
Slow Magnetic Relaxation in {[CoCxAPy)] 2.15 H2O}n MOF Built from Ladder-Structured 2D Layers with Dimeric SMM Rungs

 ,
,  ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Synthesis
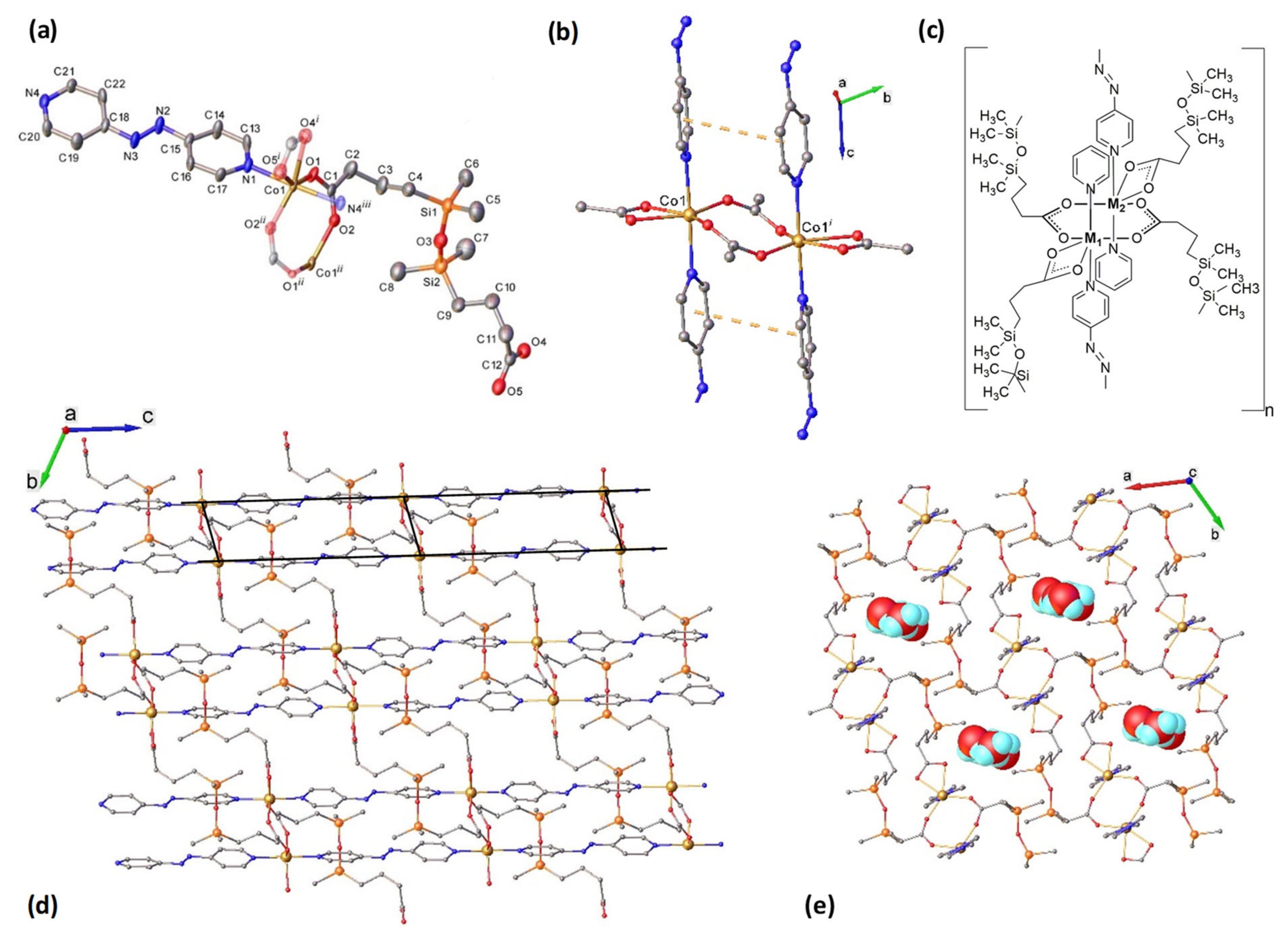
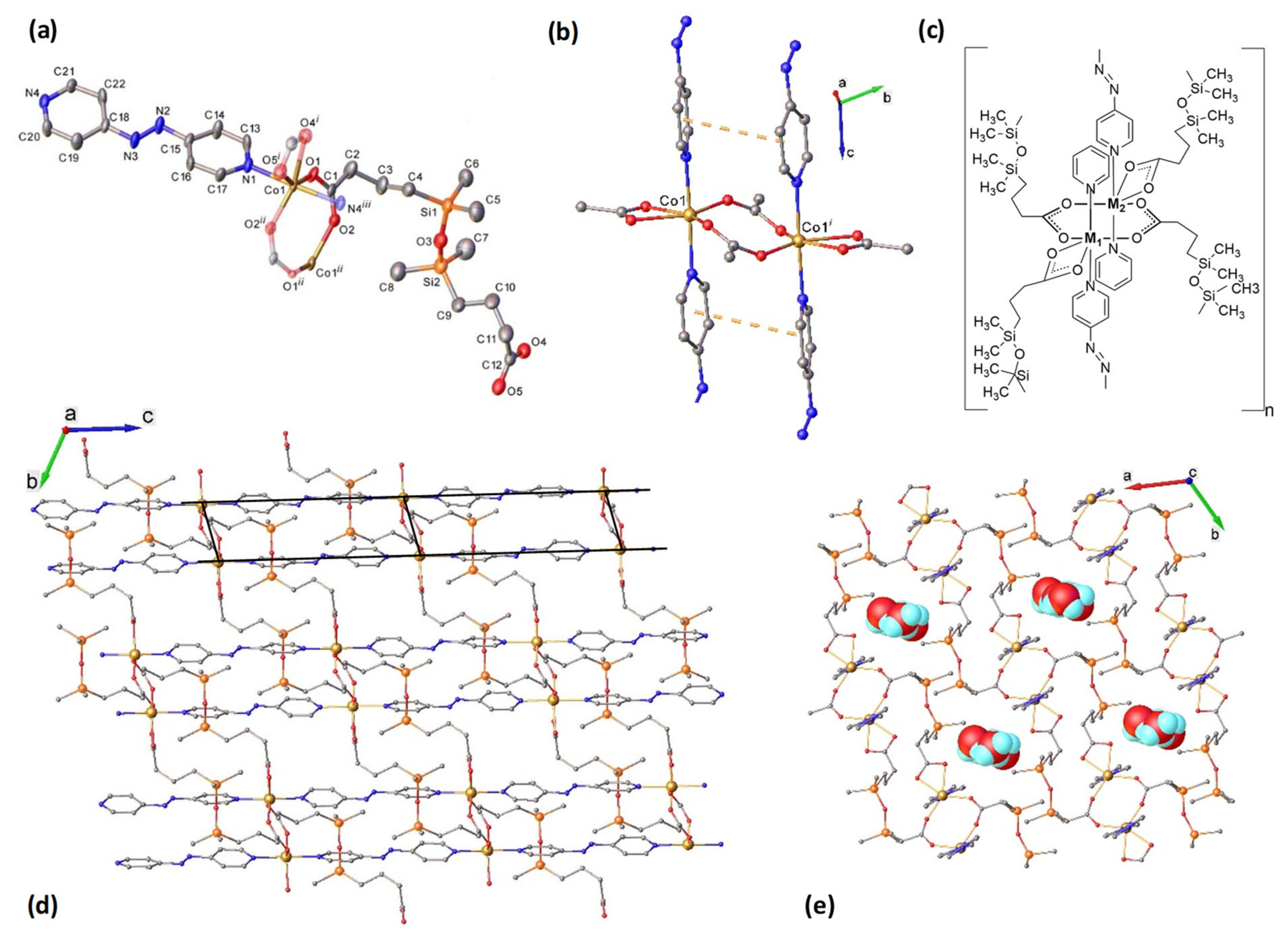
2.2. Structural Characterization
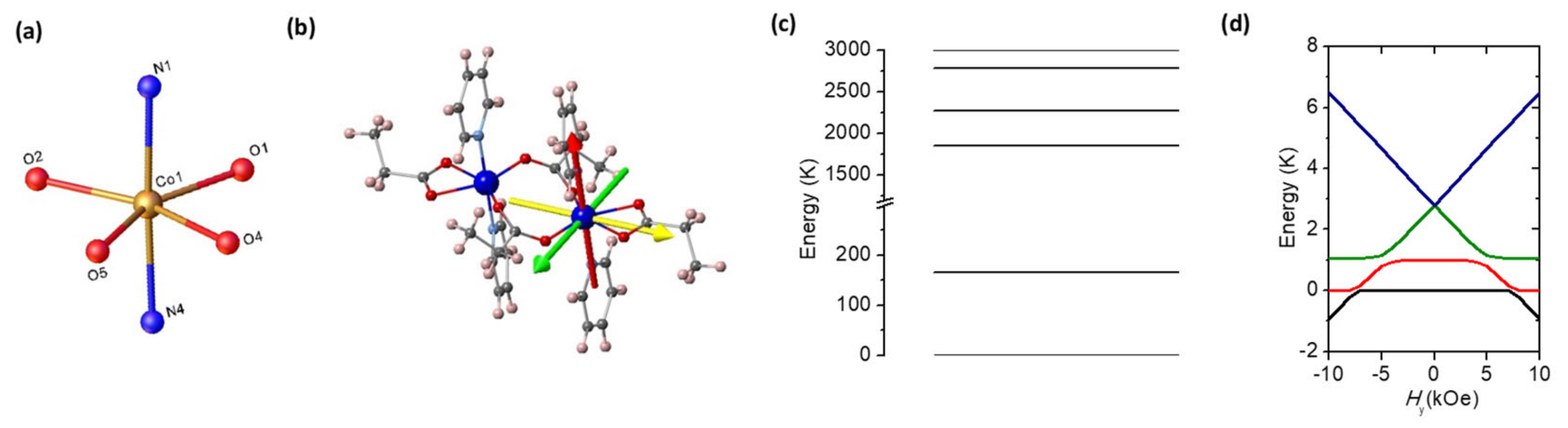
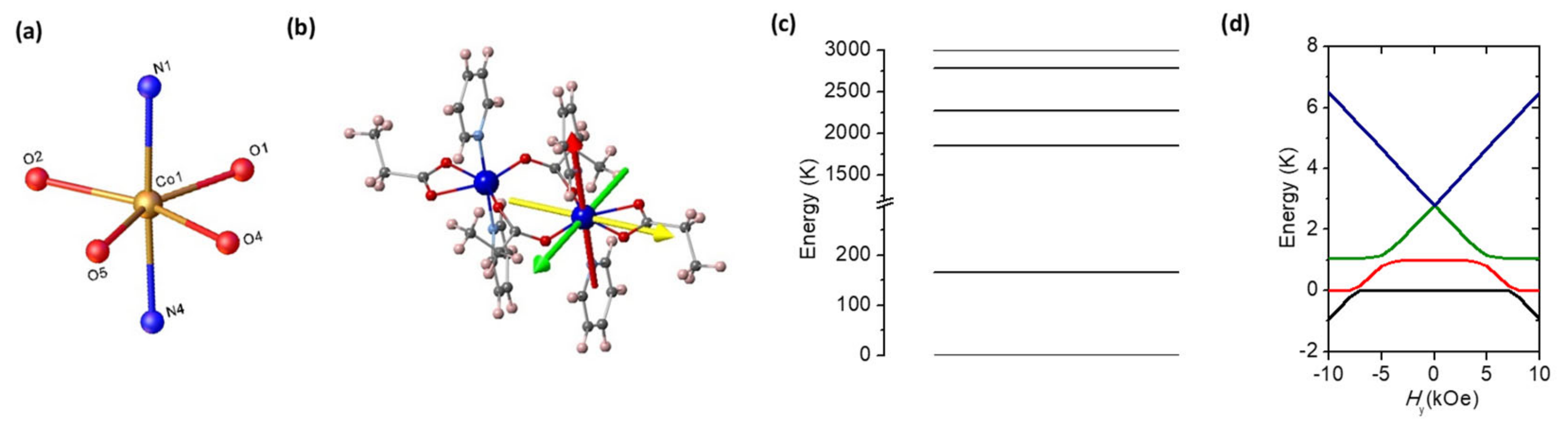
2.3. Theoretical Model and Ab Initio Calculations
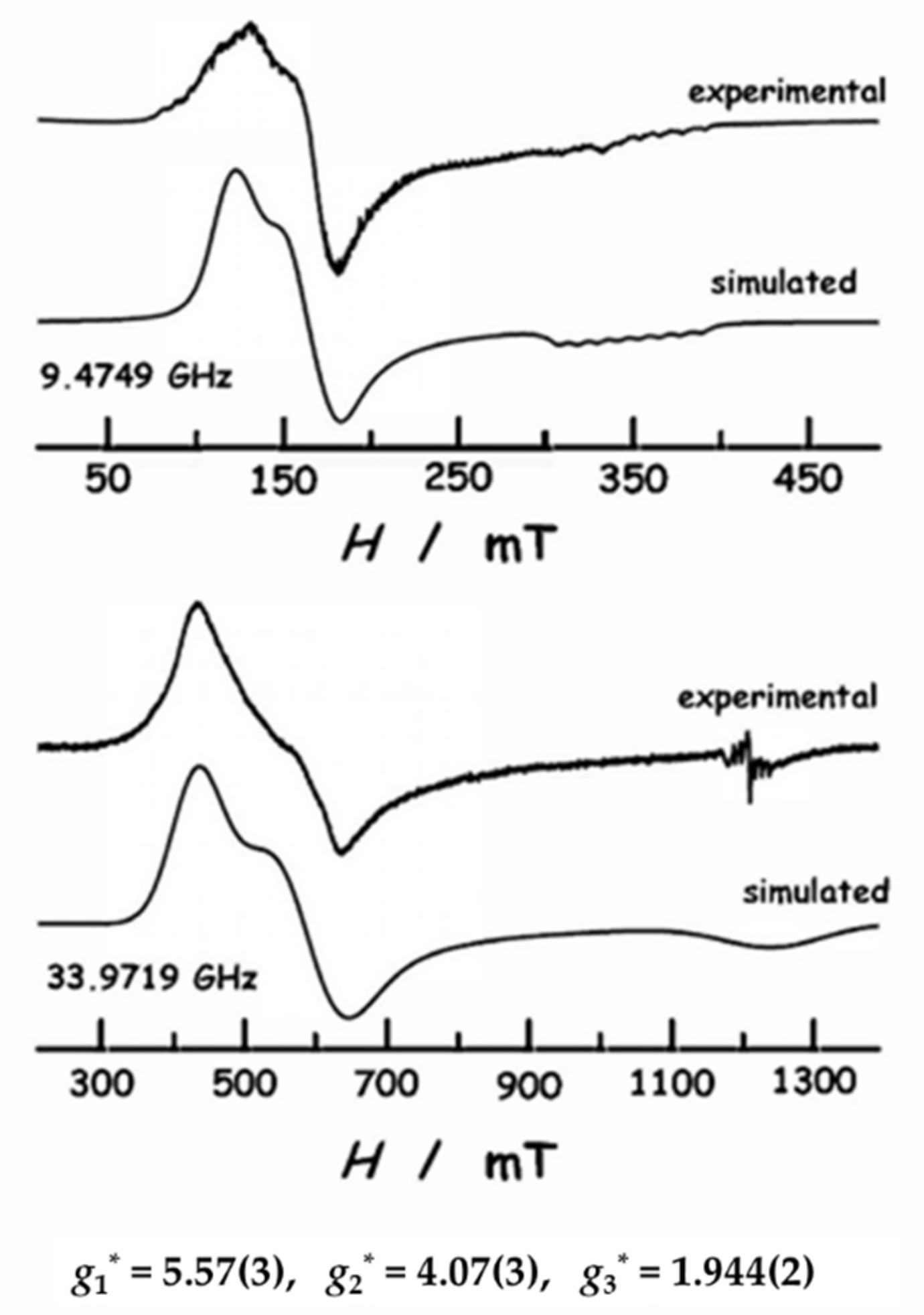
2.4. EPR Spectroscopy
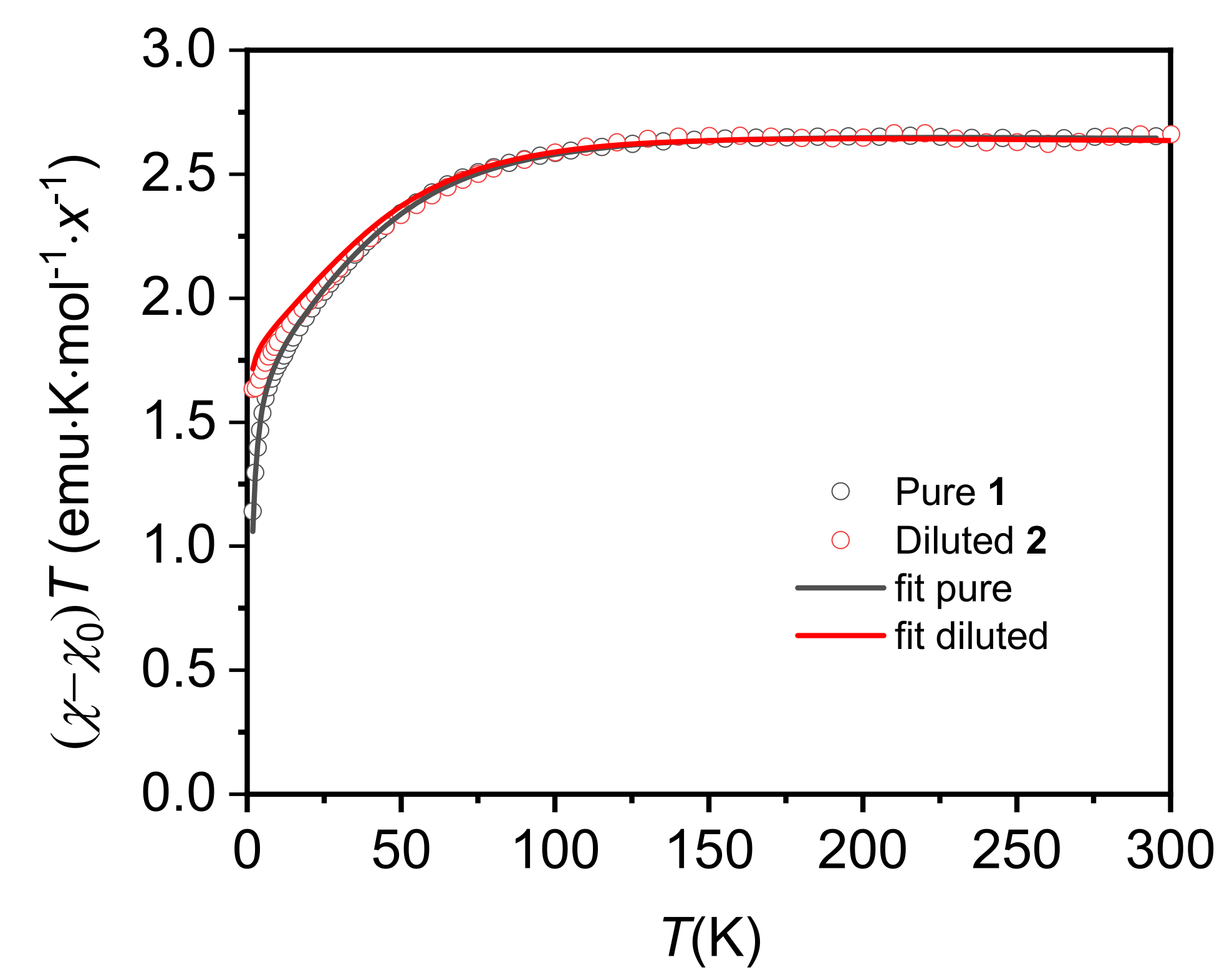
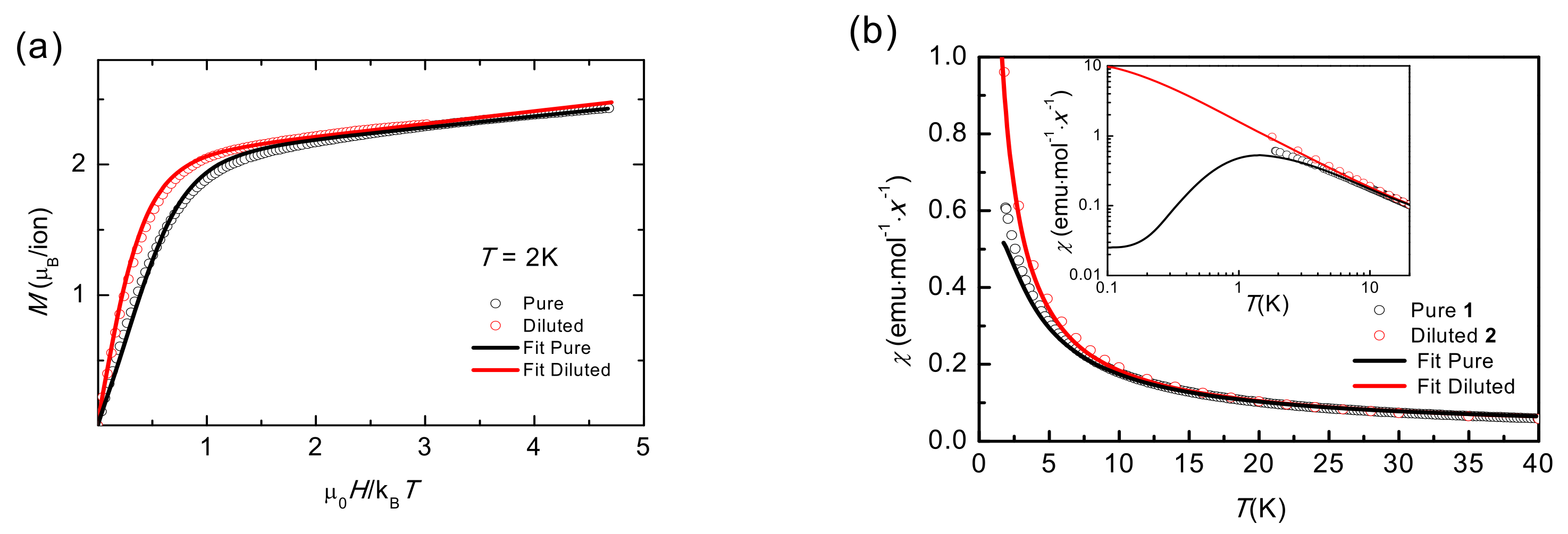
2.5. Static Magnetic Properties (Up to Room T)
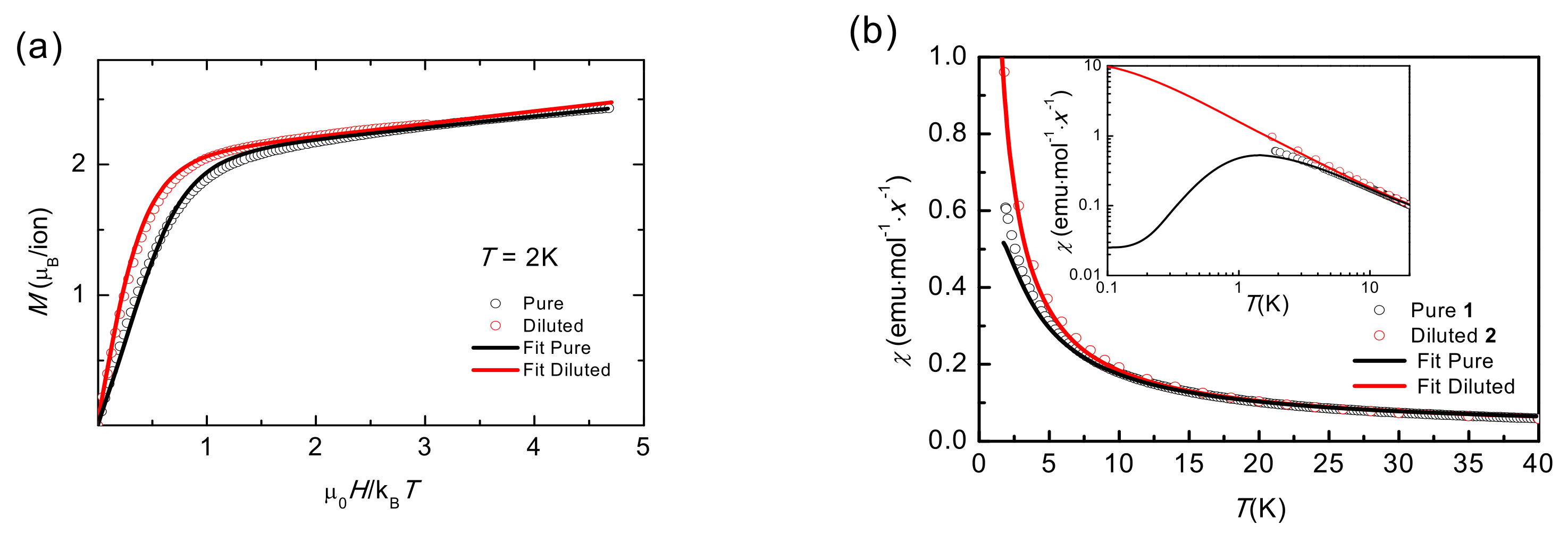
2.6. Static Magnetic Properties (T < 20 K)
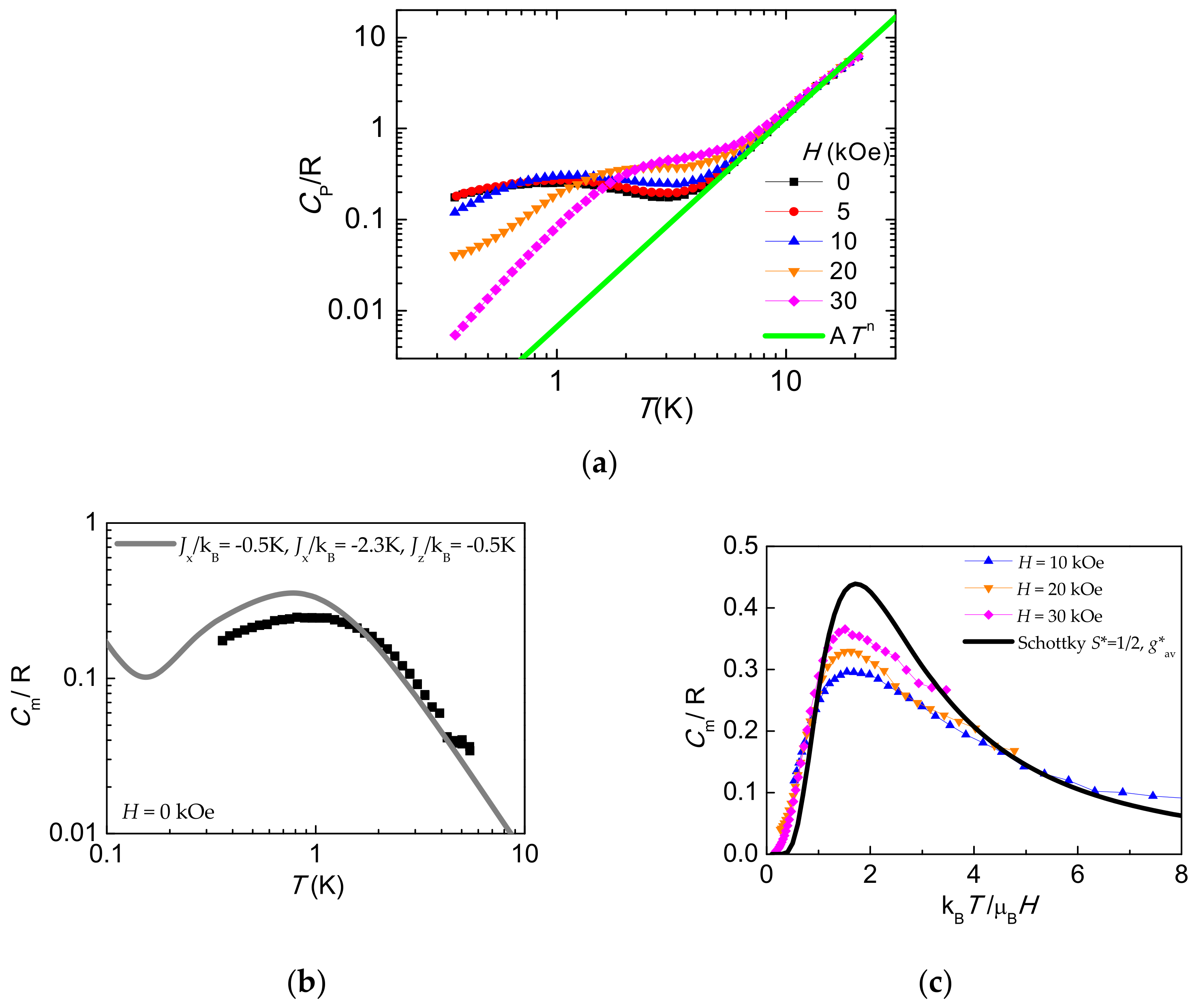
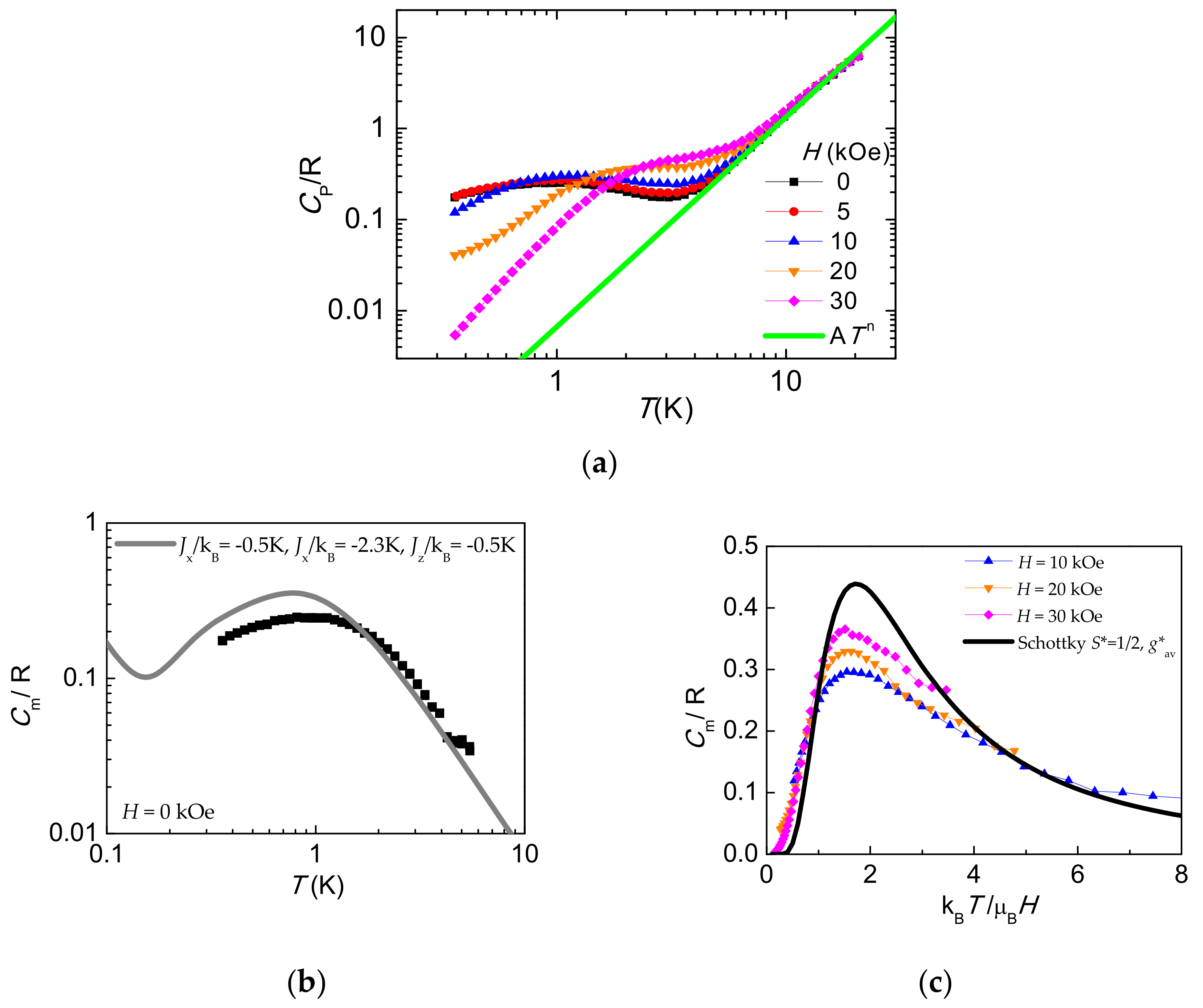
2.7. Heat Capacity
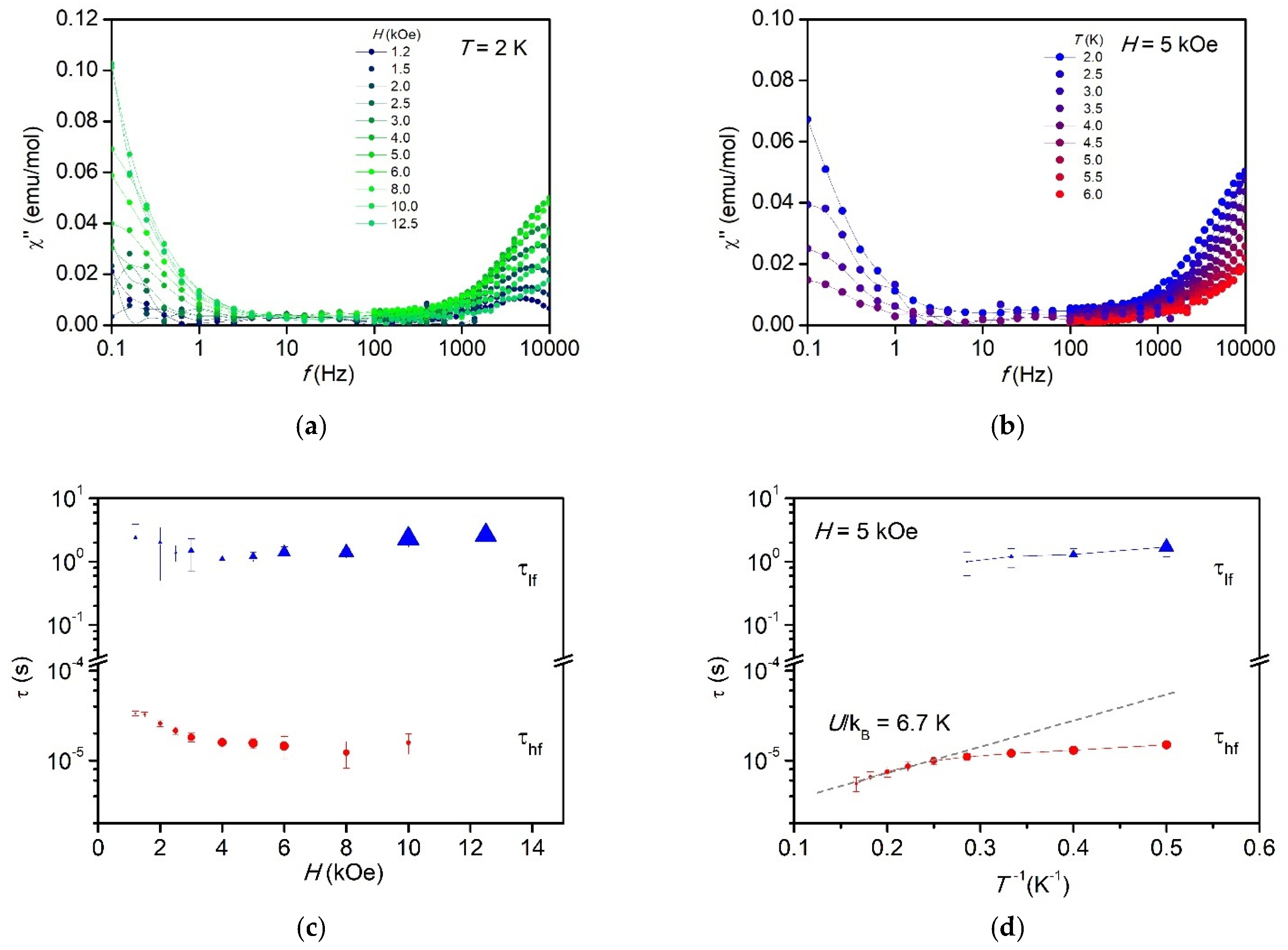
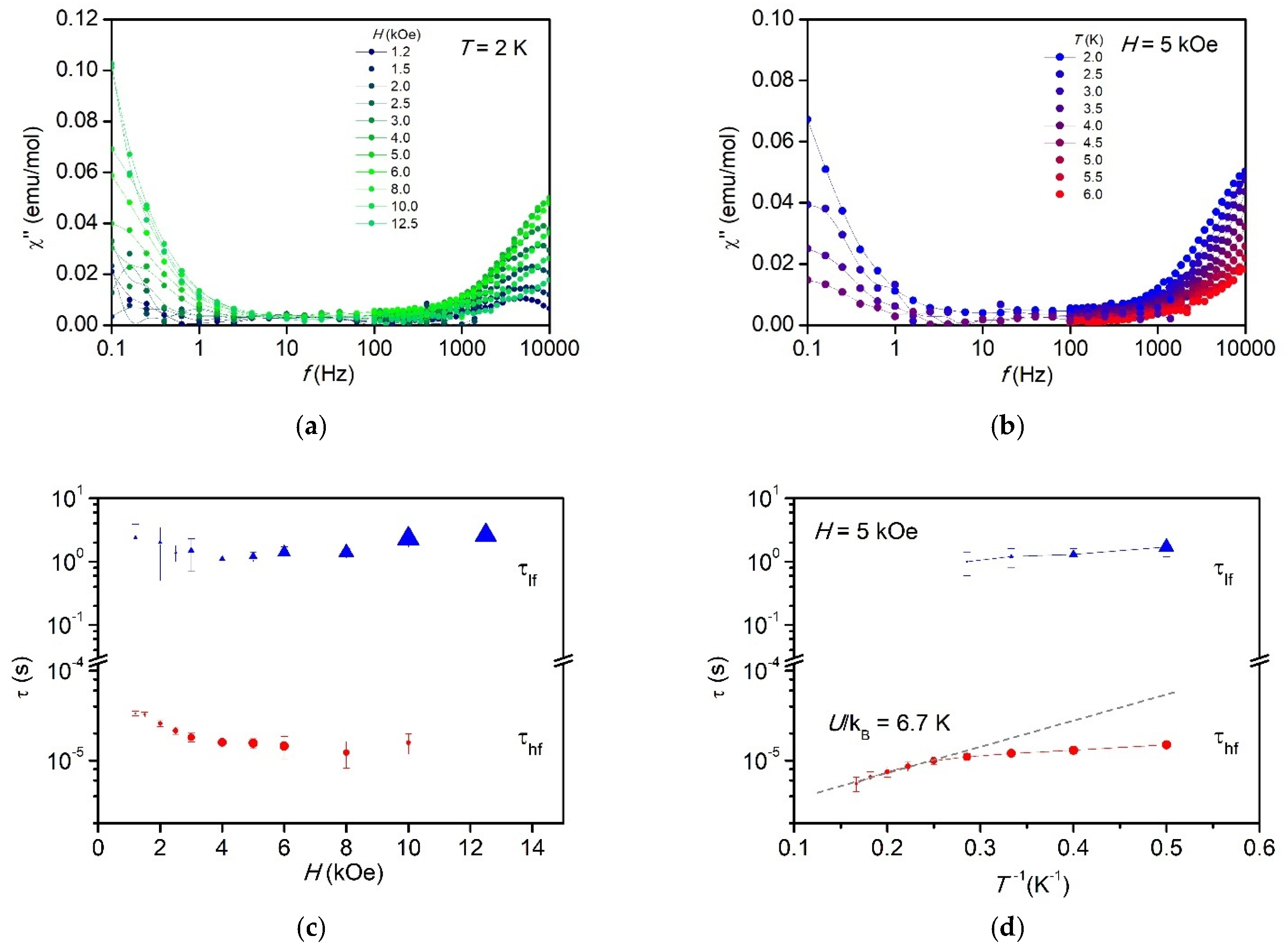
2.8. Dynamic Magnetic Properties
3. Discussion
4. Materials and Methods
4.1. Experimental Techniques
4.2. Materials
4.2.1. Preparation of {[CoCxAPy)]·2.15 H2O}n (1)
4.2.2. Preparation of {[Zn0.80Co0.2CxAPy)]·1.5 CH3OH}n (2)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suh, M.P.; Park, H.J.; Prasad, T.K.; Lim, D.-W. Hydrogen storage in metal-organic frameworks. Chem. Rev. 2012, 112, 782–835. [Google Scholar] [CrossRef]
- Sumida, K.; Rogow, D.L.; Mason, J.A.; McDonald, T.M.; Bloch, E.D.; Herm, Z.R.; Bae, T.H.; Long, J.R. Carbon dioxide capture in metal-organic frameworks. Chem. Rev. 2012, 112, 724–781. [Google Scholar] [CrossRef] [PubMed]
- Halder, G.J.; Kepert, C.J.; Moubaraki, B.; Murray, K.S.; Cashion, J.D. Guest-dependent spin crossover in a nanoporous molecular framework material. Science 2002, 298, 1762–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.R.; Sculley, J.; Zhou, H.-C. Metal–Organic Frameworks for Separations. Chem. Rev. 2012, 112, 869–932. [Google Scholar] [CrossRef]
- Yoon, M.; Srirambalaji, R.; Kim, K. Homochiral Metal–Organic Frameworks for Asymmetric Heterogeneous Catalysis. Chem. Rev. 2011, 112, 1196–1231. [Google Scholar] [CrossRef]
- Bartolomé, E.; Alonso, P.J.; Arauzo, A.; Luzón, J.; Bartolomé, J.; Racles, C.; Turta, C. Magnetic properties of the seven-coordinated nanoporous framework material Co(bpy)1.5(NO3)2 (bpy = 4,4′-bipyridine). Dalton Trans. 2012, 41, 10382. [Google Scholar] [CrossRef]
- Sun, L.; Campbell, M.G.; Dincă, M. Electrically Conductive Porous Metal-Organic Frameworks. Angew. Chem. Int. Ed. 2016, 55, 3566–3579. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.-N.; Wang, G.; Poelman, D.; Van Der Voort, P. Luminescent Lanthanide MOFs: A Unique Platform for Chemical Sensing. Materials 2018, 11, 572. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Xu, W.; Cui, X.; Wang, Y. Recent Progress in Metal-Organic Frameworks and Their Derived Nanostructures for Energy and Environmental Applications. ChemSusChem 2017, 10, 1645–1663. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jin, J.; Ding, B.; Han, X.; Han, A.; Liu, J. General Approach to Metal-Organic Framework Nanosheets With Controllable Thickness by Using Metal Hydroxides as Precursors. Front. Mater. 2020, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Espallargas, G.M.; Coronado, E. Magnetic functionalities in MOFs: From the framework to the pore. Chem. Soc. Rev. 2017, 47, 533–557. [Google Scholar] [CrossRef] [Green Version]
