
Directed Evolution Methods for Enzyme Engineering

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Generation of Diversity to Explore Sequence Space
3. Library Screening Methods
3.1. Optical Methods
3.1.1. Optical Methods > Micro-Scale Methods
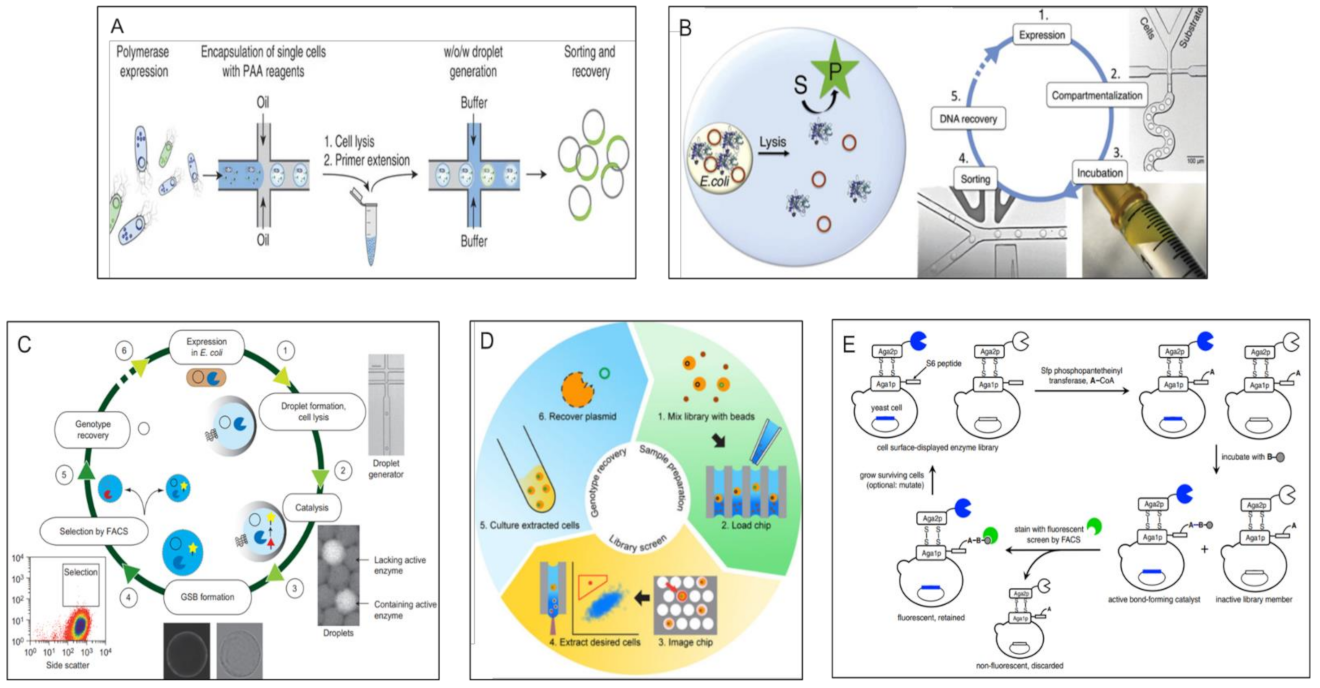
Optical Methods > Micro Scale Methods > FACS Based Double Emulsion Methods
Optical Methods > Micro Scale Methods > Water-in-Oil Emulsion Sorting Methods
Optical Methods > Micro Scale Methods > Matrix Capture Followed by FACS
Optical Methods > Micro Scale Methods > Well Arrays Combined with Microscopy
Optical Methods > Micro Scale Methods > Direct FACS Sorting of Cells

3.1.2. Optical Methods > Macro-Scale Methods
Optical Methods > Macro Scale Methods > Well Plate-Based Methods
Optical Methods > Macro Scale Methods > Culture Plate Based Screening

3.2. Survival//Retrieval Based Methods
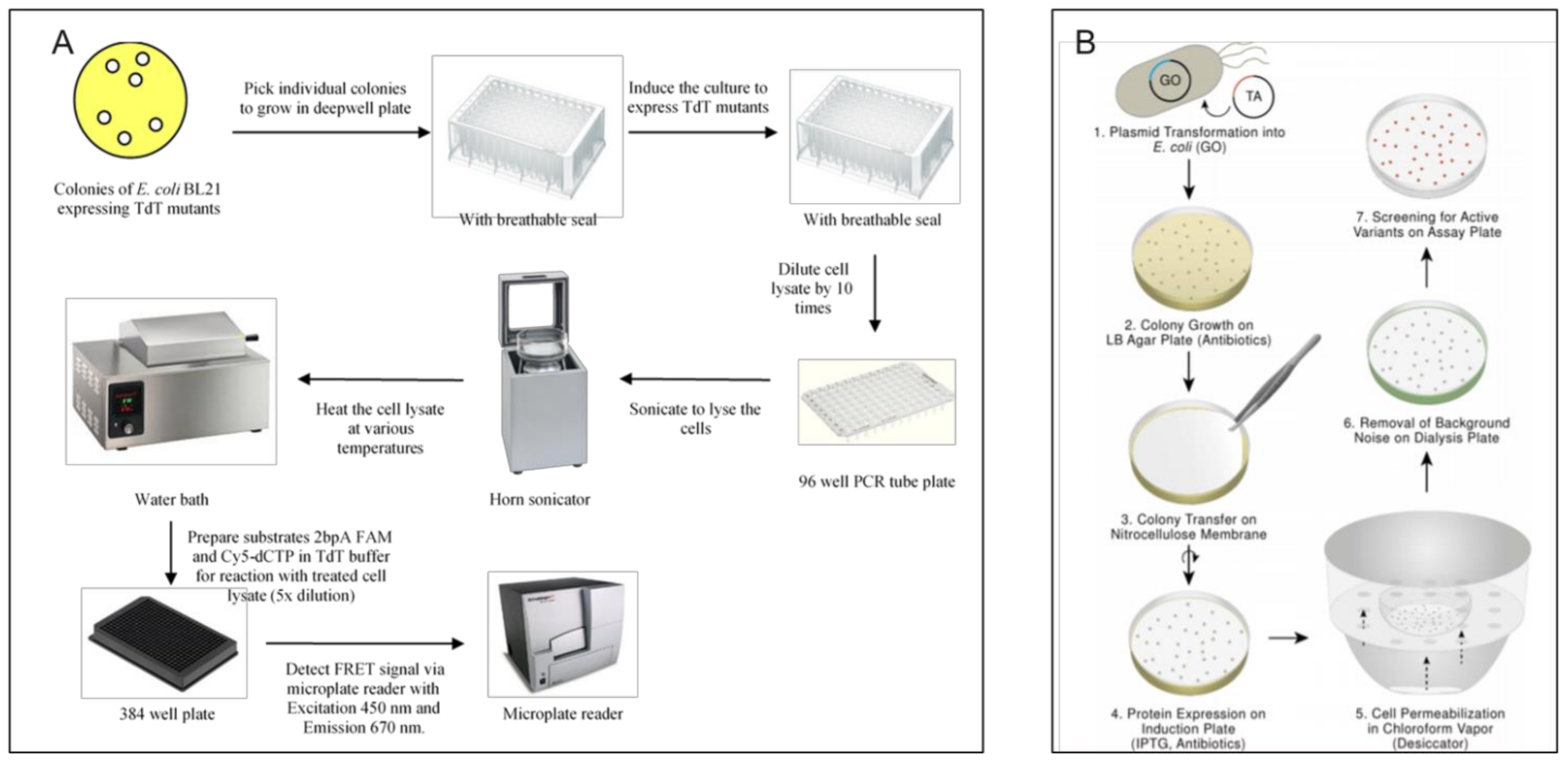
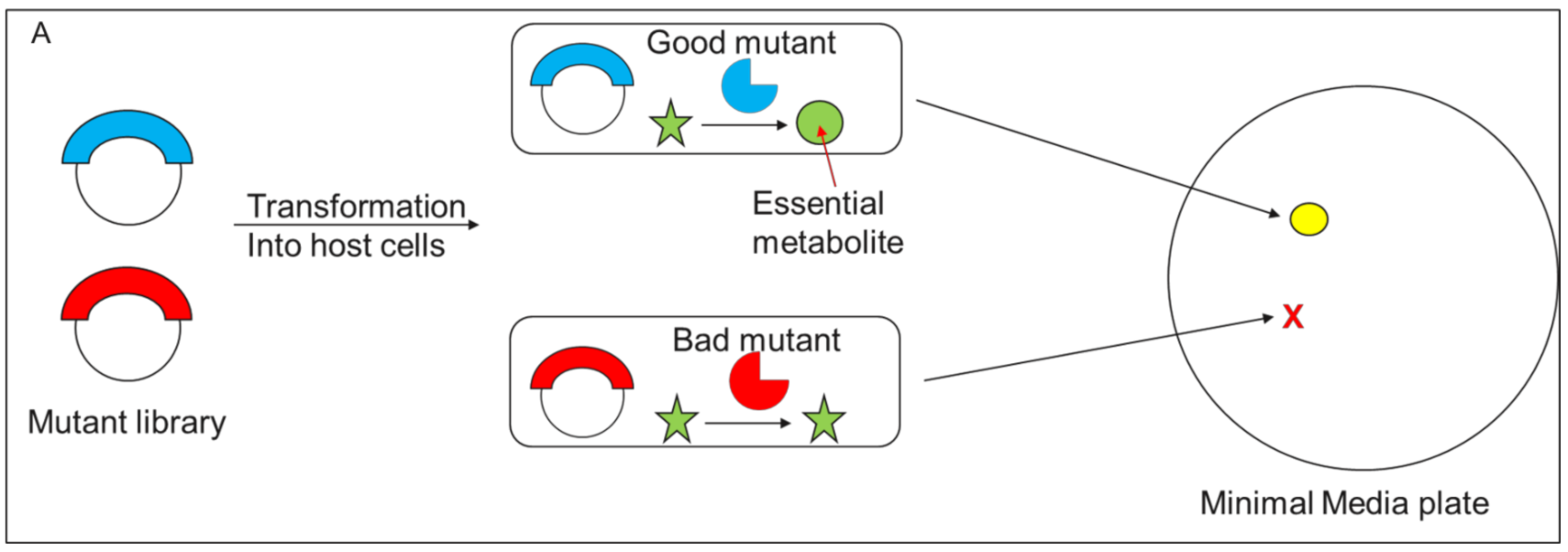
3.2.1. Survival/Retrieval Based Methods > Cell Survival Based Methods
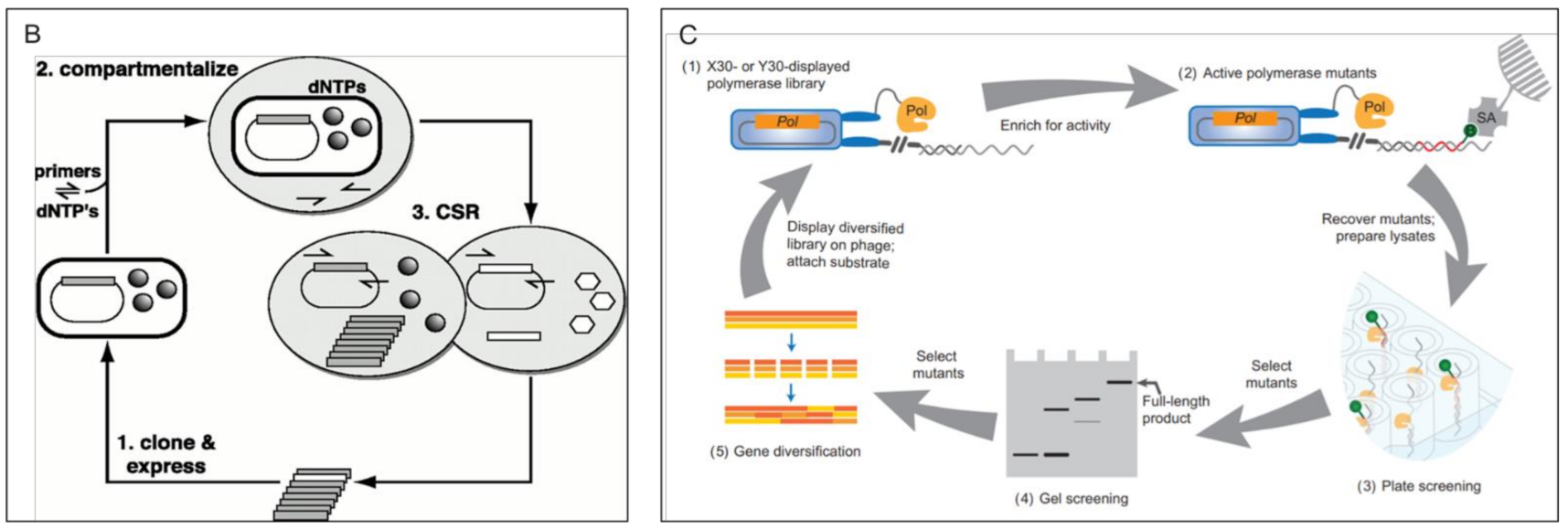
3.2.2. Survival/Retrieval Based Methods > In Vitro DNA Retrieval Based Methods
3.2.3. Survival/Retrieval Based Methods > Phage Pulldown Retrieval Methods


4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, C.-Y. DNA Polymerases Drive DNA Sequencing-by-Synthesis Technologies: Both Past and Present. Front. Microbiol. 2014, 5, 305. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, H.; Xu, Y.; Laššáková, S.; Korabečná, M.; Neužil, P. PCR Past, Present and Future. BioTechniques 2020, 69, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.Á.; Blamey, J.M. Biotechnological Applications of Archaeal Enzymes from Extreme Environments. Biol. Res. 2018, 51, 37. [Google Scholar] [CrossRef]
- Läufer, A. Starch Biorefinery Enzymes. Adv. Biochem. Eng. Biotechnol. 2019, 166, 137–152. [Google Scholar] [CrossRef]
- Thalji, N.K.; Camire, R.M. Rendering Factor Xa Zymogen-like as a Therapeutic Strategy to Treat Bleeding. Curr. Opin. Hematol. 2017, 24, 453–459. [Google Scholar] [CrossRef]
- Gurung, N.; Ray, S.; Bose, S.; Rai, V. A Broader View: Microbial Enzymes and Their Relevance in Industries, Medicine, and Beyond. BioMed Res. Int. 2013, 2013, 329121. [Google Scholar] [CrossRef] [PubMed]
- Minteer, S.D. Micellar Enzymology for Thermal, PH, and Solvent Stability. Methods Mol. Biol. Clifton NJ 2017, 1504, 19–23. [Google Scholar] [CrossRef]
- Alfaro-Chávez, A.L.; Liu, J.-W.; Porter, J.L.; Goldman, A.; Ollis, D.L. Improving on Nature’s Shortcomings: Evolving a Lipase for Increased Lipolytic Activity, Expression and Thermostability. Protein Eng. Des. Sel. PEDS 2019, 32, 13–24. [Google Scholar] [CrossRef]
- Matsumoto, T.; Yamada, R.; Ogino, H. Chemical Treatments for Modification and Immobilization to Improve the Solvent-Stability of Lipase. World J. Microbiol. Biotechnol. 2019, 35, 193. [Google Scholar] [CrossRef]
- Harris, J.L.; Craik, C.S. Engineering Enzyme Specificity. Curr. Opin. Chem. Biol. 1998, 2, 127–132. [Google Scholar] [CrossRef]
- Belsare, K.D.; Andorfer, M.C.; Cardenas, F.S.; Chael, J.R.; Park, H.J.; Lewis, J.C. A Simple Combinatorial Codon Mutagenesis Method for Targeted Protein Engineering. ACS Synth. Biol. 2017, 6, 416–420. [Google Scholar] [CrossRef]
