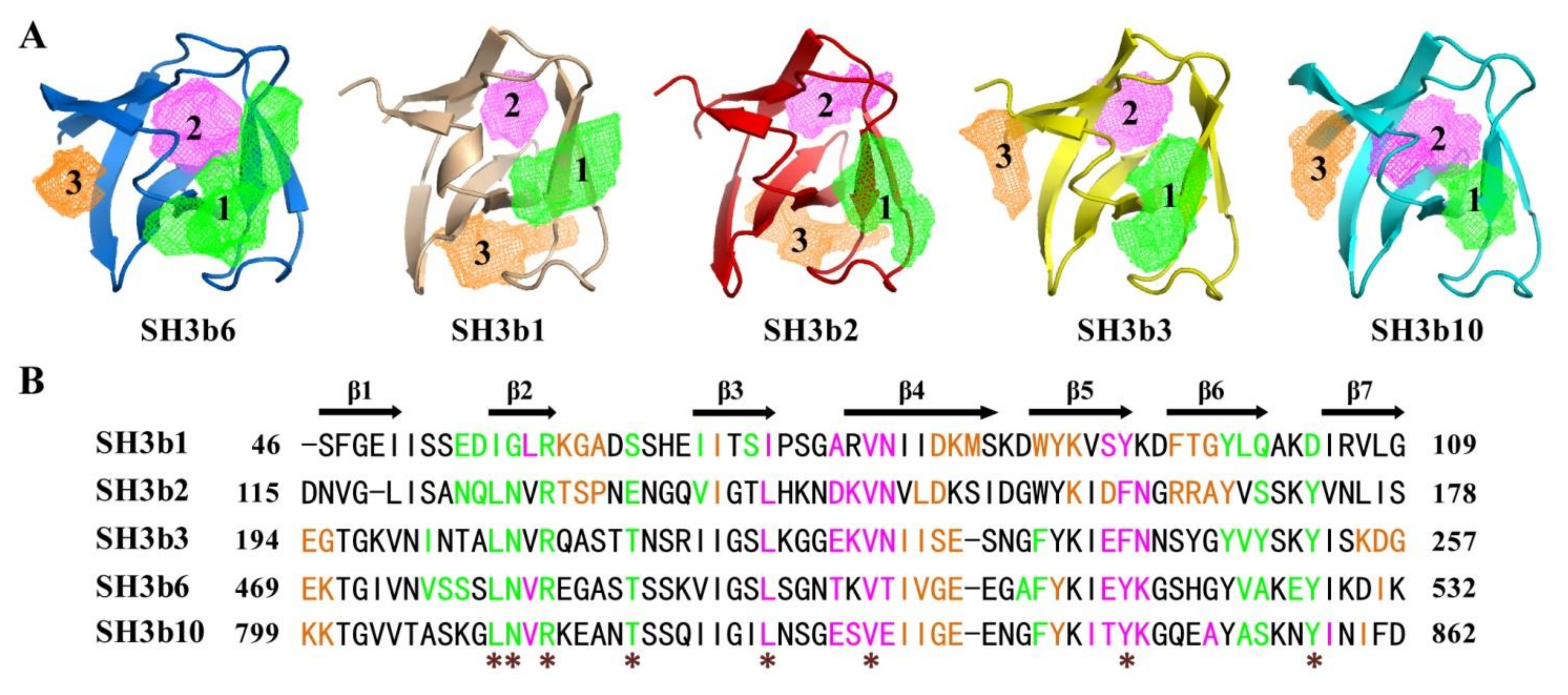
2.3. Potential Ligand-Binding Pocket of CpAcp SH3b6
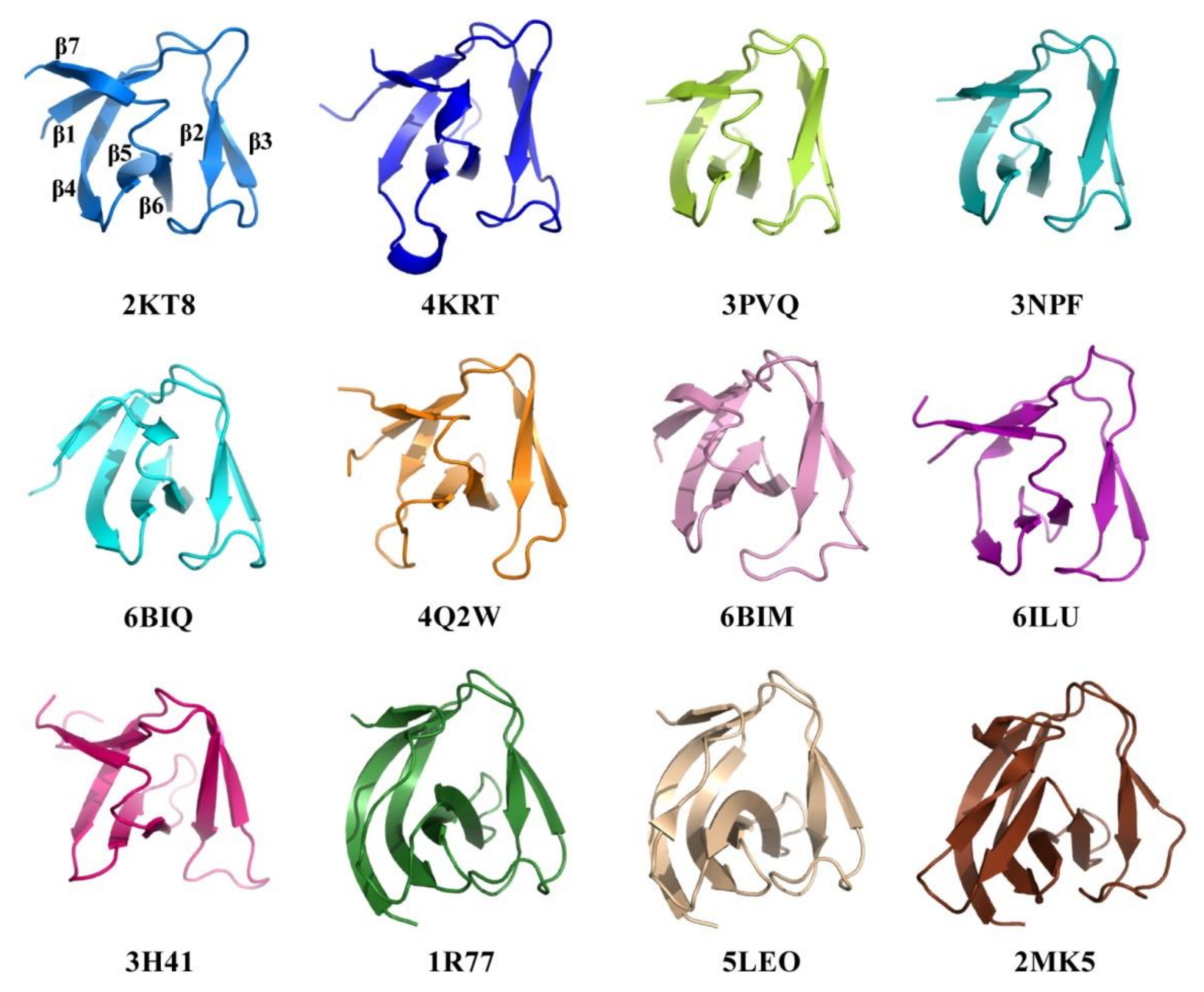
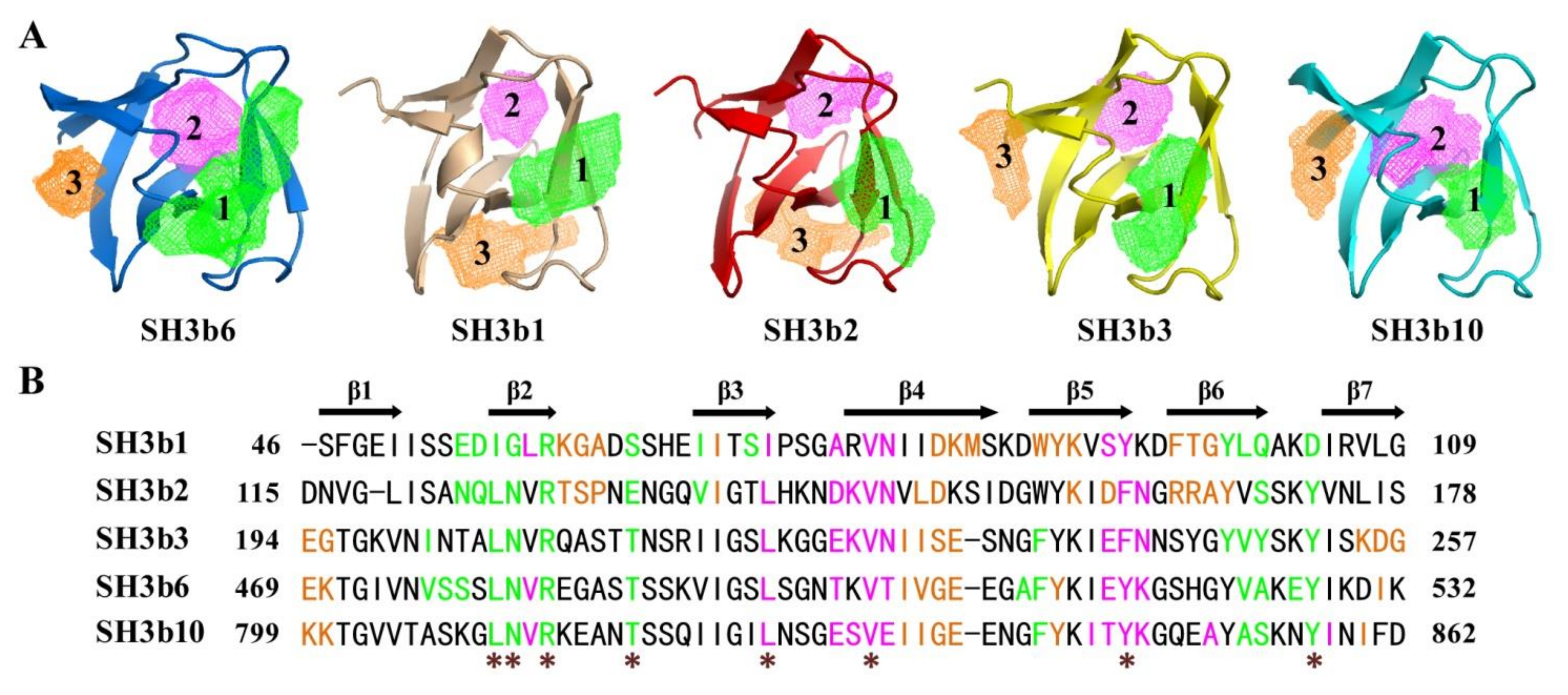
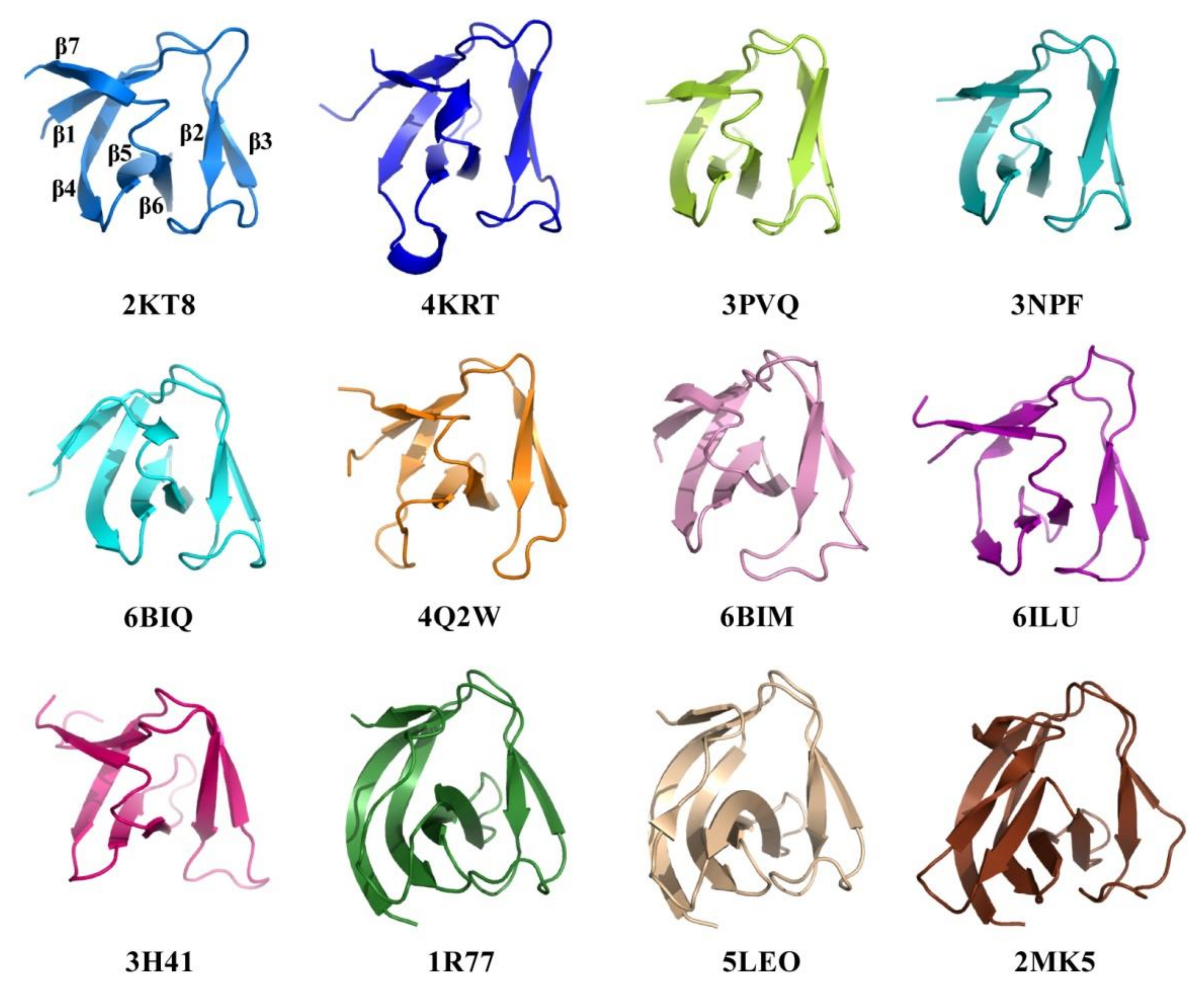
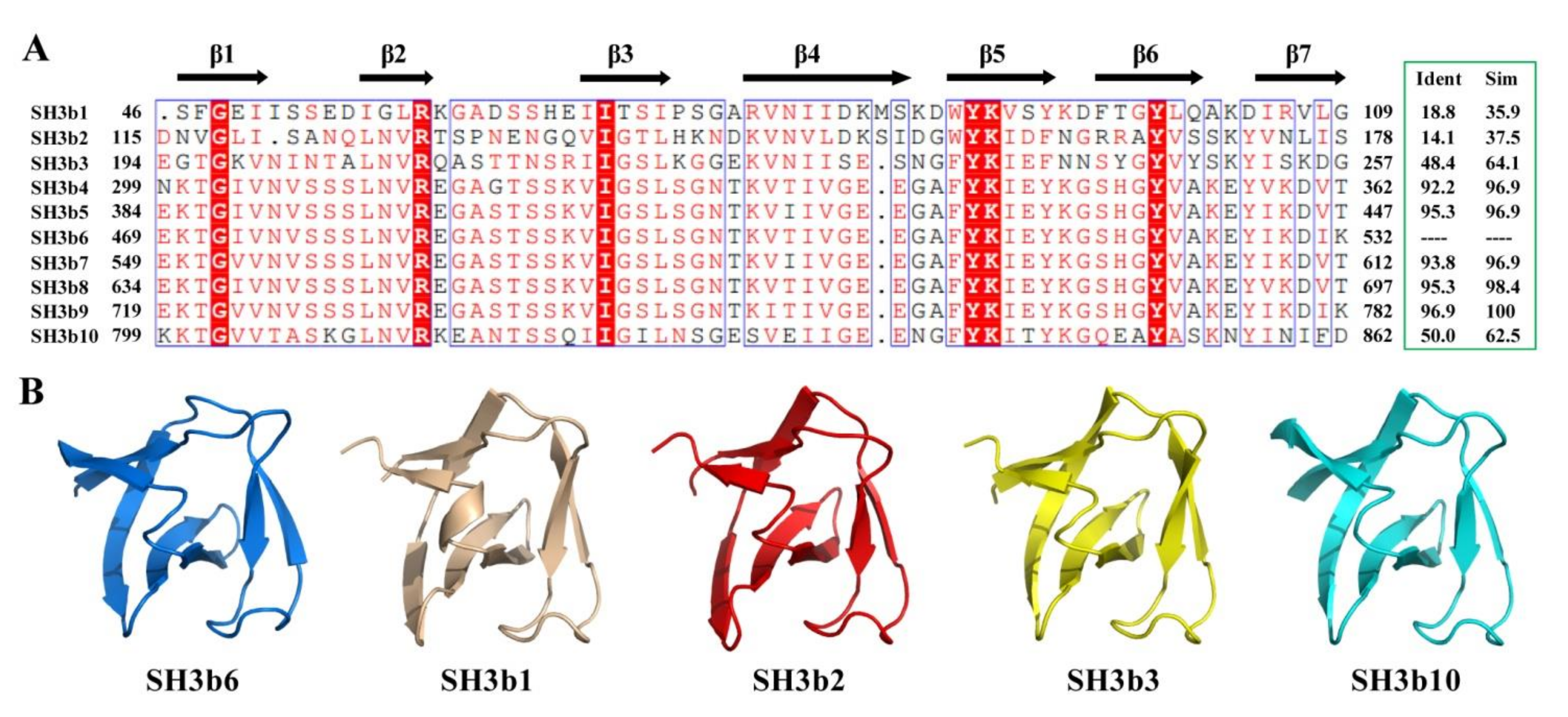
2.4. Structural Comparison of CpAcp SH3b6 with Other SH3b Proteins
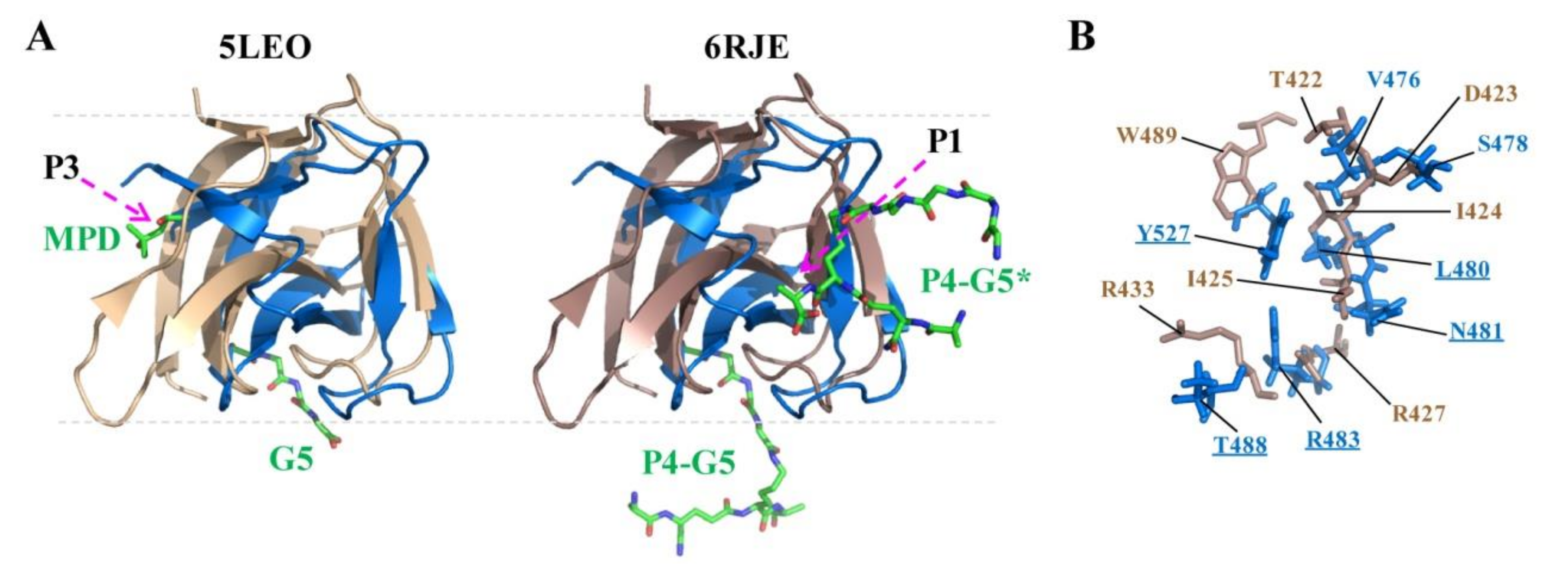
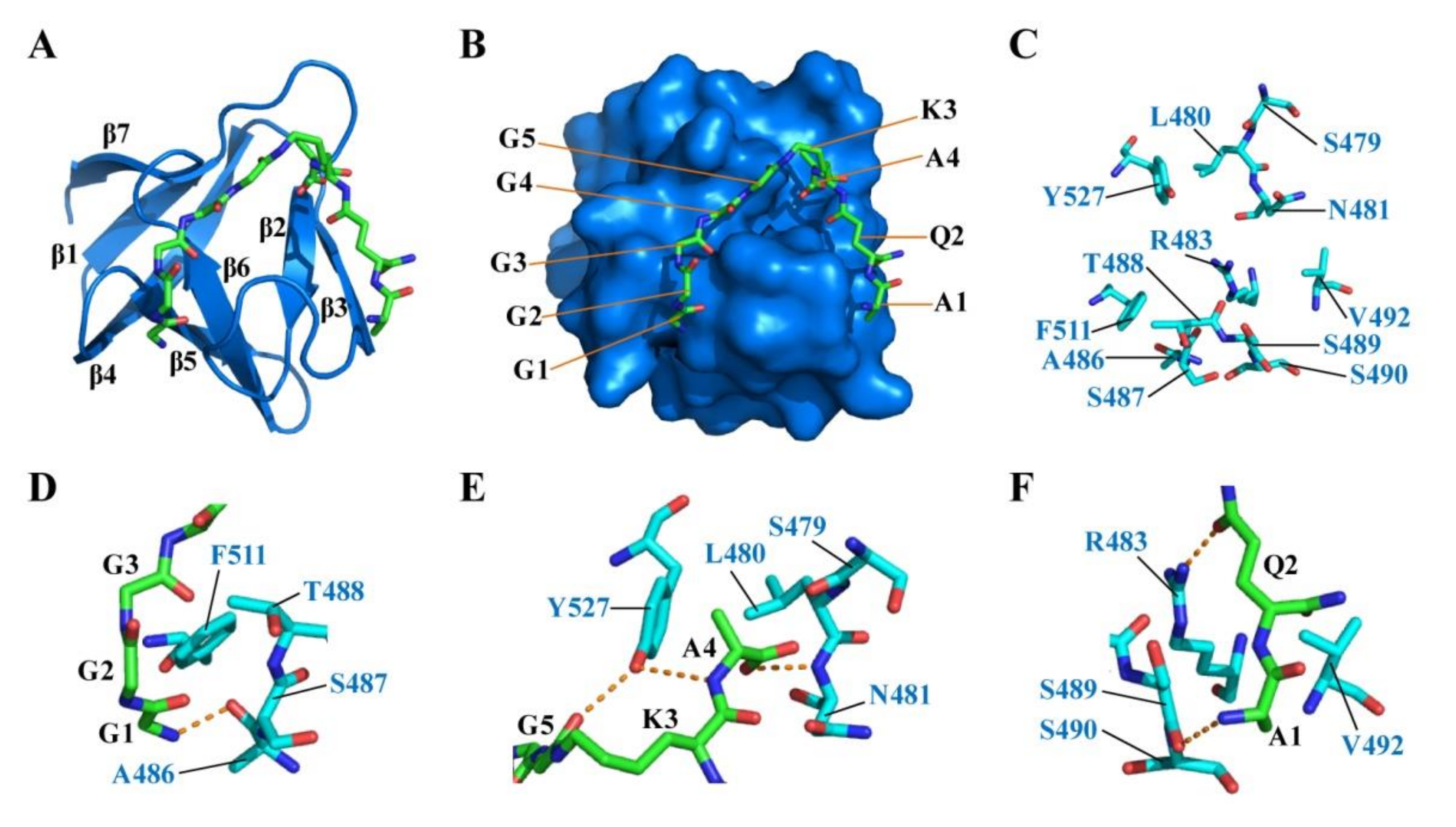
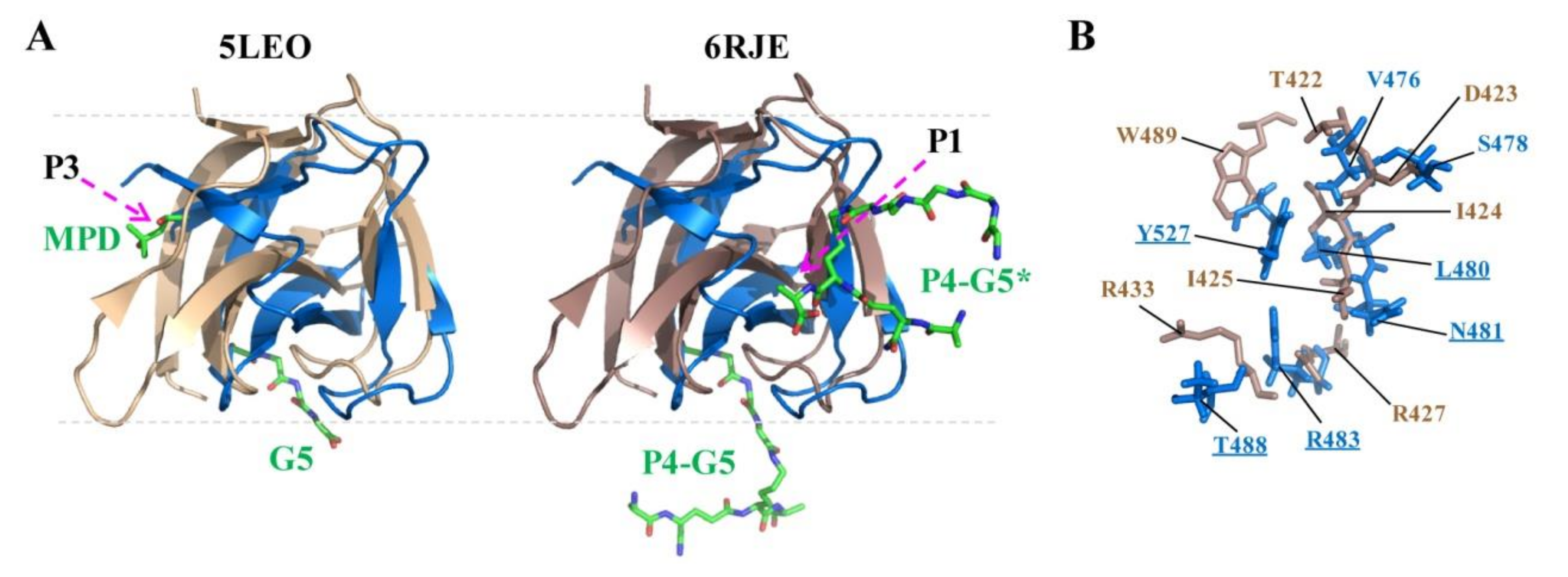
2.5. Docking of CpAcp SH3b6 with P4-G5 Peptide
2.6. Structure Simulation of CpAcp SH3b1/2/3/10
3. Materials and Methods
3.1. Protein Expression and Purification
3.2. Chemical Shift Assignments
3.3. NMR Structure Calculation
3.4. Structure Modeling
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Vollmer, W.; Blanot, D.; de Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [Green Version]
- Typas, A.; Banzhaf, M.; Gross, C.A.; Vollmer, W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 2011, 10, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Szweda, P.; Schielmann, M.; Kotlowski, R.; Gorczyca, G.; Zalewska, M.; Milewski, S. Peptidoglycan hydrolases-potential weapons against Staphylococcus aureus. Appl. Microbiol. Biotechnol. 2012, 96, 1157–1174. [Google Scholar] [CrossRef] [Green Version]
- Bhagwat, A.; Mixon, M.; Collins, C.H.; Dordick, J.S. Opportunities for broadening the application of cell wall lytic enzymes. Appl. Microbiol. Biotechnol. 2020, 104, 9019–9040. [Google Scholar] [CrossRef] [PubMed]
- Love, M.J.; Bhandari, D.; Dobson, R.C.J.; Billington, C. Potential for bacteriophage endolysins to supplement or replace antibiotics in food production and clinical care. Antibiotics Basel 2018, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, K.; Nagahama, M. Clostridium: Food poisoning by Clostridium perfringens. In Encyclopedia of Food and Health; Caballero, B., Finglas, P.M., Toldrá, F., Eds.; Academic Press: Oxford, UK, 2016; pp. 149–154. [Google Scholar]
