
Quantitative Structure–Activity Relationships of Natural-Product-Inspired, Aminoalkyl-Substituted 1-Benzopyrans as Novel Antiplasmodial Agents



Abstract
:1. Introduction
2. Results and Discussion
2.1. 3D-QSAR Modeling
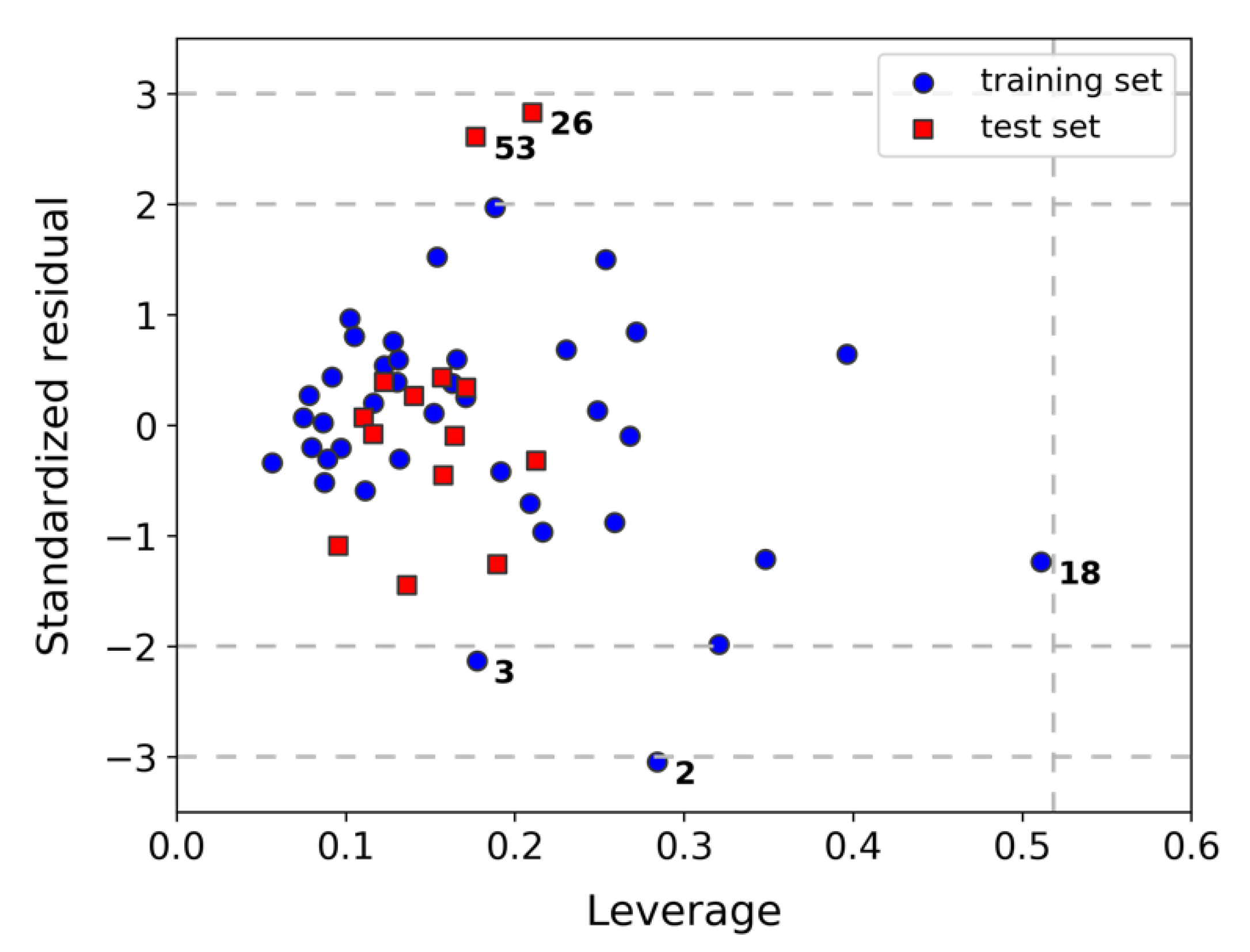
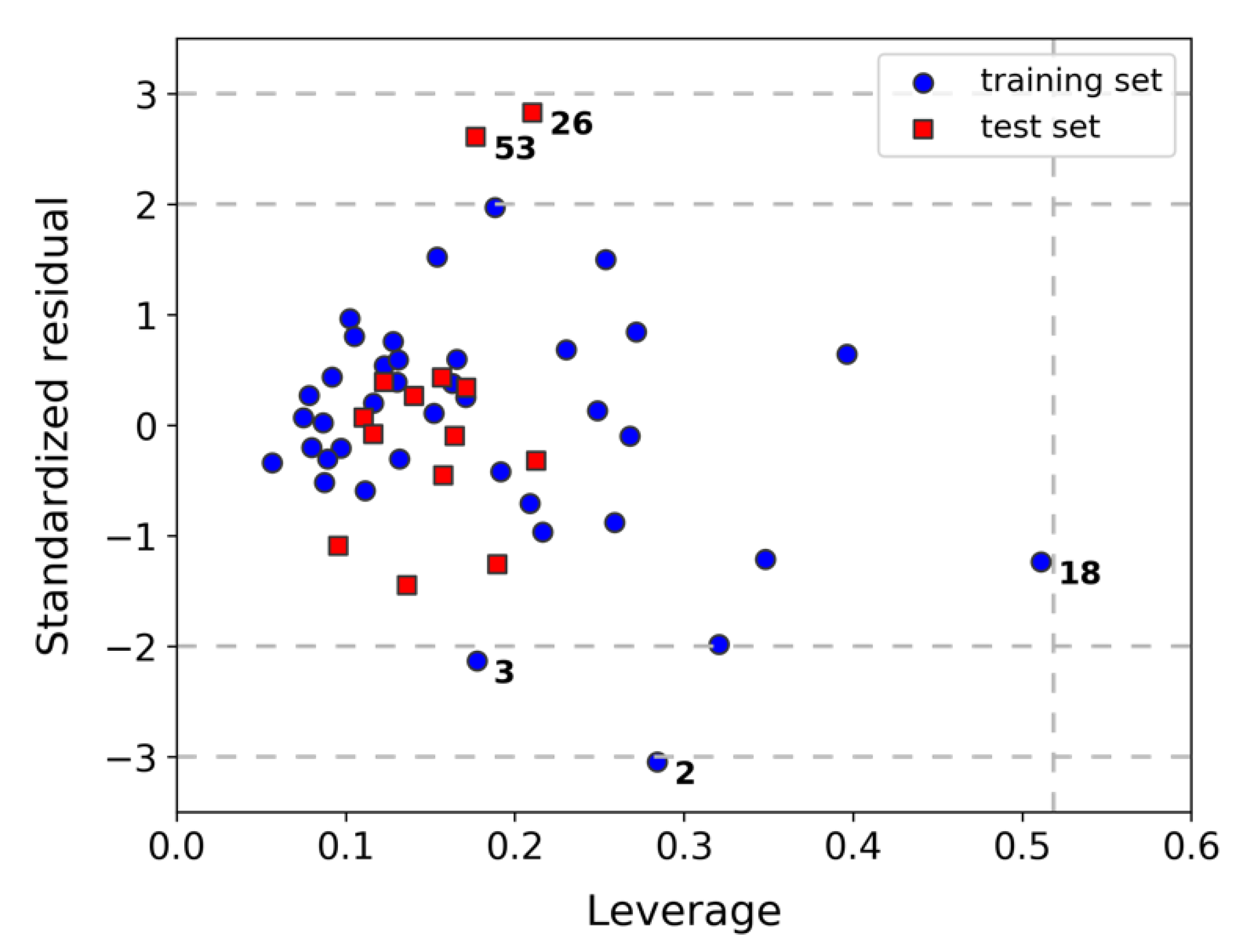
2.2. Assessment of Model Robustness and Applicability Domain (AD)
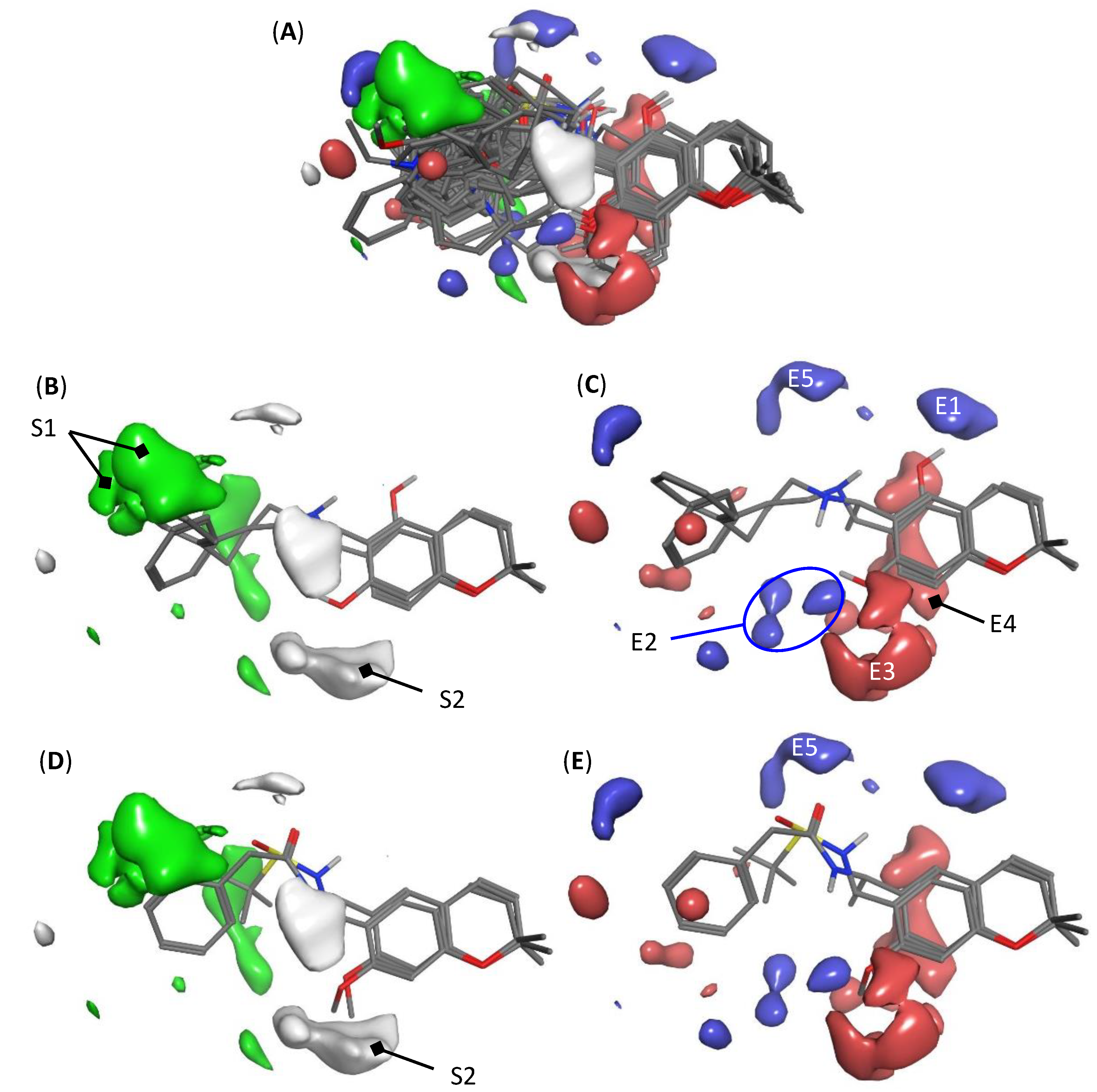
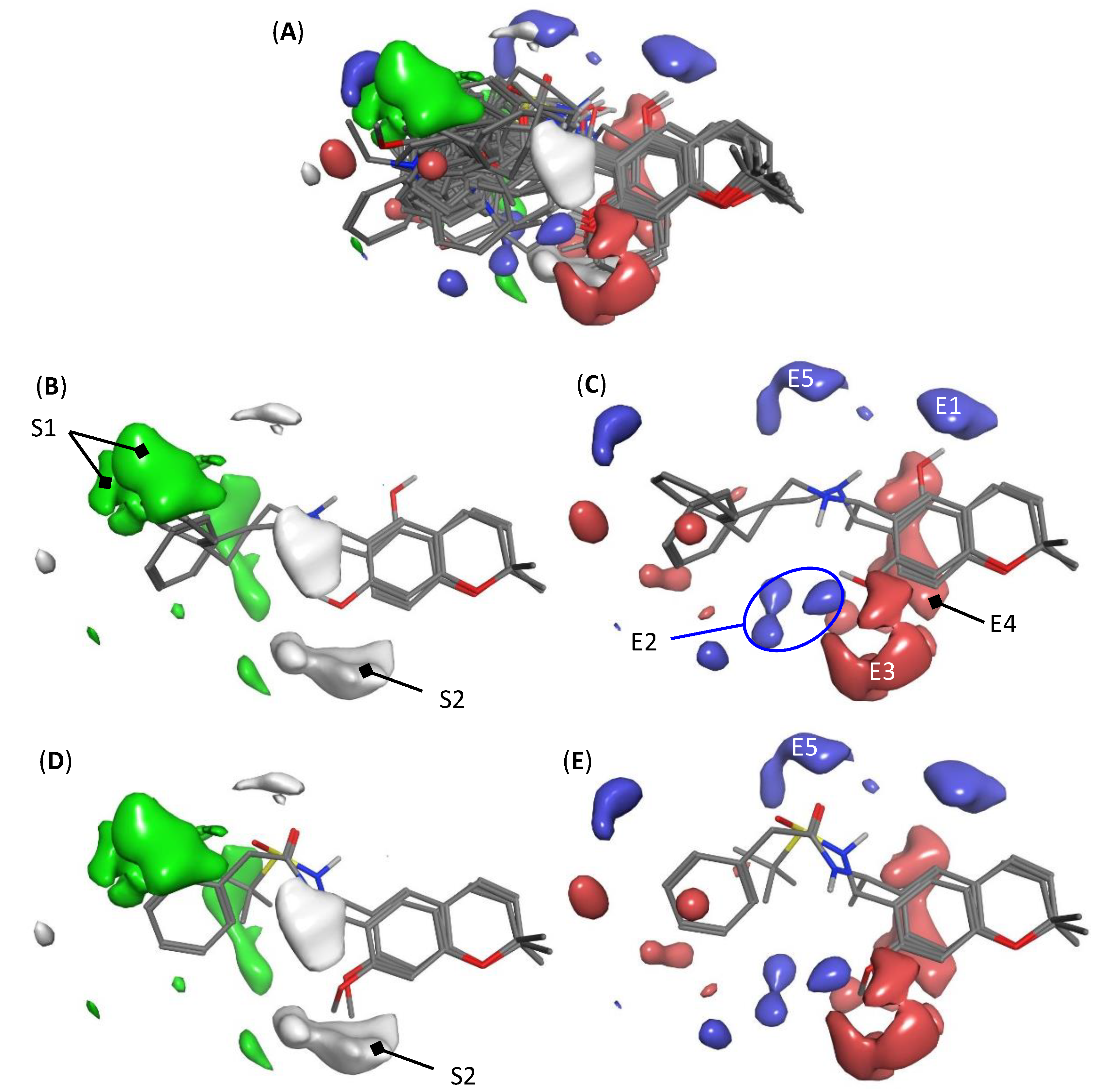