- Zheng, L.; Baumann, U.; Reymond, J.-L. An Efficient One-Step Site-Directed and Site-Saturation Mutagenesis Protocol. Nucleic Acids Res. 2004, 32, e115. [Google Scholar] [CrossRef]
- Wrenbeck, E.E.; Klesmith, J.R.; Stapleton, J.A.; Adeniran, A.; Tyo, K.E.J.; Whitehead, T.A. Plasmid-Based One-Pot Saturation Mutagenesis. Nat. Methods 2016, 13, 928–930. [Google Scholar] [CrossRef]
- Randolph, J.; Yagodkin, A.; Lamaitre, M.; Azhayev, A.; Mackie, H. Codon Based Mutagenesis Using Trimer Phosphoramidites. Nucleic Acids Symp. Ser. 2008, 52, 479. [Google Scholar] [CrossRef]
- McCullum, E.O.; Williams, B.A.R.; Zhang, J.; Chaput, J.C. Random Mutagenesis by Error-Prone PCR. Methods Mol. Biol. Clifton NJ 2010, 634, 103–109. [Google Scholar] [CrossRef]
- Badran, A.H.; Liu, D.R. Development of Potent in Vivo Mutagenesis Plasmids with Broad Mutational Spectra. Nat. Commun. 2015, 6, 8425. [Google Scholar] [CrossRef] [PubMed]
- Muteeb, G.; Sen, R. Random Mutagenesis Using a Mutator Strain. Methods Mol. Biol. 2010. [Google Scholar] [CrossRef]
- Labrou, N.E. Random Mutagenesis Methods for in Vitro Directed Enzyme Evolution. Curr. Protein Pept. Sci. 2010, 11, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, D.S.; Griffiths, A.D. Man-Made Cell-like Compartments for Molecular Evolution. Nat. Biotechnol. 1998, 16, 652–656. [Google Scholar] [CrossRef]
- Bernath, K.; Hai, M.; Mastrobattista, E.; Griffiths, A.D.; Magdassi, S.; Tawfik, D.S. In Vitro Compartmentalization by Double Emulsions: Sorting and Gene Enrichment by Fluorescence Activated Cell Sorting. Anal. Biochem. 2004, 325, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Mastrobattista, E.; Taly, V.; Chanudet, E.; Treacy, P.; Kelly, B.T.; Griffiths, A.D. High-Throughput Screening of Enzyme Libraries: In Vitro Evolution of a β-Galactosidase by Fluorescence-Activated Sorting of Double Emulsions. Chem. Biol. 2005, 12, 1291–1300. [Google Scholar] [CrossRef]
- Aharoni, A.; Amitai, G.; Bernath, K.; Magdassi, S.; Tawfik, D.S. High-Throughput Screening of Enzyme Libraries: Thiolactonases Evolved by Fluorescence-Activated Sorting of Single Cells in Emulsion Compartments. Chem. Biol. 2005, 12, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, E.; Gibbs, M.; Reeves, R.; Bergquist, P. Directed Evolution of a Thermophilic β-Glucosidase for Cellulosic Bioethanol Production. Appl. Biochem. Biotechnol. 2010, 161, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Zinchenko, A.; Devenish, S.R.A.; Kintses, B.; Colin, P.-Y.; Fischlechner, M.; Hollfelder, F. One in a Million: Flow Cytometric Sorting of Single Cell-Lysate Assays in Monodisperse Picolitre Double Emulsion Droplets for Directed Evolution. Anal. Chem. 2014, 86, 2526–2533. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.C.; Dunn, M.R.; Hatch, A.; Sau, S.P.; Youngbull, C.; Chaput, J.C. A General Strategy for Expanding Polymerase Function by Droplet Microfluidics. Nat. Commun. 2016, 7, 11235. [Google Scholar] [CrossRef]
- Ma, F.; Fischer, M.; Han, Y.; Withers, S.G.; Feng, Y.; Yang, G.-Y. Substrate Engineering Enabling Fluorescence Droplet Entrapment for IVC-FACS-Based Ultrahigh-Throughput Screening. Anal. Chem. 2016, 88, 8587–8595. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Kerbage, C.; Hunt, T.P.; Westervelt, R.M.; Link, D.R.; Weitz, D.A. Dielectrophoretic Manipulation of Drops for High-Speed Microfluidic Sorting Devices. Appl. Phys. Lett. 2006, 88, 024104. [Google Scholar] [CrossRef]