- Miyasaka, H.; Nakata, K.; Sugiura, K.-i.; Yamashita, M.; Clerac, R. A three-dimensional ferrimagnet composed of mixed-valence Mn4 clusters linked by an [Mn[N(CN)2]6]4- unit. Angew. Chem. Int. Ed. 2004, 43, 707–711. [Google Scholar] [CrossRef]
- Oyarzabal, I.; Fernández, B.; Cepeda, J.; Gómez-Ruiz, S.; Calahorro, A.J.; Seco, J.M.; Rodríguez-Diéguez, A. Slow relaxation of magnetization in 3D-MOFs based on dysprosium dinuclear entities bridged by dicarboxylic linkers. CrystEngComm 2016, 18, 3055–3063. [Google Scholar] [CrossRef]
- Vallejo, J.; Fortea-Pérez, F.R.; Pardo, E.; Benmansour, S.; Castro, I.; Krzystek, J.; Armentano, D.; Cano, J. Guest-dependent single-ion magnet behaviour in a cobalt(ii) metal–organic framework. Chem. Sci. 2015, 7, 2286–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurmoo, M. Magnetic metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1353–1379. [Google Scholar] [CrossRef] [PubMed]
- Rechkemmer, Y.; Breitgoff, F.D.; Van Der Meer, M.; Atanasov, M.; Hakl, M.; Orlita, M.; Neugebauer, P.; Neese, F.; Sarkar, B.; Van Slageren, J. A four-coordinate cobalt(II) single-ion magnet with coercivity and a very high energy barrier. Nat. Commun. 2016, 7, 10467. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhao, W.; Yi, G.; Zhou, J.; Yuan, A. Single-Ion Magnets based on 3d metals. Prog. Chem. 2019, 31, 337–350. [Google Scholar]
- Kostakis, G.E.; Perlepes, S.P.; Blatov, V.A.; Proserpiod, D.; Powell, A.K. High-nuclearity cobalt coordination clusters: Synthetic, topological and magnetic aspects. Coord. Chem. Rev. 2012, 256, 1246–1278. [Google Scholar] [CrossRef] [Green Version]
- Konieczny, P.; Gonzalez-Guillén, A.B.; Luberda-Durnaś, K.; Čižmár, E.; Pełka, R.; Oszajca, M.; Łasocha, W. 1D coordination polymer (OPD)2CoIISO4 showing SMM behaviour and multiple relaxation modes. Dalton Trans. 2019, 48, 7560–7570. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gou, X.; Shi, W.; Cheng, P. Single-chain magnets assembled in cobalt(ii) metal–organic frameworks. Chem. Commun. 2019, 55, 11000–11012. [Google Scholar] [CrossRef]
- Brunet, G.; Safin, D.A.; Jover, J.; Ruiz, E.; Murugesu, M. Single-molecule magnetism arising from cobalt(ii) nodes of a crystalline sponge. J. Mater. Chem. C 2016, 5, 835–841. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-H.; Tsai, H.-L.; Lu, Y.-L.; Wen, Y.-S.; Wang, J.-C.; Lu, K.-L. Assembly of a Robust, Thermally Stable Porous Cobalt(II) Nicotinate Framework Based on a Dicobalt Carboxylate Unit. Inorg. Chem. 2001, 40, 6426–6431. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.-L.; Kitagawa, S.; Chang, H.-C.; Ohba, M. Temperature-controlled hydrothermal synthesis of a 2D ferromagnetic coordination bilayered polymer and a novel 3D network with inorganic Co3(OH)2ferrimagnetic chains. Chem. Commun. 2004, 4, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Goswani, S.; Leitus, G.; Tripuramallu, B.K.; Goldberg, I. MnII and CoII coordination polymers showing field-dependent magnetism and slow magnetic relaxation behavior. Cryst. Growth Des. 2017, 17, 4393–4404. [Google Scholar] [CrossRef]
- Mariano, L.D.S.; Rosa, I.M.; De Campos, N.R.; Doriguetto, A.C.; Dias, D.F.; do Pim, W.D.; Valdo, A.K.S.; Martins, F.T.; Ribeiro, M.A.; De Paula, E.E.; et al. Polymorphic derivatives of NiII and CoII mesocates with 3D netwrorks and ‘Brick and ortar’ structures: Preparation, structural characterization, and cryomagnetic investigation of new single molecule magnets. Cryst. Growth Des. 2020, 20, 2462–2476. [Google Scholar] [CrossRef]