- Camiade, E.; Peltier, J.; Bourgeois, I.; Couture-Tosi, E.; Courtin, P.; Antunes, A.; Chapot-Chartier, M.P.; Dupuy, B.; Pons, J.L. Characterization of Acp, a peptidoglycan hydrolase of Clostridium perfringens with N-acetylglucosaminidase activity that is implicated in cell separation and stress-induced autolysis. J. Bacteriol. 2010, 192, 2373–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamai, E.; Sekiya, H.; Goda, E.; Makihata, N.; Maki, J.; Yoshida, H.; Kamitori, S. Structural and biochemical characterization of the Clostridium perfringens autolysin catalytic domain. FEBS Lett. 2017, 591, 231–239. [Google Scholar] [CrossRef]
- Grundling, A.; Schneewind, O. Cross-linked peptidoglycan mediates lysostaphin binding to the cell wall envelope of Staphylococcus aureus. J. Bacteriol. 2006, 188, 2463–2472. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.Z.Q.; Fujiwara, T.; Komatsuzawa, H.; Sugai, M.; Sakon, J. Cell wall-targeting domain of glycylglycine endopeptidase distinguishes among peptidoglycan cross-bridges. J. Biol. Chem. 2006, 281, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Osipovitch, D.C.; Griswold, K.E. Fusion with a cell wall binding domain renders autolysin LytM a potent anti-Staphylococcus aureus agent. FEMS Microbiol. Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef]
- Mitkowski, P.; Jagielska, E.; Nowak, E.; Bujnicki, J.M.; Stefaniak, F.; Niedzialek, D.; Bochtler, M.; Sabala, I. Structural bases of peptidoglycan recognition by lysostaphin SH3b domain. Sci. Rep.-UK 2019, 9, 5965. [Google Scholar] [CrossRef] [Green Version]