- Baret, J.-C.; Miller, O.J.; Taly, V.; Ryckelynck, M.; El-Harrak, A.; Frenz, L.; Rick, C.; Samuels, M.L.; Hutchison, J.B.; Agresti, J.J.; et al. Fluorescence-Activated Droplet Sorting (FADS): Efficient Microfluidic Cell Sorting Based on Enzymatic Activity. Lab. Chip 2009, 9, 1850–1858. [Google Scholar] [CrossRef]
- Agresti, J.J.; Antipov, E.; Abate, A.R.; Ahn, K.; Rowat, A.C.; Baret, J.-C.; Marquez, M.; Klibanov, A.M.; Griffiths, A.D.; Weitz, D.A. Ultrahigh-Throughput Screening in Drop-Based Microfluidics for Directed Evolution. Proc. Natl. Acad. Sci. USA 2010, 107, 4004–4009. [Google Scholar] [CrossRef] [PubMed]
- Kintses, B.; Hein, C.; Mohamed, M.F.; Fischlechner, M.; Courtois, F.; Lainé, C.; Hollfelder, F. Picoliter Cell Lysate Assays in Microfluidic Droplet Compartments for Directed Enzyme Evolution. Chem. Biol. 2012, 19, 1001–1009. [Google Scholar] [CrossRef]
- Obexer, R.; Godina, A.; Garrabou, X.; Mittl, P.R.E.; Baker, D.; Griffiths, A.D.; Hilvert, D. Emergence of a Catalytic Tetrad during Evolution of a Highly Active Artificial Aldolase. Nat. Chem. 2017, 9, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Nikoomanzar, A.; Vallejo, D.; Yik, E.J.; Chaput, J.C. Programmed Allelic Mutagenesis of a DNA Polymerase with Single Amino Acid Resolution. ACS Synth. Biol. 2020, 9, 1873–1881. [Google Scholar] [CrossRef]
- van Loo, B.; Heberlein, M.; Mair, P.; Zinchenko, A.; Schüürmann, J.; Eenink, B.D.G.; Holstein, J.M.; Dilkaute, C.; Jose, J.; Hollfelder, F.; et al. High-Throughput, Lysis-Free Screening for Sulfatase Activity Using Escherichia Coli Autodisplay in Microdroplets. ACS Synth. Biol. 2019, 8, 2690–2700. [Google Scholar] [CrossRef] [PubMed]
- Gielen, F.; Hours, R.; Emond, S.; Fischlechner, M.; Schell, U.; Hollfelder, F. Ultrahigh-Throughput-Directed Enzyme Evolution by Absorbance-Activated Droplet Sorting (AADS). Proc. Natl. Acad. Sci. USA 2016, 113, E7383–E7389. [Google Scholar] [CrossRef] [PubMed]
- Holstein, J.M.; Gylstorff, C.; Hollfelder, F. Cell-Free Directed Evolution of a Protease in Microdroplets at Ultrahigh Throughput. ACS Synth. Biol. 2021, 10, 252–257. [Google Scholar] [CrossRef]
- Weng, L.; Spoonamore, J.E. Droplet Microfluidics-Enabled High-Throughput Screening for Protein Engineering. Micromachines 2019, 10, 734. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.D.; Tawfik, D.S. Directed Evolution of an Extremely Fast Phosphotriesterase by in Vitro Compartmentalization. EMBO J. 2003, 22, 24–35. [Google Scholar] [CrossRef]
- Levy, M.; Griswold, K.E.; Ellington, A.D. Direct Selection of Trans-Acting Ligase Ribozymes by in Vitro Compartmentalization. RNA 2005, 11, 1555–1562. [Google Scholar] [CrossRef]
- Gianella, P.; Snapp, E.L.; Levy, M. An in Vitro Compartmentalization-Based Method for the Selection of Bond-Forming Enzymes from Large Libraries. Biotechnol. Bioeng. 2016, 113, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Fischlechner, M.; Schaerli, Y.; Mohamed, M.F.; Patil, S.; Abell, C.; Hollfelder, F. Evolution of Enzyme Catalysts Caged in Biomimetic Gel-Shell Beads. Nat. Chem. 2014, 6, 791–796. [Google Scholar] [CrossRef]
- Ma, C.; Tan, Z.L.; Lin, Y.; Han, S.; Xing, X.; Zhang, C. Gel Microdroplet–Based High-Throughput Screening for Directed Evolution of Xylanase-Producing Pichia Pastoris. J. Biosci. Bioeng. 2019, 128, 662–668. [Google Scholar] [CrossRef]
