
Rapid Identification of Potential Drug Candidates from Multi-Million Compounds’ Repositories. Combination of 2D Similarity Search with 3D Ligand/Structure Based Methods and In Vitro Screening

 , ,
, ,
Abstract
:
1. Introduction: The Emergence of Virtual Screening
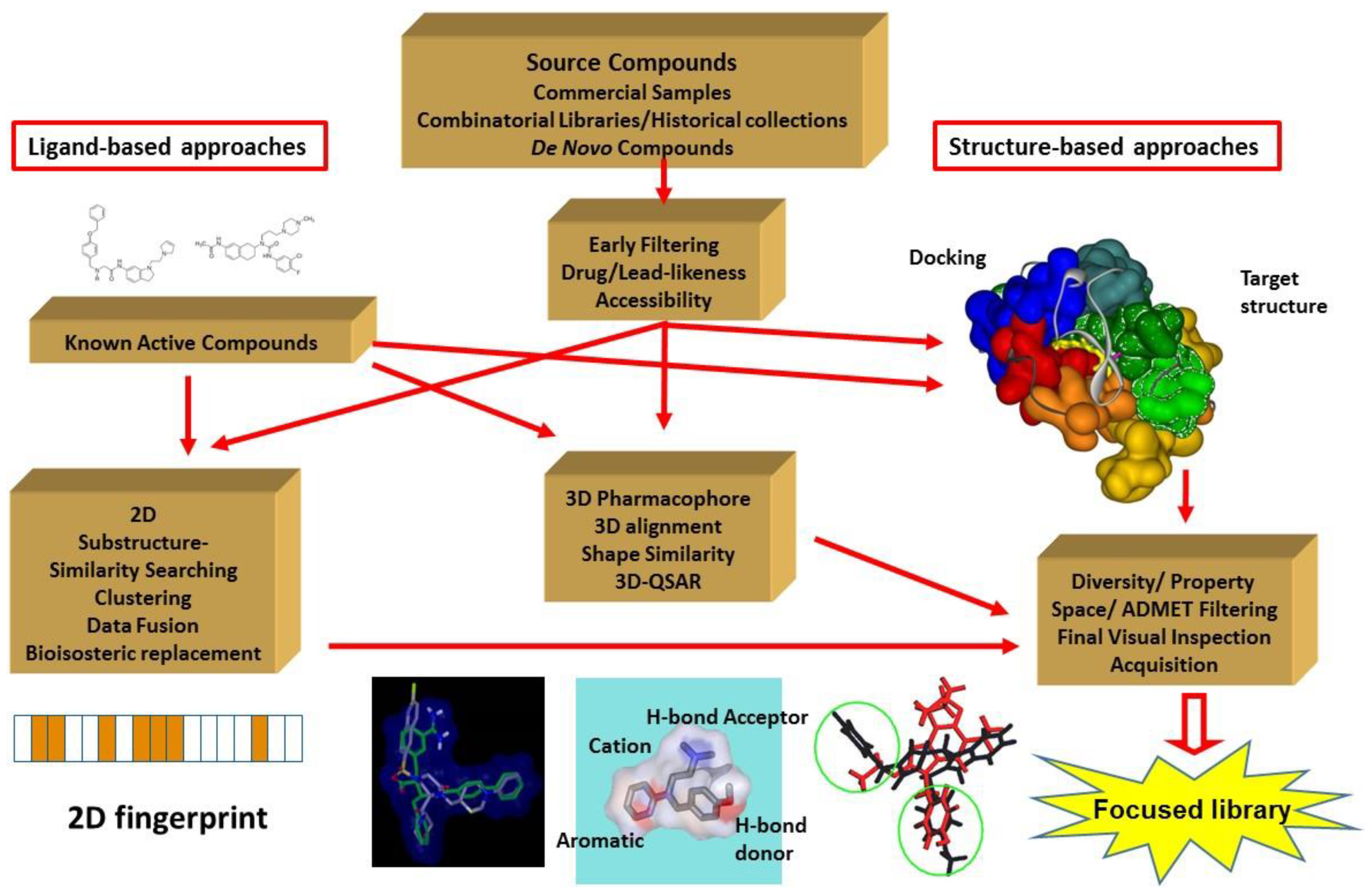
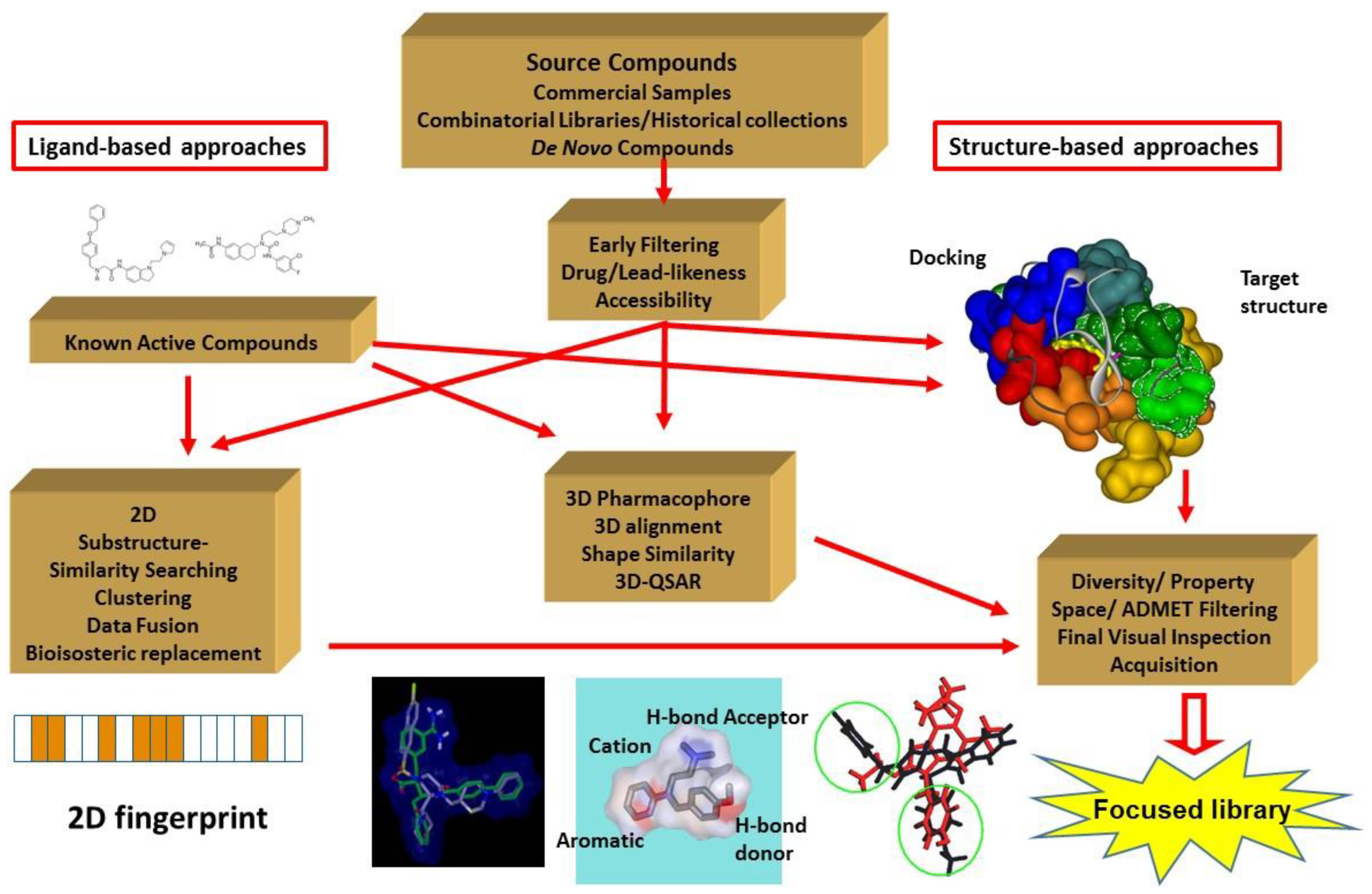
2. The Major Principles of the Integrated Virtual Screening Approaches
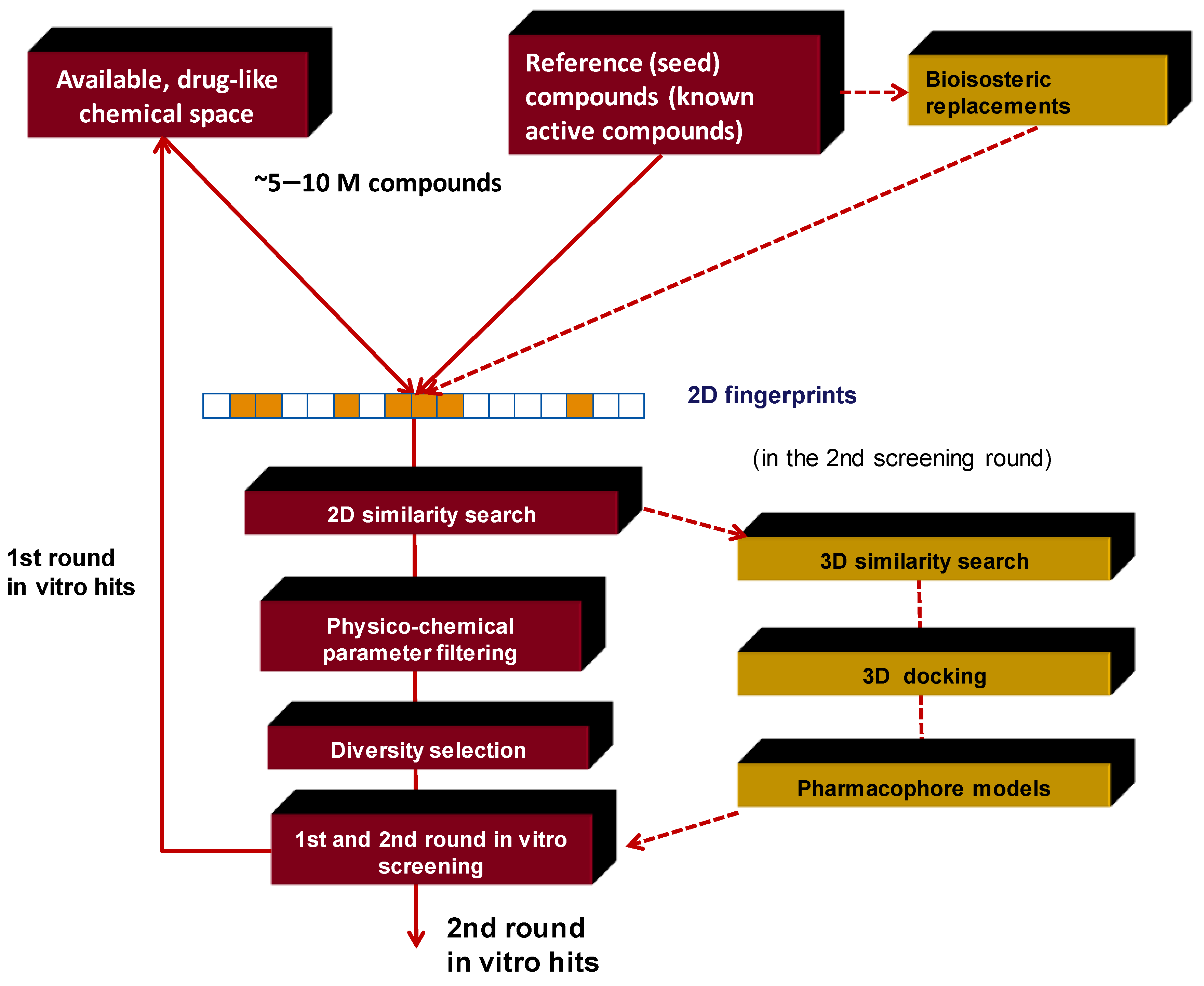
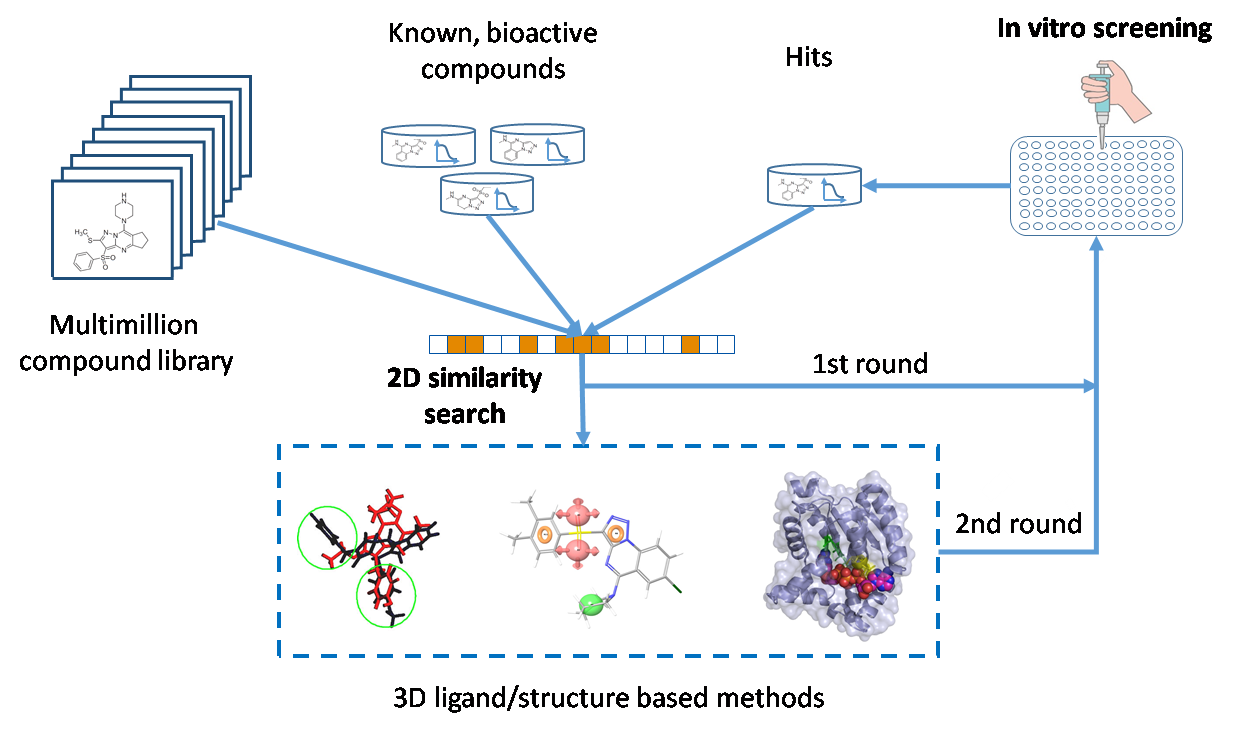
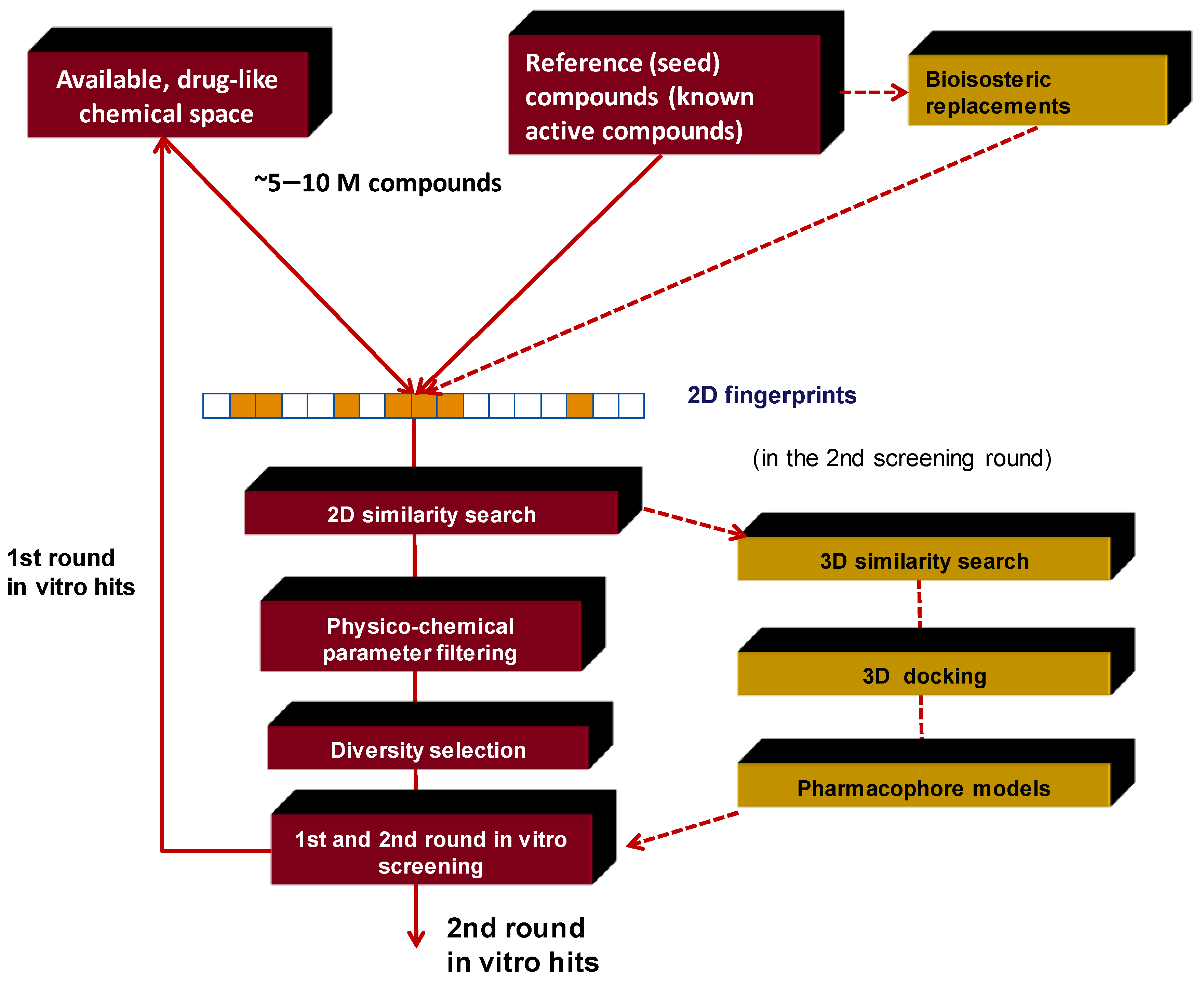
3. Integration Involving In Silico and In Vitro Screening. Our Approach
3.1. Elements of the 2D Ligand Similarity-Driven Virtual Screening
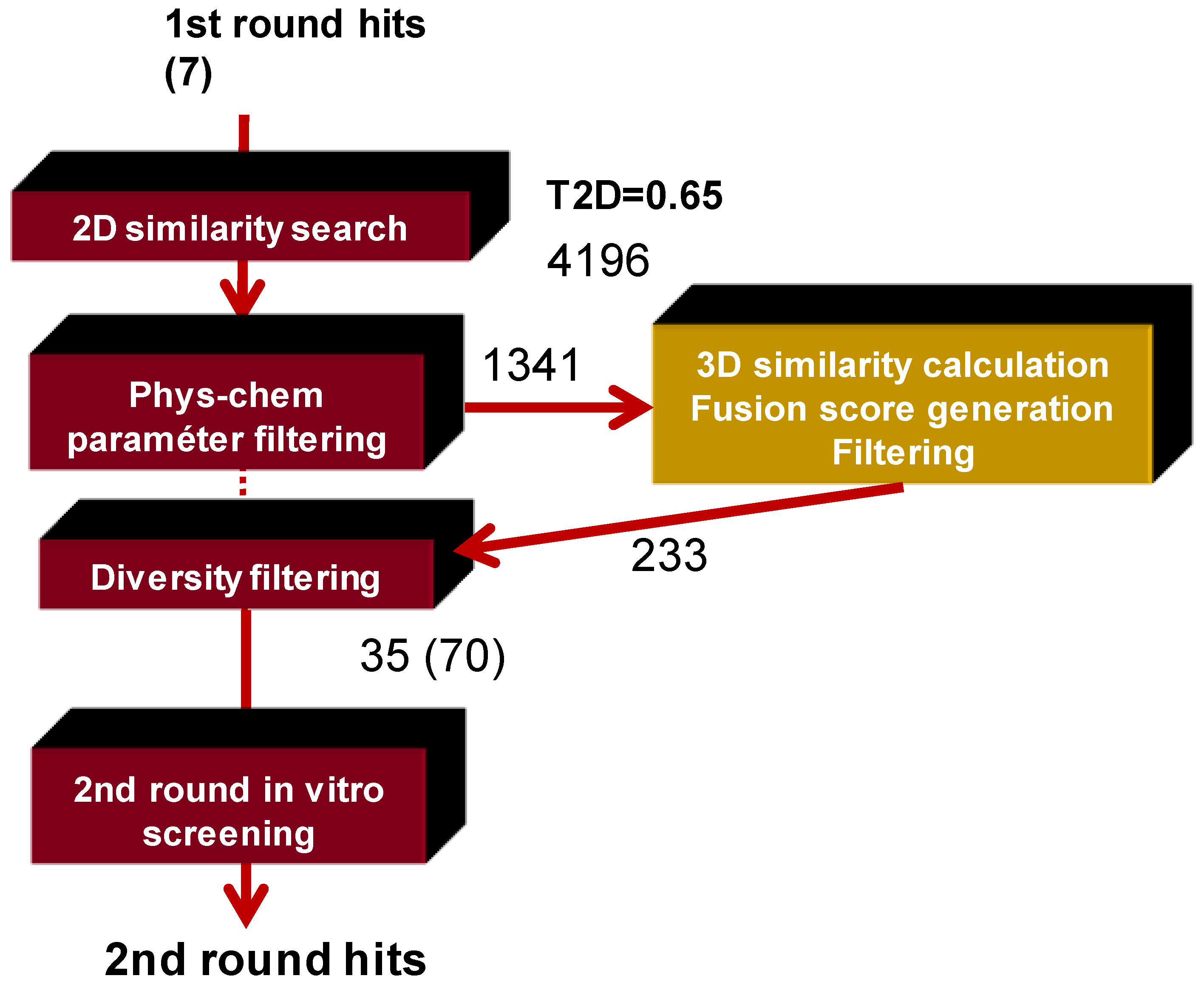
3.2. Filtering the Initial 2D Similarity Search Results
4. Case Studies for Integration of 2D/3D In Silico/In Vitro Approaches
4.1. Sequential Combination: 2D/3D Ligand-Based Similarity Approaches and In Vitro Screening: PDE4/PDE5 Inhibitors
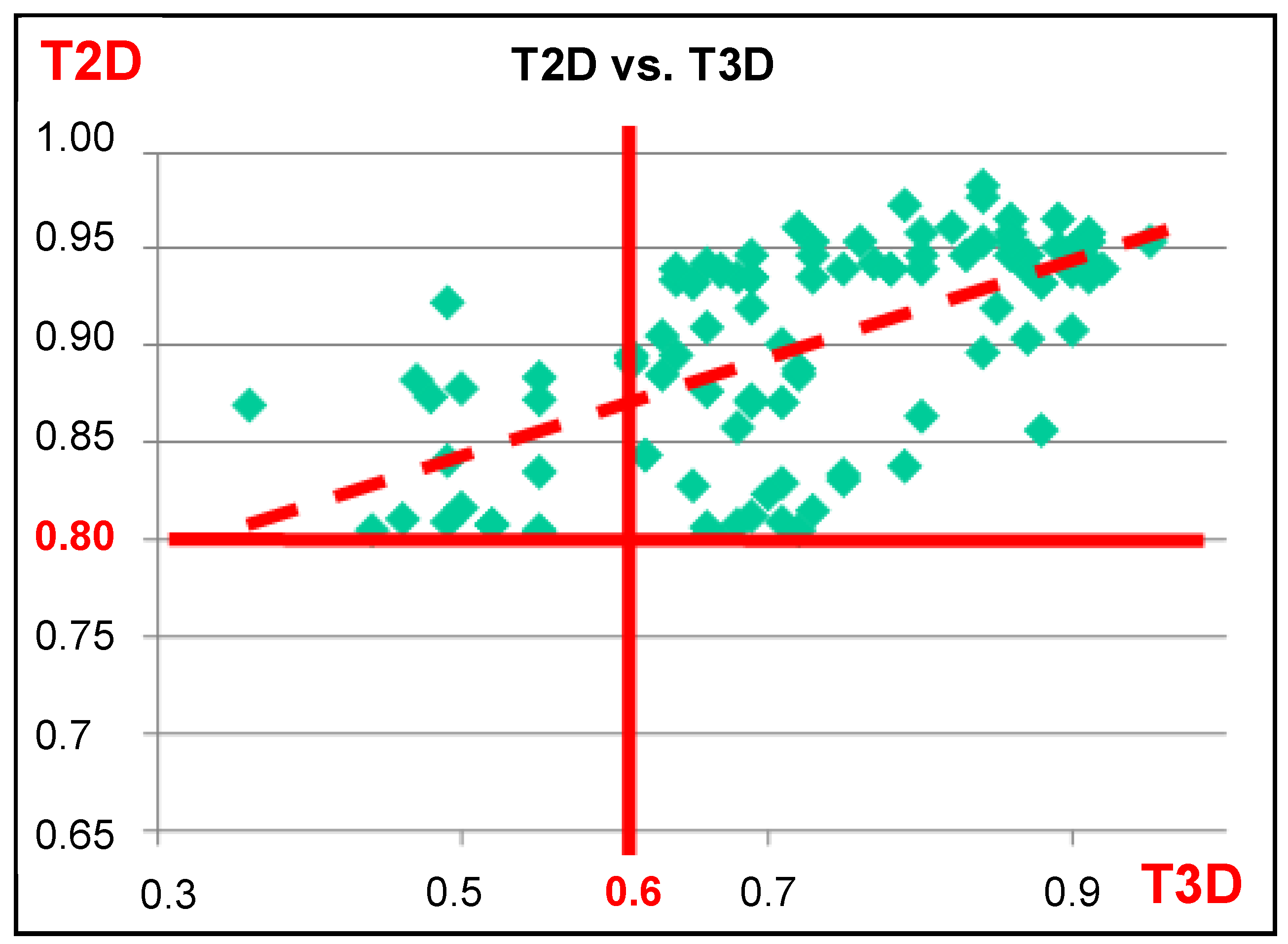
4.1.1. 2D/3D Similarity Correlation Analysis of Previously Generated PDE5 Focused Libraries
4.1.2. Sequential (or Fused) Application of 2D/3D Similarity Methods, Selection of PDE4B Libraries
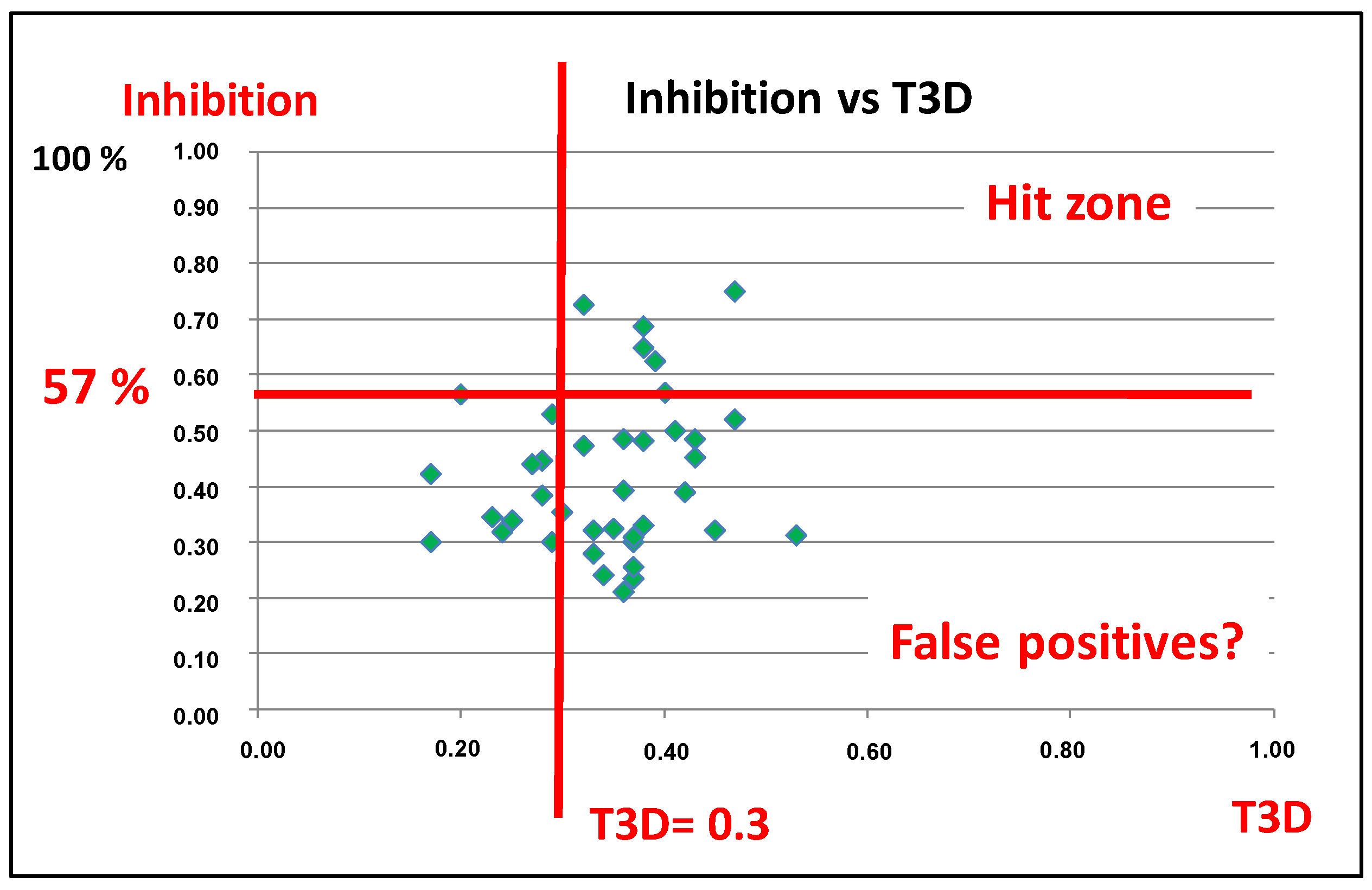
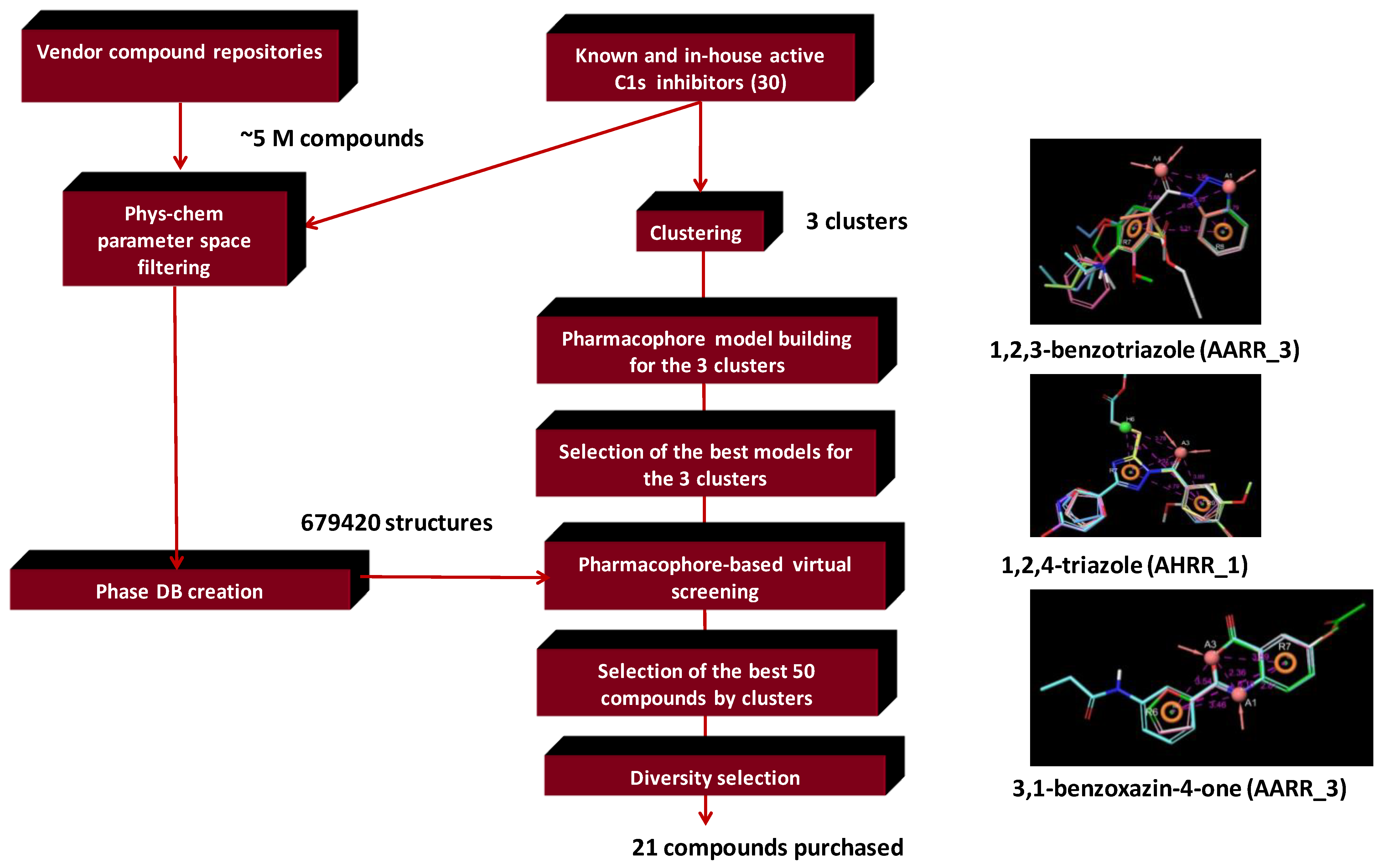
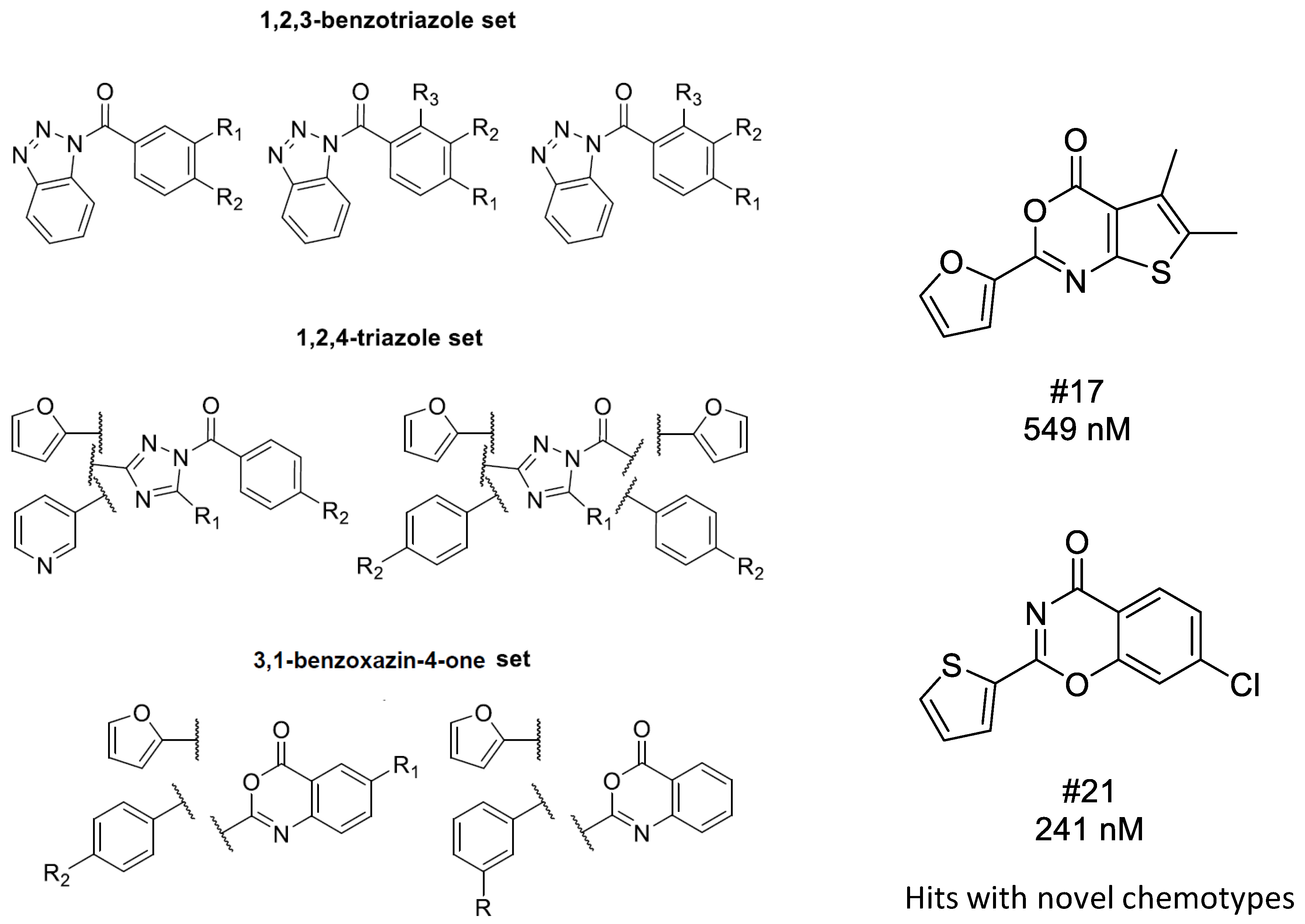
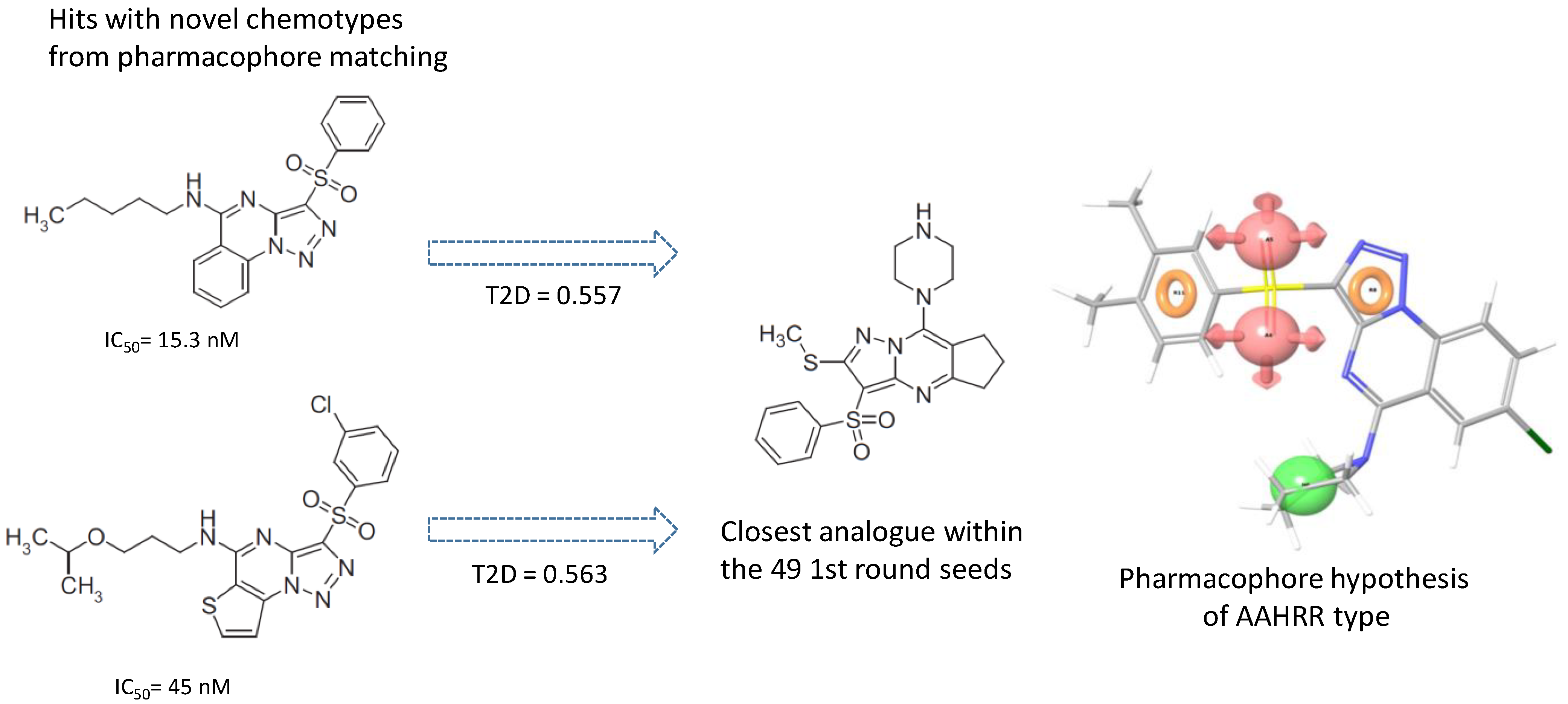
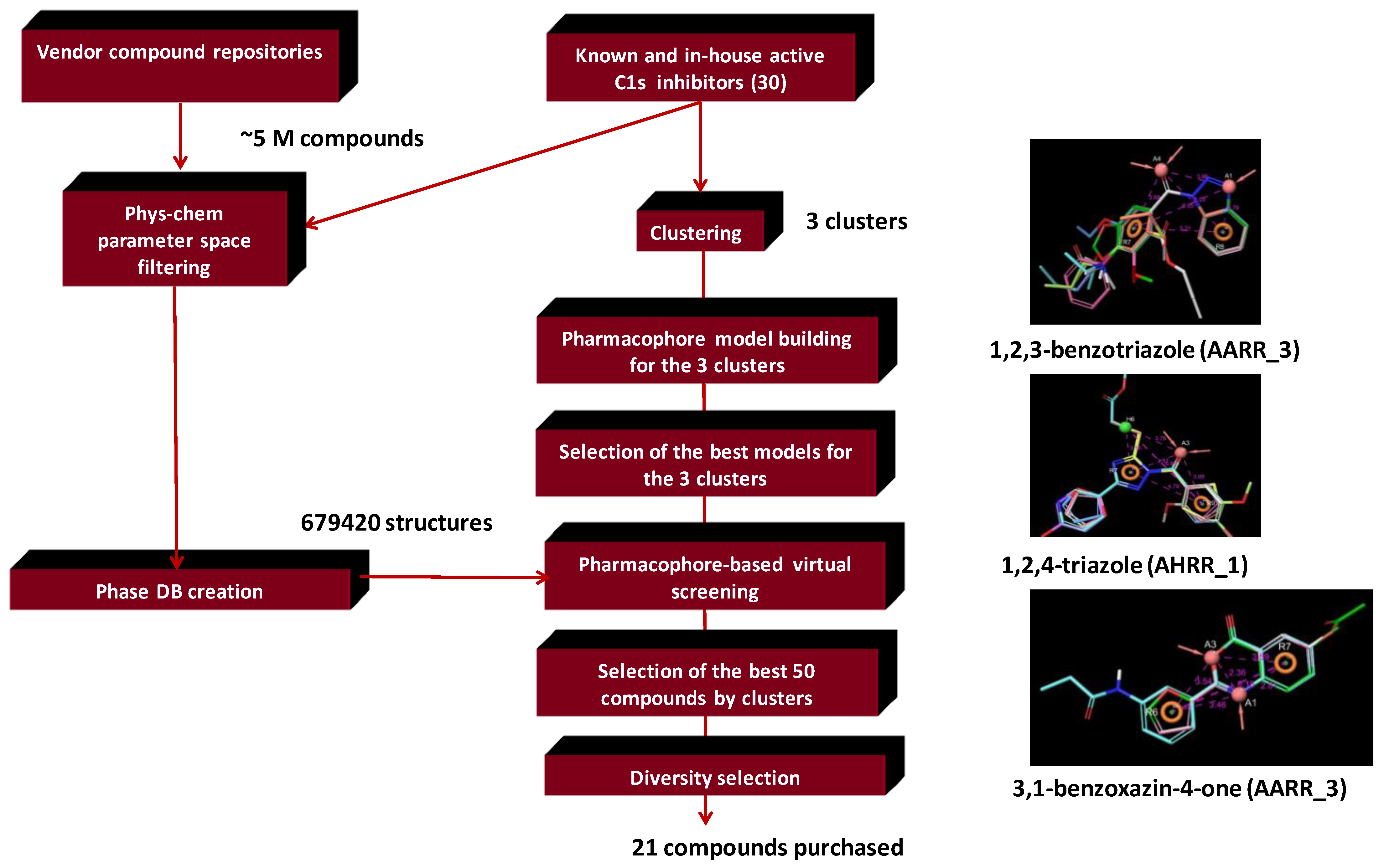
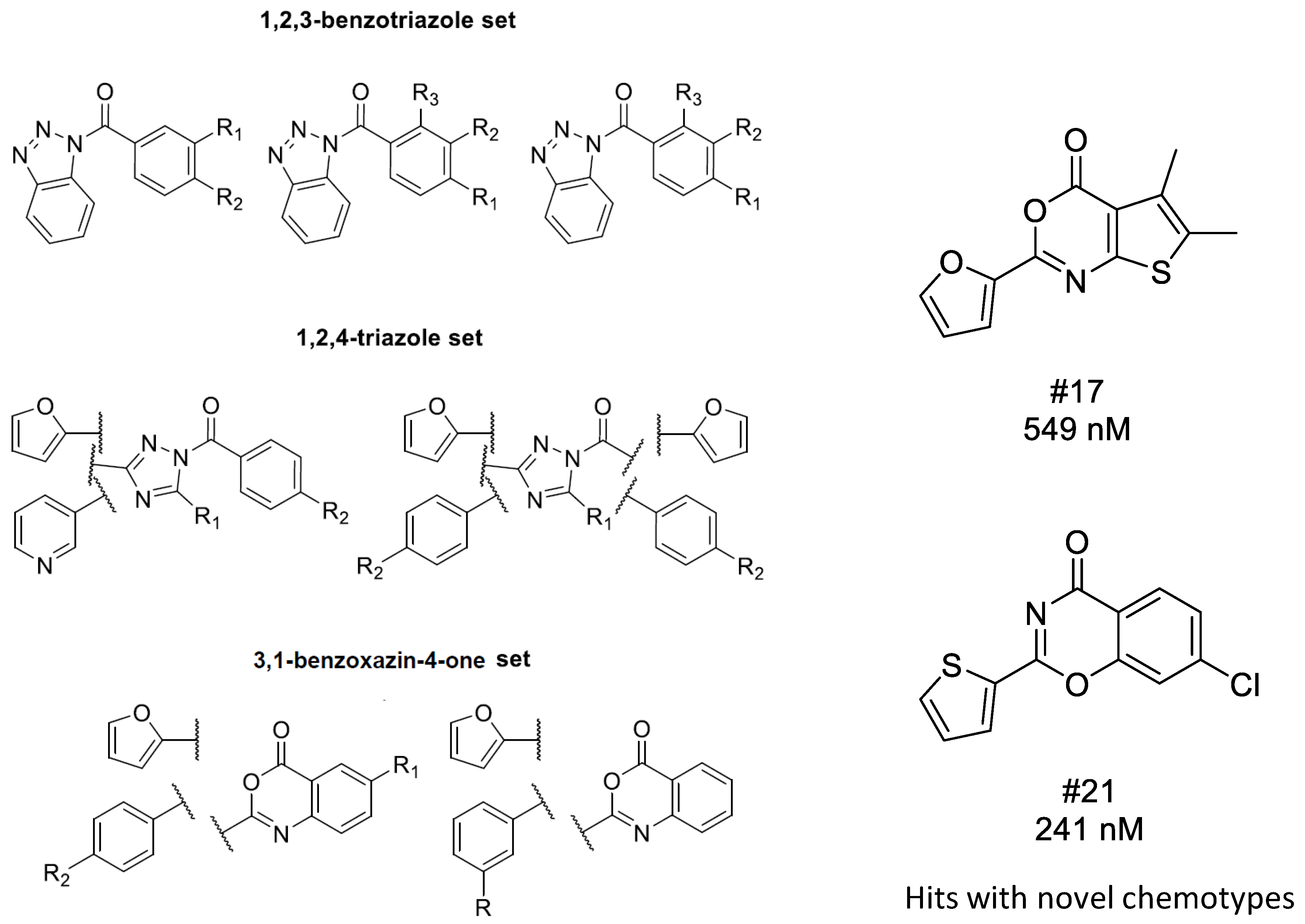
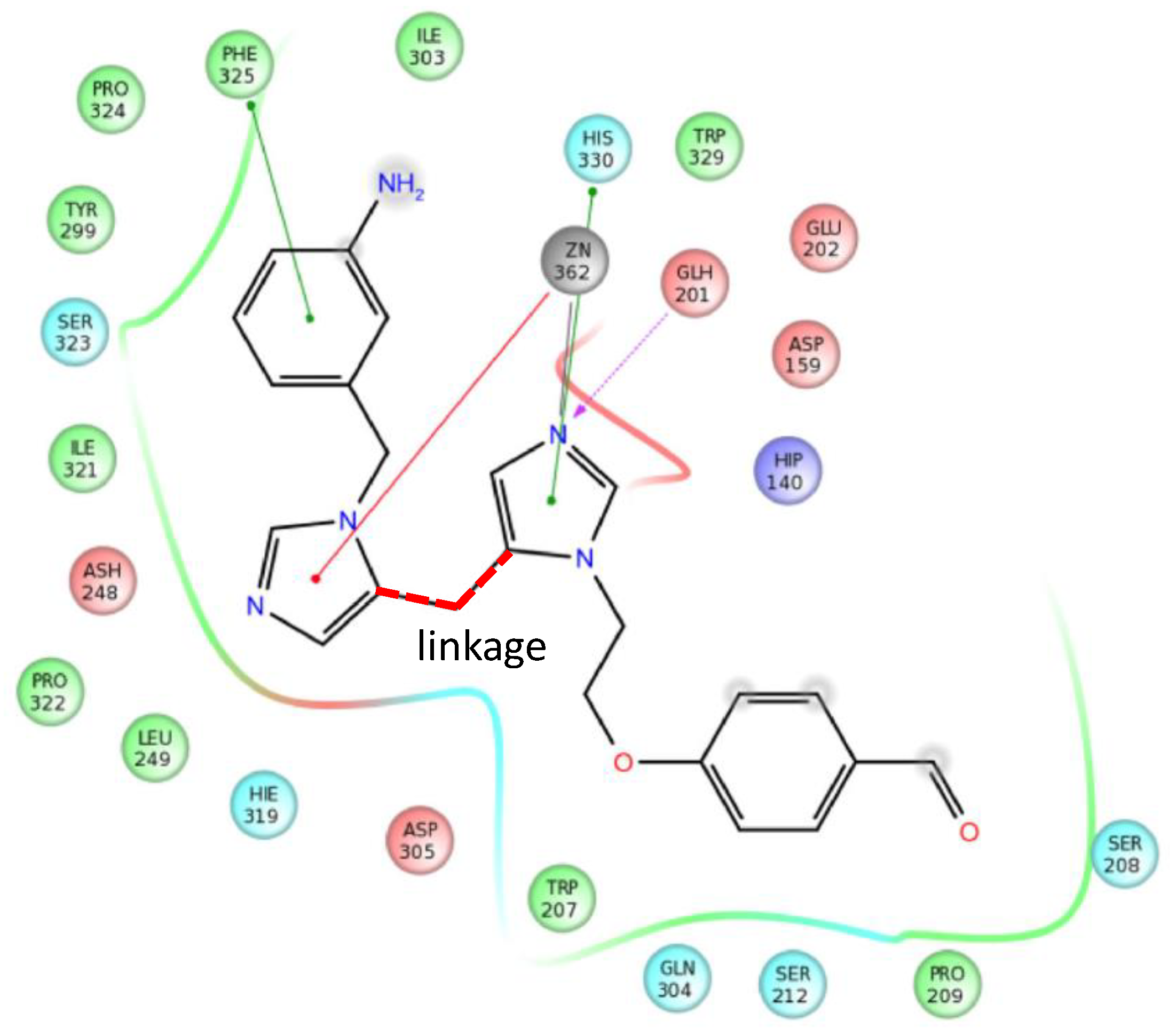
4.2. Bioisoster Extended Sequential Combination of 2D Similarity/Pharmacophore Model Search and In Vitro Screening: Inhibitors of a Serine Protease, the Complement Component C1s
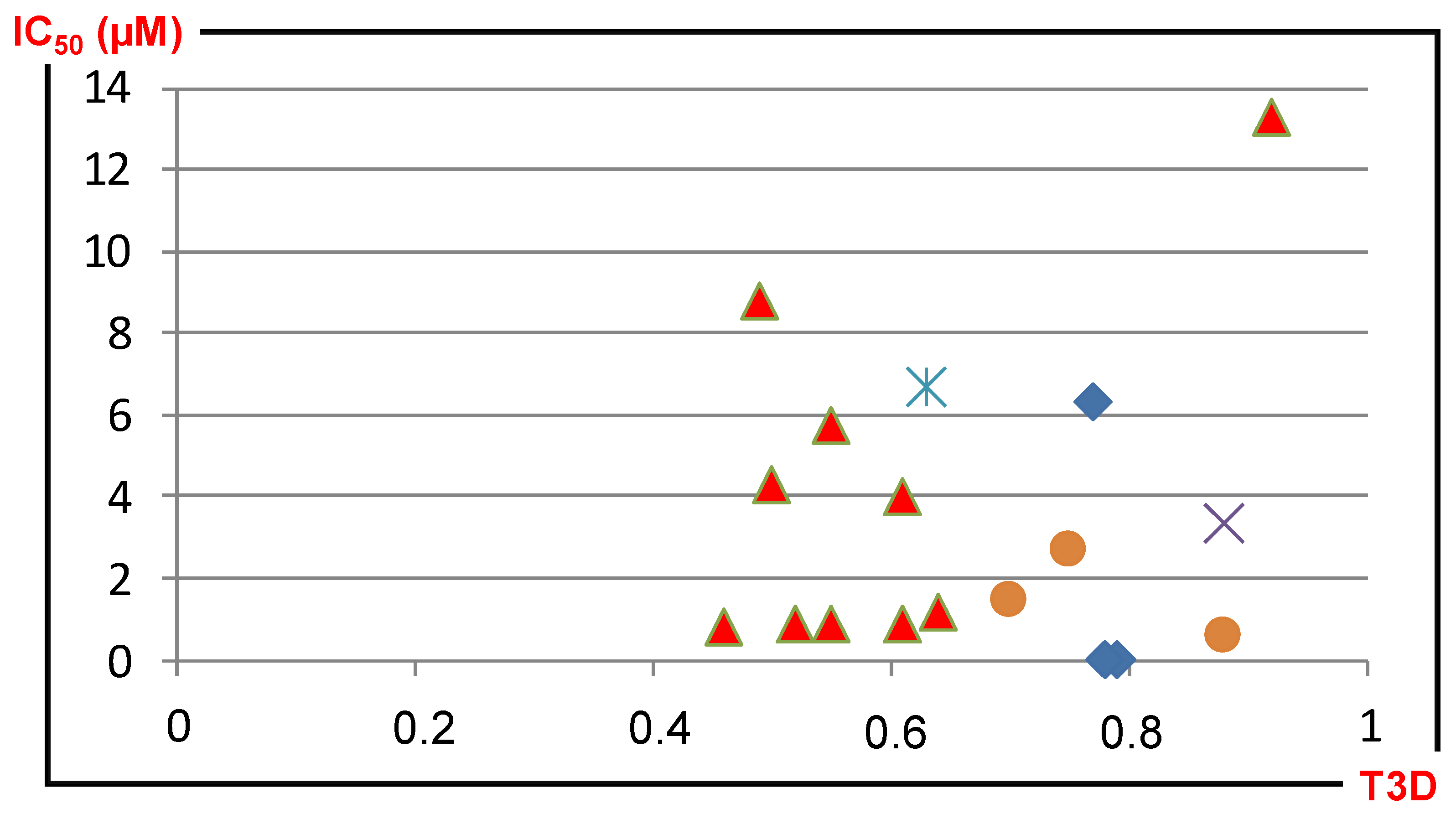
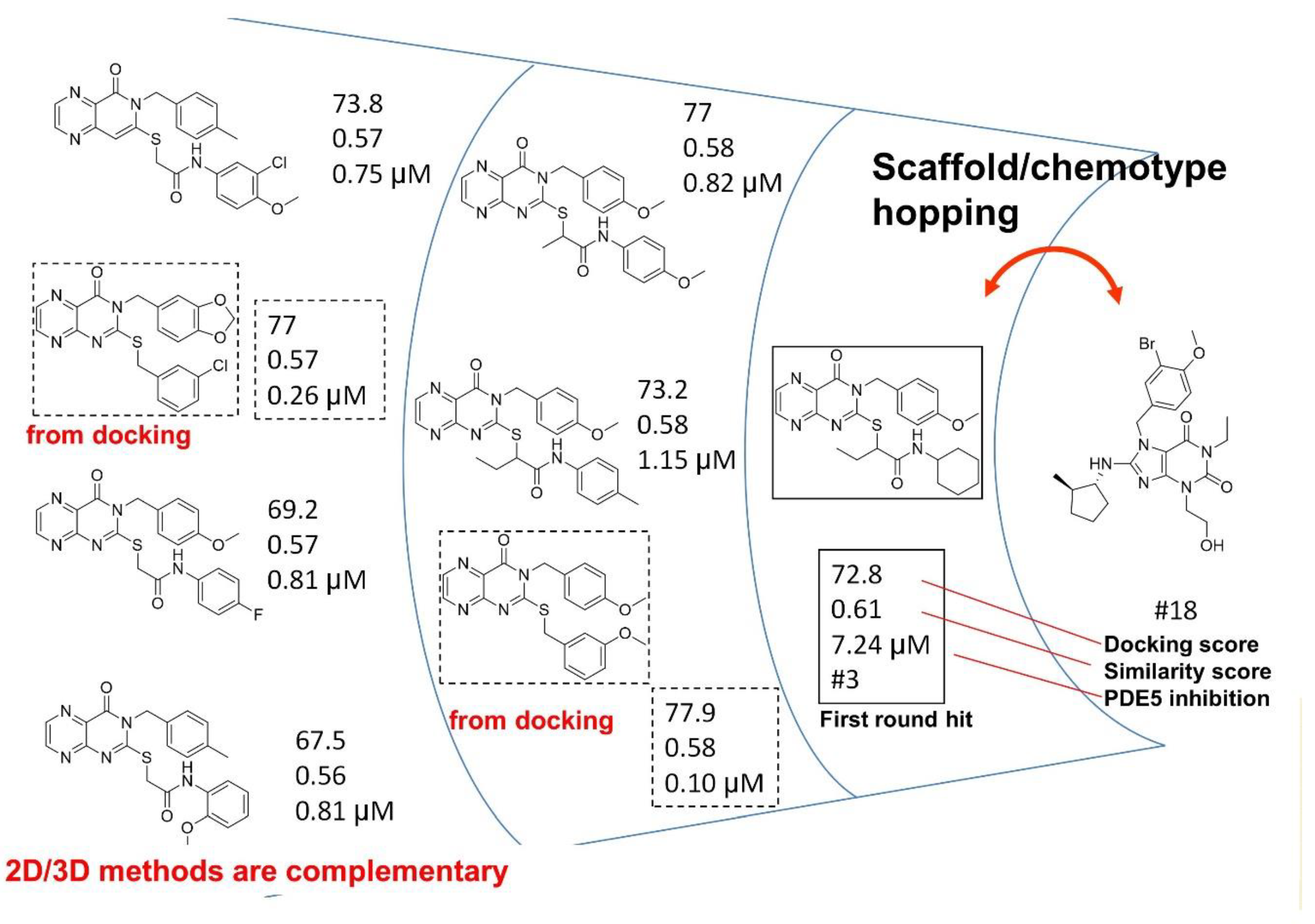
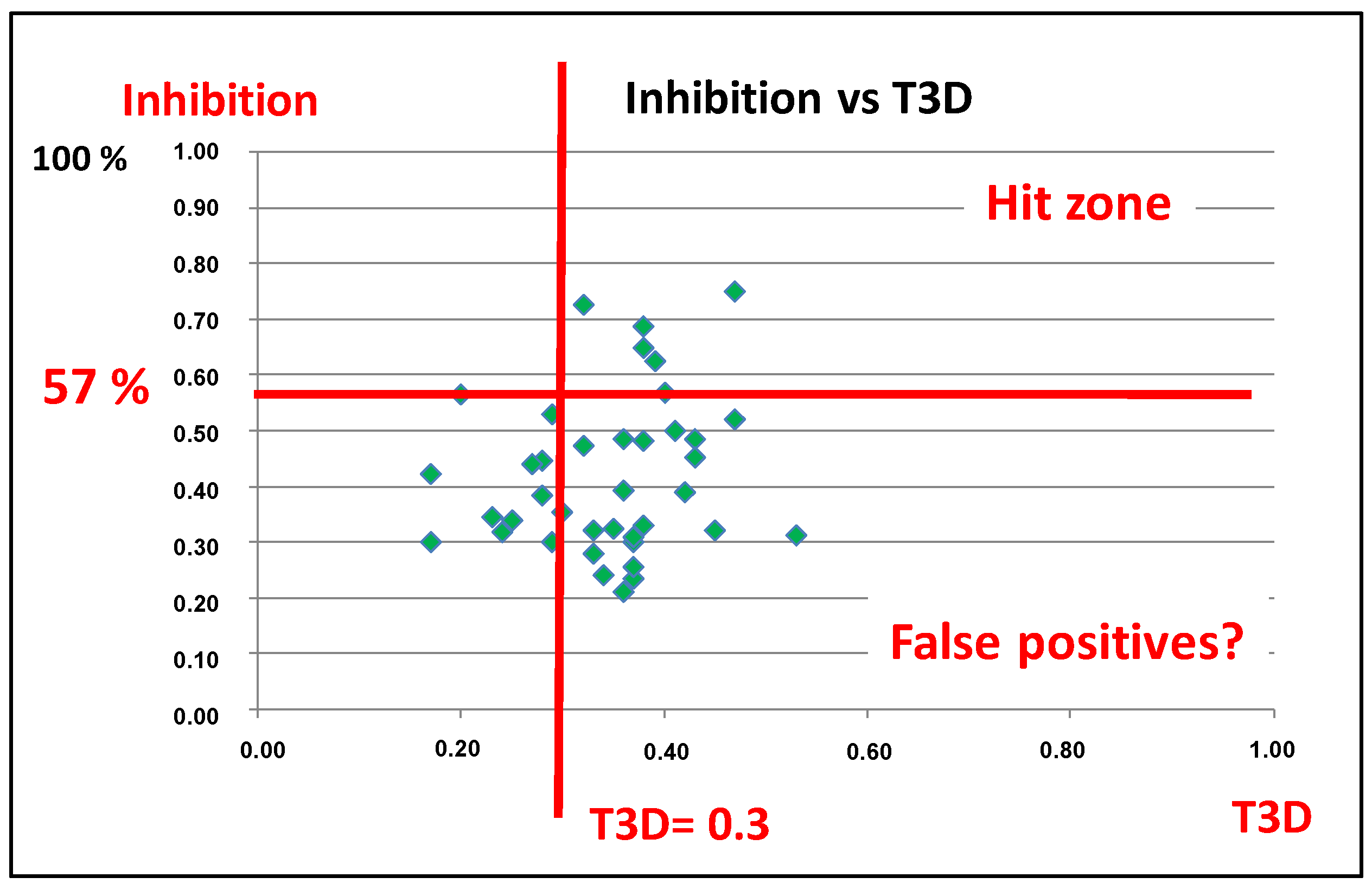
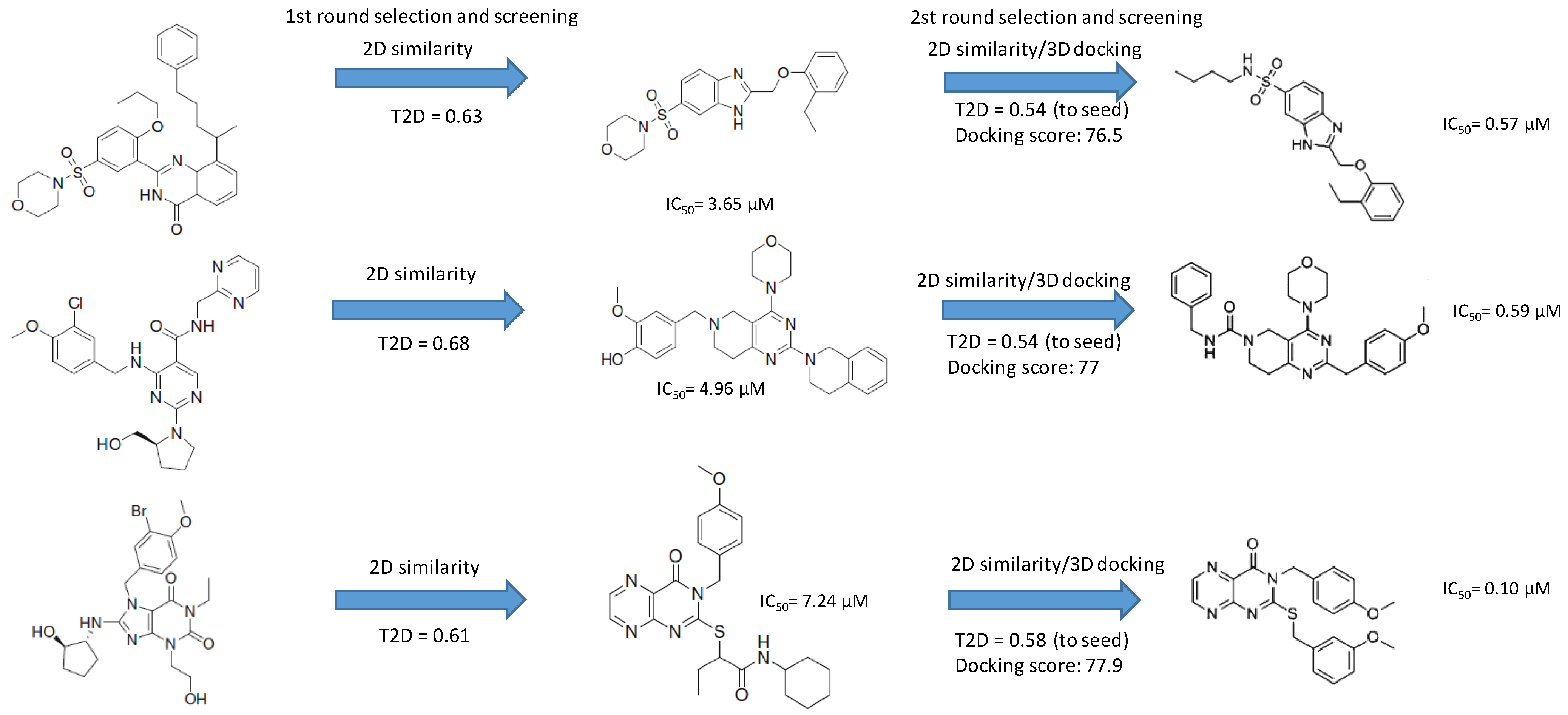
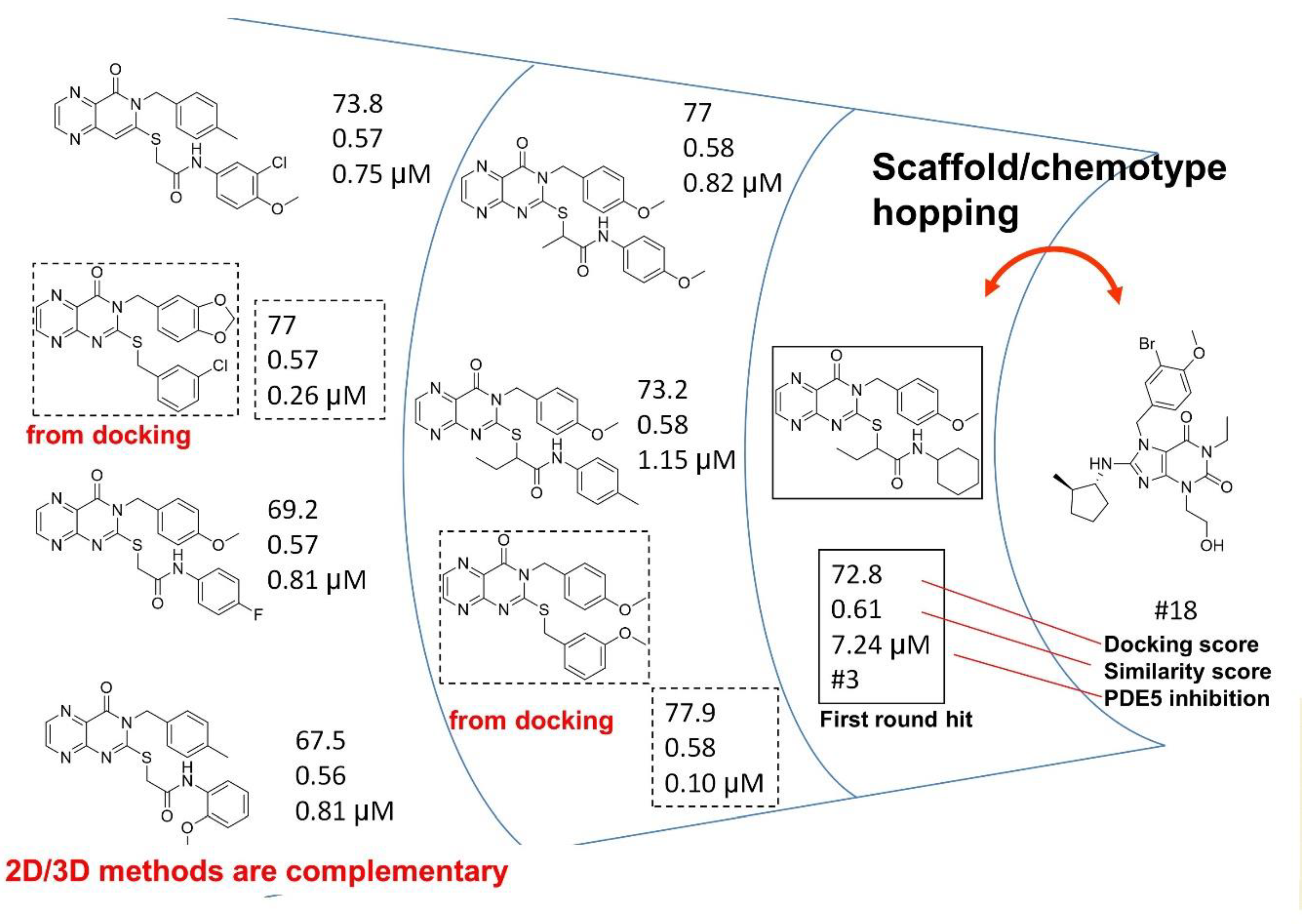