- Cazacu, M.; Vlad, A.; Turta, C.; Lisa, G. New iron-cobalt clusters with silicon-containing dicarboxylic acids. Open Chem. 2012, 10, 1079–1086. [Google Scholar] [CrossRef]
- Vlad, A.; Cazacu, M.; Zaltariov, M.F.; Shova, S.; Turta, C.; Airinei, A. Metallopolymeric structures containing highly flexible siloxane sequence. Polymer 2013, 54, 43–53. [Google Scholar] [CrossRef]
- Nitschke, J. Construction, Substitution, and Sorting of Metallo-organic Structures via Subcomponent Self-Assembly. Acc. Chem. Res. 2006, 40, 103–112. [Google Scholar] [CrossRef]
- Vlad, A.; Cazacu, M.; Zaltariov, M.F.; Bargan, A.; Shova, S.; Turta, C. A 2D metal-organic framework based on dizinc coordination units bridged through both flexible and rigid ligands. J. Mol. Struct. 2014, 1060, 94–101. [Google Scholar] [CrossRef]
- Shova, S.; Vlad, A.; Damoc, M.; Tiron, V.; Dascalu, M.; Novitchi, G.; Ursu, C.; Cazacu, M. Nanoscale Coordination Polymer of Dimanganese(II) as Infinite, Flexible Nanosheets with Photo-Switchable Morphology. Eur. J. Inorg. Chem. 2020, 2020, 2043–2054. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds. In Handbook of Vibrational Spectroscopy; Griffiths, P.R., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2006. [Google Scholar]
- Marnadu, R.; Chandrasekaran, J.; Vivek, P.; Balasubramani, V.; Maruthamuthu, S. Impact of Phase Transformation in WO3 Thin Films at Higher Temperature and its Compelling Interfacial Role in Cu/WO3/p–Si Structured Schottky Barrier Diodes. Z. Phys. Chem. 2020, 234, 355–379. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, G.; Haibibu, A.; Han, Z.; Wang, Q. High cyclic stability of electrocaloric effect in relaxor poly(vinylidene fluoride-trifluoroethylene-chlorofluoroethylene) terpolymers in the absence of ferroelectric phase transition. J. Appl. Phys. 2019, 126, 234102. [Google Scholar] [CrossRef]
- Osaki, K.; Uryû, N. Anisotropic g-Factors of Co2+ Ion in Complex Salts. J. Phys. Soc. Jpn. 1976, 40, 1575–1583. [Google Scholar] [CrossRef]
- Bocca, R. Zero-field splitting in metal complexes. Coord. Chem. Rev. 2004, 248, 757–815. [Google Scholar] [CrossRef]
- Palii, A.; Tsukerblat, B.; Clemente-Juan, J.M.; Coronado, E. Magnetic exchange between metal ions with unquenched orbital angular momenta: Basic concepts and relevance to molecular magnetism. Int. Rev. Phys. Chem. 2010, 29, 135–230. [Google Scholar] [CrossRef]
- Malmqvist, P.; Roos, B.O. The CASSCF state interaction method. Chem. Phys. Lett. 1989, 155, 189–194. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.-P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. J. Comput. Chem. 2003, 24, 1740–1747. [Google Scholar] [CrossRef]
- Izsak, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef] [PubMed]
- Heß, B.A.; Marian, C.M.; Wahlgren, U.; Gropen, O. A mean-field spin-orbit method applicable to correlated wavefunctions. Chem. Phys. Lett. 1996, 251, 365–371. [Google Scholar] [CrossRef]
- Neese, F. Efficient and accurate approximations to the molecular spin-orbit coupling operator and their use in molecular g-tensor calculations. J. Chem. Phys. 2005, 122, 034107. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Pavlov, A.A.; Nehrkorn, J.; Pankratova, Y.A.; Ozerov, M.; Mikhalyova, E.A.; Polezhaev, A.V.; Nelyubina, Y.V.; Novikov, V.V. Detailed electronic structure of a high-spin cobalt(ii) complex determined from NMR and THz-EPR spectroscopy. Phys. Chem. Chem. Phys. 2019, 21, 8201–8204. [Google Scholar] [CrossRef] [PubMed]