- Chen, B.; Lim, S.; Kannan, A.; Alford, S.C.; Sunden, F.; Herschlag, D.; Dimov, I.K.; Baer, T.M.; Cochran, J.R. High-Throughput Analysis and Protein Engineering Using Microcapillary Arrays. Nat. Chem. Biol. 2016, 12, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Longwell, C.K.; Labanieh, L.; Cochran, J.R. High-Throughput Screening Technologies for Enzyme Engineering. Curr. Opin. Biotechnol. 2017, 48, 196–202. [Google Scholar] [CrossRef]
- Zhang, Y.; Minagawa, Y.; Kizoe, H.; Miyazaki, K.; Iino, R.; Ueno, H.; Tabata, K.V.; Shimane, Y.; Noji, H. Accurate High-Throughput Screening Based on Digital Protein Synthesis in a Massively Parallel Femtoliter Droplet Array. Sci. Adv. 2019, 5, eaav8185. [Google Scholar] [CrossRef]
- Olsen, M.J.; Stephens, D.; Griffiths, D.; Daugherty, P.; Georgiou, G.; Iverson, B.L. Function-Based Isolation of Novel Enzymes from a Large Library. Nat. Biotechnol. 2000, 18, 1071–1074. [Google Scholar] [CrossRef]
- Varadarajan, N.; Gam, J.; Olsen, M.J.; Georgiou, G.; Iverson, B.L. Engineering of Protease Variants Exhibiting High Catalytic Activity and Exquisite Substrate Selectivity. Proc. Natl. Acad. Sci. USA 2005, 102, 6855–6860. [Google Scholar] [CrossRef]
- Varadarajan, N.; Rodriguez, S.; Hwang, B.-Y.; Georgiou, G.; Iverson, B.L. Highly Active and Selective Endopeptidases with Programmed Substrate Specificities. Nat. Chem. Biol. 2008, 4, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Denard, C.A.; Paresi, C.; Yaghi, R.; McGinnis, N.; Bennett, Z.; Yi, L.; Georgiou, G.; Iverson, B.L. YESS 2.0, a Tunable Platform for Enzyme Evolution, Yields Highly Active TEV Protease Variants. ACS Synth. Biol. 2021, 10, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Pitzler, C.; Wirtz, G.; Vojcic, L.; Hiltl, S.; Böker, A.; Martinez, R.; Schwaneberg, U. A Fluorescent Hydrogel-Based Flow Cytometry High-Throughput Screening Platform for Hydrolytic Enzymes. Chem. Biol. 2014, 21, 1733–1742. [Google Scholar] [CrossRef]
- Chen, I.; Dorr, B.M.; Liu, D.R. A General Strategy for the Evolution of Bond-Forming Enzymes Using Yeast Display. Proc. Natl. Acad. Sci. USA 2011, 108, 11399–11404. [Google Scholar] [CrossRef]
- Dorr, B.M.; Ham, H.O.; An, C.; Chaikof, E.L.; Liu, D.R. Reprogramming the Specificity of Sortase Enzymes. Proc. Natl. Acad. Sci. USA 2014, 111, 13343–13348. [Google Scholar] [CrossRef]
- Aharoni, A.; Thieme, K.; Chiu, C.P.C.; Buchini, S.; Lairson, L.L.; Chen, H.; Strynadka, N.C.J.; Wakarchuk, W.W.; Withers, S.G. High-Throughput Screening Methodology for the Directed Evolution of Glycosyltransferases. Nat. Methods 2006, 3, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Rich, J.R.; Gilbert, M.; Wakarchuk, W.W.; Feng, Y.; Withers, S.G. Fluorescence Activated Cell Sorting as a General Ultra-High-Throughput Screening Method for Directed Evolution of Glycosyltransferases. J. Am. Chem. Soc. 2010, 132, 10570–10577. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.D.; Garruss, A.S.; Moretti, R.; Chan, S.; Arbing, M.A.; Cascio, D.; Rogers, J.K.; Isaacs, F.J.; Kosuri, S.; Baker, D.; et al. Engineering an Allosteric Transcription Factor to Respond to New Ligands. Nat. Methods 2016, 13, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, D.; van Beek, H.L.; Syberg, F.; Schallmey, M.; Tobola, F.; Cormann, K.U.; Schlicker, C.; Baumann, P.T.; Krumbach, K.; Sokolowsky, S.; et al. Engineering and Application of a Biosensor with Focused Ligand Specificity. Nat. Commun. 2020, 11, 4851. [Google Scholar] [CrossRef]