2.3. Interpretation of 3D-QSAR Model K
3. Materials and Methods
3.1. 3D Molecular Models
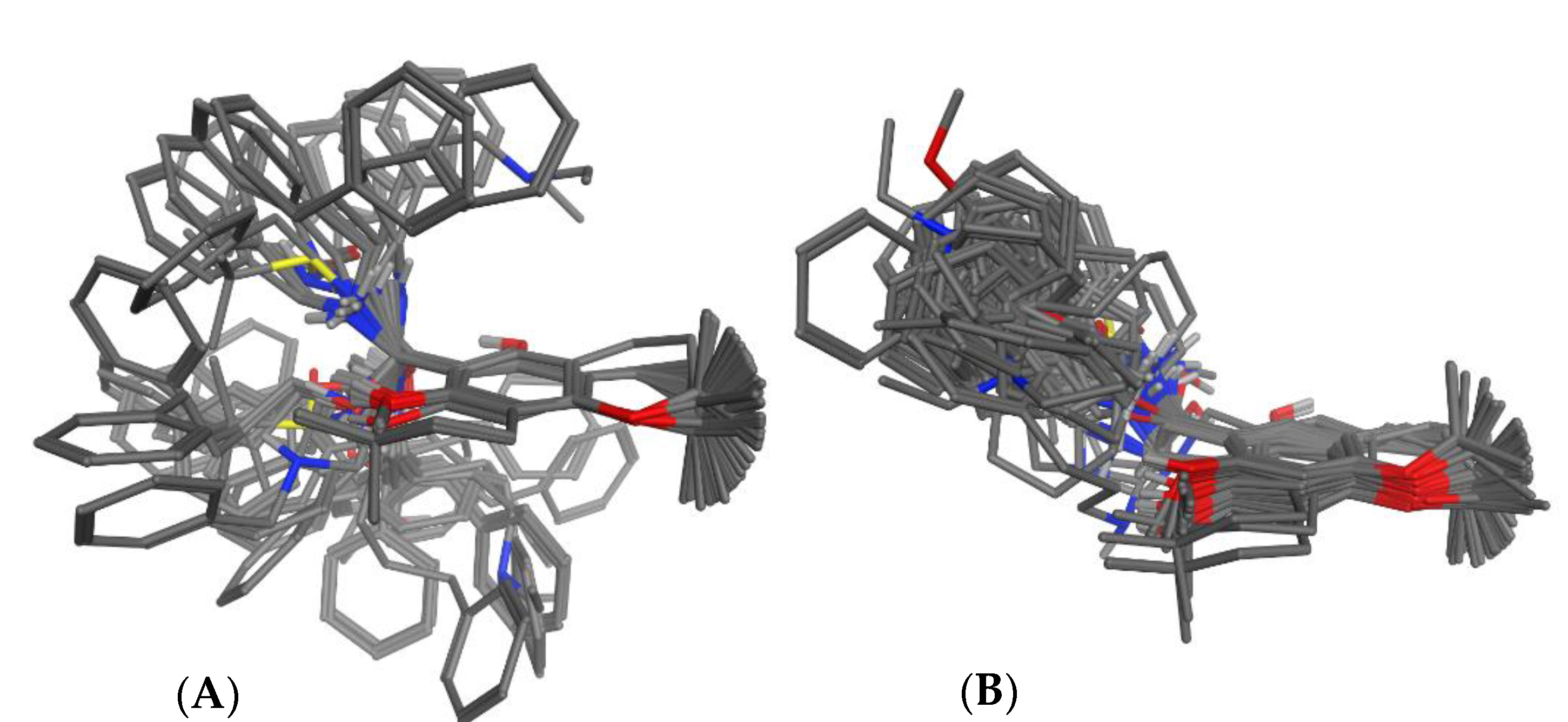
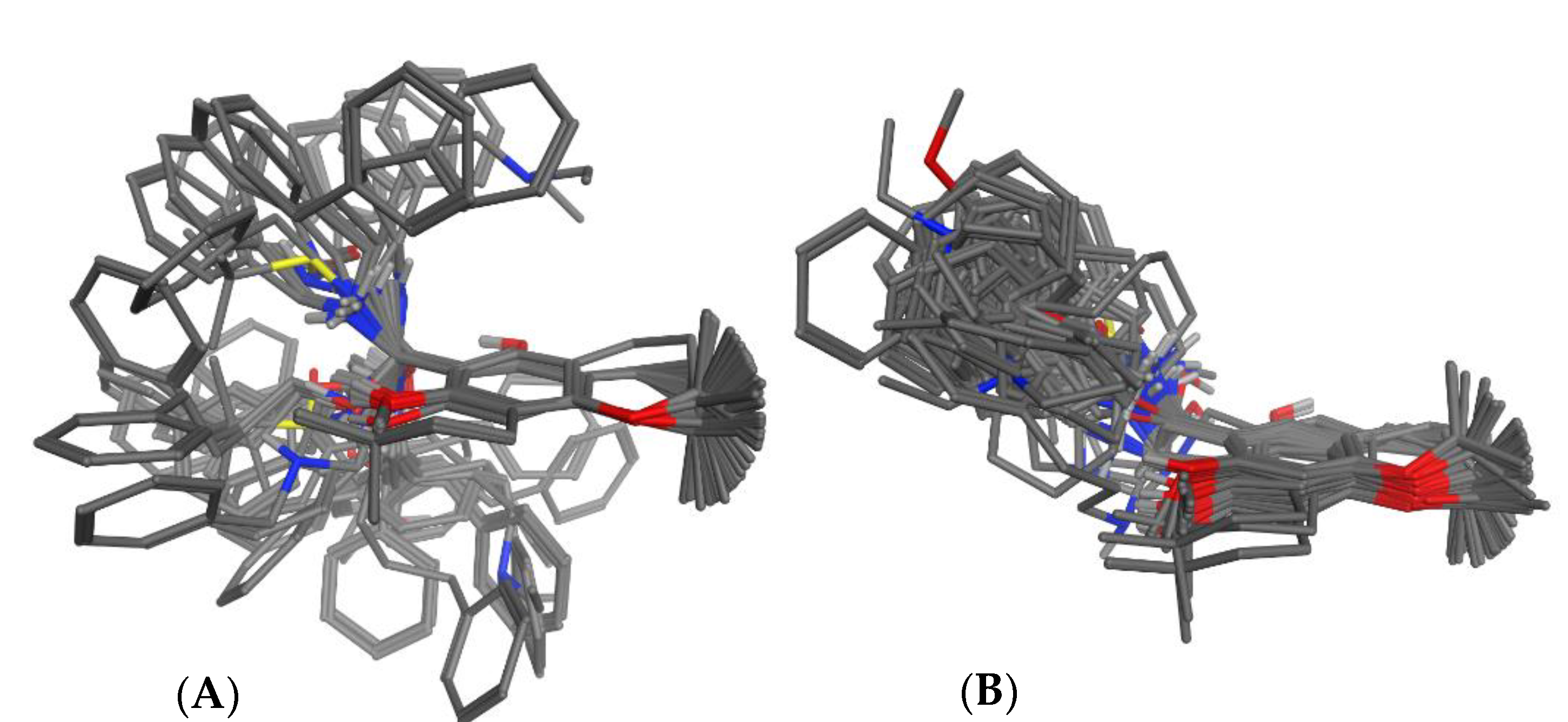
3.2. Superposition of Molecular Models
3.2.1. Manual Superposition
3.2.2. Automatic Superposition Based on Pharmacophoric Properties
3.3. 3D-QSAR Modeling
3.4. Model Validation
3.5. Assessment of the Applicability Domain (AD) of Model K
3.6. Model Visualization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization, 2019. World Malaria Report 2020: 20 Years of Global Progress and Challenges. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2020/ (accessed on 28 August 2021).