- Lazarescu, A.; Shova, S.; Bartolome, J.; Alonso, P.J.; Arauzo, A.; Balu, A.M.; Simonov, Y.A.; Gdaniec, M.; Turta, C.; Filoti, G.; et al. Heteronuclear (Co–Ca, Co–Ba) 2,3-pyridinedicarboxylate complexes: Synthesis, structure and physico-chemical properties. Dalton Trans. 2011, 40, 463–471. [Google Scholar] [CrossRef]
- Pilbrow, J.R. Effective g values for S = 3/2 and S = 5/2. J. Magn. Reson. 1978, 31, 479–490. [Google Scholar] [CrossRef]
- Bartolomé, E.; Arauzo, A.; Luzón, J.; Bartolomé, J.; Bartolomé, F. Magnetic Relaxation of Lanthanide-Based Molecular Magnets. In Handbook of Magnetic Materials; Elsevier B.V.: Amsterdam, The Netherlands, 2017; Volume 26, pp. 1–289. [Google Scholar]
- Arauzo, A.; Bartolomé, E.; Benniston, A.C.; Melnic, S.; Shova, S.; Luzón, J.; Alonso, P.J.; Barra, A.-L.; Bartolomé, J. Slow magnetic relaxation in a dimeric Mn 2 Ca 2 complex enabled by the large Mn( iii ) rhombicity. Dalton Trans. 2016, 46, 720–732. [Google Scholar] [CrossRef] [Green Version]
- Bleaney, B.; Ingram, D.J.E. Hyperfine structure of the paramagnetic resonance spectrum of divalent cobalt: Experimental. Nature 1949, 164, 116–117. [Google Scholar] [CrossRef]
- Algra, H.A.; Greidanus, F.J.A.M.; de Jongh, L.J.; Huiskamp, W.J.; Reedijk, J. Hyperfine specific heats of two paramagnetic, octahedrally coordinated Co2+ compounds. Phys. B+C 1976, 83, 85–91. [Google Scholar] [CrossRef]
- Tang, L.; Wang, H.H.; Fu, Y.H.; Wang, Y.T.; Wang, J.; Hou, X. Three cobalt-based coordination polymers with tripodal carboxylate and imidazole-containing ligands: Syntheses, structures, properties and DFT studies. RSC Adv. 2019, 9, 38902–38911. [Google Scholar] [CrossRef] [Green Version]
- Pakula, R.J.; Berry, J.F. Cobalt complexes of the chelating dicarboxylate ligand “esp”: A paddlewheel-type dimer and a heptanuclear coordination cluster. Dalton Trans. 2018, 47, 13887–13893. [Google Scholar] [CrossRef]
- Ostrovsky, S.M.; Falk, K.; Pelikán, J.; Brown, D.A.; Tomkowicz, Z.; Haase, W. Orbital Angular Momentum Contribution to the Magneto-Optical Behavior of a Binuclear Cobalt(II) Complex. Inorg. Chem. 2006, 45, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Wakimoto, S.; Kakurai, K.; Honda, Z.; Yamada, K. Magnetic excitations from the singlet dimerized state inNa2Co2(C2O4)3(H2O)2. Phys. Rev. B 2007, 75, 012405. [Google Scholar] [CrossRef]
- Konar, S.; Zangrando, E.; Drew, M.G.B.; Ribas, J.; Chaudhuri, N.R. Synthesis, structural analysis, and magnetic behaviour of three fumarate bridged coordination polymers: Five-fold interpenetrated diamond-like net of NiII, sheets of NiIIand CoII. Dalton Trans. 2004, 2, 260–266. [Google Scholar] [CrossRef]
- Talismanova, M.O.; Sidorov, A.A.; Novotortsev, V.M.; Aleksandrov, G.G.; Nefedov, S.E.; Eremenko, I.L.; Moiseev, I.I. Unusual transformation of the urea molecule giving rise to the NCO– anion as a bridging ligand between two CoII atoms. Russ. Chem. Bull. 2001, 50, 2251–2253. [Google Scholar] [CrossRef]
- Rueff, J.-M.; Masciocchi, N.; Rabu, P.; Sironi, A.; Skoulios, A. Structure and Magnetism of a Polycrystalline Transition Metal Soap − CoII[OOC(CH2)10COO](H2O)2. Eur. J. Inorg. Chem. 2001, 2001, 2843. [Google Scholar] [CrossRef]