- Meister, S.W.; Hendrikse, N.M.; Löfblom, J. Directed Evolution of the 3C Protease from Coxsackievirus Using a Novel Fluorescence-Assisted Intracellular Method. Biol. Chem. 2019, 400, 405–415. [Google Scholar] [CrossRef]
- Michener, J.K.; Smolke, C.D. High-Throughput Enzyme Evolution in Saccharomyces Cerevisiae Using a Synthetic RNA Switch. Metab. Eng. 2012, 14, 306–316. [Google Scholar] [CrossRef]
- Yeom, S.-J.; Kim, M.; Kwon, K.K.; Fu, Y.; Rha, E.; Park, S.-H.; Lee, H.; Kim, H.; Lee, D.-H.; Kim, D.-M.; et al. A Synthetic Microbial Biosensor for High-Throughput Screening of Lactam Biocatalysts. Nat. Commun. 2018, 9, 5053. [Google Scholar] [CrossRef]
- Kwon, K.K.; Lee, D.-H.; Kim, S.J.; Choi, S.-L.; Rha, E.; Yeom, S.-J.; Subhadra, B.; Lee, J.; Jeong, K.J.; Lee, S.-G. Evolution of Enzymes with New Specificity by High-Throughput Screening Using DmpR-Based Genetic Circuits and Multiple Flow Cytometry Rounds. Sci. Rep. 2018, 8, 2659. [Google Scholar] [CrossRef]
- Xiao, H.; Bao, Z.; Zhao, H. High Throughput Screening and Selection Methods for Directed Enzyme Evolution. Ind. Eng. Chem. Res. 2015, 54, 4011–4020. [Google Scholar] [CrossRef]
- Chua, J.P.S.; Go, M.K.; Osothprarop, T.; Mcdonald, S.; Karabadzhak, A.G.; Yew, W.S.; Peisajovich, S.; Nirantar, S. Evolving a Thermostable Terminal Deoxynucleotidyl Transferase. ACS Synth. Biol. 2020, 9, 1725–1735. [Google Scholar] [CrossRef]
- Williams, G.J.; Thorson, J.S. A High-Throughput Fluorescence-Based Glycosyltransferase Screen and Its Application in Directed Evolution. Nat. Protoc. 2008, 3, 357–362. [Google Scholar] [CrossRef]
- García-Ruiz, E.; Maté, D.; Ballesteros, A.; Martinez, A.T.; Alcalde, M. Evolving Thermostability in Mutant Libraries of Ligninolytic Oxidoreductases Expressed in Yeast. Microb. Cell Factories 2010, 9, 17. [Google Scholar] [CrossRef]
- Listwan, P.; Pédelacq, J.-D.; Lockard, M.; Bell, C.; Terwilliger, T.C.; Waldo, G.S. The Optimization of in Vitro High-Throughput Chemical Lysis of Escherichia Coli. Application to ACP Domain of the Polyketide Synthase PpsC from Mycobacterium Tuberculosis. J. Struct. Funct. Genom. 2010, 11, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Santos-Aberturas, J.; Dörr, M.; Waldo, G.S.; Bornscheuer, U.T. In-Depth High-Throughput Screening of Protein Engineering Libraries by Split-GFP Direct Crude Cell Extract Data Normalization. Chem. Biol. 2015, 22, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Knapp, A.; Ripphahn, M.; Volkenborn, K.; Skoczinski, P.; Jaeger, K.-E. Activity-Independent Screening of Secreted Proteins Using Split GFP. J. Biotechnol. 2017, 258, 110–116. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Altenbuchner, J.; Meyer, H.H. Directed Evolution of an Esterase for the Stereoselective Resolution of a Key Intermediate in the Synthesis of Epothilones. Biotechnol. Bioeng. 1998, 58, 554–559. [Google Scholar] [CrossRef]
- Alexeeva, M.; Enright, A.; Dawson, M.J.; Mahmoudian, M.; Turner, N.J. Deracemization of α-Methylbenzylamine Using an Enzyme Obtained by In Vitro Evolution. Angew. Chem. Int. Ed. 2002, 41, 3177–3180. [Google Scholar] [CrossRef]
