
Variational Solutions for Resonances by a Finite-Difference Grid Method

 ,
,
Abstract
:1. Introduction
2. Methodology
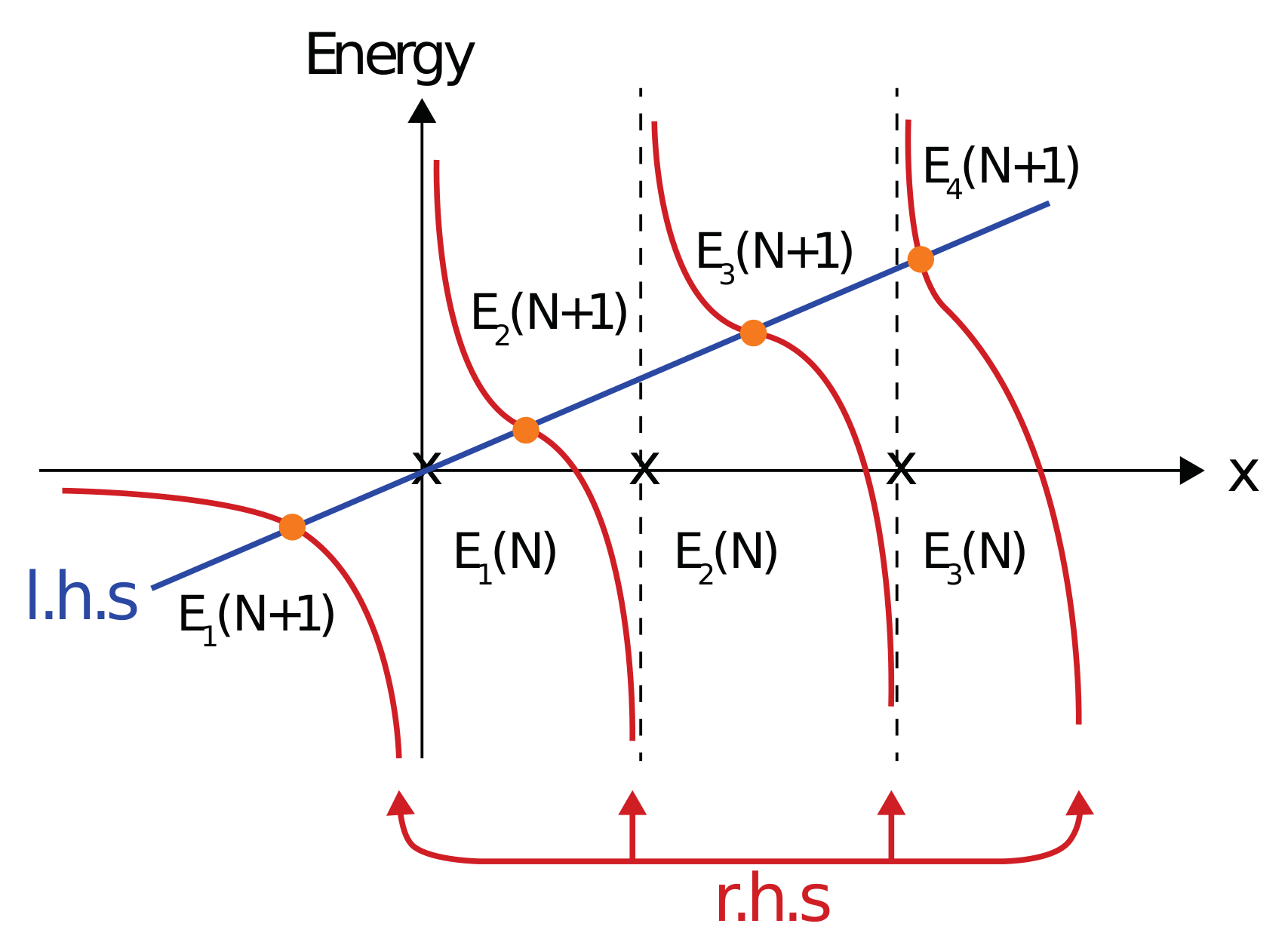
3. The Variational Principle for Finite Difference Method
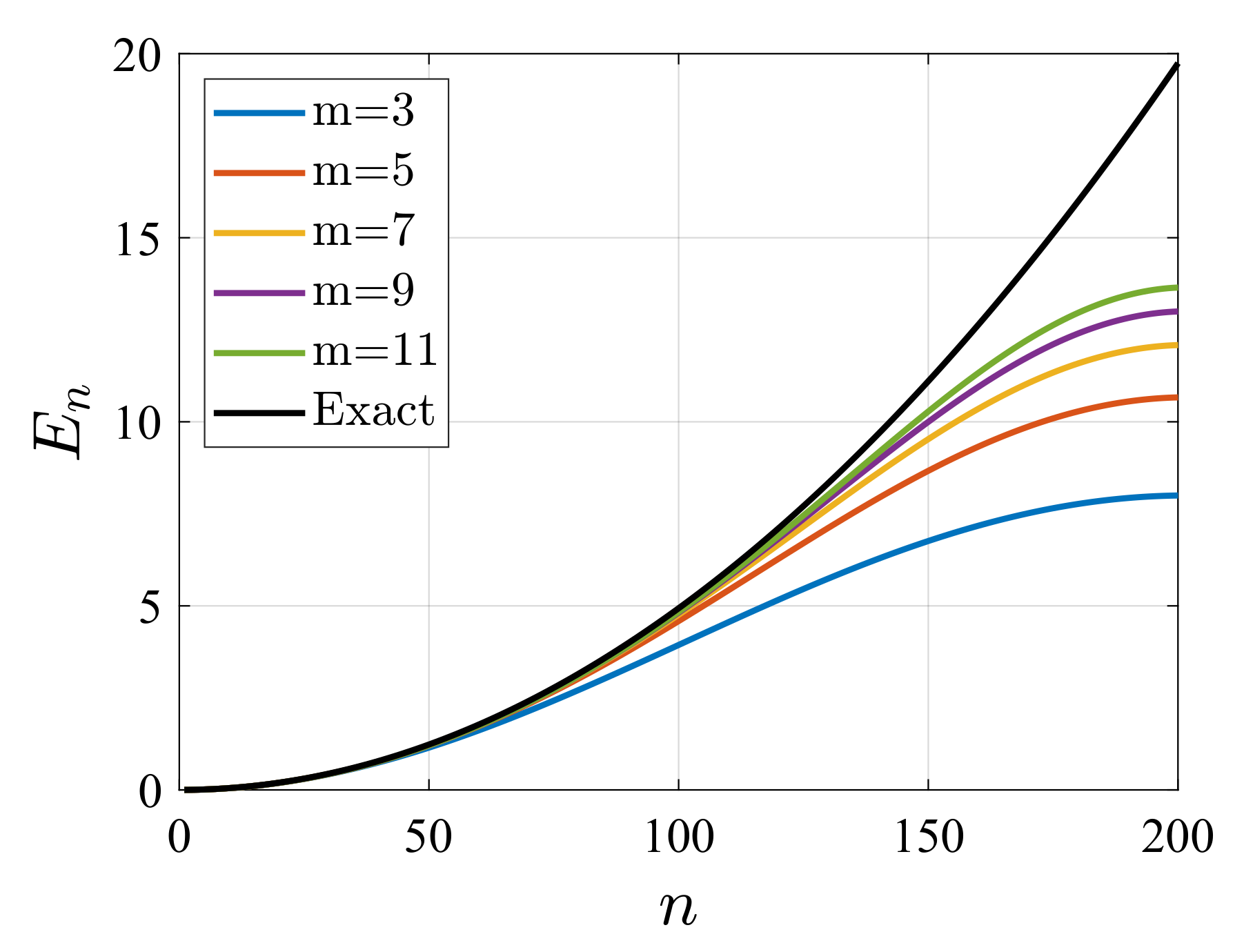
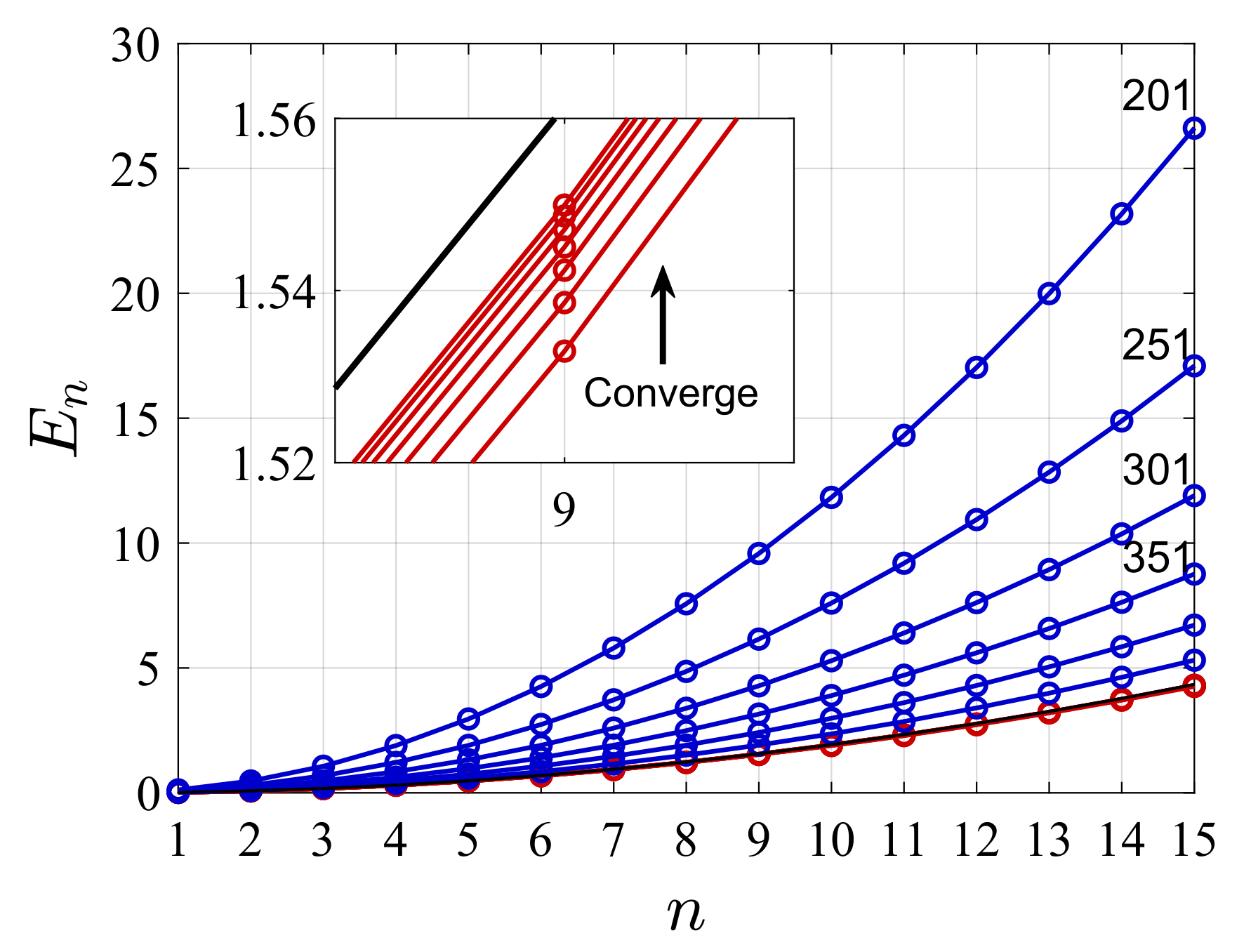
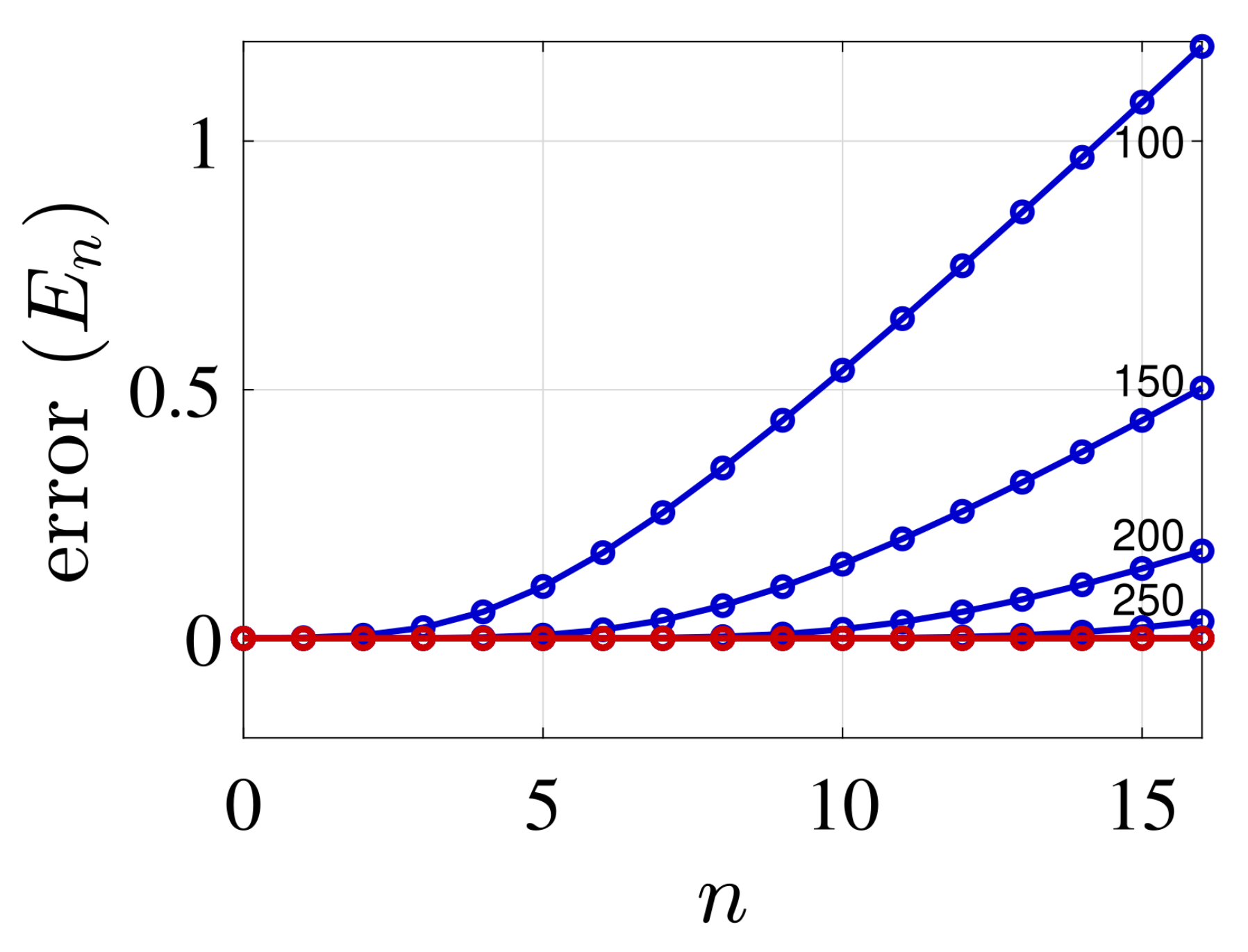
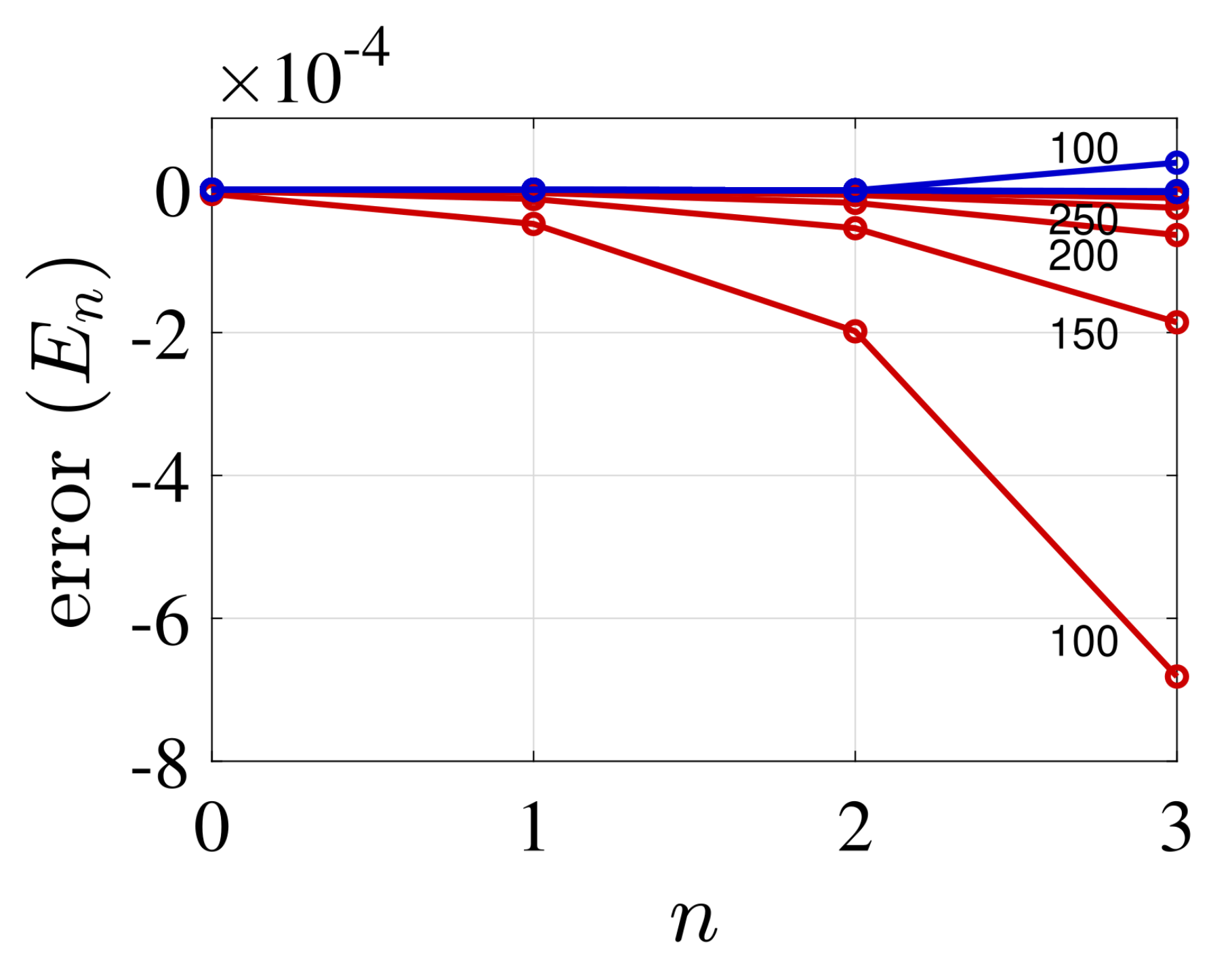
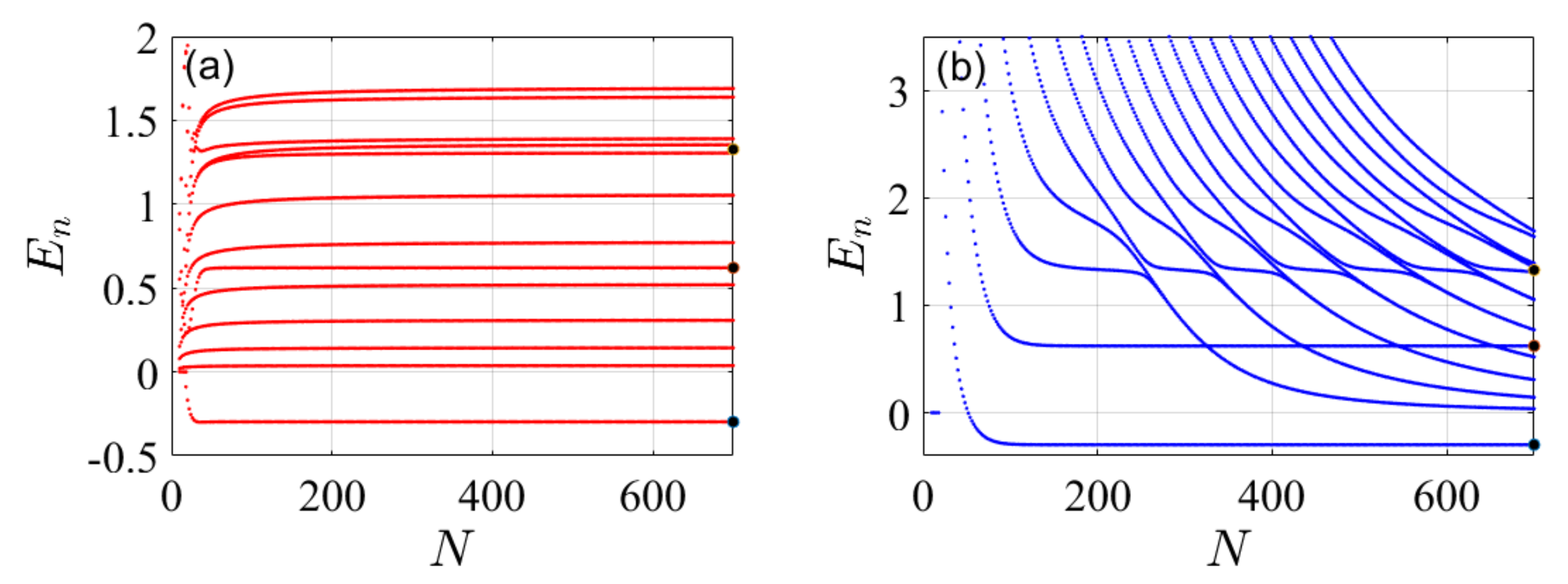
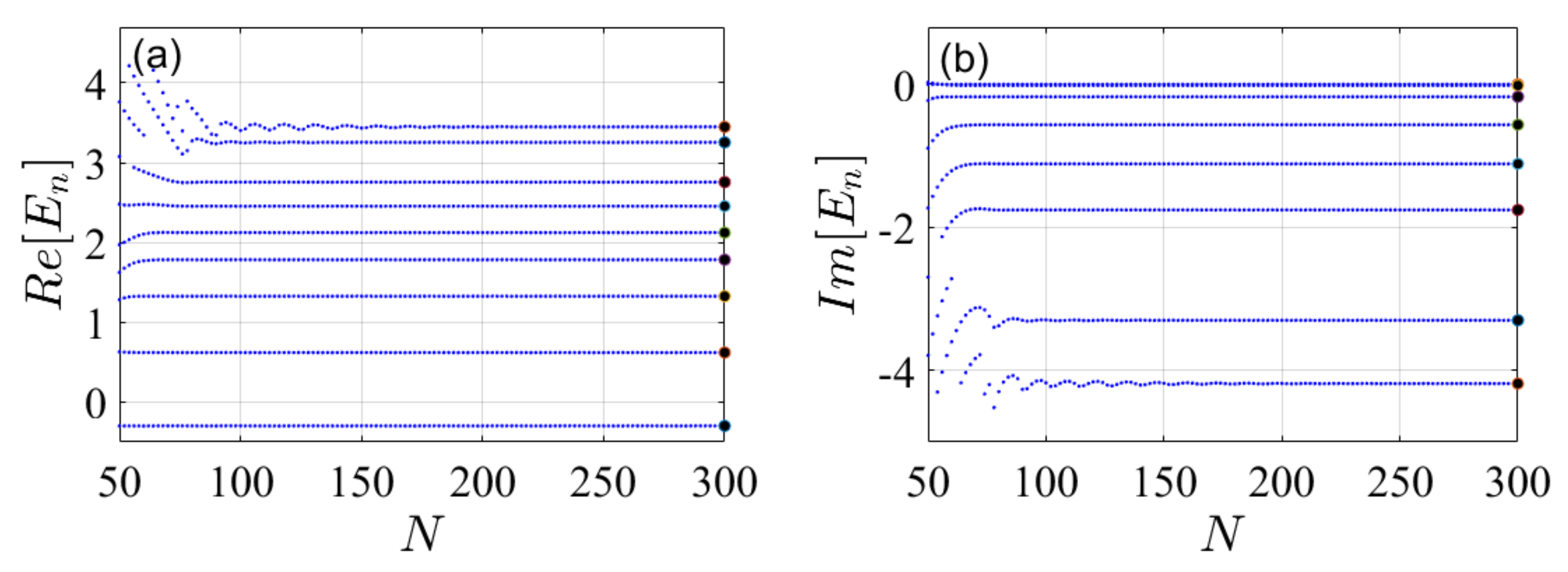
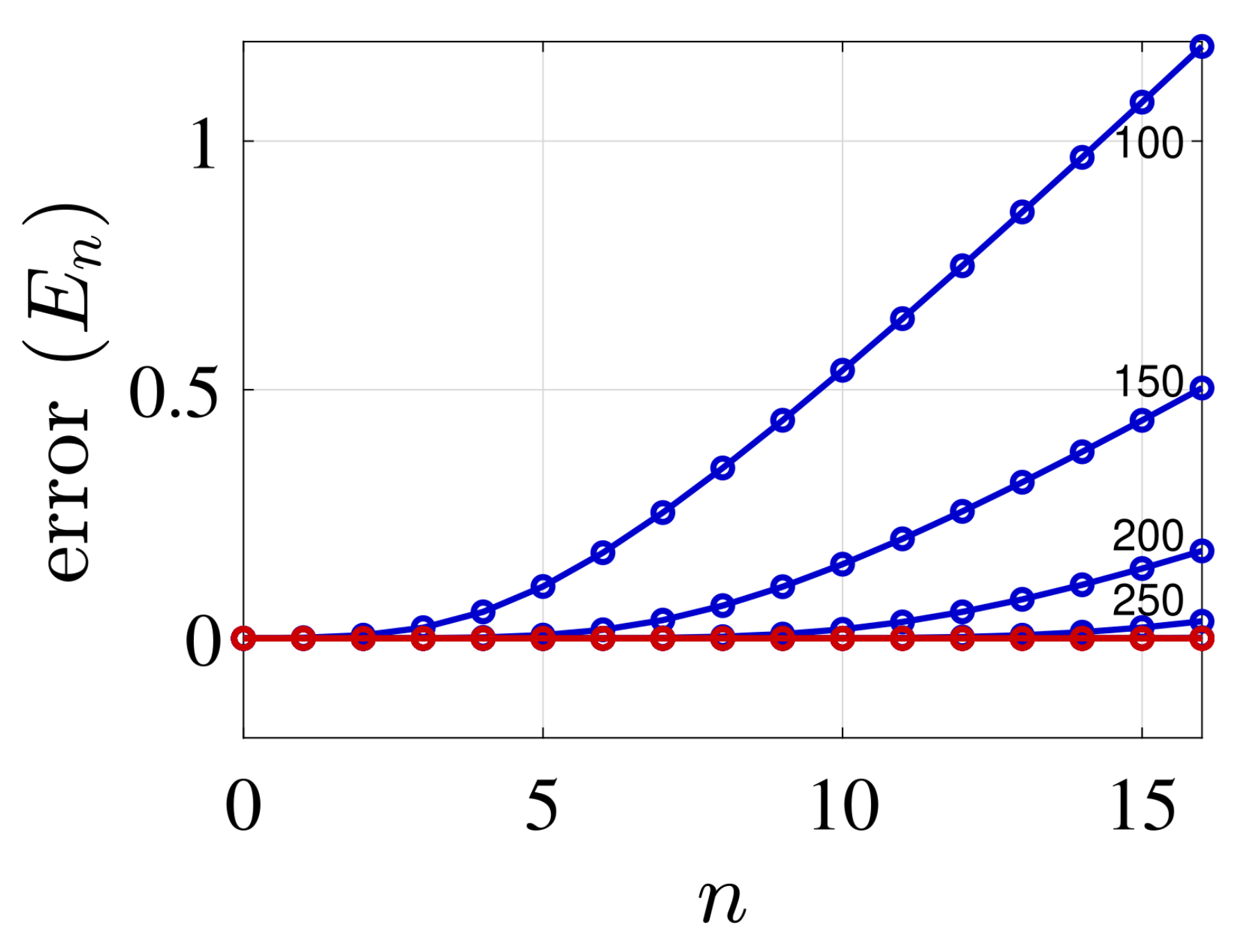
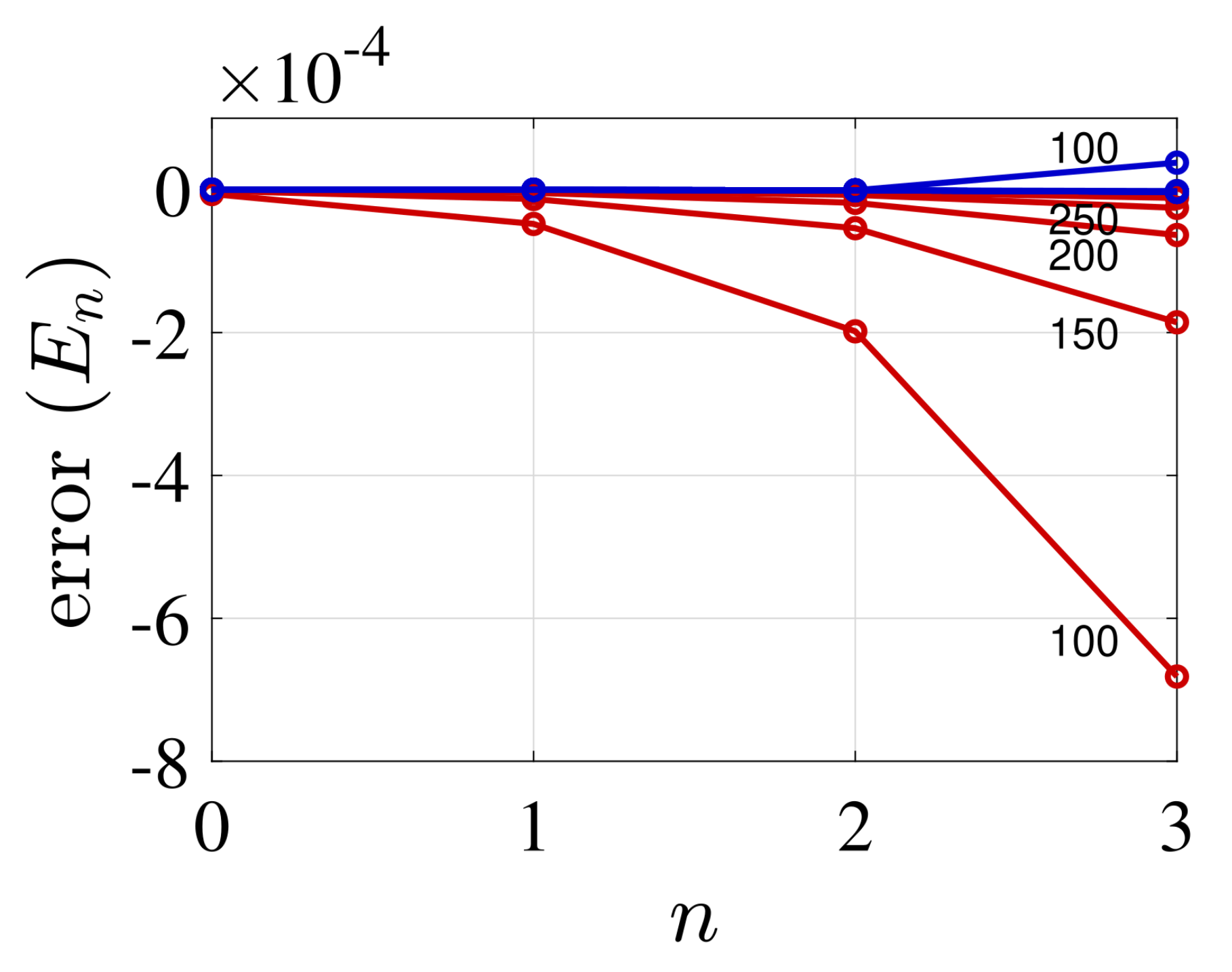
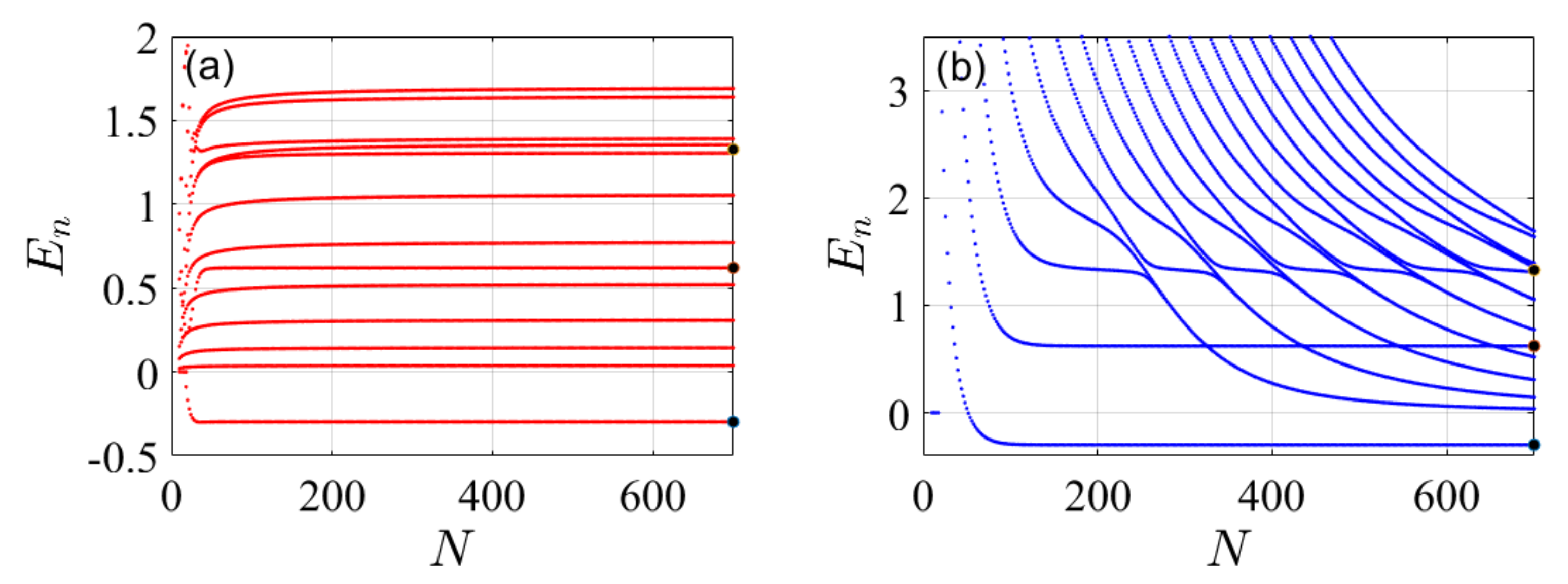
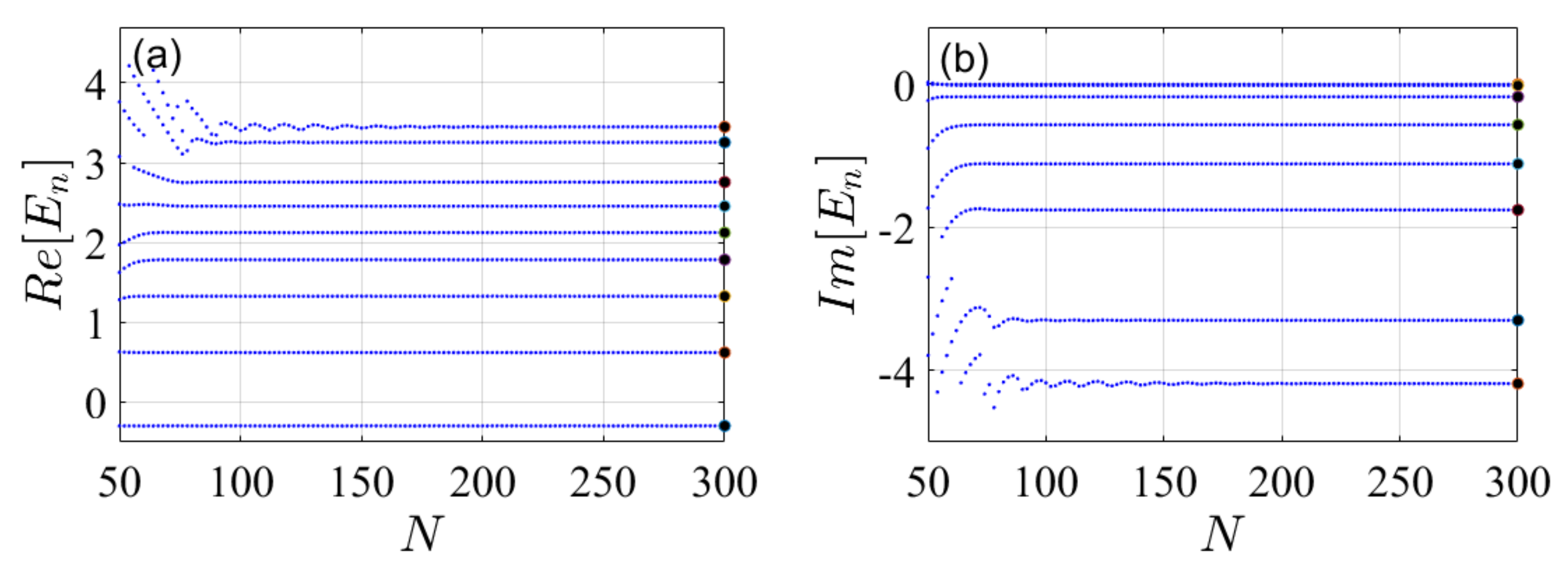
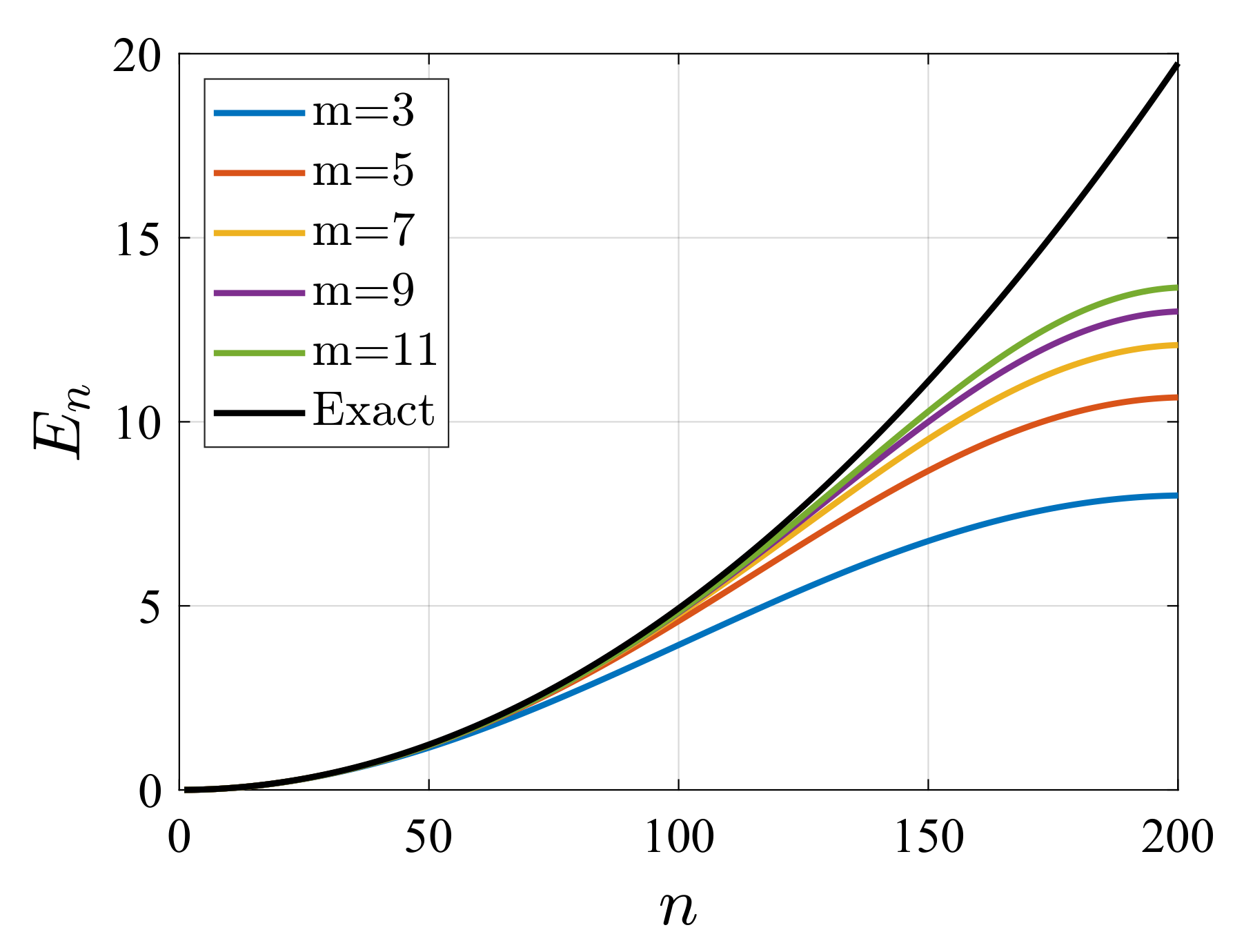
4. Illustrative Numerical Examples for the Calculations of Energies and Widths of Resonances
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| QM | Quantum mechanics |
| FDM | Finite difference method |
Appendix A. Derivation of the Representation of the Laplacian by the Standard Finite Difference Method
