
The Antagonist pGlu-βGlu-Pro-NH2 Binds to an Allosteric Site of the Thyrotropin-Releasing Hormone Receptor




Abstract
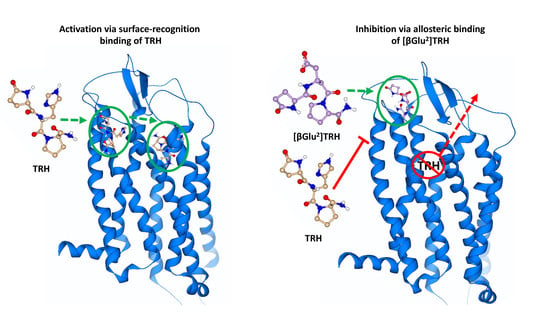
:
1. Introduction
2. Results and Discussion
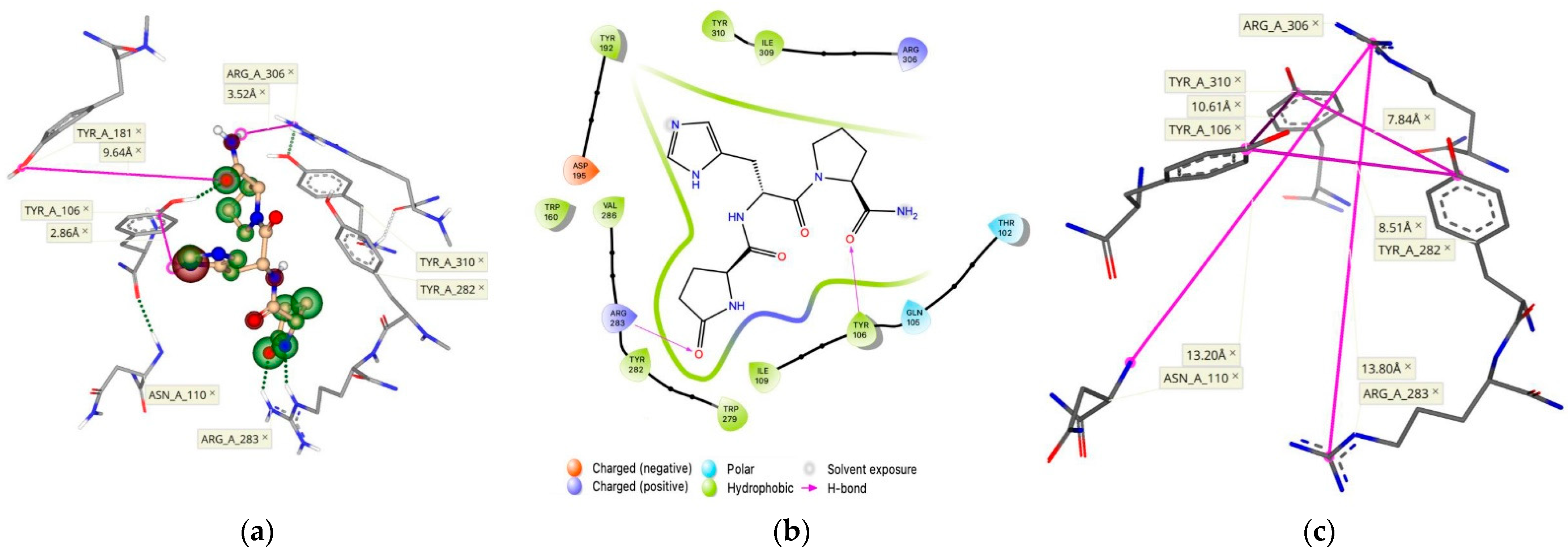
2.1. Refining the Active Binding Site of the hTRH-R
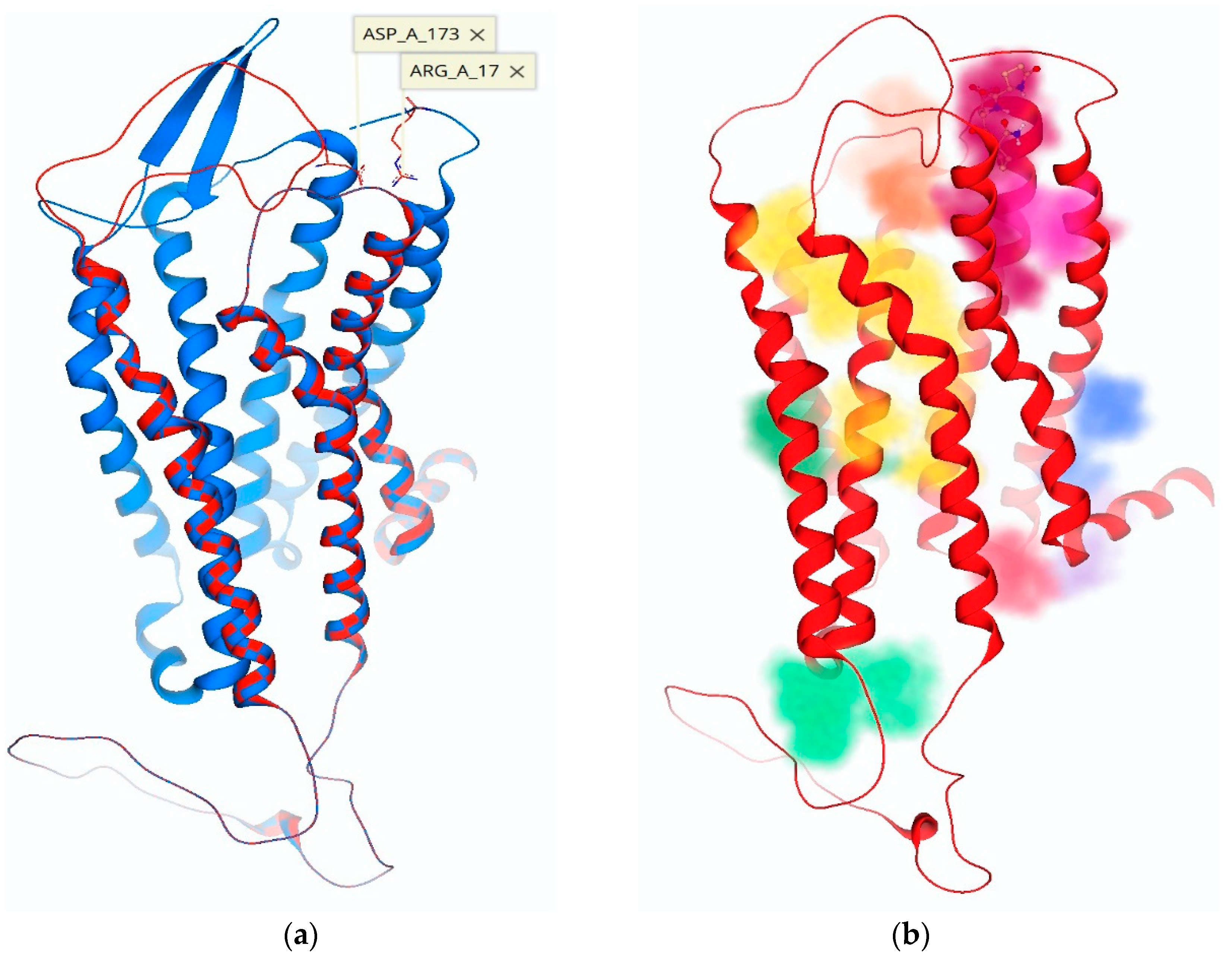
2.2. ECD Modeling of the hTRH-R
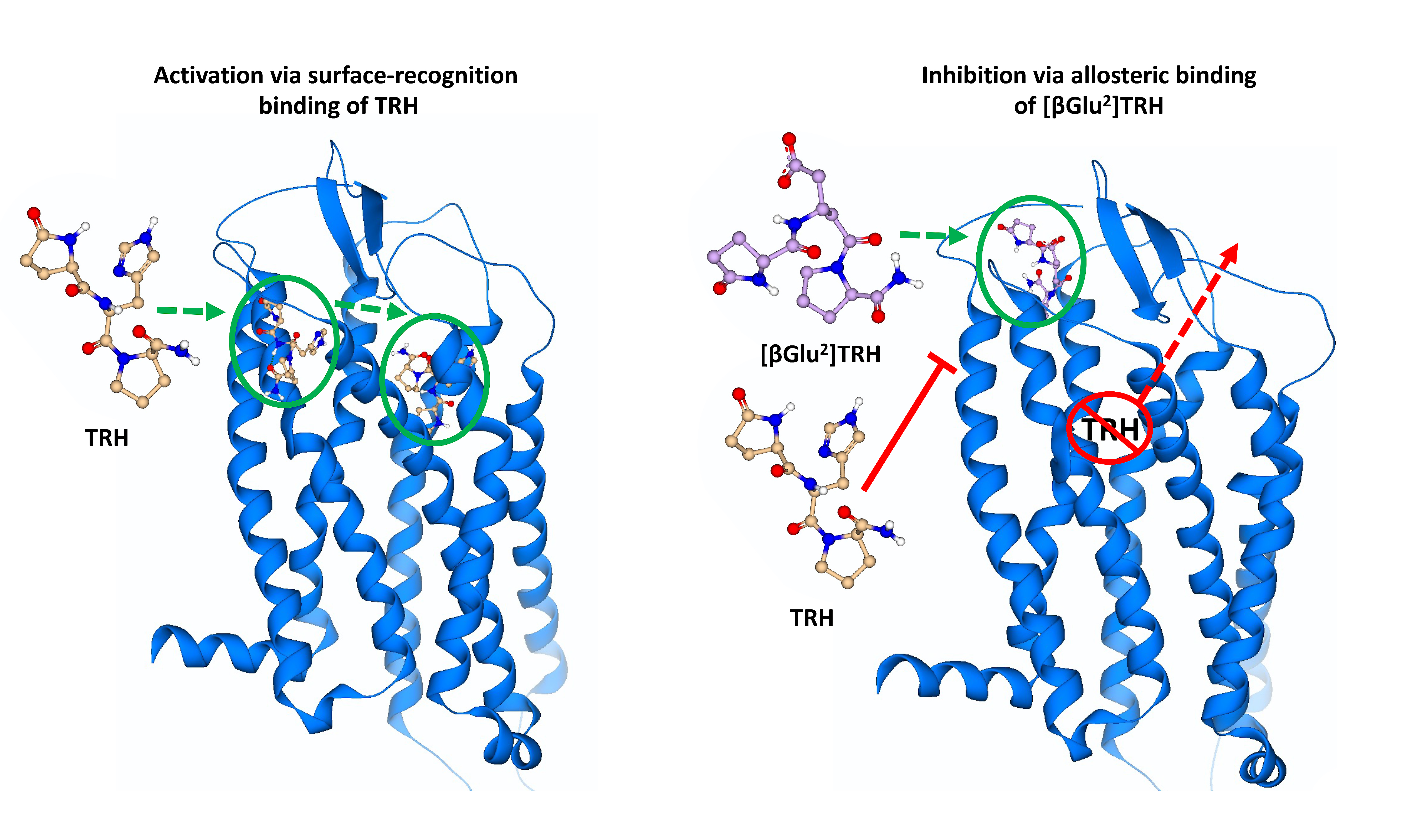
2.3. Surface-Recognition Binding Site of the hTRH-R
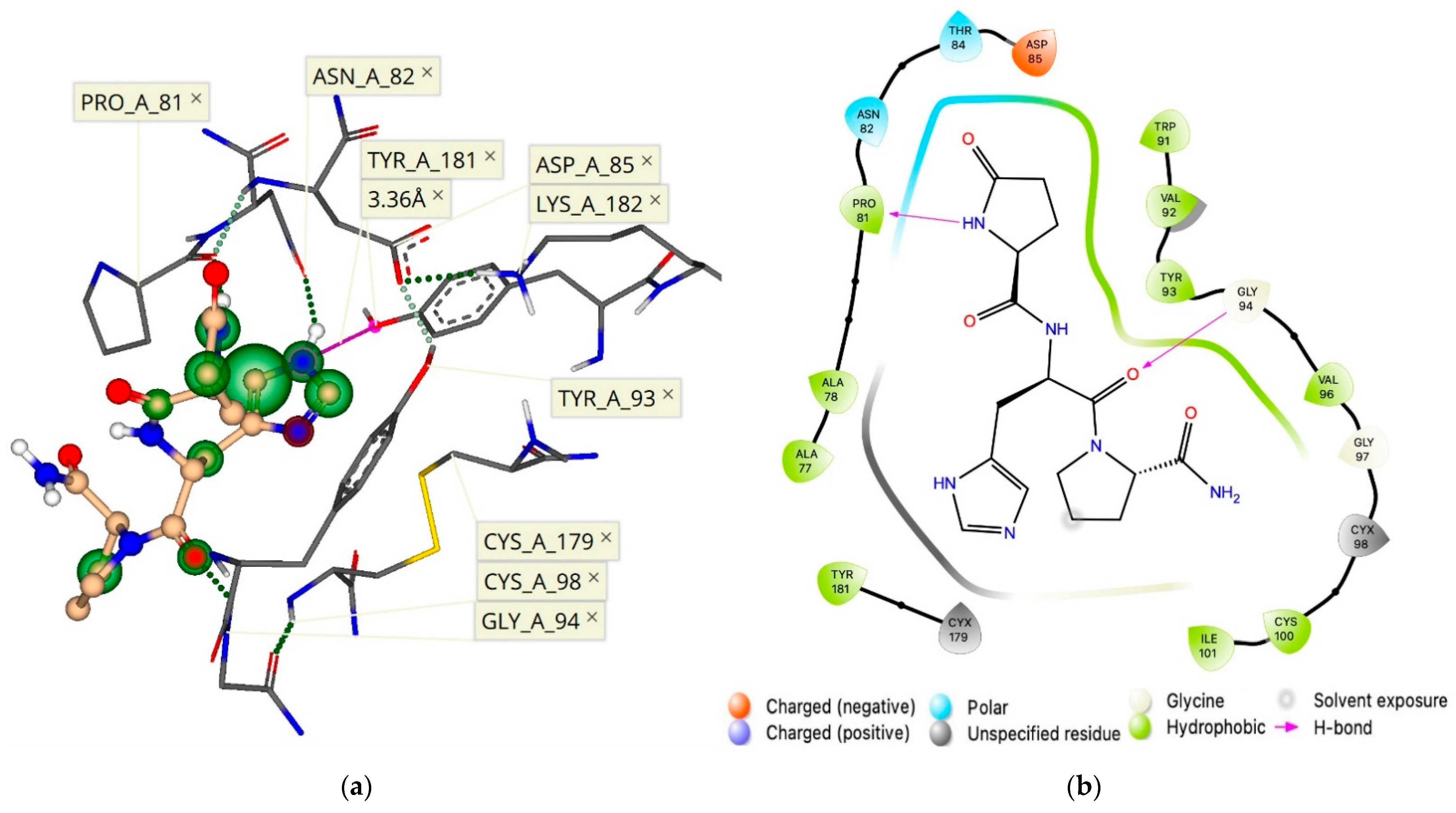
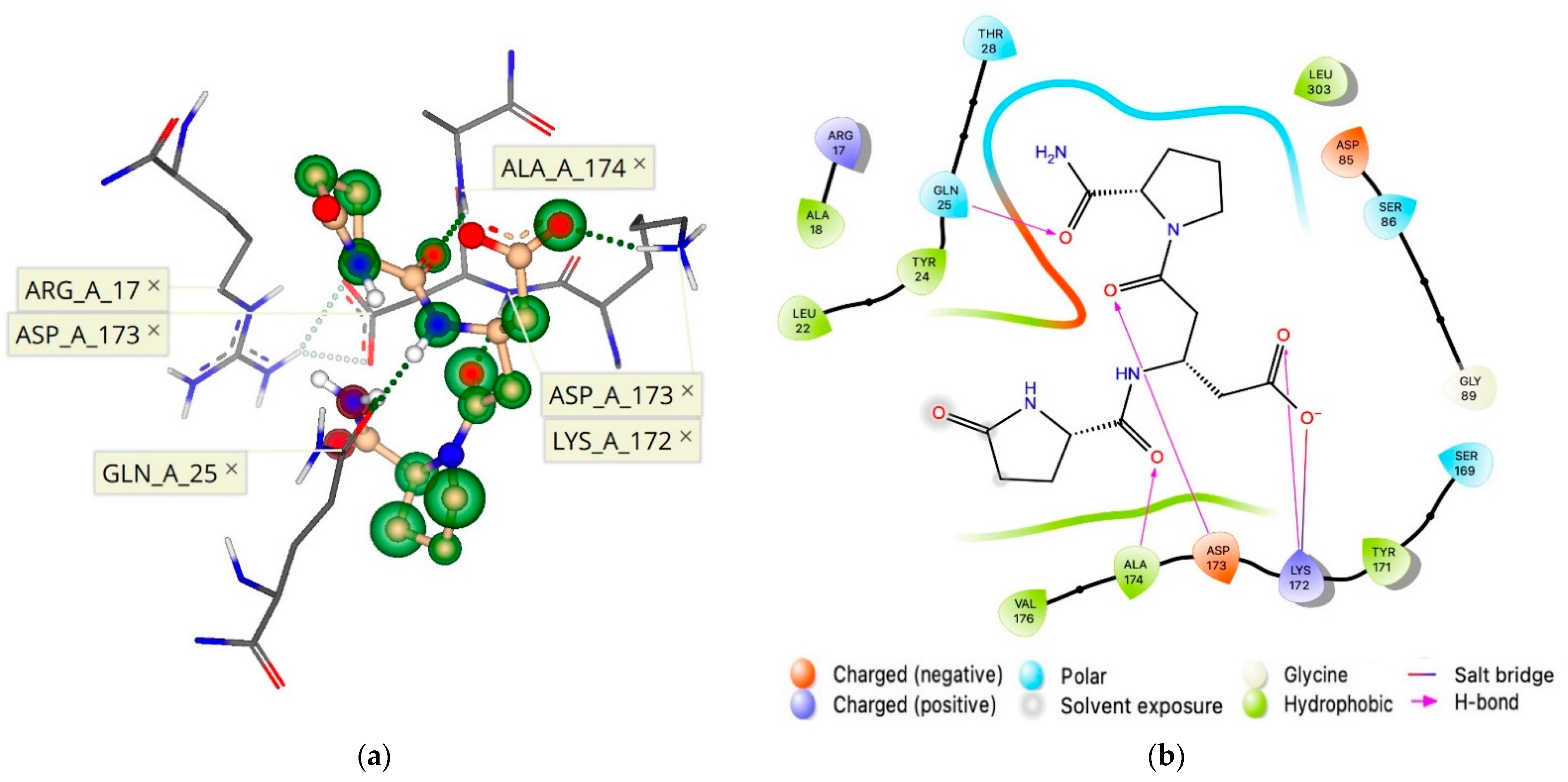
2.4. Allosteric Binding Site of the hTRH-R
3. Materials and Methods
3.1. hTRH-R Homology Model
3.1.1. Template Identification
3.1.2. Homology Model Building
3.2. ECD Modeling
3.3. Docking
Ligand and Protein Setup, Docking Protocol, and Identification of Binding Sites