- Zoll, S.; Schlag, M.; Shkumatov, A.V.; Rautenberg, M.; Svergun, D.I.; Gotz, F.; Stehle, T. Ligand-binding properties and conformational dynamics of autolysin repeat domains in Staphylococcal cell wall recognition. J. Bacteriol. 2012, 194, 3789–3802. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.H.; Chen, H.J.; Jiang, Y.L.; Wen, Z.S.; Huang, Y.B.; Cheng, W.; Li, Q.; Qi, L.; Zhang, J.R.; Chen, Y.X.; et al. Structure of Pneumococcal peptidoglycan hydrolase LytB reveals insights into the bacterial cell wall remodeling and pathogenesis. J. Biol. Chem. 2014, 289, 23403–23416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saksela, K.; Permi, P. SH3 domain ligand binding: What’s the consensus and where’s the specificity? FEBS Lett. 2012, 586, 2609–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponting, C.P.; Aravind, L.; Schultz, J.; Bork, P.; Koonin, E.V. Eukaryotic signalling domain homologues in archaea and bacteria. Ancient ancestry and horizontal gene transfer. J. Mol. Biol. 1999, 289, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Delgado, L.S.; Walters-Morgan, H.; Salamaga, B.; Robertson, A.J.; Hounslow, A.M.; Jagielska, E.; Sabala, I.; Williamson, M.P.; Lovering, A.L.; Mesnage, S. Two-site recognition of Staphylococcus aureus peptidoglycan by lysostaphin SH3b. Nat. Chem. Biol. 2020, 16, 24–30. [Google Scholar] [CrossRef]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef]
- Zhu, W.K.; Li, Y.; Liu, M.L.; Zhu, J.; Yang, Y.H. Uncorrelated effect of interdomain contact on Pin1 isomerase activity reveals positive catalytic cooperativity. J. Phys. Chem. Lett. 2019, 10, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Tamai, E.; Yoshida, H.; Sekiya, H.; Nariya, H.; Miyata, S.; Okabe, A.; Kuwahara, T.; Maki, J.; Kamitori, S. X-ray structure of a novel endolysin encoded by episomal phage phiSM101 of Clostridium perfringens. Mol. Microbiol. 