- Harel, D.; Schepmann, D.; Prinz, H.; Brun, R.; Schmidt, T.J.; Wünsch, B. Natural product derived antiprotozoal agents: Synthesis, biological evaluation and structure-activity relationships of novel chromene and chromane derivatives. J. Med. Chem. 2013, 56, 7442–7448. [Google Scholar] [CrossRef] [PubMed]
- Harel, D.; Schepmann, D.; Prinz, H.; Brun, R.; Schmidt, T.J.; Wünsch, B. Enantioselective synthesis of encecaline-derived potent antimalarial agents. Org. Biomol. Chem. 2013, 11, 7342–7349. [Google Scholar] [CrossRef] [PubMed]
- Uth, J.-F.; Börgel, F.; Lehmkuhl, K.; Schepmann, D.; Kaiser, M.; Nonato, M.C.; Krauth-Siegel, L.; Schmidt, T.J.; Wünsch, B. Synthesis and biological evaluation of natural product-inspired, aminoalkyl substituted 1-benzopyrans as novel antiplasmodial agents. J. Med. Chem. 2021, 64, 6397–6409. [Google Scholar] [CrossRef] [PubMed]
- Nour, A.M.M.; Khalid, S.A.; Brun, R.; Kaiser, M.; Schmidt, T.J. The antiprotozoal activity of methylated flavonoids from Ageratum conyzoides L. J. Ethnopharmacol. 2010, 129, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Harel, D.; Khalid, S.A.; Kaiser, M.; Brun, R.; Wünsch, B.; Schmidt, T.J. Encecalol angelate, an unstable chromene from Ageratum conyzoides L.: Total Synthesis and Investigation of its Antiprotozoal Activity. J. Ethnopharmacol. 2011, 137, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Tosco, P.; Balle, T. Open3DQSAR: A new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J. Mol. Modeling 2011, 17, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.D.; Fox, P.C. Statistical variation in progressive scrambling. J. Comp. Aided Mol. Des. 2004, 18, 563–576. [Google Scholar] [CrossRef] [PubMed]
- OECD Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models; OECD Series on Testing and Assessment; OECD Publishing: Paris, France, 2007; ISBN 9789264085442.
- Netzeva, T.I.; Worth, A.P.; Aldenberg, T.; Benigni, R.; Cronin, M.T.D.; Gramatica, P.; Jaworska, J.S.; Kahn, S.; Klopman, G.; Marchant, C.A.; et al. Current Status of Methods for Defining the Applicability Domain of (Quantitative) Structure-Activity Relationships—The Report and Recommendations of ECVAM Workshop 52. Altern. Lab. Anim. 2005, 33, 1–19. [Google Scholar]
- Gadaleta, D.; Mangiatordi, G.F.; Catto, M.; Carotti, A.; Nicolotti, O. Applicability Domain for QSAR Models. Int. J. Quant. Struct. Relatsh. 2016, 1, 45–63. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Gramatica, P. On the development and validation of QSAR models. In Computational Toxicology. Methods in Molecular Biology; Reisfeld, B., Mayeno, A., Eds.; Humana Press: Totowa, NJ, USA, 2013; Volume 930, pp. 499–526. ISBN 9781627030588. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
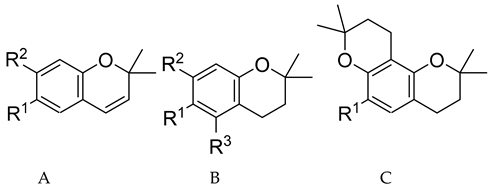
| Compound Number | Number in [Reference] | Type | R1 | R2 | R3 | pIC50 |
|---|---|---|---|---|---|---|
| 1 | 15b [2] | A | Ph(CH2)3NHCH(CH3)‒ | OH | - | 8.00 |
| 2 | 8b [4] | B | Ph(CH2)2NHCH2‒ | - | OH | 8.00 |
| 3 | 19c [4] | B | Ph(CH2)3NHCH2CH(OH)‒ | OH | H | 7.77 |