4.3. Parallel and Sequential Combination of 2D Similarity/3D Docking and In Vitro Screening: PDE5 Inhibitors
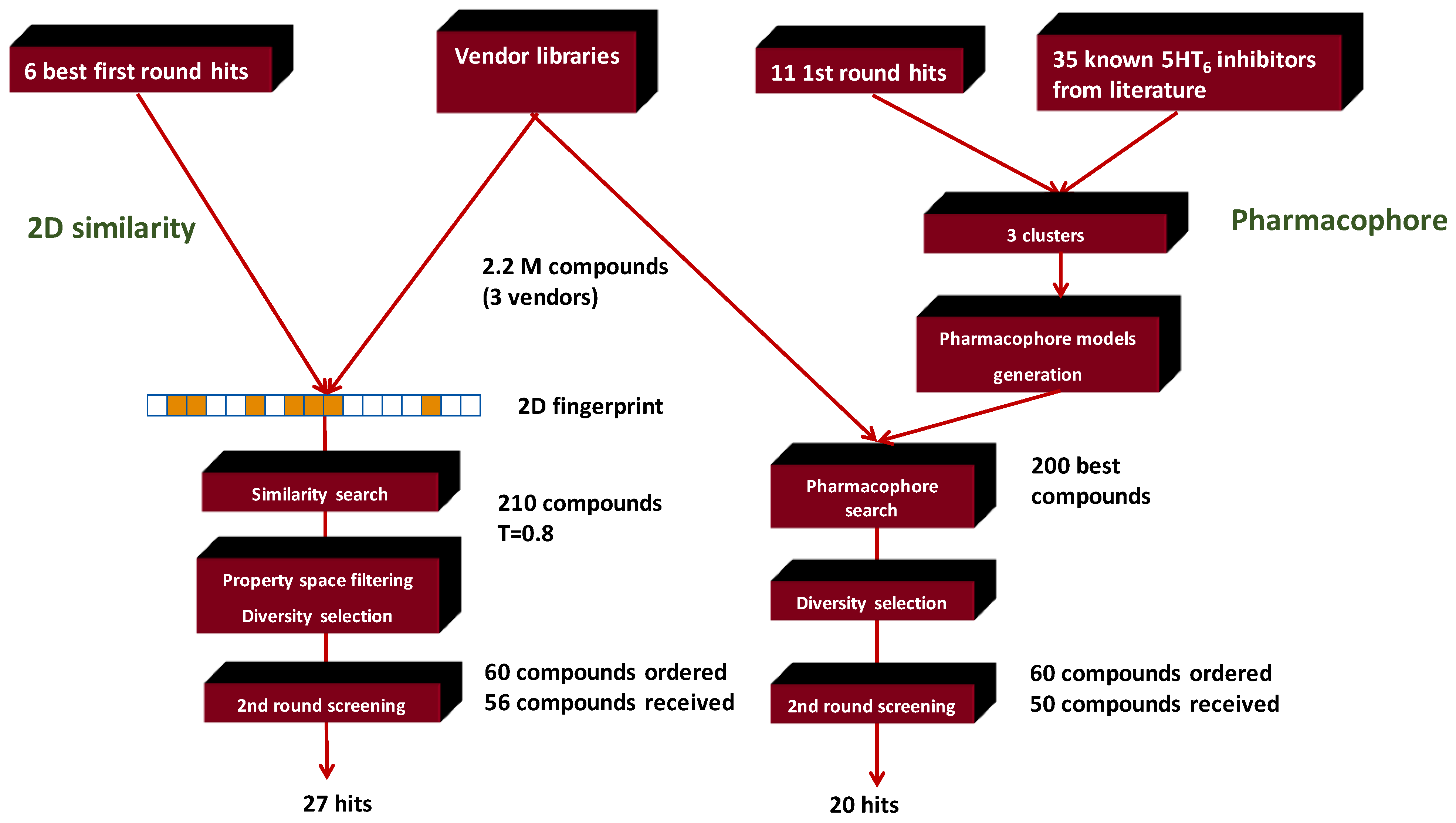
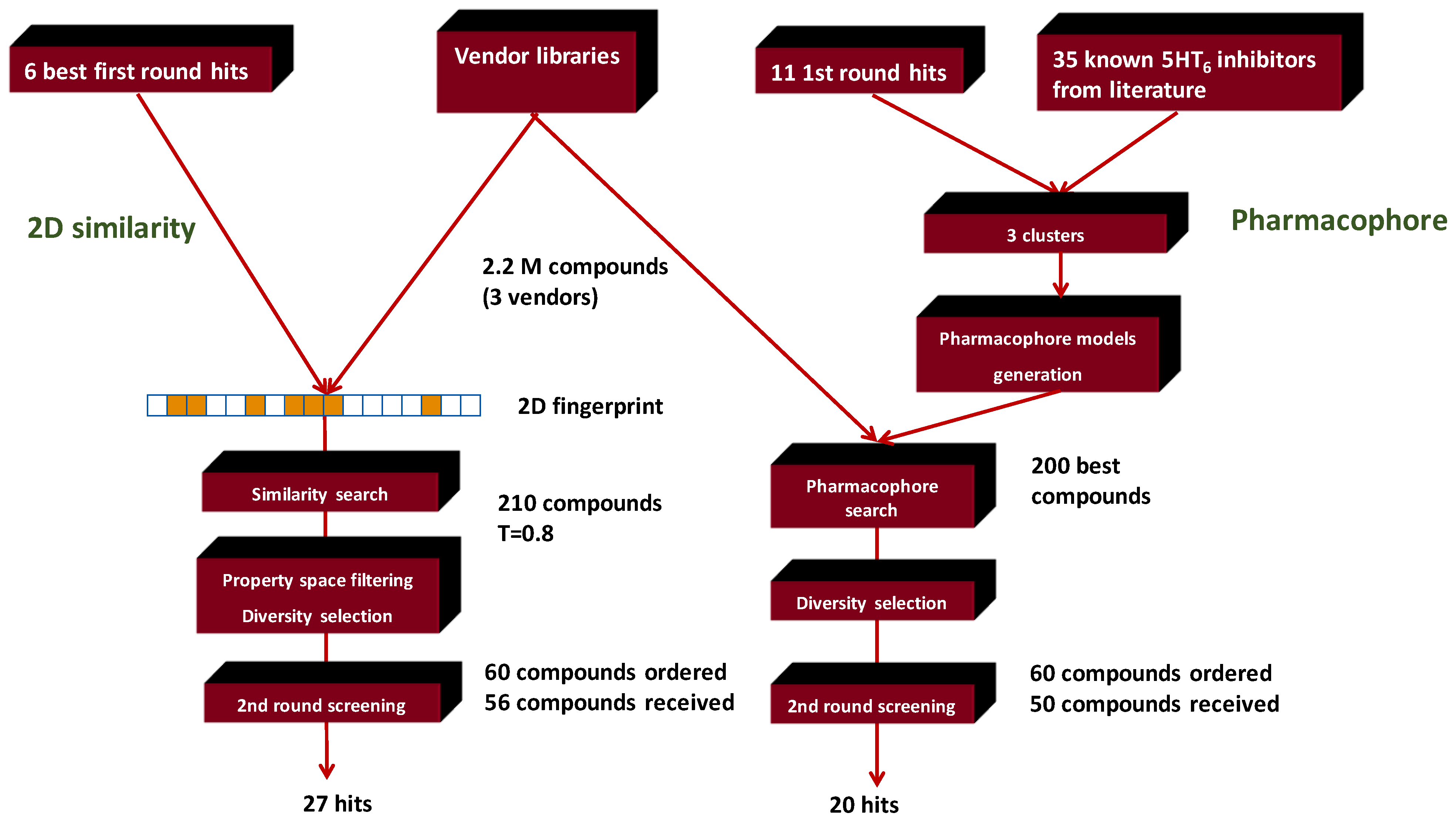
4.4. Parallel Combination of 2D Similarity/Pharmacophore Model Search and In Vitro Screening: 5HT6 Antagonists
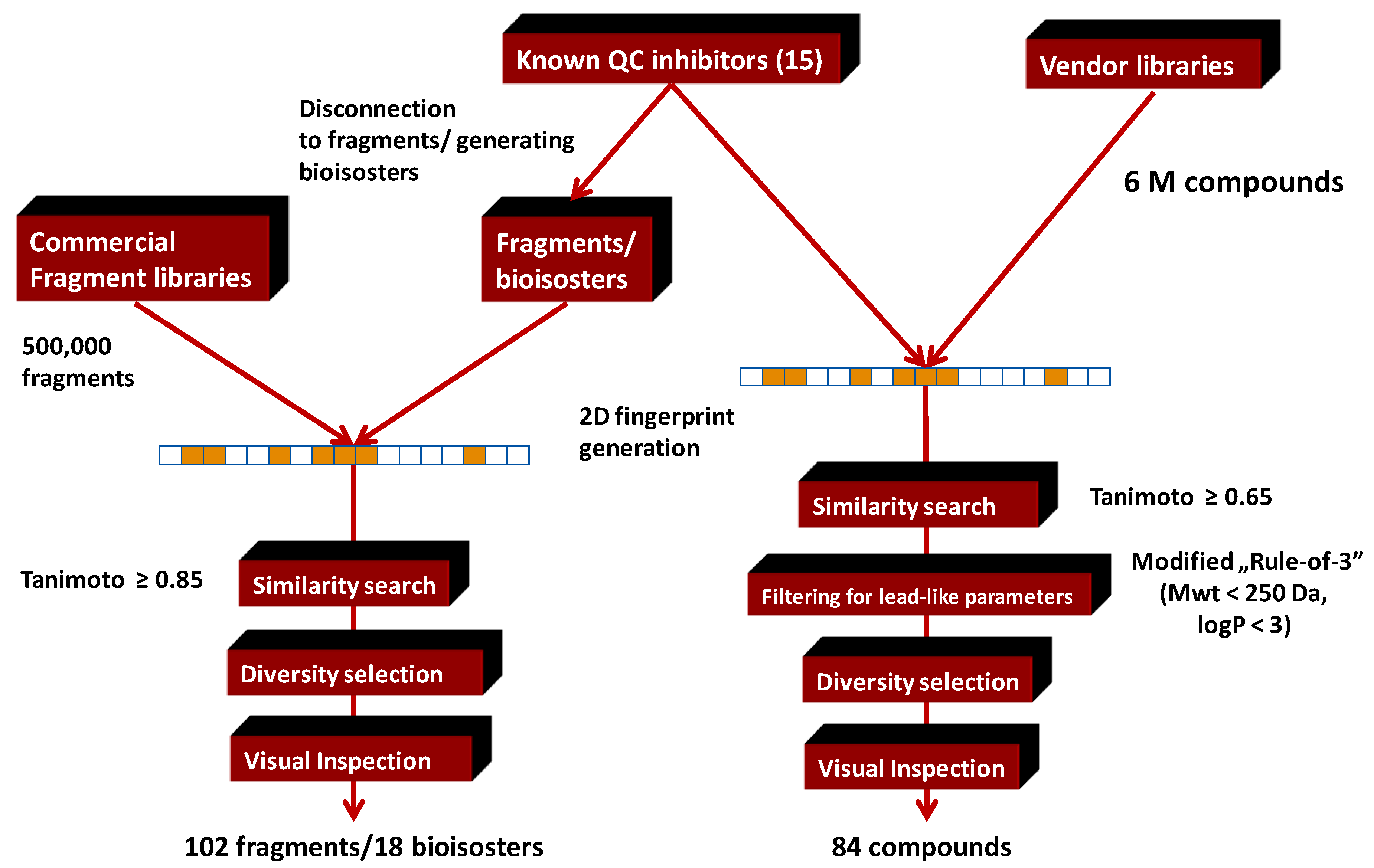
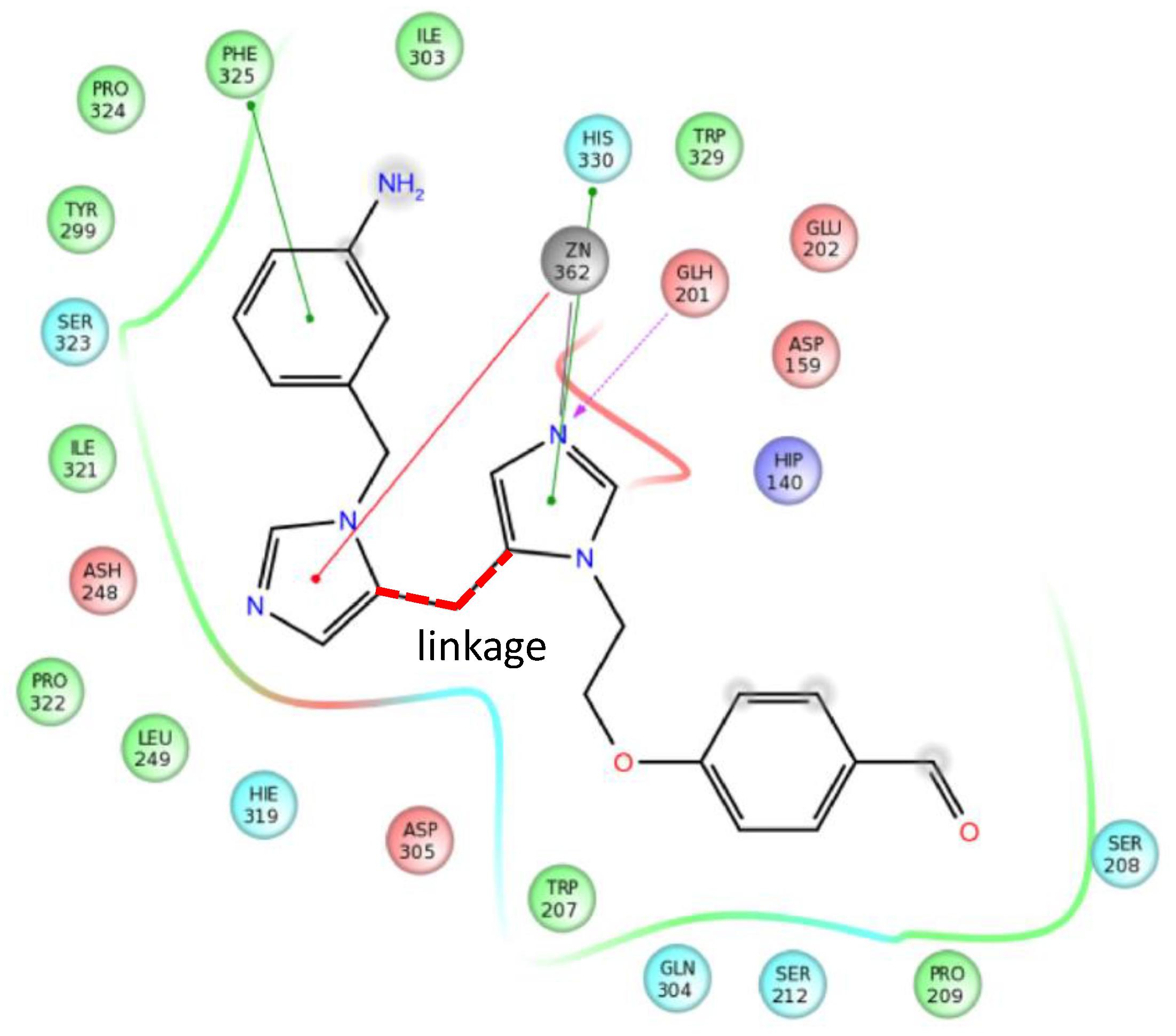
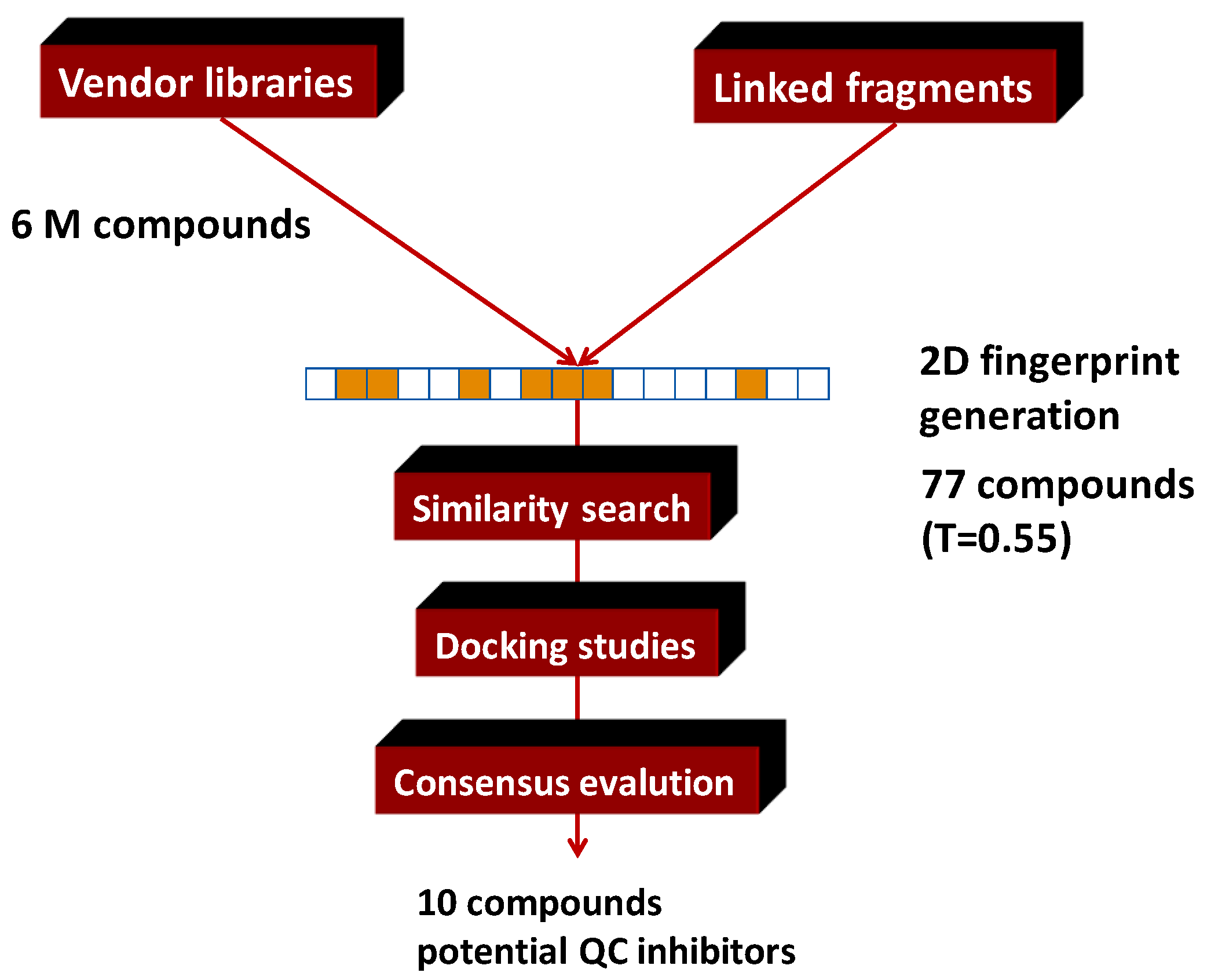
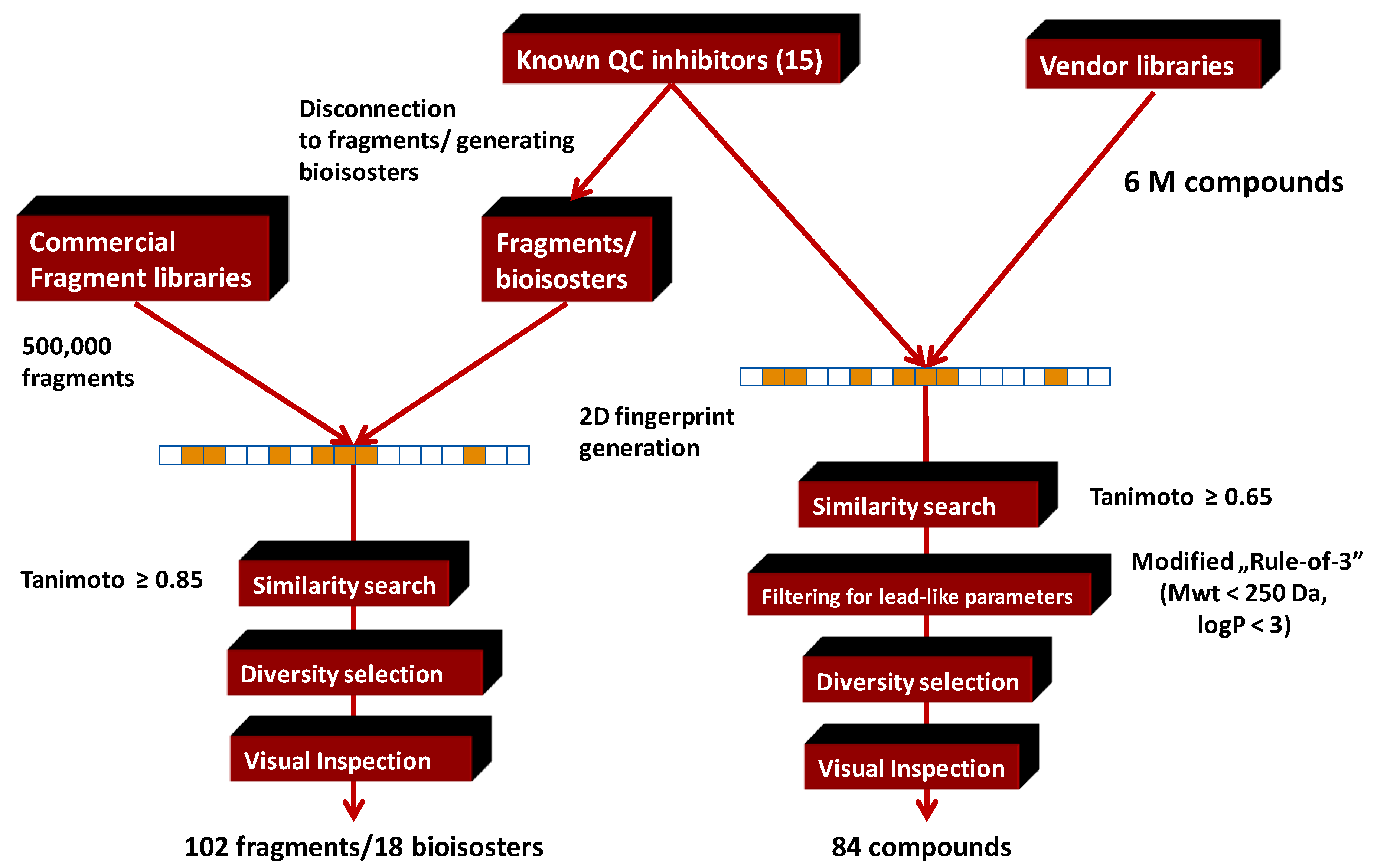
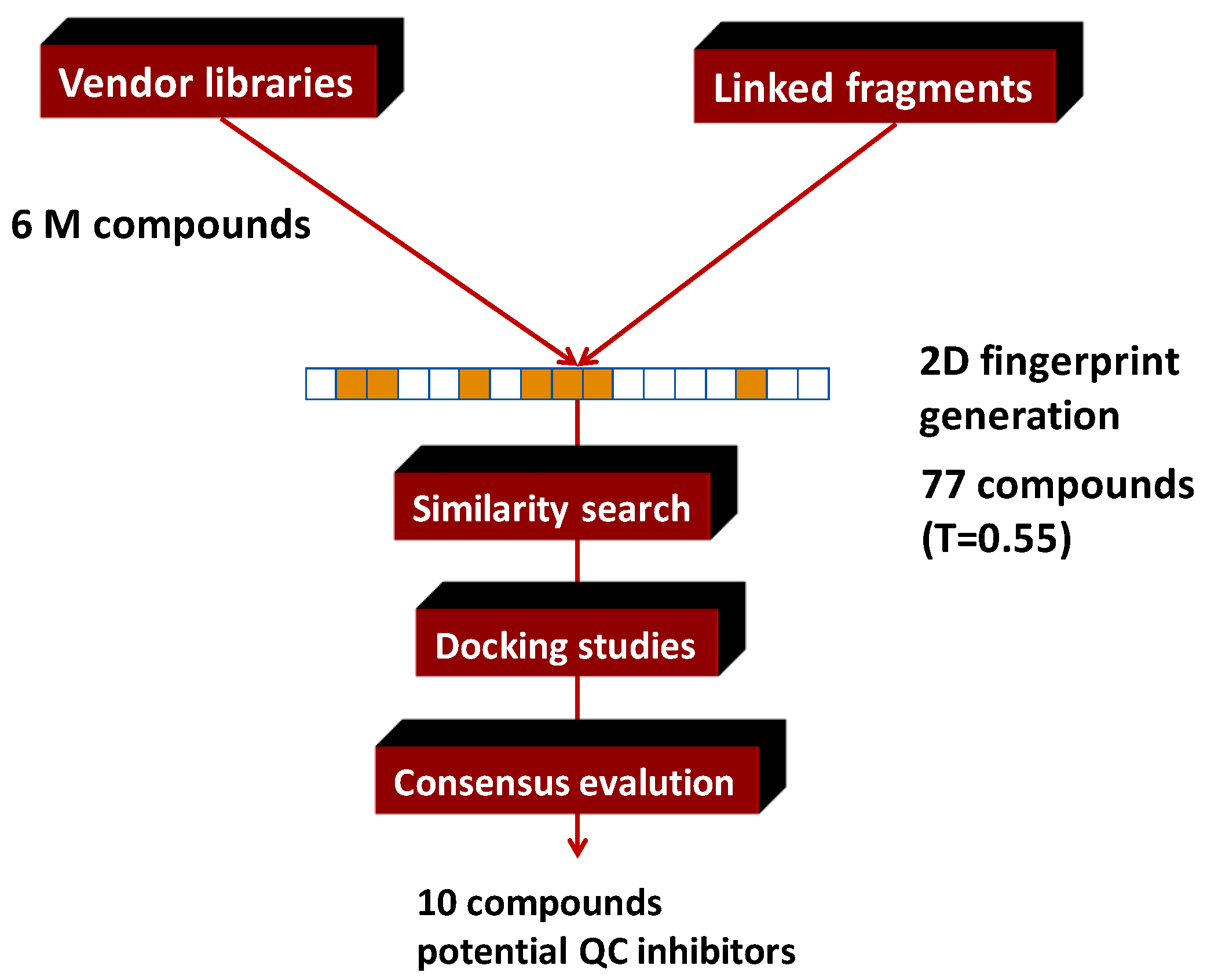
4.5. Parallel Combination of 2D Similarity Search/Fragment-Based Design, 3D Docking, and In Vitro Screening: Glutaminyl Cyclase (QC) Inhibitors
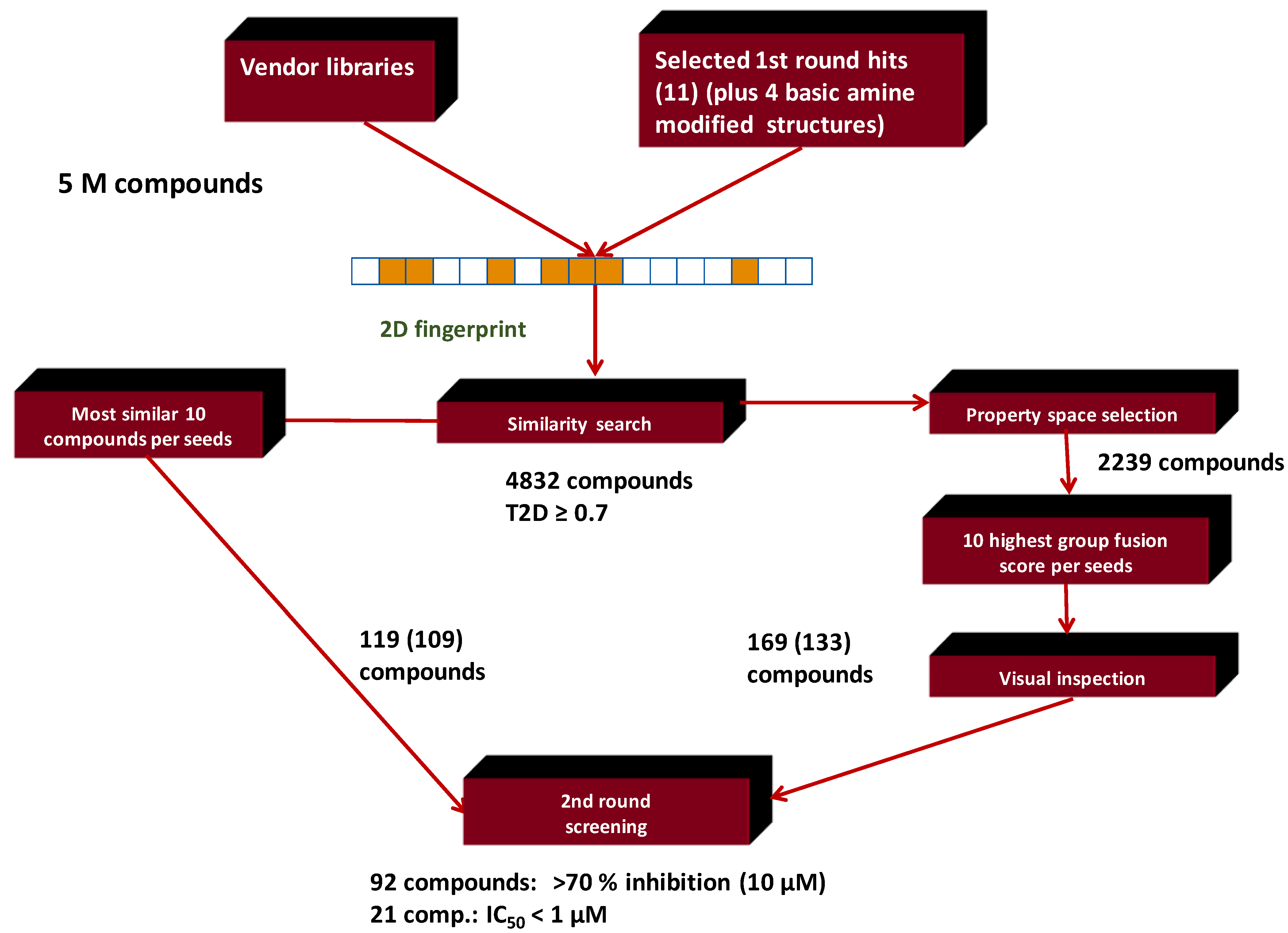
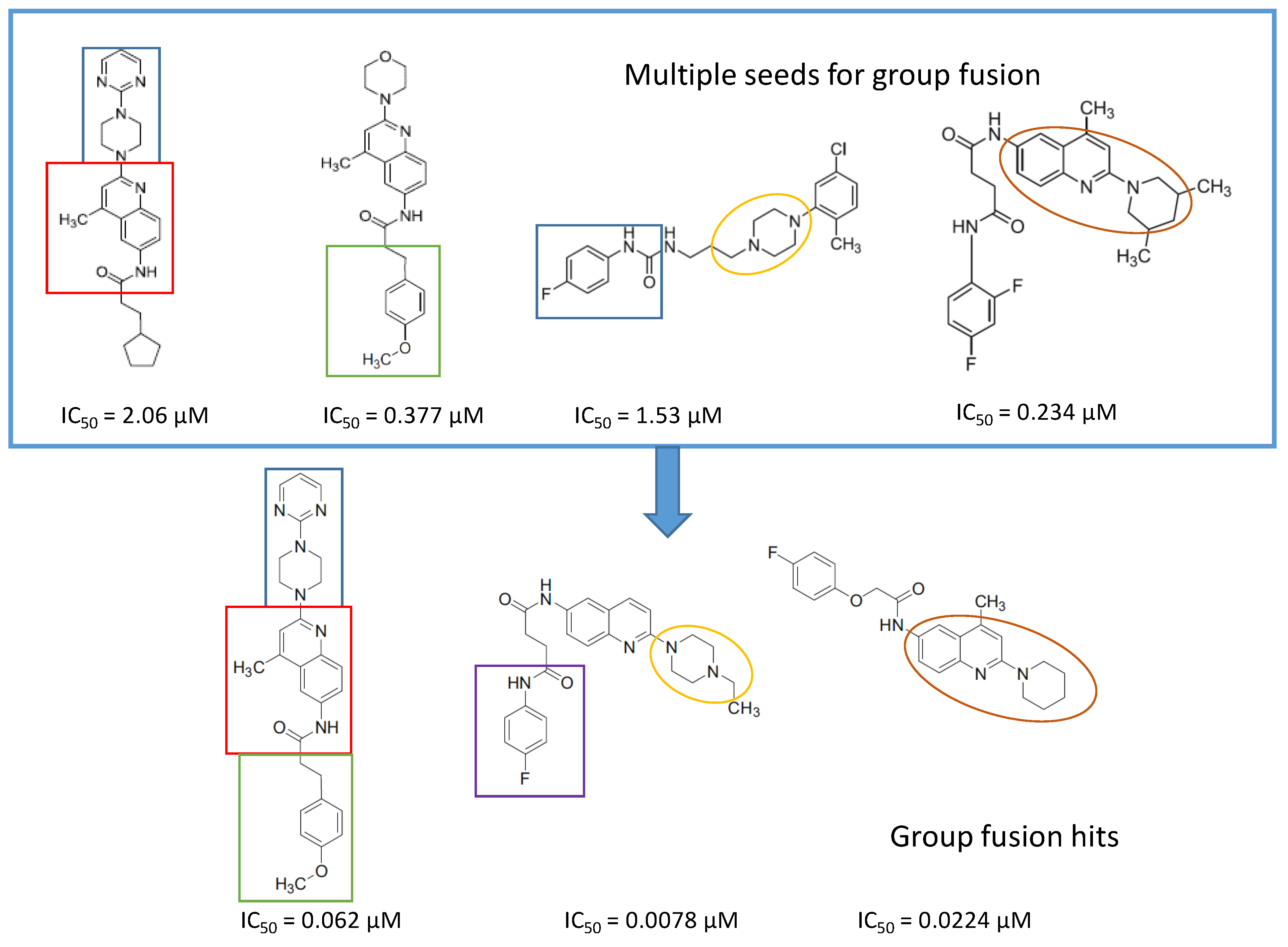
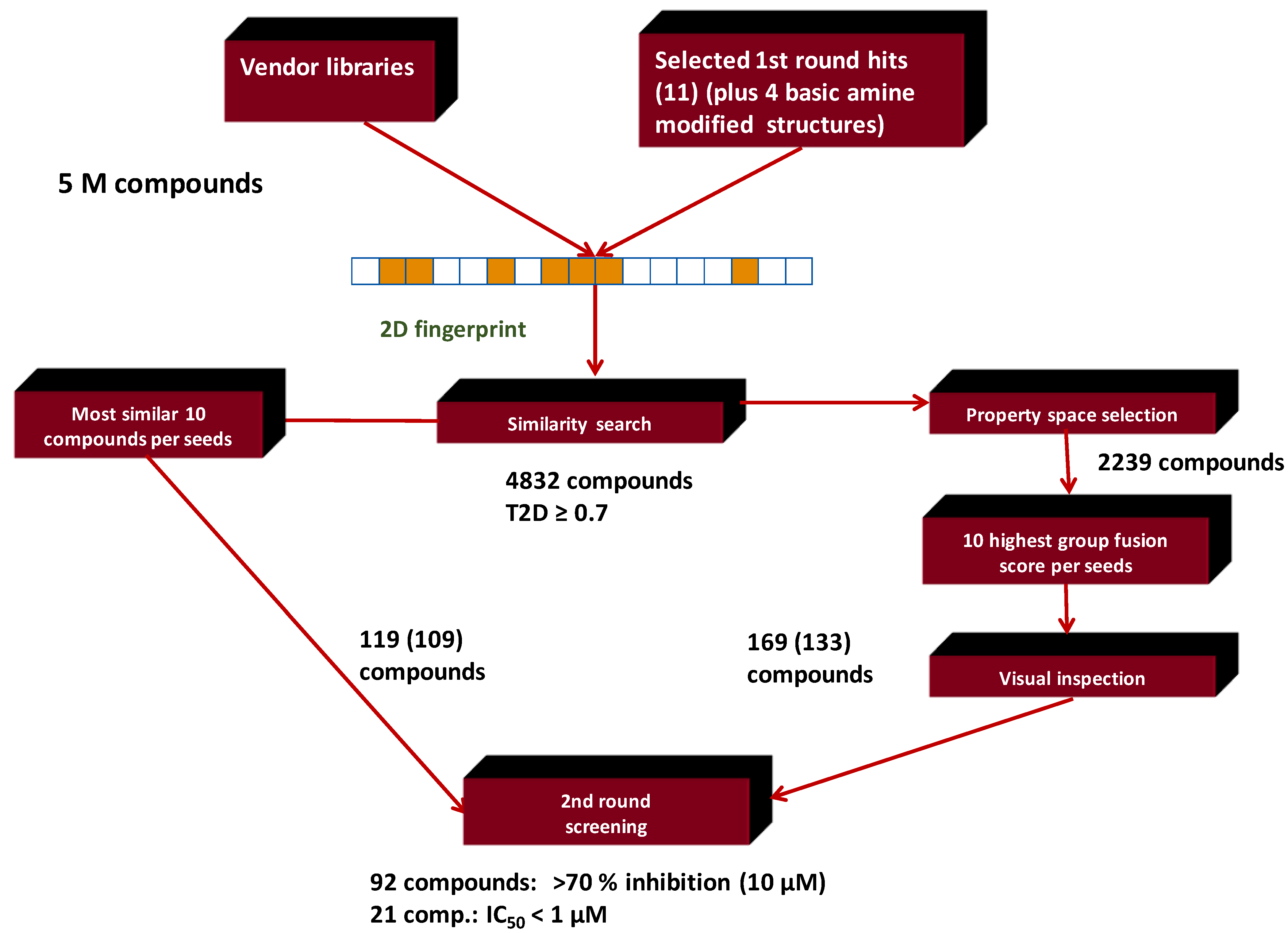
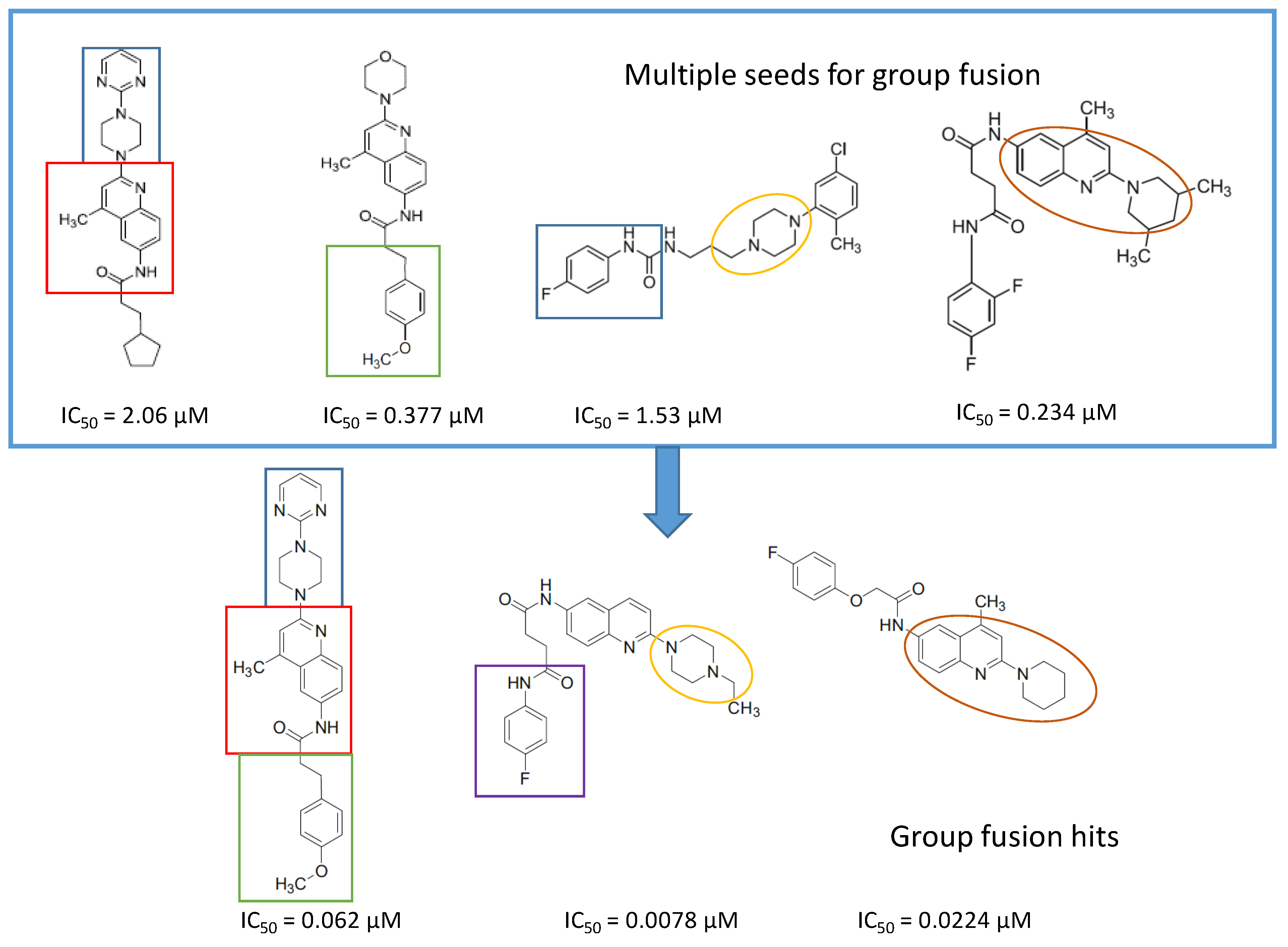
4.6. Parallel 2D Similarity Selection/Group Fusion Aproach, Combination with In Vitro Screening: Melanin-Concentrating Hormone Receptor-1 (MCHR1)
5. Summary, Conclusions, and Additional Application Areas
Additional, Emerging Application Areas for 2D Similarity Search
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- McInnes, C. Virtual screening strategies in drug discovery. Curr. Opin. Biol. 2007, 11, 494–502. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; K Vassilatis, D.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Edwards, B.S.; Bologa, C.; Young, S.M.; Balakin, K.V.; Prossnitz, E.R.; Savchuck, N.P.; Sklar, L.A.; Oprea, T.I. Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol. Pharmacol. 2005, 68, 1301–1310. [Google Scholar] [CrossRef]
- Murgueitio, M.S.; Bermudez, M.; Mortier, J.; Wolber, G. In silico virtual screening approaches for anti-viral drug discovery. Drug Discov. Today Technologies 2012, 9, e219–e225. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massague, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallve, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef]