- Rueff, J.-M.; Masciocchi, N.; Rabu, P.; Sironi, A.; Skoulios, A. Synthesis, Structure and Magnetism of Homologous Series of Polycrystalline Cobalt Alkane Mono- and Dicarboxylate Soaps. Chem.-A Eur. J. 2002, 8, 1813–1820. [Google Scholar] [CrossRef]
- Wei, Y.; Yu, Y.; Sa, R.; Li, Q.; Wu, K. Two cobalt(II) coordination polymers [Co2(H2O) 4(Hbidc)2]n and [Co(Hbidc)]n (Hbidc = 1H-benzimidazole-5,6-dicarboxylate): Syntheses, crystal structures, and magnetic properties. CrystEngComm 2009, 11, 1054–1060. [Google Scholar] [CrossRef]
- Demessence, A.; Rogez, G.; Welter, R.; Rabu, P. Structure and magnetic properties of a new cobalt(II) thiophenedicarboxylate coordination polymer showing unprecedented coordination. Inorg. Chem. 2007, 46, 3423–3425. [Google Scholar] [CrossRef] [PubMed]
- Fomina, I.G.; Sidorov, A.A.; Aleksandrov, G.G.; Zhilov, V.; Ikorskii, V.N.; Novotortsev, V.M.; Eremenko, I.L.; Moiseev, I.I. Unsymmetrical dinuclear cobalt and nickel trimethylacetate complexes. Russ. Chem. Bull. 2004, 53, 118–126. [Google Scholar] [CrossRef]
- Brown, D.A.; Errington, W.; Glass, W.K.; Haase, W.; Kemp, T.J.; Nimir, H.; Ostrovsky, S.; Werner, R. Magnetic, Spectroscopic, and Structural Studies of Dicobalt Hydroxamates and Model Hydrolases‖. Inorg. Chem. 2001, 40, 5962–5971. [Google Scholar] [CrossRef] [PubMed]
- Ostrovsky, S.; Werner, R.; Brown, D.A.; Haase, W. Magnetic properties of dinuclear cobalt complexes. Chem. Phys. Lett. 2002, 353, 290–294. [Google Scholar] [CrossRef]
- Turpeinen, U.; Hämäläinen, R.; Reedijk, J. The dinuclear unit μ-aqua-bis(μ-carboxylato)dimetal. X-ray structure and magnetism of cobalt and nickel(II) compounds containing bridging carboxylato groups and a bridging water molecule. Polyhedron 1987, 6, 1603–1610. [Google Scholar] [CrossRef]
- Pruchnik, F.P.; Dawid, U.; Kochel, A. Structure and properties of the dinuclear complex [Co2(μ-OAc)2(OAc)2(μ-H2O)(phen)2]. Polyhedron 2006, 25, 3647–3652. [Google Scholar] [CrossRef]
- Manna, S.C.; Zangrando, E.; Ribas, J.; Chaudhuri, N.R. Cobalt(ii)–(dpyo)–dicarboxylate networks: Unique H-bonded assembly and rare bridging mode of dpyo in one of them [dpyo = 4,4′-dipyridyl N, N′-dioxide]. J. Chem. Soc. Dalton Trans. 2007, 14, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Kashiwagi, T.; Yamaguchi, H.; Kimura, S.; Honda, Z.; Yamada, K.; Kindo, K.; Hagiwara, M. High Field ESR and Magnetization in Na2Co2(C2O4)3(H2O)2. J. Phys. Soc. Jpn. 2006, 75, 124708. [Google Scholar] [CrossRef]
- Price, D.J.; Powell, A.K.; Wood, P.T. Hydrothermal synthesis, structure, stability and magnetism of Na2Co2(C2O4)3(H2O)2: A new metal oxalate ladder. J. Chem. Soc. Dalton Trans. 2000, 20, 3566–3569. [Google Scholar] [CrossRef]
- Honda, Z.; Katsumata, K.; Kikkawa, A.; Yamada, K. Thermodynamic Properties in the Approach to the Quantum Critical Point of the Spin-Ladder MaterialNa2Co2(C2O4)3(H2O)2. Phys. Rev. Lett. 2005, 95, 087204. [Google Scholar] [CrossRef]
- Lazarov, N.; Spasojević, V.; Kusigerski, V.; Matić, V.; Milic, M. Magnetic susceptibility calculation of the dinuclear cobalt complex 6Co2(ox)tpmc9(ClO4)2a3H2O. J. Magn. Magn. Mater. 2004, 272, 1065–1066. [Google Scholar] [CrossRef]
- Lazarov, N.D.; Spasojević, V.; Matic, V.M.; Kusigerski, V.; Guillot, M. Magnetic properties of asymmetric Co ( II ) dimer at low temperatures. Rev. Roum. Chim. 2007, 52, 1027–1031. [Google Scholar]