2014, 92, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.P.; Abdubek, P.; Astakhova, T.; Axelrod, H.L.; Bakolitsa, C.; Cai, X.H.; Carlton, D.; Chen, C.; Chiu, H.J.; Chiu, M.; et al. Structure of the gamma-D-glutamyl-L-diamino acid endopeptidase YkfC from Bacillus cereus in complex with L-Ala-gamma-D-Glu: Insights into substrate recognition by NlpC/P60 cysteine peptidases. Acta Crystallogr. F 2010, 66, 1354–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.O.; Kong, M.; Kim, I.; Bai, J.; Cha, S.; Kim, B.; Ryu, K.S.; Ryu, S.; Suh, J.Y. Structural basis for cell-wall recognition by bacteriophage PBC5 endolysin. Structure 2019, 27, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [Green Version]
- Holm, L.; Laakso, L.M. Dali server update. Nucleic Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.P.; Mengin-Lecreulx, D.; Liu, X.Q.W.; Patin, D.; Farr, C.L.; Grant, J.C.; Chiu, H.J.; Jaroszewski, L.; Knuth, M.W.; Godzik, A.; et al. Insights into substrate specificity of NlpC/P60 cell wall hydrolases containing bacterial SH3 domains. mBio 2015, 6, e02327–e14. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, J.; Biboy, J.; Vollmer, W.; Hirt, R.P.; Keown, J.R.; Artuyants, A.; Black, M.M.; Goldstone, D.C.; Simoes-Barbosa, A. The protozoan Trichomonas vaginalis targets bacteria with laterally acquired NlpC/P60 peptidoglycan hydrolases. mBio 2018, 9, e01784-18. [Google Scholar] [CrossRef] [Green Version]
- Tossavainen, H.; Raulinaitis, V.; Kauppinen, L.; Pentikainen, U.; Maaheimo, H.; Permi, P. Structural and functional insights into lysostaphin-substrate interaction. Front. Mol. Biosci. 2018, 5, 60. [Google Scholar] [CrossRef]