- Rognan, D. The impact of in silico screening in the discovery of novel and safer drug candidates. Pharmacol. Ther. 2017, 175, 47–66. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [Green Version]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef]
- Drwal, M.N.; Griffith, R. Combination of ligand-and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef]
- Mannhold, R.; Kubinyi, H.; Folkers, G. Virtual Screening: Principles, Challenges, and Practical Guidelines; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; Volume 48. [Google Scholar]
- Vázquez, J.; López, M.; Gibert, E.; Herrero, E.; Luque, F.J. Merging ligand-based and structure-based methods in drug discovery: An overview of combined virtual screening approaches. Molecules 2020, 25, 4723. [Google Scholar] [CrossRef]
- Tan, L.; Geppert, H.; Sisay, M.T.; Gutschow, M.; Bajorath, J. Integrating structure- and ligand-based virtual screening: Comparison of individual, parallel, and fused molecular docking and similarity search calculations on multiple targets. ChemMedChem 2008, 3, 1566–1571. [Google Scholar] [CrossRef]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A. Machine-learning approaches in drug discovery: Methods and applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, K.A.; Huang, X. Machine learning-based virtual screening and its applications to Alzheimer’s drug discovery: A review. Curr. Pharm. Des. 2018, 24, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Jasial, S.; Gilberg, E.; Blaschke, T.; Bajorath, J. Machine learning distinguishes with high accuracy between pan-assay interference compounds that are promiscuous or represent dark chemical matter. J. Med. Chem. 2018, 61, 10255–10264. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, E.; Ebejer, J.-P. Applying machine learning to ultrafast shape recognition in ligand-based virtual screening. Front. Pharmacol. 2020, 10, 1675. [Google Scholar] [CrossRef] [PubMed]