- Turner, N.J. Agar Plate-Based Assays. In Enzyme Assays; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2005; pp. 137–161. [Google Scholar] [CrossRef]
- Carr, R.; Alexeeva, M.; Enright, A.; Eve, T.S.C.; Dawson, M.J.; Turner, N.J. Directed Evolution of an Amine Oxidase Possessing Both Broad Substrate Specificity and High Enantioselectivity. Angew. Chem. Int. Ed. 2003, 42, 4807–4810. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, T.L.; Greene, D.N.; Matsumura, I. Diversification and Specialization of HIV Protease Function During In Vitro Evolution. Mol. Biol. Evol. 2006, 23, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Teather, R.M.; Wood, P.J. Use of Congo Red-Polysaccharide Interactions in Enumeration and Characterization of Cellulolytic Bacteria from the Bovine Rumen. Appl. Environ. Microbiol. 1982, 43, 777–780. [Google Scholar] [CrossRef]
- Kasana, R.C.; Salwan, R.; Dhar, H.; Dutt, S.; Gulati, A. A Rapid and Easy Method for the Detection of Microbial Cellulases on Agar Plates Using Gram’s Iodine. Curr. Microbiol. 2008, 57, 503–507. [Google Scholar] [CrossRef]
- Heinis, C.; Melkko, S.; Demartis, S.; Neri, D. Two General Methods for the Isolation of Enzyme Activities by Colony Filter Screening. Chem. Biol. 2002, 9, 383–390. [Google Scholar] [CrossRef]
- Asial, I.; Cheng, Y.X.; Engman, H.; Dollhopf, M.; Wu, B.; Nordlund, P.; Cornvik, T. Engineering Protein Thermostability Using a Generic Activity-Independent Biophysical Screen inside the Cell. Nat. Commun. 2013, 4, 2901. [Google Scholar] [CrossRef] [PubMed]
- Kermekchiev, M.B.; Tzekov, A.; Barnes, W.M. Cold-Sensitive Mutants of Taq DNA Polymerase Provide a Hot Start for PCR. Nucleic Acids Res. 2003, 31, 6139–6147. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weiß, M.S.; Pavlidis, I.V.; Vickers, C.; Höhne, M.; Bornscheuer, U.T. Glycine Oxidase Based High-Throughput Solid-Phase Assay for Substrate Profiling and Directed Evolution of (R)- and (S)-Selective Amine Transaminases. Anal. Chem. 2014, 86, 11847–11853. [Google Scholar] [CrossRef]
- Hecky, J.; Müller, K.M. Structural Perturbation and Compensation by Directed Evolution at Physiological Temperature Leads to Thermostabilization of β-Lactamase. Biochemistry 2005, 44, 12640–12654. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-S.; Nam, S.-H.; Lee, J.K.; Yoon, C.N.; Mannervik, B.; Benkovic, S.J.; Kim, H.-S. Design and Evolution of New Catalytic Activity with an Existing Protein Scaffold. Science 2006, 311, 535–538. [Google Scholar] [CrossRef]
- Jürgens, C.; Strom, A.; Wegener, D.; Hettwer, S.; Wilmanns, M.; Sterner, R. Directed Evolution of a (Βα)8-Barrel Enzyme to Catalyze Related Reactions in Two Different Metabolic Pathways. Proc. Natl. Acad. Sci. USA 2000, 97, 9925–9930. [Google Scholar] [CrossRef] [PubMed]
- Leopoldseder, S.; Claren, J.; Jürgens, C.; Sterner, R. Interconverting the Catalytic Activities of (Βα)8-Barrel Enzymes from Different Metabolic Pathways: Sequence Requirements and Molecular Analysis. J. Mol. Biol. 2004, 337, 871–879. [Google Scholar] [CrossRef]
- Claren, J.; Malisi, C.; Höcker, B.; Sterner, R. Establishing Wild-Type Levels of Catalytic Activity on Natural and Artificial (Βα)8-Barrel Protein Scaffolds. Proc. Natl. Acad. Sci. USA 2009, 106, 3704–3709. [Google Scholar] [CrossRef]