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sun, Y.; Lu, X.; Gershengorn, M.C. G-protein-coupled receptor signaling in neuroendocrine systems. Thyrotropin-releasing hormone receptors—Similarities and differences. J. Mol. Endocrinol. 2003, 30, 87–97. [Google Scholar] [CrossRef] [Green Version]
- O’dowd, B.F.; Lee, D.K.; Huang, W.; Nguyen, T.; Cheng, R.; Liu, Y.; Wang, B.; Gershengorn, M.C.; George, S.R. TRH-R2 exhibits similar binding and acute signaling but distinct regulation and anatomic distribution compared with TRH-R1. Mol. Endocrinol. 2000, 14, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Heuer, H.; Schäfer, M.K.H.; O’Donnell, D.; Walker, P.; Bauer, K. Expression of thyrotropin-releasing hormone receptor 2 (TRH-R2) in the central nervous system of rats. J. Comput. Neurol. 2000, 428, 319–336. [Google Scholar] [CrossRef]
- Hinkle, P.M.; Pekary, A.E.; Senanayaki, S.; Sattin, A. Role of TRH receptors as possible mediators of analeptic actions of TRH-like peptides. Brain Res. 2002, 935, 59–64. [Google Scholar] [CrossRef]
- Gloriam, D.E.; Fredriksson, R.; Schiöth, H.B. The G protein-coupled receptor subset of the rat genome. BMC Genom. 2007, 8, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredriksson, R.; Schiöth, H.B. The repertoire of G-protein–coupled receptors in fully sequenced genomes. Mol. Pharm. 2005, 67, 1414–1425. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Shi, L.; Javitch, J.A. Structural mimicry in G protein-coupled receptors: Implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol. Pharm. 