- Gu, J.M.; Feng, Y.G.; Feng, X.; Sun, C.J.; Lei, L.C.; Ding, W.; Niu, F.F.; Jiao, L.Y.; Yang, M.; Li, Y.; et al. Structural and biochemical characterization reveals LysGH15 as an unprecedented “EF-Hand-Like” calcium-binding phage lysin. PLoS Pathog. 2014, 10, e1004109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.Y.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Xiao, R.; Anderson, S.; Aramini, J.; Belote, R.; Buchwald, W.A.; Ciccosanti, C.; Conover, K.; Everett, J.K.; Hamilton, K.; Huang, Y.J.; et al. The high-throughput protein sample production platform of the Northeast Structural Genomics Consortium. J. Struct. Biol. 2010, 172, 21–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, D.; Szyperski, T.; Otting, G.; Senn, H.; Wuthrich, K. Stereospecific nuclear magnetic-resonance assignments of the methyl-groups of valine and leucine in the DNA-binding domain of the 434-repressor by biosynthetically directed fractional C-13 labeling. Biochemistry 1989, 28, 7510–7516. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe—A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [Green Version]
- Bahrami, A.; Assadi, A.H.; Markley, J.L.; Eghbalnia, H.R. Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. Plos Comput. Biol. 2009, 5, e1000307. [Google Scholar] [CrossRef]
- Guntert, P.; Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 2015, 62, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Delaglio, F.; Cornilescu, G.; Bax, A. TALOS plus: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Powers, R.; Montelione, G.T. Protein NMR recall, precision, and F-measure scores (RPF scores): Structure