- Banerjee, A.; Banerjee, S.; García, C.J.G.; Benmansour, S.; Chattopadhyay, S. Field-induced single molecule magnet behavior of a dinuclear cobalt(ii) complex: A combined experimental and theoretical study. Dalton Trans. 2020, 49, 16778–16790. [Google Scholar] [CrossRef]
- Herchel, R.; Dvořák, Z.; Trávníček, Z.; Mikuriya, M.; Louka, F.R.; Mautner, F.A.; Massoud, S.S. Cobalt(II) and copper(II) covalently and non-covalently dichlorido-bridged complexes of an unsymmetrical tripodal pyrazolyl-pyridyl amine ligand: Structures, magnetism and cytotoxicity. Inorg. Chim. Acta 2016, 451, 102–110. [Google Scholar] [CrossRef]
- Rajnák, C.; Titiš, J.; Miklovič, J.; Kostakis, G.E.; Fuhr, O.; Ruben, M.; Boča, R. Five mononuclear pentacoordinate Co(II) complexes with field-induced slow magnetic relaxation. Polyhedron 2017, 126, 174–183. [Google Scholar] [CrossRef]
- Świtlicka, A.; Machura, B.; Penkala, M.; Bieńko, A.; Bieńko, D.C.; Titiš, J.; Rajnák, C.; Boča, R.; Ozarowski, A. Slow magnetic relaxation in hexacoordinated cobalt(ii) field-induced single-ion magnets. Inorg. Chem. Front. 2020, 7, 2637–2650. [Google Scholar] [CrossRef]
- Gómez-Coca, S.; Urtizberea, A.; Cremades, E.; Alonso, P.J.; Camón, A.; Ruiz, E.; Luis, F. Origin of slow magnetic relaxation in Kramers ions with non-uniaxial anisotropy. Nat. Commun. 2014, 5, 4300. [Google Scholar] [CrossRef] [Green Version]
- Haldane, F.D.M. O(3) nonlinear model and the topological distinction between integer- and half-integer-spin antiferromagnets in two dimensions. Phys. Rev. Lett. 1988, 61, 1029–1032. [Google Scholar] [CrossRef]
- Dagotto, E.; Rice, T.M. Surprises on the Way from One- to Two-Dimensional Quantum Magnets: The Ladder Materials. Science 1996, 271, 618–623. [Google Scholar] [CrossRef] [Green Version]
- Aristov, D.N.; Brünger, C.; Assaad, F.F.; Kiselev, M.N.; Weichselbaum, A.; Capponi, S.; Alet, F. Asymmetric spin- 1 2 two-leg ladders: Analytical studies supported by exact diagonalization, DMRG, and Monte Carlo simulations. Phys. Rev. B Condens. Matter Mater. Phys. 2010, 82, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Shelton, D.; Nersesyan, A. Antiferromagnetic spin ladders: Crossover between spin S=1/2 and S=1 chains. Phys. Rev. B-Condens. Matter Mater. Phys. 1996, 53, 8521–8532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CrysAlis RED; Version 1.171.36.32; Oxford Diffr. Ltd.: Abingdon, UK, 2003.
- Sheldrick, G.M. SHELXT– Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazacu, M.; Marcu, M.; Vlad, A.; Caraiman, D.; Racles, C. Synthesis of functional telechelic polydimethylsiloxanes by ion-exchangers catalysis. Eur. Polym. J. 1999, 35, 1629–1635. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |
|---|---|---|
| CCDC | 2005545 | 2005606 |
| Empirical formula | C22H36.3CoN4O7.15Si2 | C23.5H38Co0.2N4O6.5Si2Zn0.80 |
| Formula weight | 586.36 | 600.81 |
| Temperature/K | 200 | 200 |
| Crystal system | triclinic | triclinic |
| Space group | P-1 | P-1 |
| a/Å | 11.8234(13) | 11.7865(9) |
| b/Å | 11.8324(13) | 11.8192(4) |
| c/Å | 13.3262(11) | 13.3333(5) |
| α/° | 110.597(8) | 110.367(4) |
| β/° | 96.107(8) | 96.376(5) |
| γ/° | 114.090(10) | 114.261(5) |
| V/Å3 | 1523.5 | 1515.77(15) |
| Z | 2 | 2 |
| Dcalc/mg/mm3 | 1.278 | 1.316 |
| μ/mm−1 | 0.685 | 0.882 |
| Crystal size/mm3 | 0.15 × 0.10 × 0.10 | 0.30 × 0.10 × 0.05 |