2001, 60, 1–19. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Nguyen, V.; Prokai, L. [β-Glu2]TRH is a functional antagonist of thyrotropin-releasing hormone (TRH) in the rodent brain. Int. J. Mol. Sci. 2021, 22, 6230. [Google Scholar] [CrossRef] [PubMed]
- Bilek, R.; Starka, L. The computer modelling of human TRH receptor, TRH and TRH-like peptides. Physiol. Res. 2005, 54, 141–150. [Google Scholar] [PubMed]
- Palczewski, K.; Kumasaka, T.; Hori, T.; Behnke, C.A.; Motoshima, H.; Fox, B.A.; Le Trong, I.; Teller, D.C.; Okada, T.; Stenkamp, R.E.; et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000, 289, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Woolley, M.J.; Conner, A.C. Understanding the common themes and diverse roles of the second extracellular loop (ECL2) of the GPCR super-family. Mol. Cell. Endocrinol. 2017, 449, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Vaidehi, N.; Floriano, W.B.; Trabanino, R.; Hall, S.E.; Freddolino, P.; Choi, E.J.; Zamanakos, G.; Goddard, W.A. Prediction of structure and function of G protein-coupled receptors. Proc. Nat. Acad. Sci. USA 2002, 99, 12622–12627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheatley, M.; Wootten, D.; Conner, M.T.; Simms, J.; Kendrick, R.; Logan, R.T.; Poyner, D.R.; Barwell, J. Lifting the lid on GPCRs: The role of extracellular loops. Br. J. Pharm. 2012, 165, 1688–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, S.; Gershengorn, M.C. Thyrotropin-releasing hormone and its receptors—A hypothesis for binding and receptor activation. Pharm. Ther. 2007, 113, 410–419. [Google Scholar] [CrossRef]
- Colson, A.O.; Perlman, J.H.; Smolyar, A.; Gershengorn, M.C.; Osman, R. Static and dynamic roles of extracellular loops in G-protein-coupled receptors: A mechanism for sequential binding of thyrotropin-releasing hormone to its receptor. Biophys. J. 1998, 74, 1087–1100. [Google Scholar] [CrossRef] [Green Version]
- Kristiansen, K. Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: Molecular modeling and mutagenesis approaches to receptor structure and function. Pharm. Ther. 2004, 103, 21–80. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- SeeSAR. Version 11.0.2. BioSolveIT GmbH, Sankt Augustin, Germany. 2021. Available online: www.biosolveit.de/SeeSAR (accessed on 13 June 2021).