quality assessment measures based on information retrieval statistics. J. Am. Chem. Soc. 2005, 127, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Tejero, R.; Montelione, G.T. Evaluating protein structures determined by structural genomics consortia. Proteins 2007, 66, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.M.; Zhang, D.; Zhou, P.; Li, B.T.; Huang, S.Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conformationally Restricting Constraints 2 | |
|---|---|
| NOE-based distance constraints | |
| Total | 822 |
| Intra-residue (i = j) | 168 |
| Sequential (|i − j| = 1) | 222 |
| Medium-range (1< |i − j| <5) | 69 |
| Long-range (|i − j| ≥ 5) | 363 |
| Hydrogen bond constraints | |
| Long-range (|i − j| ≥ 5)/total | 46/54 |
| Dihedral angle constraints | 50 |
| Residue Constraint Violations 2 | |
| Average number of distance violations per structure | |
| 0.1–0.2 Å | 3.05 |
| 0.2–0.5 Å | 0.35 |
| >0.5 Å | 0 |
| Average RMS distance violation/constraint (Å) | 0.01 |
| Maximum distance violation (Å) | 0.36 |
| Average number of dihedral angle violations per structure | |
| 1–10° | 0.25 |
| >10° | 0 |
| Average RMS dihedral angle violation/constraint (degree) | 0.12 |
| Maximum dihedral angle violation (degree) | 1.90 |
| RMSD from average coordinates 2,3 | |
| Backbone/heavy atoms (Å) | 0.3/0.9 |
| Structure Quality Factors | |
| Ramachandran plot statistics 2,3 | |
| Most favored/allowed regions (%) | 98.0/2.0 |
| Disallowed regions (%) | 0.0 |
| Global quality scores(raw/Z-score) 2 | |
| Verify3D | 0.47/0.16 |
| Prosall | 0.31/−1.41 |
| Procheck(phi-psi) 3 | −0.36/−1.10 |
| Procheck(all) 3 | −0.13/−0.77 |
| Molprobity clash | 17.01/−1.39 |
| RPF Scores 4 | |
| Recall/precision | 0.999/0.943 |