- Ai, G.; Tian, C.; Deng, D.; Fida, G.; Chen, H.; Ma, Y.; Ding, L.; Gu, Y. A combination of 2D similarity search, pharmacophore, and molecular docking techniques for the identification of vascular endothelial growth factor receptor-2 inhibitors. Anticancer Drugs 2015, 26, 399–409. [Google Scholar] [CrossRef]
- Staroń, J.; Kurczab, R.; Warszycki, D.; Satała, G.; Krawczyk, M.; Bugno, R.; Lenda, T.; Popik, P.; Hogendorf, A.S.; Hogendorf, A. Virtual screening-driven discovery of dual 5-HT6/5-HT2A receptor ligands with pro-cognitive properties. Eur. J. Med. Chem. 2020, 185, 111857. [Google Scholar] [CrossRef]
- Pant, S.; Singh, M.; Ravichandiran, V.; Murty, U.S.N.; Srivastava, H.K. Peptide-like and small-molecule inhibitors against Covid-19. J. Biomol. Struct. Dyn. 2021, 39, 2904–2913. [Google Scholar] [CrossRef] [Green Version]
- Dou, X.; Jiang, L.; Wang, Y.; Jin, H.; Liu, Z.; Zhang, L. Discovery of new GSK-3beta inhibitors through structure-based virtual screening. Bioorg. Med. Chem. Lett. 2018, 28, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Schyman, P.; Liu, R.; Wallqvist, A. General purpose 2D and 3D similarity approach to identify hERG blockers. J. Chem. Inf. Model. 2016, 56, 213–222. [Google Scholar] [CrossRef]
- Gancia, E.; De Groot, M.; Burton, B.; Clark, D.E. Discovery of LRRK2 inhibitors by using an ensemble of virtual screening methods. Bioorg. Med. Chem Lett 2017, 27, 2520–2527. [Google Scholar] [CrossRef]
- Gagic, Z.; Ruzic, D.; Djokovic, N.; Djikic, T.; Nikolic, K. In silico methods for design of kinase inhibitors as anticancer drugs. Front. Chem. 2020, 7, 873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velnati, S.; Ruffo, E.; Massarotti, A.; Talmon, M.; Varma, K.S.S.; Gesu, A.; Fresu, L.G.; Snow, A.L.; Bertoni, A.; Capello, D.; et al. Identification of a novel DGKalpha inhibitor for XLP-1 therapy by virtual screening. Eur. J. Med. Chem. 2019, 164, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tian, J.Y.; Ye, F.; Xiao, Z. Identification of natural products as selective PTP1B inhibitors via virtual screening. Bioorg. Chem. 2020, 98, 103706. [Google Scholar] [CrossRef] [PubMed]