- Meena, C.L.; Thakur, A.; Nandekar, P.P.; Sharma, S.S.; Sangamwar, A.T.; Jain, R. Synthesis and biology of ring-modified l-Histidine containing thyrotropin-releasing hormone (TRH) analogues. Eur. J. Med. Chem. 2016, 111, 72–83. [Google Scholar] [CrossRef]
- Perlman, J.H.; Laakkonen, L.J.; Guarnieri, F.; Osman, R.; Gershengorn, M.C. A refined model of the thyrotropin-releasing hormone (TRH) receptor binding pocket. Experimental analysis and energy minimization of the complex between TRH and TRH receptor. Biochemistry 1996, 35, 7643–7650. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.M.; Du, Q.S.; Meng, J.Z.; Pang, Z.W.; Huang, R.B. The multiple roles of histidine in protein interactions. Chem. Cen. J. 2013, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Srinivasan, Y.; Arlow, D.H.; Fung, J.J.; Palmer, D.; Green, H.F.; Pandey, A.; Dror, R.O.; Shaw, D.E.; Weis, W.I. High-resolution crystal structure of human protease-activated receptor 1. Nature 2012, 492, 377–392. [Google Scholar] [CrossRef] [Green Version]
- Perlman, J.H.; Colson, A.O.; Jain, R.; Czyzewski, B.; Cohen, L.A.; Osman, R.; Gershengorn, M.C. Role of the extracellular loops of the thyrotropin-releasing hormone receptor: Evidence for an initial interaction with thyrotropin-releasing hormone. Biochemistry 1997, 36, 15670–15676. [Google Scholar] [CrossRef]
- Gershengorn, M.C.; Osman, R. Minireview: Insights into G protein-coupled receptor function using molecular models. Endocrinology 2001, 142, 2–10. [Google Scholar] [CrossRef]
- Huang, W.; Osman, R.; Gershengorn, M.C. Agonist-induced conformational changes in thyrotropin-releasing hormone receptor type I: Disulfide cross-linking and molecular modeling approaches. Biochemistry 2005, 44, 2419–2431. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.G.; Sali, A. Modeling of loops in protein structures. Prot. Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M. Analyzing the topology of active sites: On the prediction of pockets and subpockets. J. Chem. Inf. Mod. 2010, 50, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarbrough, G.G. Thyrotropin releasing hormone and CNS cholinergic neurons. Life Sci. 1983, 33, 111–118. [Google Scholar] [CrossRef]
- SWISS-MODEL. Available online: http://swissmodel.expasy.org (accessed on 13 June 2021).
- Universal Protein Resource. Available online: http://www.uniprot.org (accessed on 10 June 2021).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studer, G.; Tauriello, G.; Bienert, S.; Biasini, M.; Johner, N.; Schwede, T. ProMod3—A versatile homology modelling toolbox. PLoS Comput. Biol. 2021, 17, 1008667. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Han, S.; Keller, M.; Kaiser, A.; Bender, B.J.; Bosse, M.; Burkert, K.; Kögler, L.M.; Wifling, D.; Bernhardt, G.; et al. Structural basis of ligand binding modes at the neuropeptide YY 1 receptor. Nature 2018, 556, 520–524. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2017, 45, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2021-2: Maestro; Schrödinger, LLC: New York, NY, USA, 2021.
- Schneider, N.; Lange, G.; Hindle, S.; Klein, R.; Rarey, M. A consistent description of HYdrogen bond and DEhydration energies in protein–ligand complexes: Methods behind the HYDE scoring function. J. Comput. Mol. Des. 2013, 27, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Binding Affinity (ΔG, kcal/mol) | Kd |
|---|---|---|
| TRH (1a) | – 1 | – 1 |
| [βGlu2]TRH (1b) | −8.8 ± 2.5 | nM |
| [Glu2]]TRH (1c) | −6.1 ± 2.5 | nM–µM |
| [β-homoGlu2]TRH (1d) | −5.6 ± 2.5 | μM–mM |
| [Asp2]TRH (1e) | −5.4 ± 2.5 | µM–mM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De La Cruz, D.L.; Prokai, L.; Prokai-Tatrai, K. The Antagonist pGlu-βGlu-Pro-NH2 Binds to an Allosteric Site of the Thyrotropin-Releasing Hormone Receptor. Molecules 2021, 26, 5397. https://doi.org/10.3390/molecules26175397
De La Cruz DL, Prokai L, Prokai-Tatrai K. The Antagonist pGlu-βGlu-Pro-NH2 Binds to an Allosteric Site of the Thyrotropin-Releasing Hormone Receptor. Molecules. 2021; 26(17):5397. https://doi.org/10.3390/molecules26175397
Chicago/Turabian StyleDe La Cruz, Daniel L., Laszlo Prokai, and Katalin Prokai-Tatrai. 2021. "The Antagonist pGlu-βGlu-Pro-NH2 Binds to an Allosteric Site of the Thyrotropin-Releasing Hormone Receptor" Molecules 26, no. 17: 5397. https://doi.org/10.3390/molecules26175397
APA StyleDe La Cruz, D. L., Prokai, L., & Prokai-Tatrai, K. (2021). The Antagonist pGlu-βGlu-Pro-NH2 Binds to an Allosteric Site of the Thyrotropin-Releasing Hormone Receptor. Molecules, 26(17), 5397. https://doi.org/10.3390/molecules26175397