- Flores, H.; Lin, S.; Contreras-Ferrat, G.; Cronan, J.E.; Morett, E. Evolution of a New Function in an Esterase: Simple Amino Acid Substitutions Enable the Activity Present in the Larger Paralog, BioH. Protein Eng. Des. Sel. 2012, 25, 387–395. [Google Scholar] [CrossRef]
- Saab-Rincón, G.; Olvera, L.; Olvera, M.; Rudiño-Piñera, E.; Benites, E.; Soberón, X.; Morett, E. Evolutionary Walk between (β/α) 8 Barrels: Catalytic Migration from Triosephosphate Isomerase to Thiamin Phosphate Synthase. J. Mol. Biol. 2012, 416, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Schultz, P.G. Expanding the Genetic Code. Angew. Chem. Int. Ed. 2005, 44, 34–66. [Google Scholar] [CrossRef]
- Pirman, N.L.; Barber, K.W.; Aerni, H.R.; Ma, N.J.; Haimovich, A.D.; Rogulina, S.; Isaacs, F.J.; Rinehart, J. A Flexible Codon in Genomically Recoded Escherichia Coli Permits Programmable Protein Phosphorylation. Nat. Commun. 2015, 6, 8130. [Google Scholar] [CrossRef]
- Lang, K.; Chin, J.W. Cellular Incorporation of Unnatural Amino Acids and Bioorthogonal Labeling of Proteins. Chem. Rev. 2014, 114, 4764–4806. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Z.; Brock, A.; Schultz, P.G. Addition of the Keto Functional Group to the Genetic Code of Escherichia Coli. Proc. Natl. Acad. Sci. USA 2003, 100, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.W.; Cropp, T.A.; Anderson, J.C.; Mukherji, M.; Zhang, Z.; Schultz, P.G. An Expanded Eukaryotic Genetic Code. Science 2003, 301, 964–967. [Google Scholar] [CrossRef]
- Wang, L.; Schultz, P.G. A General Approach for the Generation of Orthogonal TRNAs. Chem. Biol. 2001, 8, 883–890. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of A•T to G•C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Ghadessy, F.J.; Ong, J.L.; Holliger, P. Directed Evolution of Polymerase Function by Compartmentalized Self-Replication. Proc. Natl. Acad. Sci. USA 2001, 98, 4552–4557. [Google Scholar] [CrossRef]
- Arezi, B.; McKinney, N.; Hansen, C.; Cayouette, M.; Fox, J.; Chen, K.; Lapira, J.; Hamilton, S.; Hogrefe, H. Compartmentalized Self-Replication under Fast PCR Cycling Conditions Yields Taq DNA Polymerase Mutants with Increased DNA-Binding Affinity and Blood Resistance. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Kermekchiev, M.B.; Barnes, W.M. Use of Whole Blood in Pcr Reactions. WO2005113829A2, 1 December 2005. [Google Scholar]
- Ramsay, N.; Jemth, A.-S.; Brown, A.; Crampton, N.; Dear, P.; Holliger, P. CyDNA: Synthesis and Replication of Highly Cy-Dye Substituted DNA by an Evolved Polymerase. J. Am. Chem. Soc. 2010, 132, 5096–5104. [Google Scholar] [CrossRef]
- Ong, J.L.; Loakes, D.; Jaroslawski, S.; Too, K.; Holliger, P. Directed Evolution of DNA Polymerase, RNA Polymerase and Reverse Transcriptase Activity in a Single Polypeptide. J. Mol. Biol. 2006, 361, 537–550. [Google Scholar] [CrossRef]
- Pinheiro, V.B.; Arangundy-Franklin, S.; Holliger, P. Compartmentalized Self-Tagging for In Vitro-Directed Evolution of XNA Polymerases. Curr. Protoc. Nucleic Acid Chem. 2014, 57, 9. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, V.B.; Taylor, A.I.; Cozens, C.; Abramov, M.; Renders, M.; Zhang, S.; Chaput, J.C.; Wengel, J.; Peak-Chew, S.-Y.; McLaughlin, S.H.; et al. Synthetic Genetic Polymers Capable of Heredity and Evolution. Science 2012, 336, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Arangundy-Franklin, S.; Taylor, A.I.; Porebski, B.T.; Genna, V.; Peak-Chew, S.; Vaisman, A.; Woodgate, R.; Orozco, M.; Holliger, P. A Synthetic Genetic Polymer with an Uncharged Backbone Chemistry Based on Alkyl Phosphonate Nucleic Acids. Nat. Chem. 2019, 11, 533–542. [Google Scholar] [CrossRef]