| F-measure/DP-score | 0.971/0.875 |
| No. | Volume (Å3) | Surface (Å2) | Hydrogen Bond Donors/Acceptors | Hydrophobic Interactions | Amino Acid Ratio | |||
|---|---|---|---|---|---|---|---|---|
| Nonpolar | Polar | Positive | Negative | |||||
| 1 | 317.38 | 738.87 | 19/36 | 28 | 0.38 | 0.47 | 0.10 | 0.05 |
| 2 | 210.82 | 372.21 | 4/10 | 19 | 0.36 | 0.37 | 0.18 | 0.09 |
| 3 | 125.25 | 372.62 | 4/14 | 17 | 0.44 | 0.22 | 0.11 | 0.22 |
| PDB | Uniprot | Z-Score | RMSD | Ident% | Organism | Description | Ref. |
|---|---|---|---|---|---|---|---|
| 4KRT | Q0SPG7 | 8.6 | 1.8 | 27 | Clostridium phage phiSM101 | Endolysin, Psm | [21] |
| 3PVQ | Q8A860 | 8.3 | 2.1 | 19 | Bacteroides thetaiotaomicron | NlpC/P60 family, YkfC | [26] |
| 3NPF | A7LS31 | 8.2 | 2.8 | 17 | Bacteroides ovatus | NlpC/P60 family, YkfC | [26] |
| 6BIQ | A2DC48 | 8.0 | 2.8 | 11 | Trichomonas vaginalis | NlpC/P60 family, NlpC_A2 | [27] |
| 4Q2W | P59205 | 8.0 | 3.5 | 16 | Streptococcus pneumoniae | N-acetylglucosaminidase, LytB | [14] |
| 6BIM | A2D7D7 | 7.7 | 3.8 | 11 | Trichomonas vaginalis | NlpC/P60 family, NlpC_A1 | [27] |
| 1R77 | O05156 | 7.5 | 1.6 | 15 | Staphylococcus capitis | Lysostaphin, ALE-1 | [10] |
| 5LEO | P10547 | 7.5 | 1.5 | 15 | Staphylococcus simulans | Lysostaphin | [12] |
| 6RK4 | P10547 | 7.5 | 1.5 | 15 | Staphylococcus simulans | Lysostaphin | [17] |
| 6ILU | A0A218KCJ1 | 7.4 | 1.9 | 15 | Bacillus phage PBC5 | Endolysin, LysPBC5 | [23] |
| 6RJE | P10547 | 7.4 | 1.5 | 15 | Staphylococcus simulans | Lysostaphin | [17] |
| 5NMY | P10547 | 7.2 | 1.6 | 15 | Staphylococcus simulans | Lysostaphin | [28] |
| 2MK5 | D6QY02 | 6.9 | 1.9 | 10 | Staphylococcus phage G15 | Endolysin, LysGH15 | [29] |
| 3H41 | Q736M3 | 6.7 | 5.0 | 13 | Bacillus cereus | NlpC/P60 family, YkfC | [22] |