- Ghamari, N.; Zarei, O.; Reiner, D.; Dastmalchi, S.; Stark, H.; Hamzeh-Mivehroud, M. Histamine H3 receptor ligands by hybrid virtual screening, docking, molecular dynamics simulations, and investigation of their biological effects. Chem. Biol. Drug. Des. 2019, 93, 832–843. [Google Scholar] [CrossRef]
- Divsalar, D.N.; Simoben, C.V.; Schonhofer, C.; Richard, K.; Sippl, W.; Ntie-Kang, F.; Tietjen, I. Novel Histone Deacetylase Inhibitors and HIV-1 Latency-Reversing Agents Identified by Large-Scale Virtual Screening. Front. Pharmacol. 2020, 11, 905. [Google Scholar] [CrossRef]
- Polgár, T.; Keseru, G.M. Integration of virtual and high throughput screening in lead discovery settings. Comb. Chem. High. Throughput Screen. 2011, 14, 889–897. [Google Scholar] [CrossRef]
- Polgár, T.; Baki, A.; Szendrei, G.I.; Keserű, G.M. Comparative virtual and experimental high-throughput screening for glycogen synthase kinase-3β inhibitors. J. Med. Chem. 2005, 48, 7946–7959. [Google Scholar] [CrossRef]
- Stahura, F.L.; Bajorath, J. Virtual screening methods that complement HTS. Comb. Chem. High. Throughput Screen. 2004, 7, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dou, X.; Jiang, L.; Jin, H.; Zhang, L.; Zhang, L.; Liu, Z. Discovery of novel glycogen synthase kinase-3alpha inhibitors: Structure-based virtual screening, preliminary SAR and biological evaluation for treatment of acute myeloid leukemia. Eur. J. Med. Chem. 2019, 171, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Cabrera, Á.; Murcia, P.A.S.; Morreale, A.; Gago, F. Ligand-Based Drug Discovery and Design. In In Silico Drug Discovery and Design; CRC Press: Boca Raton, FL, USA, 2015; pp. 116–139. [Google Scholar]