Appendix B. Convergence of the Kinetic Energy Representation by the Standard Finite Difference Method
References
- Moiseyev, N.; Certain, P.; Weinhold, F. Resonance properties of complex-rotated hamiltonians. Mol. Phys. 1978, 36, 1613–1630. [Google Scholar] [CrossRef]
- Bravaya, K.B.; Zuev, D.; Epifanovsky, E.; Krylov, A.I. Complex-scaled equation-of-motion coupled-cluster method with single and double substitutions for autoionizing excited states: Theory, implementation, and examples. J. Chem. Phys. 2013, 138, 124106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagau, T.C.; Krylov, A.I. Complex absorbing potential equation-of-motion coupled-cluster method yields smooth and internally consistent potential energy surfaces and lifetimes for molecular resonances. J. Phys. Chem. Lett. 2014, 5, 3078–3085. [Google Scholar] [CrossRef]
- Zuev, D.; Jagau, T.C.; Bravaya, K.B.; Epifanovsky, E.; Shao, Y.; Sundstrom, E.; Head-Gordon, M.; Krylov, A.I. Complex absorbing potentials within EOM-CC family of methods: Theory, implementation, and benchmarks. J. Chem. Phys. 2014, 141, 024102. [Google Scholar] [CrossRef] [Green Version]
- Jagau, T.C.; Zuev, D.; Bravaya, K.B.; Epifanovsky, E.; Krylov, A.I. A fresh look at resonances and complex absorbing potentials: Density matrix-based approach. J. Phys. Chem. Lett. 2014, 5, 310–315. [Google Scholar] [CrossRef]
- Jagau, T.C.; Dao, D.B.; Holtgrewe, N.S.; Krylov, A.I.; Mabbs, R. Same but different: Dipole-stabilized shape resonances in CuF−and AgF−. J. Phys. Chem. Lett. 2015, 6, 2786–2793. [Google Scholar] [CrossRef] [Green Version]
- Kunitsa, A.A.; Bravaya, K.B. First-principles calculations of the energy and width of the 2Au shape resonance in p-benzoquinone: A gateway state for electron transfer. J. Phys. Chem. Lett. 2015, 6, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Jagau, T.C. Investigating tunnel and above-barrier ionization using complex-scaled coupled-cluster theory. J. Chem. Phys. 2016, 145, 204115. [Google Scholar] [CrossRef] [PubMed]