| θmin, θ max(°) | 4.938 to 50.048 | 3.97 to 50.052 |
| Reflections collected | 10714 | 11780 |
| Independent reflections | 5309 [Rint = 0.0576] | 5329 [Rint = 0.0300] |
| Data/restraints/parameters | 5309/0/339 | 5329/0/350 |
| R1a(I > 2σ(I) | 0.0829 | 0.0438 |
| wR2b(all data) | 0.2221 | 0.1198 |
| GOF c | 1.022 | 1.058 |
| Largest diff. peak/hole/e Å−3 | 1.21/−0.56 | 1.01/−0.30 |
| Ab initio/Fit Compound 1 | Ab initio Compound 2 | Experimental | ||
|---|---|---|---|---|
| S = 3/2 | D/kB(K) | 82.0 | 83.0 | |
| η = E/D | 0.13 | 0.20 | 0.165(5) | |
| J/kB(K) | −0.32 | −0.44 | ||
| gx gy gz | 2.40 2.58 1.96 | 2.37 2.64 1.95 | 2.74(3) 2.28(2) 2.11(1) | |
| S* = 1/2 | gx* gy* gz* | 3.82 6.05 1.91 | 3.28 6.58 1.79 | 4.07(3) 5.57(3) 1.944(2) |
| Pure 1 | Diluted 2 | |
|---|---|---|
| χ0 (emu·mol−1) | −7.0·10−4 | −1.7·10−3 |
| C (emu·K/mol) | 2.672 ± 0.008 | 2.67 ± 0.01 |
| θ (K) | −1.1 ± 0.7 | −0.1 ± 0.3 |
| Ref. | Coord. | Exch. Path | Co-Co (Å) | C (emu·K/mol dimer) | θ (K) | g | Model Spin | J/kB (K) | D/kB (K) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Co-Co Carboxylate Exchange Paths | ||||||||||
| Co2(esp)2(EtOH)2 (dimer) | [55] | CoO4L L-outwards | (4) syn-syn | 2.7245 | ∼4.4 | g⊥ = 2.10 g‖ = 3.5 | HH 3/2 | −9.49 AF | −79 | |
| {[CoCxAPy]·2.15 H2O}n (2LL dimer) | This work. | CoN2O4 apical | (2) syn-syn | 3.9684 | 5.35 | −1.1 | gx = 2.40 gy = 2.58 gz = 1.96 | HH 3/2 | −0.32 | 82 η = 0.13 |
| “ | “ | “ | “ | g*x = 4.07 g*y = 5.57 g*z = 1.94 | HAN 1/2 | −2.3 ex = 0.2 ey = 0.2 | ||||
| [Co(Htatb)(bimbp)]·DMF (dimer) | [54] | CoN2O4 apical | (2) syn-syn | 4.156 | 3.73 | −7.21 | 2.28 | HH 3/2 | −6.65 AF | |
| [Co(Htatb)(1,3-bimyb)] (dimer) | [54] | CoN2O4 apical | (2) syn-syn | 4.176 | 3.69 | −8.44 | 2.4 | HH 3/2 | −5.23 AF | |
| [{Co(phen)}2(fum)2] (dimer) | [58] | CoN2O4 adjacent | (2) distorted syn-syn | 4.464 | 5.93 | g*‖ = 6.0 g*⊥ = 3.5 | HAN 1/2 | <1K Ferro | ||
| dipy2Co2(μ-OOCCMe3)2 (OOCCMe3)2 (dimer) | [59] | CoN2O4 CoO5 adjacent | (2) syn-syn | 4.383 | 5.688 | HH 3/2 | ||||
| [Co(3,4-pyda)(H2O)2]n·nH2O (bilayer-dimer) | [23] | CoNO5 CoO5 | (1) syn-anti | 4.773 | 4.39 | 8.3 | HH 3/2 | |||
| [Co{OOC(CH2)n-2-COO}(H2O)2] d-12Co (chain) | [60,61] | CoO6 | (1) anti-anti | 5.93 | 6.0 | −19.7 | HI 1/2 | −1.02 | ||
| [Co{CH3(CH2)n-2COO}2(H2O)2] m-12Co, m-20Co (chain) | [61] | CoO6 | (1) anti-anti | 6.3 | 7.0 | −17.4 −31.8 | HI 1/2 | −0.65 | ||
| Co-Co Carboxylate and Oxygen Mediated Exchange Paths | ||||||||||
| [Co2(H2O)4(Hbidc)2]n (chain) | [62] | CoNO5 outwards | (2) syn-syn (2) Co-O-Co | 3.114 | 5.35 | 1 | 2.39 | HH 3/2 | 1.21 Ferro | |
| {Co((κ1-κ1)-( κ1-μ2)-μ4TDC)(μ2H2O)0.5(H2O)}n (dimer) | [63] | CoO6 | (2) syn-syn (1) Co-O-Co | 3.212 | 7.06 | 2.63 | g* = 5.2 | HI 1/2 | ||
| bpyCo2(μ2-O,η2-OOCCMe3) (μ2-O,O’-OOCCMe3)2 (η2-OOCCMe3) (unsymmetrical dimer) | [64] | CoNO5 CoO5 | (2) syn-syn (1) Co-O-Co | 3.272 | Ferro | |||||
| [Co2(μ-OAc)3(urea)(tmen)2]- [OTf] (dimer) | [56] | CoN2O4 adjacent | (2) syn-syn (1) Co-O-Co | 3.4813 | 5.59 | 4.73 (dimer) (2 × 2.63) | HH L = 1 | 26 | 57 | |
| [Co2(μOAc)2(μ-AA)(urea) (tmen)2][OTf] (3) (dimer) | [65,66] | CoN2O4 adjacent | (2) syn-syn (1) Co-O-Co | 3.4316 | 6.48 | g‖ = 3.0 g⊥ = 2.6 gav = 2.66 | HH 3/2 | −5.2 | 43 | |
| [Co2(μ-H2O)(μOAc)2(OAc)2 (tmen)2] (1) (dimer) | [65,66,67] | CoN2O4 adjacent | (2) syn-syn (1) Co-O-Co | 3.597 | 6.42 | −8.5 | g‖ =2.9 g⊥ = 2.5 gav =2.64 | HH 3/2 | −1 | 64.3 |