- Koeppen, H.; Kriegl, J.; Lessel, U.; Tautermann, C.S.; Wellenzohn, B. Ligand-Based Virtual Screening. In Virtual Screening: Principles, Challenges, and Practical Guidelines; Wiley-VCH: New York, NY, USA; Weinheim, Germany, 2011; pp. 61–85. [Google Scholar]
- Maggiora, G.M.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular similarity in medicinal chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef] [PubMed]
- Maggiora, G.M.; Shanmugasundaram, V. Molecular similarity measures. In Chemoinformatics and Computational Chemical Biology; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2011; Volume 672, pp. 39–100. [Google Scholar]
- Martin, Y.C.; Kofron, J.L.; Traphagen, L.M. Do structurally similar molecules have similar biological activity? J. Med. Chem. 2002, 45, 4350–4358. [Google Scholar] [CrossRef]
- Scior, T.; Medina-Franco, J.; Do, Q.-T.; Martínez-Mayorga, K.; Yunes Rojas, J.; Bernard, P. How to recognize and workaround pitfalls in QSAR studies: A critical review. Curr. Med. Chem. 2009, 16, 4297–4313. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The light and dark sides of virtual screening: What is there to know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [Green Version]
- Medina-Franco, J.L.; Martínez-Mayorga, K.; Bender, A.; Marín, R.M.; Giulianotti, M.A.; Pinilla, C.; Houghten, R.A. Characterization of activity landscapes using 2D and 3D similarity methods: Consensus activity cliffs. J. Chem. Inf. Model. 2009, 49, 477–491. [Google Scholar] [CrossRef]
- Peltason, L.; Bajorath, J. SAR index: Quantifying the nature of structure− activity relationships. J. Med. Chem. 2007, 50, 5571–5578. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC− a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- ChemSpider. Available online: http://www.chemspider.com (accessed on 17 April 2017).