| No. | SH3b6 | SH3b1 | SH3b2 | SH3b3 | SH3b10 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | |
| Volume (Å3) | 317.4 | 210.8 | 125.3 | 204.7 | 102.5 | 166.1 | 147.6 | 121.9 | 167.9 | 208.6 | 142.1 | 116.2 | 160.5 | 180.7 | 135.2 |
| Surface (Å2) | 738.9 | 372.2 | 372.6 | 387.6 | 231.6 | 296.2 | 322.6 | 309.9 | 440.5 | 376.5 | 312.4 | 362.3 | 321.3 | 361.9 | 320.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shan, Y.; He, X.; Wang, Z.; Yue, X.; Zhu, J.; Yang, Y.; Liu, M. Structural Investigations on the SH3b Domains of Clostridium perfringens Autolysin through NMR Spectroscopy and Structure Simulation Enlighten the Cell Wall Binding Function. Molecules 2021, 26, 5716. https://doi.org/10.3390/molecules26185716
Shan Y, He X, Wang Z, Yue X, Zhu J, Yang Y, Liu M. Structural Investigations on the SH3b Domains of Clostridium perfringens Autolysin through NMR Spectroscopy and Structure Simulation Enlighten the Cell Wall Binding Function. Molecules. 2021; 26(18):5716. https://doi.org/10.3390/molecules26185716
Chicago/Turabian StyleShan, Yubao, Xiaoling He, Zi Wang, Xiali Yue, Jiang Zhu, Yunhuang Yang, and Maili Liu. 2021. "Structural Investigations on the SH3b Domains of Clostridium perfringens Autolysin through NMR Spectroscopy and Structure Simulation Enlighten the Cell Wall Binding Function" Molecules 26, no. 18: 5716. https://doi.org/10.3390/molecules26185716
APA StyleShan, Y., He, X., Wang, Z., Yue, X., Zhu, J., Yang, Y., & Liu, M. (2021). Structural Investigations on the SH3b Domains of Clostridium perfringens Autolysin through NMR Spectroscopy and Structure Simulation Enlighten the Cell Wall Binding Function. Molecules, 26(18), 5716. https://doi.org/10.3390/molecules26185716