| [Co2(μ-OAc)2(OAc)2(μ-H2O) (phen)2] (dimer) | [68] | CoN2O4 adjacent | 3.57 | 6.5 | ||||||
| [Co3(OH)2(3,4-pyda)2(H2O)2]n | [23] | CoNO5 CoO6 | (1) syn-syn (1) Co-O-Co | 2.988 | HH 3/2 | −8.3 | ||||
| Co2(H2O)(C6H4O2N)4·0.5CH3- CH2OH·0.5H2O (1) (dimer) | [22] | CoN2O4 | (1) syn-syn (1) Co-O-Co | 3.17 | −31.2 | 2.53 | HH 3/2 | −7.9 | ||
| Co2(H2O)(C6H4O2N)4·C6H5CH2OH (dimer) | [22] | CoN2O4 | (1) syn-syn (1) Co-O-Co | 3.42 | −33.1 | 2.63 | HH 3/2 | −8.3 | ||
| {[Co(dpyo)(tp)(H2O)2]· [Co(H2O)6]·(tp)·(H2O)}n (chain) | [69] | CoO6 | (1) syn-syn (1) Co-O-Co | 3.742 | 6.56 | HI 1/2 | −12.4 | |||
| Co-Co Oxalate Exchange Path | ||||||||||
| Na2Co2(C2O4)3(H2O)2 (2 LL-dimer) | [57], [70,71,72] | CoO6 | (2) oxalate Co-O-C-O-C | 5.393 | 4.86 | −36 | gx* = 2.8 gy* = 3.3 gz* = 5.75 | HAN 1/2 | −30.5 ex = 0.31 ey = 038 | |
| [Co2(ox)tpmc](ClO4)2.3H2O (asymmetric dimer) | [73] | CoN4O2 | (2) oxalate | ga = 3.42 gb = 4.02 | HH 3/2 | −12.9 | 6.6 | |||
| “ | [74] | “ | “ | ga* = 4.43 gb* = 3.12 | HI 1/2 | −16 |
| Compound | Ref. | Coord. | Bridges | Co-Co (Å) | J/kB (K) | D/kB (K) | H (kOe) | τo (s) | Ueff/kB (K) | n | C/K-n (s−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| [(dmso)CoL2(μ-(m-NO2) C6H4COO)Co(NCS)] (2) symmetric | [75] | CoN2O4 CoNO5 | (2)Co-O-Co (1)Nitrobenzoate | 2.98(2) | 6.52 | 11.7 −1.86 | 3 | 6.7 × 10−13 | 31.3 | ||
| [Co2(bedmpzp)2(μ-Cl)2] (PF6)2 | [76] | CoN4Cl2 | (2) Co-Cl-Co | 4.0061 | 2.0 | 273 | 5 | Yes No data | Yes No data | ||
| [CoCl2LC7] (2) | [77] | CoN3Cl2 | π-π contacts | 5.662 | 1.03 | 219 | 2 | 1.07 × 10−7 | 14.6 | ||
| [CoCl2LC14] (5) | [77] | CoN3Cl2 | π-π contacts | 4.515 | 0.76 | 125 | 2 | 5.96 × 10−8 | 40.5 | ||
| [Co2(pypz)2(μ1,1-N3)2 (N3)2]·2CH3OH | [78] | CoN6 | (2) Co-N-Co | 3.3417 | 12.8 | 2.14 | 3 | HF 5.15 × 10−15 LF 1.5 | 95 | 1.6 | 412 |
| {[CoCxAPy)]·2.15 H2O}n (2 LL-dimer) | This work | CoN2O4 apical | (2) syn-syn carboxylate | 3.9684 | −0.45 | 82 E/D = 0.13 | 5 | HF 1.9 × 10−6 LF 2.0 | 6.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arauzo, A.; Bartolomé, E.; Luzón, J.; Alonso, P.J.; Vlad, A.; Cazacu, M.; Zaltariov, M.F.; Shova, S.; Bartolomé, J.; Turta, C. Slow Magnetic Relaxation in {[CoCxAPy)] 2.15 H2O}n MOF Built from Ladder-Structured 2D Layers with Dimeric SMM Rungs. Molecules 2021, 26, 5626. https://doi.org/10.3390/molecules26185626
Arauzo A, Bartolomé E, Luzón J, Alonso PJ, Vlad A, Cazacu M, Zaltariov MF, Shova S, Bartolomé J, Turta C. Slow Magnetic Relaxation in {[CoCxAPy)] 2.15 H2O}n MOF Built from Ladder-Structured 2D Layers with Dimeric SMM Rungs. Molecules. 2021; 26(18):5626. https://doi.org/10.3390/molecules26185626
Chicago/Turabian StyleArauzo, Ana, Elena Bartolomé, Javier Luzón, Pablo J. Alonso, Angelica Vlad, Maria Cazacu, Mirela F. Zaltariov, Sergiu Shova, Juan Bartolomé, and Constantin Turta. 2021. "Slow Magnetic Relaxation in {[CoCxAPy)] 2.15 H2O}n MOF Built from Ladder-Structured 2D Layers with Dimeric SMM Rungs" Molecules 26, no. 18: 5626. https://doi.org/10.3390/molecules26185626
APA StyleArauzo, A., Bartolomé, E., Luzón, J., Alonso, P. J., Vlad, A., Cazacu, M., Zaltariov, M. F., Shova, S., Bartolomé, J., & Turta, C. (2021). Slow Magnetic Relaxation in {[CoCxAPy)] 2.15 H2O}n MOF Built from Ladder-Structured 2D Layers with Dimeric SMM Rungs. Molecules, 26(18), 5626. https://doi.org/10.3390/molecules26185626