| 4 | R-2 [3] | A | PhCH2NHC*H(CH3)‒ [*(R)] | OH | - | 7.59 |
| 5 | 9c [4] | B | Ph(CH2)3NHCH2‒ | OH | H | 7.42 |
| 6 | 8a [4] | B | PhCH2NHCH2‒ | - | OH | 7.28 |
| 7 | 9d [4] | B | Ph(CH2)4NHCH2‒ | OH | H | 7.15 |
| 8 | 10b [4] | A | Ph(CH2)2NHCH2‒ | OH | H | 7.12 |
| 9 | 10d [4] | A | Ph(CH2)4NHCH2‒ | OH | H | 7.10 |
| 10 | 10c [4] | A | Ph(CH2)3NHCH2‒ | OH | H | 7.09 |
| 11 | 9b [4] | B | Ph(CH2)2NHCH2‒ | OH | H | 7.09 |
| 12 | 9a [4] | B | PhCH2NHCH2‒ | OH | H | 7.00 |
| 13 | 19a [4] | B | PhCH2NHCH2CH(OH)‒ | OH | H | 7.00 |
| 14 | 8c [4] | B | Ph(CH2)3NHCH2‒ | H | OH | 6.82 |
| 15 | 10a [4] | A | PhCH2NHCH2‒ | OH | H | 6.80 |
| 16 | 8d [4] | B | Ph(CH2)4NHCH2‒ | H | OH | 6.75 |
| 17 | 12d [4] | C | PhCH2NHCH2‒ | - | - | 6.68 |
| 18 | 12b [4] | C | Ph(CH2)2NHCH2‒ | - | - | 6.62 |
| 19 | R-1 [3] | A | PhCH2NHC*H(CH3)‒ [*(R)] | OCH3 | - | 6.55 |
| 20 | 12c [4] | C | Ph(CH2)3NHCH2‒ | - | - | 6.43 |
| 21 | 12g [2] | A | PhC*H(CH3)NHCH(CH3)‒ [*(S)] | OCH3 | - | 6.43 |
| 22 | 12a [4] | C | PhCH2NHCH2‒ | - | - | 6.33 |
| 23 | 12f [2] | A | PhC*H(CH3)NHCH(CH3)‒ [*(R)] | OCH3 | - | 6.14 |
| 24 | S-1 [3] | A | PhCH2NHC*H(CH3)‒ [*(S)] | OCH3 | - | 6.13 |
| 25 | 12c [2] | A | PhCH2NHCH(CH3)‒ | OCH3 | - | 6.07 |
| 26 | S-2 [3] | A | PhCH2NHC*H(CH3)‒ [*(S)] | OH | - | 6.05 |
| 27 | 12e [2] | A | p-OCH3-PhCH2NHCH(CH3)‒ | OCH3 | - | 6.03 |
| 28 | 25 [4] | B | PhCH2NH(CH2)3‒ | OH | H | 6.01 |
| 29 | 12o [2] | A | Ph(CH2)3N(CH)3CH(CH3)‒ | OCH3 | - | 5.91 |
| 30 | R-10 [3] | A | PhCH2N(CH3)C*H(CH3)‒ [*(R)] | OCH3 | - | 5.91 |
| 31 | S-10 [3] | A | PhCH2N(CH3)C*H(CH3)‒ [*(S)] | OCH3 | - | 5.89 |
| 32 | 14d [2] | B | Ph(CH2)4NHCH(CH3)‒ | OCH3 | H | 5.87 |
| 33 | 12d [2] | A | Ph(CH2)4NHCH(CH3)‒ | OCH3 | - | 5.85 |
| 34 | 14a [2] | B | PhCH2NHCH(CH3)‒ | OCH3 | H | 5.82 |
| 35 | 14c [2] | B | Ph(CH2)3NHCH(CH3)‒ | OCH3 | H | 5.67 |
| 36 | 12k [2] a | A | (C2H5)2N(CH2)3C*(CH3)NHC**H(CH3)‒ [*(S), **(R)] | OCH3 | - | 5.62 |
| 37 | 12k [2] a | A | (C2H5)2N(CH2)3C*(CH3)NHC**H(CH3)‒ [*(R), **(R)] | OCH3 | - | 5.62 |
| 38 | 12b [2] | A | Ph(CH2)2NHCH(CH3)‒ | OCH3 | - | 5.62 |
| 39 | (S,R)-8 [3] | A | tertC4H9S+*(O−)NHC**H(CH3)‒ [*(S), **(R)] | OCH3 | - | 5.61 |
| 40 | 12l [2] | A | cyclo-C6H11-CH2NHCH(CH3)‒ | OCH3 | - | 5.61 |
| 41 | 14b [2] | B | Ph(CH2)2NHCH(CH3)‒ | OCH3 | H | 5.60 |
| 42 | 14e [2] | B | CH3(CH2)3NHCH(CH3)‒ | OCH3 | H | 5.49 |
| 43 | 12j [2] | A | CH3(CH2)3NHCH(CH3)‒ | OCH3 | - | 5.38 |
| 44 | 12h [2] | A | Ph(E)CH=CHCH2NHCH(CH3)‒ | OCH3 | - | 5.36 |
| 45 | 12i [2] | A | CH3NHCH(CH3)‒ | OCH3 | - | 5.09 |
| 46 | 9a [2] | A | PhCH2OCH(CH3)‒ | OCH3 | - | 5.02 |
| 47 | 12m [2] | A | N-morpholino-CH(CH3)‒ | OCH3 | - | 5.00 |
| 48 | 11a [2] | A | PhCONHCH(CH3)‒ | OCH3 | - | 4.94 |
| 49 | (R,S)-8 [3] | A | tertC4H9S+*(O−)NHC**H(CH3)‒ [*(R),**(S)] | OCH3 | - | 4.80 |
| 50 | 12n [2] | A | N-pyrrolidino-CH(CH3)‒ | OCH3 | - | 4.79 |
| 51 | 1 [6] | A | ageloyloxy-CH(CH3)‒ | OCH3 | - | 4.72 |
| 52 | 11b [2] | A | PhCH2CONHCH(CH3)‒ | OCH3 | - | 4.51 |
| 53 | 9b [2] | A | Ph(CH2)2OCH(CH3)‒ | OCH3 | - | 4.22 |