- Esvelt, K.M.; Carlson, J.C.; Liu, D.R. A System for the Continuous Directed Evolution of Biomolecules. Nature 2011, 472, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Badran, A.H.; Guzov, V.M.; Huai, Q.; Kemp, M.M.; Vishwanath, P.; Kain, W.; Nance, A.M.; Evdokimov, A.; Moshiri, F.; Turner, K.H.; et al. Continuous Evolution of Bacillus Thuringiensis Toxins Overcomes Insect Resistance. Nature 2016, 533, 58–63. [Google Scholar] [CrossRef]
- Bryson, D.I.; Fan, C.; Guo, L.-T.; Miller, C.; Söll, D.; Liu, D.R. Continuous Directed Evolution of Aminoacyl-TRNA Synthetases. Nat. Chem. Biol. 2017, 13, 1253–1260. [Google Scholar] [CrossRef]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 Variants with Broad PAM Compatibility and High DNA Specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef]
- Takahashi, N.; Kakinuma, H.; Liu, L.; Nishi, Y.; Fujii, I. In Vitro Abzyme Evolution to Optimize Antibody Recognition for Catalysis. Nat. Biotechnol. 2001, 19, 563–567. [Google Scholar] [CrossRef]
- Kather, I.; Jakob, R.P.; Dobbek, H.; Schmid, F.X. Increased Folding Stability of TEM-1 Beta-Lactamase by in Vitro Selection. J. Mol. Biol. 2008, 383, 238–251. [Google Scholar] [CrossRef]
- Chen, T.; Hongdilokkul, N.; Liu, Z.; Adhikary, R.; Tsuen, S.S.; Romesberg, F.E. Evolution of Thermophilic DNA Polymerases for the Recognition and Amplification of C2′-Modified DNA. Nat. Chem. 2016, 8, 556–562. [Google Scholar] [CrossRef]
- Amstutz, P.; Pelletier, J.N.; Guggisberg, A.; Jermutus, L.; Cesaro-Tadic, S.; Zahnd, C.; Plückthun, A. In Vitro Selection for Catalytic Activity with Ribosome Display. J. Am. Chem. Soc. 2002, 124, 9396–9403. [Google Scholar] [CrossRef] [PubMed]
- Bieberich, E.; Kapitonov, D.; Tencomnao, T.; Yu, R.K. Protein–Ribosome–MRNA Display: Affinity Isolation of Enzyme–Ribosome–MRNA Complexes and CDNA Cloning in a Single-Tube Reaction. Anal. Biochem. 2000, 287, 294–298. [Google Scholar] [CrossRef]
- Skirgaila, R.; Pudzaitis, V.; Paliksa, S.; Vaitkevicius, M.; Janulaitis, A. Compartmentalization of Destabilized Enzyme–MRNA–Ribosome Complexes Generated by Ribosome Display: A Novel Tool for the Directed Evolution of Enzymes. Protein Eng. Des. Sel. 2013, 26, 453–461. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hall, B.G. Experimental Evolution of a New Enzymatic Function. II. Evolution of Multiple Functions for Ebg Enzyme in E. Coli. Genetics 1978, 89, 453–465. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nirantar, S.R. Directed Evolution Methods for Enzyme Engineering. Molecules 2021, 26, 5599. https://doi.org/10.3390/molecules26185599
Nirantar SR. Directed Evolution Methods for Enzyme Engineering. Molecules. 2021; 26(18):5599. https://doi.org/10.3390/molecules26185599
Chicago/Turabian StyleNirantar, Saurabh Rajendra. 2021. "Directed Evolution Methods for Enzyme Engineering" Molecules 26, no. 18: 5599. https://doi.org/10.3390/molecules26185599
APA StyleNirantar, S. R. (2021). Directed Evolution Methods for Enzyme Engineering. Molecules, 26(18), 5599. https://doi.org/10.3390/molecules26185599