- Jagau, T.C.; Krylov, A.I. Characterizing metastable states beyond energies and lifetimes: Dyson orbitals and transition dipole moments. J. Chem. Phys. 2016, 144, 054113. [Google Scholar] [CrossRef]
- Kunitsa, A.A.; Granovsky, A.A.; Bravaya, K.B. CAP-XMCQDPT2 method for molecular electronic resonances. J. Chem. Phys. 2017, 146, 184107. [Google Scholar] [CrossRef]
- Jagau, T.C.; Bravaya, K.B.; Krylov, A.I. Extending quantum chemistry of bound states to electronic resonances. Annu. Rev. Phys. Chem. 2017, 68, 525–553. [Google Scholar] [CrossRef] [Green Version]
- Benda, Z.; Jagau, T.C. Communication: Analytic gradients for the complex absorbing potential equation-of-motion coupled-cluster method. J. Chem. Phys. 2017, 146, 031101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benda, Z.; Jagau, T.C. Locating exceptional points on multidimensional complex-valued potential energy surfaces. J. Phys. Chem. Lett. 2018, 9, 6978–6984. [Google Scholar] [CrossRef]
- Jagau, T.C. Non-iterative triple excitations in equation-of-motion coupled-cluster theory for electron attachment with applications to bound and temporary anions. J. Chem. Phys. 2018, 148, 024104. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Ryszka, M.; Dawley, M.M.; Carmichael, I.; Bravaya, K.B.; Ptasińska, S. Dipole-supported electronic resonances mediate electron-induced amide bond cleavage. Phys. Rev. Lett. 2019, 122, 073002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández Vera, M.; Jagau, T.C. Resolution-of-the-identity approximation for complex-scaled basis functions. J. Chem. Phys. 2019, 151, 111101. [Google Scholar] [CrossRef]
- Hernández Vera, M.; Jagau, T.C. Resolution-of-the-identity second-order Møller–Plesset perturbation theory with complex basis functions: Benchmark calculations and applications to strong-field ionization of polyacenes. J. Chem. Phys. 2020, 152, 174103. [Google Scholar] [CrossRef] [PubMed]