| Model | PC | R2 (SDEC) | F-Test | Q2 (SDEPint) | P2 (SDEPext) | Progressive Scrambling (Critical Value: 0.85; Fit Order: 2) [8] | |
|---|---|---|---|---|---|---|---|
| Fitted Qs2* | Q02* | ||||||
| A | 5 | 0.9935 (0.0733) | 1006.9488 | 0.7847 (0.4212) | 0.4507 (0.6872) | ||
| B | 5 | 0.9820 (0.1254) | 360.5910 | 0.8099 (0.4080) | 0.3893 (0.6749) | ||
| C | 5 | 0.9779 (0.1303) | 291.6092 | 0.7531 (0.4354) | 0.6760 (0.6094) | 0.5933 | 0.6980 |
| D | 5 | 0.9750 (0.1406) | 257.3185 | 0.7241 (0.4670) | 0.7385 (0.5461) | ||
| E | 5 | 0.9801 (0.1294) | 325.3210 | 0.7680 (0.4420) | 0.6713 (0.5280) | 0.6349 | 0.7469 |
| F | 5 | 0.9926 (0.0817) | 891.0630 | 0.8849 (0.3233) | 0.2737 (0.6719) | ||
| G | 5 | 0.9796 (0.1306) | 316.6972 | 0.8126 (0.3957) | 0.6935 (0.5140) | 0.6284 | 0.7393 |
| H | 4 | 0.9737 (0.1485) | 314.1282 | 0.8029 (0.4062) | 0.5954 (0.5892) | ||
| I | 4 | 0.9610 (0.1806) | 209.2875 | 0.6316 (0.5547) | 0.7668 (0.4477) | ||
| J | 5 | 0.9805 (0.1275) | 332.5837 | 0.8040 (0.4047) | 0.6968 (0.5097) | 0.6307 | 0.7420 |
| K | 5 | 0.9806 (0.1273) | 333.7178 | 0.8139 (0.3943) | 0.7076 (0.5018) | 0.6501 | 0.7648 |
| L | 5 | 0.9797 (0.1302) | 318.8622 | 0.7008 (0.5001) | 0.6731 (0.5304) | ||
| M | 5 | 0.9806 (0.1294) | 334.3370 | 0.7922 (0.4240) | 0.6600 (0.5099) | 0.6157 | 0.7244 |
| N | 5 | 0.9802 (0.1310) | 326.0172 | 0.8126 (0.4026) | 0.5481 (0.5882) | ||
| O | 3 | 0.9186 (0.2605) | 131.6544 | 0.7038 (0.4969) | 0.5791 (0.6035) | ||
| P | 5 | 0.9791 (0.1346) | 309.5729 | 0.8096 (0.4064) | 0.4710 (0.6365) | ||
| Q | 5 | 0.9852 (0.1111) | 440.3674 | 0.7923 (0.4165) | 0.5436 (0.6282) | ||
| R | 5 | 0.9909 (0.0836) | 721.4217 | 0.8681 (0.3190) | 0.5098 (0.7076) | ||
| S | 5 | 0.9928 (0.0755) | 903.8373 | 0.8857 (0.2999) | 0.5039 (0.6936) | ||
| T | 5 | 0.9810 (0.1264) | 341.2076 | 0.7502 (0.4587) | 0.6616 (0.5324) | 0.5995 | 0.7053 |
| mean | 4.8 | 0.9782 (0.1289) | 423.3272 | 0.7853 (0.4188) | 0.5905 (0.5855) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wunsch, F.M.; Wünsch, B.; Bernal, F.A.; Schmidt, T.J. Quantitative Structure–Activity Relationships of Natural-Product-Inspired, Aminoalkyl-Substituted 1-Benzopyrans as Novel Antiplasmodial Agents. Molecules 2021, 26, 5249. https://doi.org/10.3390/molecules26175249
Wunsch FM, Wünsch B, Bernal FA, Schmidt TJ. Quantitative Structure–Activity Relationships of Natural-Product-Inspired, Aminoalkyl-Substituted 1-Benzopyrans as Novel Antiplasmodial Agents. Molecules. 2021; 26(17):5249. https://doi.org/10.3390/molecules26175249
Chicago/Turabian StyleWunsch, Friederike M., Bernhard Wünsch, Freddy A. Bernal, and Thomas J. Schmidt. 2021. "Quantitative Structure–Activity Relationships of Natural-Product-Inspired, Aminoalkyl-Substituted 1-Benzopyrans as Novel Antiplasmodial Agents" Molecules 26, no. 17: 5249. https://doi.org/10.3390/molecules26175249
APA StyleWunsch, F. M., Wünsch, B., Bernal, F. A., & Schmidt, T. J. (2021). Quantitative Structure–Activity Relationships of Natural-Product-Inspired, Aminoalkyl-Substituted 1-Benzopyrans as Novel Antiplasmodial Agents. Molecules, 26(17), 5249. https://doi.org/10.3390/molecules26175249