- eMolecules. Available online: https://reaxys.emolecules.com (accessed on 17 April 2017).
- The PubChem Project. Available online: http://pubchem.ncbi.nlm.nih.gov/ (accessed on 3 May 2017).
- CheEMBL-European Bioinformatics Institute. Available online: https://www.ebi.ac.uk/chembl/ (accessed on 3 May 2017).
- Binding DB—The Binding Database. Available online: http://www.bindingdb.org/ (accessed on 3 May 2017).
- Butina, D. Unsupervised data base clustering based on daylight’s fingerprint and Tanimoto similarity: A fast and automated way to cluster small and large data sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Willett, P. Similarity searching using 2D structural fingerprints. Methods Mol. Biol. 2011, 672, 133–158. [Google Scholar]
- Willett, P. Searching techniques for databases of two-and three-dimensional chemical structures. J. Med. Chem. 2005, 48, 4183–4199. [Google Scholar] [CrossRef]
- Stumpfe, D.; Bajorath, J. Similarity searching. WIREs Comput. Mol. Sci. 2011, 1, 260–282. [Google Scholar] [CrossRef]
- Willett, P.; Winterman, V. A Comparison of Some Measures for the Determination of Inter-Molecular Structural Similarity Measures of Inter-Molecular Structural Similarity. Quant. Struct.-Act. Rel. 1986, 5, 18–25. [Google Scholar] [CrossRef]
- Glen, R.C.; Adams, S.E. Similarity metrics and descriptor spaces–which combinations to choose? QSAR Comb. Sci. 2006, 25, 1133–1142. [Google Scholar] [CrossRef]
- Heikamp, K.; Bajorath, J. The future of virtual compound screening. Chem. Biol. Drug Des. 2013, 81, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Cabrera, A.; Gago, F.; Morreale, A. A reverse combination of structure-based and ligand-based strategies for virtual screening. J. Comput. Aid. Mol. Des. 2012, 26, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Decornez, H.; Gulyás-Forró, A.; Papp, Á.; Szabó, M.; Sármay, G.; Hajdú, I.; Cseh, S.; Dormán, G.; Kitchen, D.B. Design, selection, and evaluation of a general kinase-focused library. ChemMedChem 2009, 4, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Stahura, F.L.; Godden, J.W.; Bajorath, J. Fingerprint scaling increases the probability of identifying molecules with similar activity in virtual screening calculations. J. Chem. Inf. Comp. Sci. 2001, 41, 746–753. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Carter, G.T. Drug-like property concepts in pharmaceutical design. Curr. Pharm. Design 2009, 15, 2184–2194. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R. The influence of target family and functional activity on the physicochemical properties of pre-clinical compounds. J. Med. Chem. 2006, 49, 2969–2978. [Google Scholar] [CrossRef]
- Martin, Y.C. Molecular diversity: How we measure it? Has it lived up to its promise? Il Farmaco 2001, 56, 137–139. [Google Scholar] [CrossRef]
- Cao, Y.; Jiang, T.; Girke, T. A maximum common substructure-based algorithm for searching and predicting drug-like compounds. Bioinformatics 2008, 24, i366–i374. [Google Scholar] [CrossRef] [Green Version]
- Papp, A.; Gulyás-Forró, A.; Gulyás, Z.; Dormán, G.; Ürge, L.; Darvas, F. Explicit Diversity Index (EDI): A novel measure for assessing the diversity of compound databases. J. Chem. Inf. Model. 2006, 46, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Dobi, K.; Hajdú, I.; Flachner, B.; Fabó, G.; Szaszkó, M.; Bognár, M.; Magyar, C.; Simon, I.; Szisz, D.; Lőrincz, Z.; et al. Combination of 2D/3D ligand-based similarity search in rapid virtual screening from multimillion compound repositories. Selection and biological evaluation of potential PDE4 and PDE5 inhibitors. Molecules 2014, 19, 7008–7039. [Google Scholar] [CrossRef]
- Kalászi, A.; Szisz, D.; Imre, G.; Polgár, T. Screen3D: A novel fully flexible high-throughput shape-similarity search method. J. Chem. Inf. Model. 2014, 54, 1036–1049. [Google Scholar] [CrossRef]
- Tömöri, T.; Hajdú, I.; Barna, L.; Lőrincz, Z.; Cseh, S.; Dormán, G. Combining 2D and 3D in silico methods for rapid selection of potential PDE5 inhibitors from multimillion compounds’ repositories: Biological evaluation. Mol. Divers. 2012, 16, 59–72. [Google Scholar] [CrossRef]
- Szilágyi, K.; Hajdú, I.; Flachner, B.; Lőrincz, Z.; Balczer, J.; Gál, P.; Závodszky, P.; Pirli, C.; Balogh, B.; Mándity, I.M.; et al. Design and selection of novel C1s inhibitors by in silico and in vitro approaches. Molecules 2019, 24, 3641. [Google Scholar] [CrossRef] [Green Version]
- Dobi, K.; Flachner, B.; Pukáncsik, M.; Máthé, E.; Bognár, M.; Szaszkó, M.; Magyar, C.; Hajdú, I.; Lőrincz, Z.; Simon, I.; et al. Combination of Pharmacophore Matching, 2D Similarity Search, and In vitro Biological Assays in the Selection of Potential 5-HT6 Antagonists from Large Commercial Repositories. Chem. Biol. Drug Des. 2015, 86, 864–880. [Google Scholar] [CrossRef]
- Szaszkó, M.; Hajdú, I.; Flachner, B.; Dobi, K.; Magyar, C.; Simon, I.; Lőrincz, Z.; Kapui, Z.; Pázmány, T.; Cseh, S.; et al. Identification of potential glutaminyl cyclase inhibitors from lead-like libraries by in silico and in vitro fragment-based screening. Mol. Divers. 2017, 21, 175–186. [Google Scholar] [CrossRef]
- Flachner, B.; Tömöri, T.; Hajdú, I.; Dobi, K.; Lőrincz, Z.; Cseh, S.; Dormán, G. Rapid in silico selection of an MCHR1 antagonists’ focused library from multi-million compounds’ repositories: Biological evaluation. Med. Chem. Res. 2014, 23, 1234–1247. [Google Scholar] [CrossRef]
- Venkatraman, V.; Pérez-Nueno, V.I.; Mavridis, L.; Ritchie, D.W. Comprehensive comparison of ligand-based virtual screening tools against the DUD data set reveals limitations of current 3D methods. J. Chem. Inf. Model. 2010, 50, 2079–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuffenhauer, A.; Gillet, V.J.; Willett, P. Similarity searching in files of three-dimensional chemical structures: Analysis of the BIOSTER database using two-dimensional fingerprints and molecular field descriptors. J. Chem. Inf. Model. 2000, 40, 295–307. [Google Scholar]
- Hajdú, I.; Kardos, J.; Major, B.; Fabó, G.; Lőrincz, Z.; Cseh, S.; Dormán, G. Inhibition of the lox enzyme family members with old and new ligands. selectivity analysis revisited. Bioorg. Med. Chem. Lett. 2018, 28, 3113–3118. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Martinez-Mayorga, K.; Meurice, N. Balancing novelty with confined chemical space in modern drug discovery. Expert Opin. Drug Dis. 2014, 9, 151–165. [Google Scholar] [CrossRef]
- Dormán, G.; Flachner, B.; Hajdú, I.; András, D.C. Target identification and polypharmacology of nutraceuticals. In Nutraceuticals: Efficacy, Safety and Toxicity; Gupta, R.C., Lall, R., Srivastava, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 315–343. [Google Scholar]
- Flachner, B.; Lorincz, Z.; Carotti, A.; Nicolotti, O.; Kuchipudi, P.; Remez, N.; Sanz, F.; Tovari, J.; Szabo, M.J.; Bertok, B.; et al. A chemocentric approach to the identification of cancer targets. PLoS ONE 2012, 7, e35582. [Google Scholar] [CrossRef]
- Yera, E.R.; Cleves, A.E.; Jain, A.N. Prediction of off-target drug effects through data fusion. In Pacific Symposium on Biocomputing 2014; World Scientific: Hackensack, NJ, USA, 2014; pp. 160–171. [Google Scholar]
- Mellor, C.L.; Marchese Robinson, R.L.; Benigni, R.; Ebbrell, D.; Enoch, S.J.; Firman, J.W.; Madden, J.C.; Pawar, G.; Yang, C.; Cronin, M.T.D. Molecular fingerprint-derived similarity measures for toxicological read-across: Recommendations for optimal use. Regul. Toxicol. Pharmacol. 2019, 101, 121–134. [Google Scholar] [CrossRef]
- Coley, C.W.; Rogers, L.; Green, W.H.; Jensen, K.F. Computer-Assisted Retrosynthesis Based on Molecular Similarity. ACS Cent. Sci. 2017, 3, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Schuler, J.; Samudrala, R. Fingerprinting CANDO: Increased Accuracy with Structure- and Ligand-Based Shotgun Drug Repurposing. ACS Omega 2019, 4, 17393–17403. [Google Scholar] [CrossRef] [Green Version]