- Parravicini, V.; Jagau, T.C. Embedded equation-of-motion coupled-cluster theory for electronic excitation, ionisation, electron attachment, and electronic resonances. Mol. Phys. 2021, e1943029. [Google Scholar] [CrossRef]
- Fennimore, M.A.; Matsika, S. Core-excited and shape resonances of uracil. Phys. Chem. Chem. Phys. 2016, 18, 30536–30545. [Google Scholar] [CrossRef] [PubMed]
- Fennimore, M.A.; Matsika, S. Electronic resonances of nucleobases using stabilization methods. J. Phys. Chem. A 2018, 122, 4048–4057. [Google Scholar] [CrossRef]
- Marante, C.; Argenti, L.; Martín, F. Hybrid Gaussian–B-spline basis for the electronic continuum: Photoionization of atomic hydrogen. Phys. Rev. A 2014, 90, 012506. [Google Scholar] [CrossRef] [Green Version]
- Marante, C.; González, J.; Corral, I.; Klinker, M.; Argenti, L.; Martín, F. Merging quantum chemistry packages with B-splines for the multichannel scattering problem. J. Phys. Conf. Ser. 2015, 635, 092013. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Davidson, E.R.; Feller, D. Basis set selection for molecular calculations. Chem. Rev. 1986, 86, 681–696. [Google Scholar] [CrossRef]
- Lewars, E. Computational chemistry. In Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Springer: Berlin, Germany, 2011. [Google Scholar]
- Hinchliffe, A. Modelling Molecular Structures; J. Wiley: Hoboken, NJ, USA, 1996. [Google Scholar]
- Szabo, A.; Ostlund, N.S. Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory. 2012. Available online: https://books.google.com.hk/books?hl=zh-CN&lr=&id=KQ3DAgAAQBAJ&oi=fnd&pg=PP1&dq=Szabo,+A.%3B+Ostlund,+N.S.+Modern+Quantum+Chemistry:+Introduction+to+Advanced+Electronic+Structure+Theory&ots=P_rALVqhcF&sig=dLVyK6AAWkXw42xo6fjxLt4j5cQ&redir_esc=y#v=onepage&q&f=false (accessed on 26 July 2021).