- Hage-Melim, L.; Federico, L.B.; de Oliveira, N.K.S.; Francisco, V.C.C.; Correia, L.C.; de Lima, H.B.; Gomes, S.Q.; Barcelos, M.P.; Francischini, I.A.G.; da Silva, C. Virtual screening, ADME/Tox predictions and the drug repurposing concept for future use of old drugs against the COVID-19. Life Sci. 2020, 256, 117963. [Google Scholar] [CrossRef] [PubMed]
- Santibáñez-Morán, M.G.; López-López, E.; Prieto-Martínez, F.D.; Sánchez-Cruz, N.; Medina-Franco, J.L. Consensus virtual screening of dark chemical matter and food chemicals uncover potential inhibitors of SARS-CoV-2 main protease. RSC Adv. 2020, 10, 25089–25099. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VS Method | Input Data | Computation Demand | Hit Diversity/Novelty |
|---|---|---|---|
| 3D Docking | Protein crystal structure (preferably with bound ligand) | ↑↑ | ↑↑ |
| Pharmacophore modelling | Several known ligands or protein crystal structure with bound ligand | ↑ | ↑↑ |
| 2D/3D Ligand-based similarity search | One or several known ligands | ↓ | ↑ |
| Approach | Examples | Comments |
|---|---|---|
| Sequential | Hierarchical VS: pharmacophore screening, application of property filters (druglikeness, ADMET), docking, manual selection |
|
| Parallel | Parallel application of pharmacophores, similarity methods, docking, followed by automated selection |
|
| Hybrid | Protein-ligand pharmacophores, docking with pharmacophore constraints |
|
| Approaches | Databases Used for Studies | Target Proteins | Number of Hits Identified | Activities Types and Ranges | References |
|---|---|---|---|---|---|
| Sequential integration approaches | |||||
| Sequential combination of 2D similarity search, pharmacophore, and molecular docking | Zinc-Specs Database (441,574 compounds) | Human vascular endothelial growth factor receptor-2 (VEGFR-2) | 2 hits | Inhibitory effects on the proliferation of cancer cells (U87 and MCF-7) expressing VEGFR-2. | Ai et al. [19] |
| Sequential approach, including 2D pharmacophore-based and structural fingerprints, ADME/Tox filtering and flexible docking | Commercial and academic libraries (58 reference ligands) | 5-HT6R (serotonin 5-HT6 receptor) | 6 hits | Competition binding for human serotonin 5-HT6R, 5-HT2aR, 5-HT1aR, 5-HT7R and dopaminergic D2R | Staron et al. [20] |
| Integrated in silico screening sequence (2D similarity followed by docking) | CHEMBL database, ZINC database, FDA approved drugs; molecules under clinical trials (5M compounds) | SARS-CoV-2 main protease | 4 potential inhibitors | Inhibitory effect against SARS-CoV-2 main protease | Pant et al. [21] |
| Reverse virtual screening method: (A) VS. for identify novel scaffolds (B) 2D similarity search for hit expansion | SPECS; PKU-CNCL (Peking University) | Glycogen synthase kinase-3b (GSK-3b) | 14 hits (IC50 = 0.71–18.2 μM) | Inhibitory effect against glycogen synthase kinase-3b (GSK-3b) | Dou et al. [22] |
| Sequential virtual screening: combination of 2D fingerprint matching and 3D shape modelling | CHEMBL database, FDA approved drugs | Human ether-a-go-go-related gene (hERG) | high recovery rate, maximum sensitivity, and specificity in hERG inhibition prediction | Blocking effect on human ether-a-go-go-related gene (hERG) | Schyman et al. [23] |
| Parallel integration approaches | |||||
| Parallel screening with ligand- and structure-based and virtual screening approaches | 6.6 M commercial compounds for LB 1.3 M compound for docking | Leucine rich repeat kinase-2 (LRRK2) | 35 compounds with IC50 < 10 μM | Inhibitory effect on leucine rich repeat kinase-2 (LRRK2) | Gancia et al. [24] |
| Parallel 2D similarity and pharmacophore-based virtual screening | SPECS | Fibroblast growth factor receptor 1 (FGFR1) | 19 compounds with activity >50% at 50 μM | Inhibitory effect on fibroblast growth factor receptor 1 (FGFR1) | Gagic et a.l. [25] |
| Parallel 2D/3D similarity search | Pubchem | Diacylglycerol kinase alpha (DGKalpha) | 17 active compounds with activity >25% at 10 μM | Inhibitory effect on Diacylglycerol kinase alpha (DGKalpha) | Velnati et al. [26] |
| Parallel hierarchical protocol combining ligand-based (2D similarity/pharmacophore model) and SB approaches for VS | DUD-E | Protein tyrosine phosphatase 1B (PTP1B) | 10 compounds with IC50 at micromolar level | Inhibitory effect on protein tyrosine phosphatase 1B (PTP1B) | Yan et al. [27] |
| Hybrid integration approaches | |||||
| Hybrid integration protocol (molecular modeling methods: molecular docking, molecular dynamics simulation) | ZINC DB | Histamine H3 receptor | 3 hits within micromolar and sub micromolar Ki range | Histamine H3 receptor antagonism | Ghamari et al. [28] |
| 2D similarity searching, docking and scoring | PubChem library (100 M) | Histone deacetylase (HDAC) | 60 virtual hit compounds; 4 active compounds | Inhibitory effect on histone deacetylase | Divsalar et al. [29] |
| The Searchable Chemical Space | ||||
|---|---|---|---|---|
| Database | Unique Compounds | References | ||
| Zinc library | 35 million | [43,44] | ||
| Chemspider | 58 million | [45] | ||
| eMolecules | 5.9 million | [46] | ||
| The Biologically Relevant Chemical Space | ||||
| Database | Compounds | Bioactivity or Binding Data | Protein Targets | References |
| PubChem | 91,371,681 | 232,760,104 | n.a. | [47] |
| ChEMBL | 2,036,512 | 14,371,197 | 11.224 | [48] |
| Binding_DB | 590.985 | 1,328,228 | 6.922 | [49] |
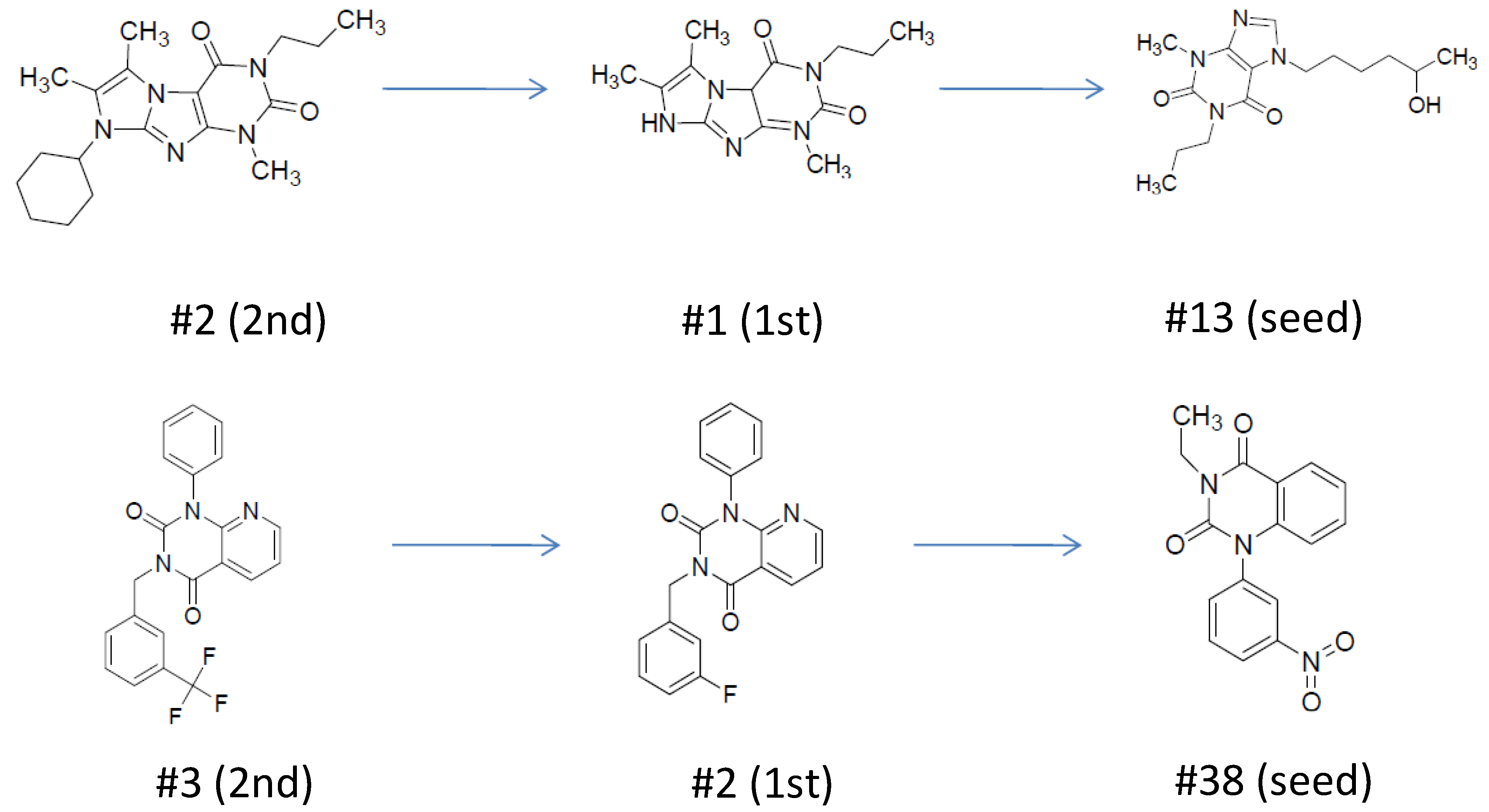
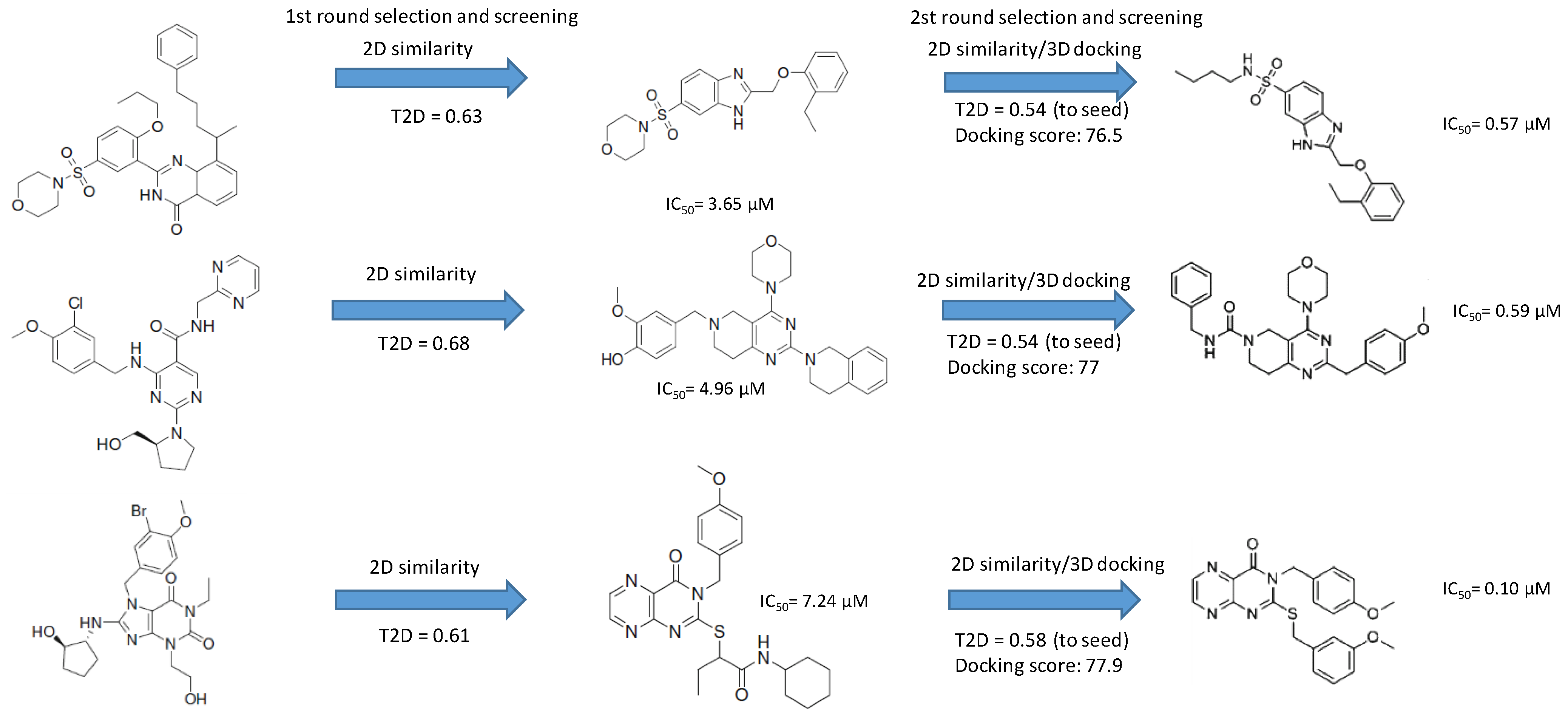
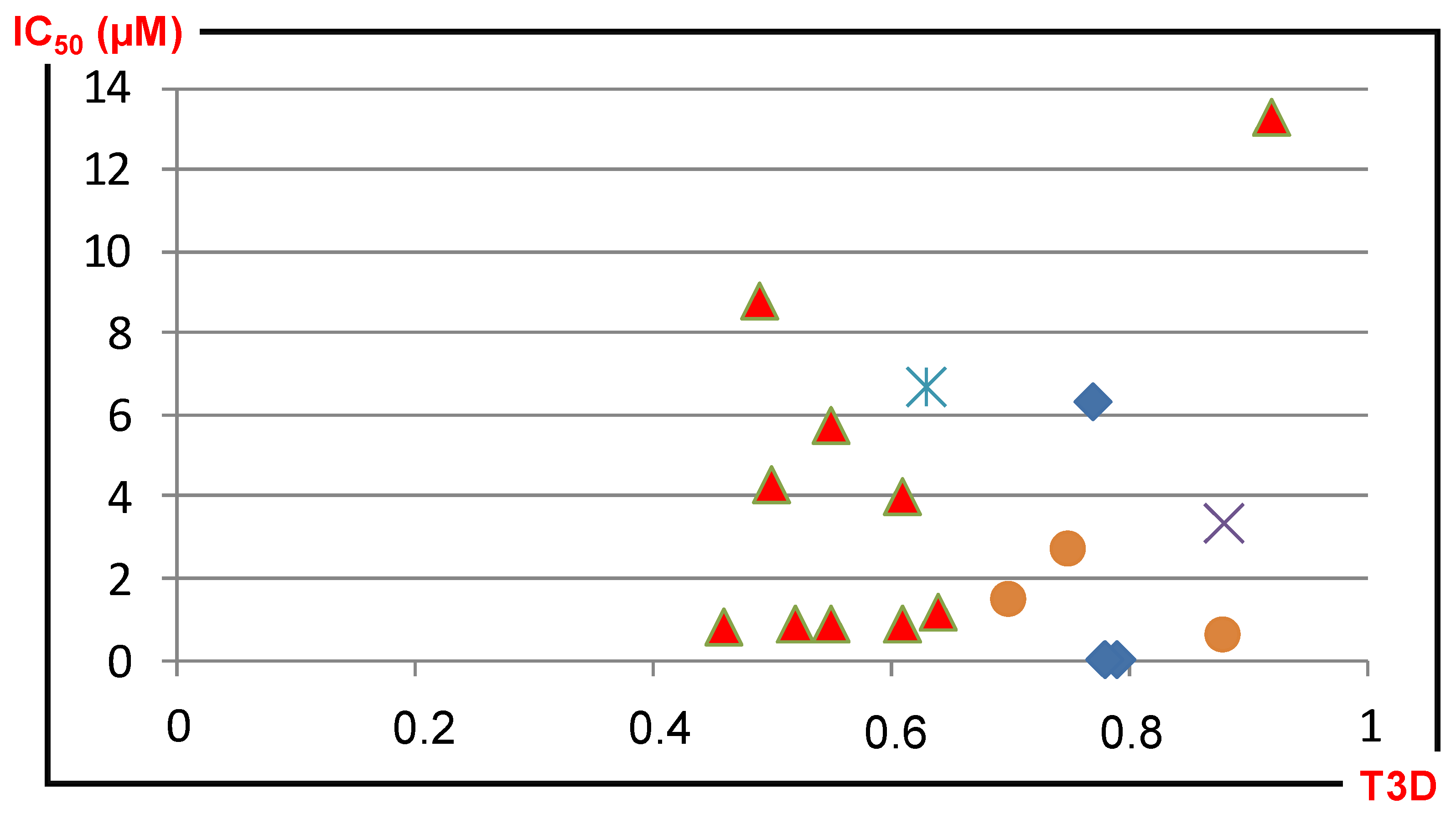
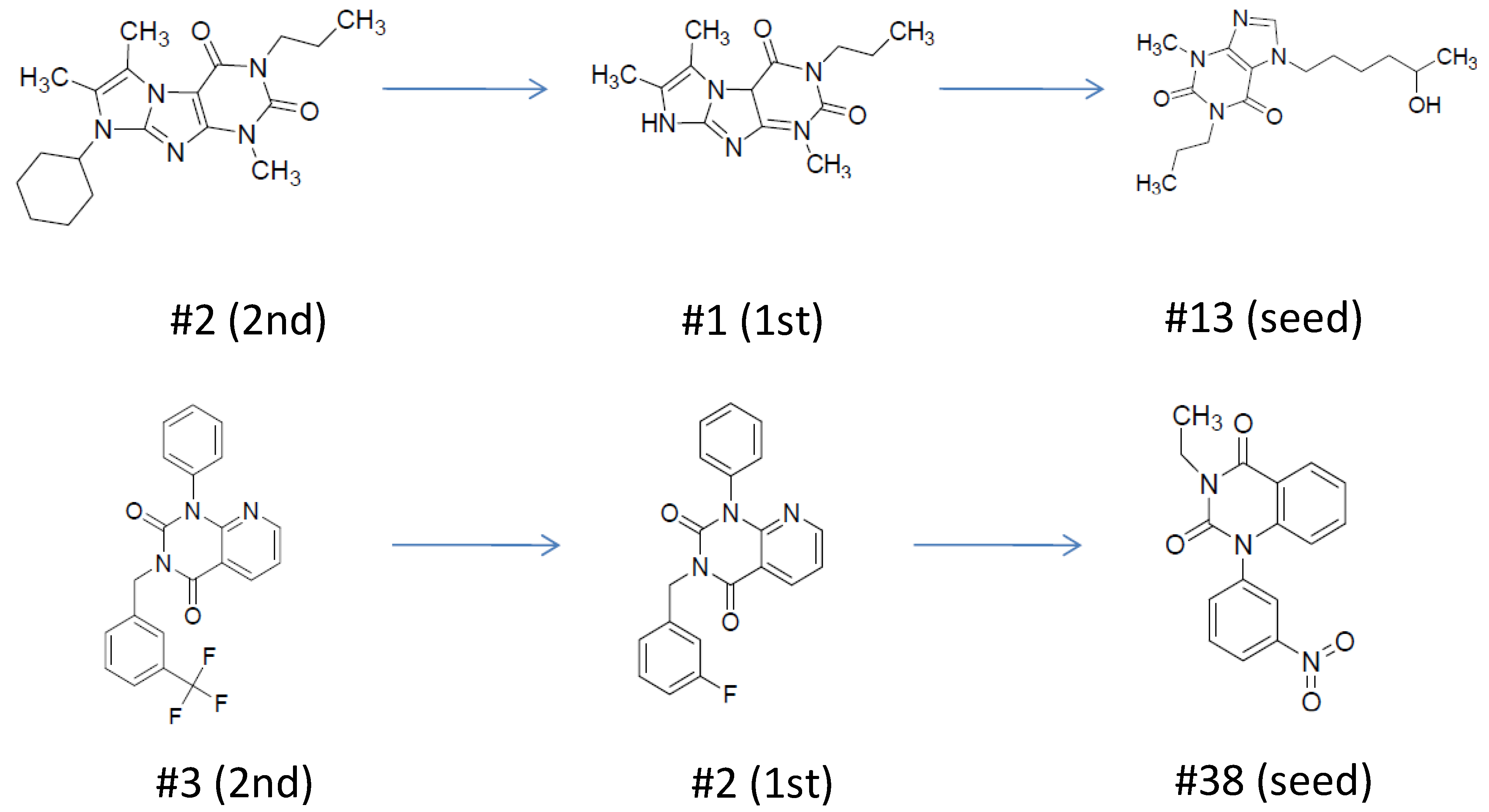
| ID | Hits | IC50 (uM) | T2D | T3D | Seeds | Seed ID |
|---|---|---|---|---|---|---|
| 1 | 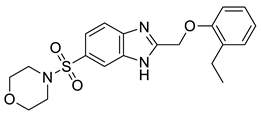 | 3.65 | 0.63 | 0.47 | 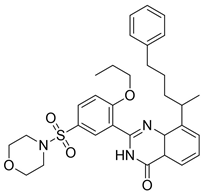 | 13 |
| 2 | 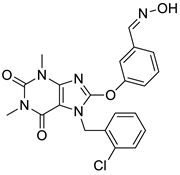 | 1.99 | 0.61 | 0.38 |  | 18 |
| 3 | 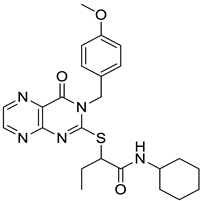 | 7.24 | 0.61 | 0.4 | 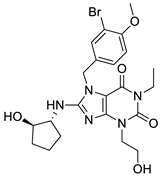 | 18 |
| 4 |  | 0.19 | 0.69 | 0.32 | 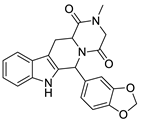 | 44 |
| 5 | 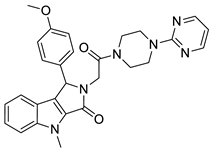 | 6.74 | 0.67 | 0.38 | 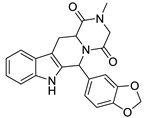 | 44 |
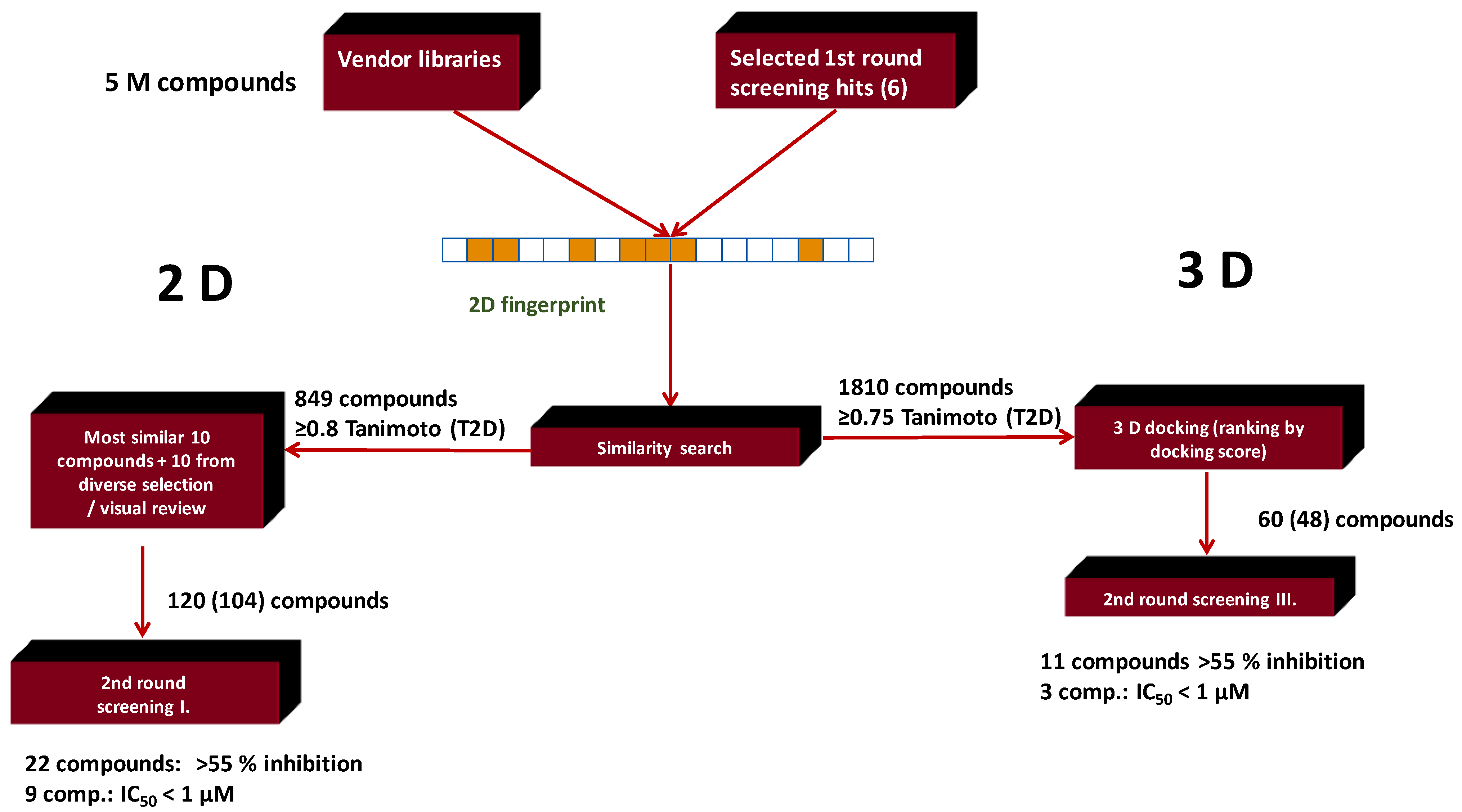
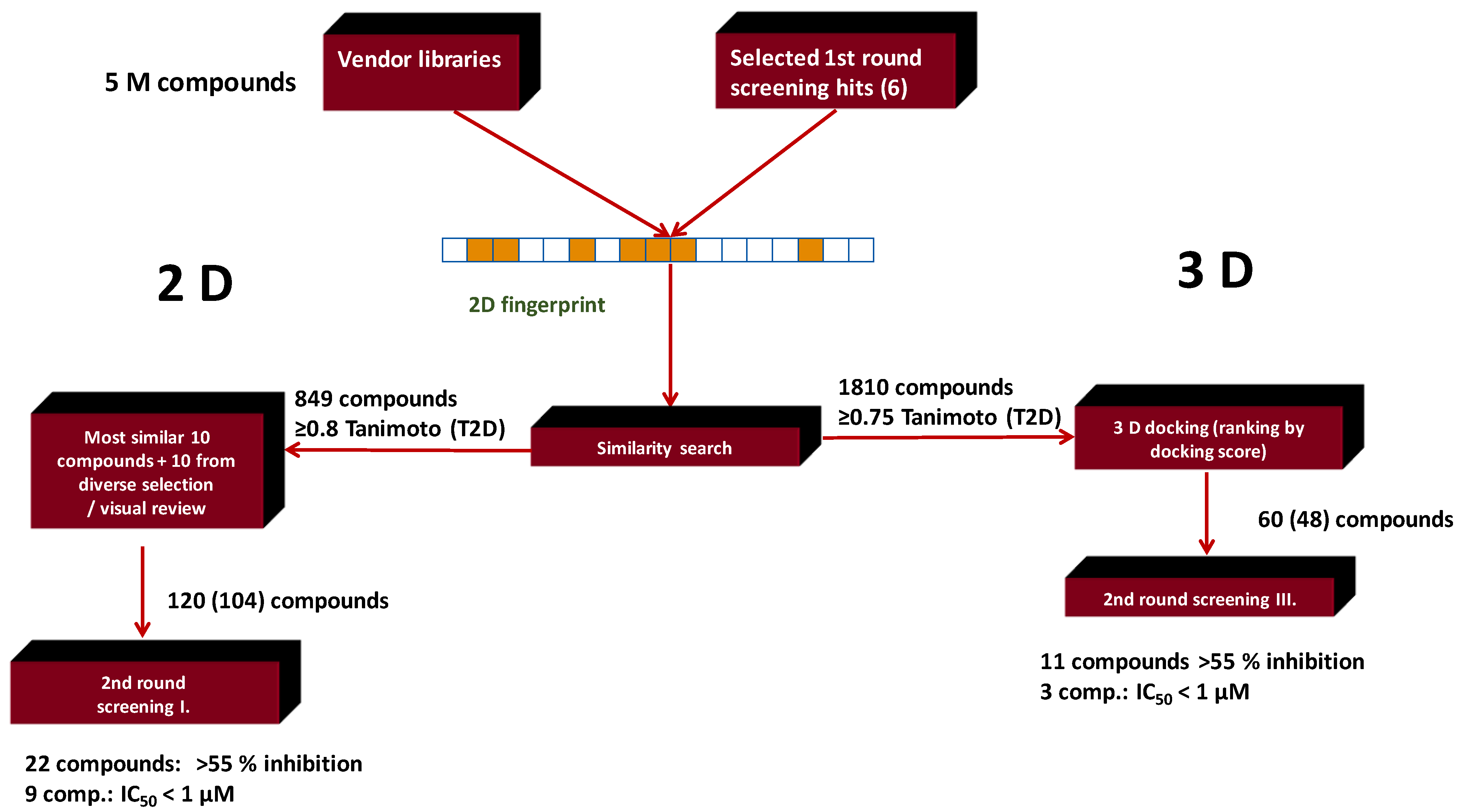
| Search Method | Measured | Hits: Inhibition > 55% (10 μM) | Hit Rate % | Hits: IC50 < 1 μM | Hit Rate % |
|---|---|---|---|---|---|
| 2D similarity search | 104 | 22 | 21.1 | 9 | 8.6 |
| 2D similarity search plus docking | 48 | 11 | 23 | 3 | 6.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szilágyi, K.; Flachner, B.; Hajdú, I.; Szaszkó, M.; Dobi, K.; Lőrincz, Z.; Cseh, S.; Dormán, G. Rapid Identification of Potential Drug Candidates from Multi-Million Compounds’ Repositories. Combination of 2D Similarity Search with 3D Ligand/Structure Based Methods and In Vitro Screening. Molecules 2021, 26, 5593. https://doi.org/10.3390/molecules26185593
Szilágyi K, Flachner B, Hajdú I, Szaszkó M, Dobi K, Lőrincz Z, Cseh S, Dormán G. Rapid Identification of Potential Drug Candidates from Multi-Million Compounds’ Repositories. Combination of 2D Similarity Search with 3D Ligand/Structure Based Methods and In Vitro Screening. Molecules. 2021; 26(18):5593. https://doi.org/10.3390/molecules26185593
Chicago/Turabian StyleSzilágyi, Katalin, Beáta Flachner, István Hajdú, Mária Szaszkó, Krisztina Dobi, Zsolt Lőrincz, Sándor Cseh, and György Dormán. 2021. "Rapid Identification of Potential Drug Candidates from Multi-Million Compounds’ Repositories. Combination of 2D Similarity Search with 3D Ligand/Structure Based Methods and In Vitro Screening" Molecules 26, no. 18: 5593. https://doi.org/10.3390/molecules26185593
APA StyleSzilágyi, K., Flachner, B., Hajdú, I., Szaszkó, M., Dobi, K., Lőrincz, Z., Cseh, S., & Dormán, G. (2021). Rapid Identification of Potential Drug Candidates from Multi-Million Compounds’ Repositories. Combination of 2D Similarity Search with 3D Ligand/Structure Based Methods and In Vitro Screening. Molecules, 26(18), 5593. https://doi.org/10.3390/molecules26185593