- Levine, I.N.; Busch, D.H.; Shull, H. Quantum Chemistry; Pearson Prentice Hall: Upper Saddle River, NJ, USA, 2009; Volume 6. [Google Scholar]
- Huey, R.; Goodsell, D.S.; Morris, G.M.; Olson, A.J. Grid-based hydrogen bond potentials with improved directionality. Lett. Drug Des. Discov. 2004, 1, 178–183. [Google Scholar] [CrossRef]
- Forsythe, G.E.; Wasow, W.R. Finite Difference Methods for Partial Differential Equations; John Wiley and Sons: New York, NY, USA, 1960. [Google Scholar]
- Perrone, N.; Kao, R. A general finite difference method for arbitrary meshes. Comput. Struct. 1975, 5, 45–57. [Google Scholar] [CrossRef]
- Liszka, T.; Orkisz, J. The finite difference method at arbitrary irregular grids and its application in applied mechanics. Comput. Struct. 1980, 11, 83–95. [Google Scholar] [CrossRef]
- Chelikowsky, J.R.; Troullier, N.; Saad, Y. Finite-difference-pseudopotential method: Electronic structure calculations without a basis. Phys. Rev. Lett. 1994, 72, 1240. [Google Scholar] [CrossRef] [PubMed]
- Özişik, M.N.; Orlande, H.R.; Colaço, M.J.; Cotta, R.M. Finite Difference Methods in Heat Transfer; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Teixeira, F.L. Time-domain finite-difference and finite-element methods for Maxwell equations in complex media. IEEE Trans. Antennas Propag. 2008, 56, 2150–2166. [Google Scholar] [CrossRef] [Green Version]
- Holland, R. Finite-difference solution of Maxwell’s equations in generalized nonorthogonal coordinates. IEEE Trans. Nucl. Sci. 1983, 30, 4589–4591. [Google Scholar] [CrossRef]
- Simos, T.; Williams, P. A finite-difference method for the numerical solution of the Schrödinger equation. J. Comput. Appl. Math. 1997, 79, 189–205. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhang, X.; Liu, Z.; Zhang, Y. A new high-order compact finite difference scheme based on precise integration method for the numerical simulation of parabolic equations. Adv. Differ. Equ. 2020, 2020, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Maragakis, P.; Soler, J.; Kaxiras, E. Variational finite-difference representation of the kinetic energy operator. Phys. Rev. B 2001, 64, 193101. [Google Scholar] [CrossRef] [Green Version]
- Hylleraas, E.A.; Undheim, B. Numerische berechnung der 2S-terme von ortho-und par-helium. Z. Phys. 1930, 65, 759–772. [Google Scholar] [CrossRef]
- MacDonald, J.K.L. Successive approximations by the Rayleigh-Ritz variation method. Phys. Rev. 1933, 43, 830. [Google Scholar] [CrossRef]
- Epstein, S. The Variation Method in Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 2012; Volume 33. [Google Scholar]
- Rosen, N.; Morse, P.M. On the vibrations of polyatomic molecules. Phys. Rev. 1932, 42, 210. [Google Scholar] [CrossRef]
- Wentzel, G. Eine Verallgemeinerung der Quantenbedingungen für die Zwecke der Wellenmechanik. Z. Phys. 1926, 38, 518–529. [Google Scholar] [CrossRef]
- Kramers, H.A. Wellenmechanik und halbzahlige Quantisierung. Z. Phys. 1926, 39, 828–840. [Google Scholar] [CrossRef]
- Brillouin, L. La mécanique ondulatoire de Schrödinger; une méthode générale de résolution par approximations successives. Compt. Rend. Hebd. Seances Acad. Sci. 1926, 183, 24–26. [Google Scholar]
- Moiseyev, N. Quantum theory of resonances: Calculating energies, widths and cross-sections by complex scaling. Phys. Rep. 1998, 302, 212–293. [Google Scholar] [CrossRef]
- Hazi, A.U.; Taylor, H.S. Stabilization method of calculating resonance energies: Model problem. Phys. Rev. A 1970, 1, 1109. [Google Scholar] [CrossRef]
- Maier, C.; Cederbaum, L.; Domcke, W. A spherical-box approach to resonances. J. Phys. B At. Mol. Phys. 1980, 13, L119. [Google Scholar] [CrossRef]
- Falcetta, M.; Jordan, K. Stabilization calculations on the. pi.* anion states of 1,4-cyclohexadiene: Confirmation of the. pi.-* below. pi.+* orbital ordering. J. Am. Chem. Soc. 1991, 113, 2903–2909. [Google Scholar] [CrossRef]
- Falcetta, M.; Choi, Y.; Jordan, K. Ab initio investigation of the temporary anion states of perfluoroethane. J. Phys. Chem. A 2000, 104, 9605–9612. [Google Scholar] [CrossRef]
- Falcetta, M.F.; DiFalco, L.A.; Ackerman, D.S.; Barlow, J.C.; Jordan, K.D. Assessment of various electronic structure methods for characterizing temporary anion states: Application to the ground state anions of N2, C2H2, C2H4, and C6H6. J. Phys. Chem. A 2014, 118, 7489–7497. [Google Scholar] [CrossRef]
- Falcetta, M.F.; Fair, M.C.; Tharnish, E.M.; Williams, L.M.; Hayes, N.J.; Jordan, K.D. Ab initio calculation of the cross sections for electron impact vibrational excitation of CO via the 2Π shape resonance. J. Chem. Phys. 2016, 144, 104303. [Google Scholar] [CrossRef] [Green Version]
- Landau, A.; Haritan, I.; Kaprálová-Zd’ánská, P.R.; Moiseyev, N. Atomic and molecular complex resonances from real eigenvalues using standard (hermitian) electronic structure calculations. J. Phys. Chem. A 2016, 120, 3098–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landau, A.; Bhattacharya, D.; Haritan, I.; Ben-Asher, A.; Moiseyev, N. Ab initio complex potential energy surfaces from standard quantum chemistry packages. Adv. Quantum Chem. 2017, 74, 321–346. [Google Scholar]
- Bhattacharya, D.; Ben-Asher, A.; Haritan, I.; Pawlak, M.; Landau, A.; Moiseyev, N. Polyatomic ab initio complex potential energy surfaces: Illustration of ultracold collisions. J. Chem. Theory Comput. 2017, 13, 1682–1690. [Google Scholar] [CrossRef]
- Gasperich, K.; Jordan, K.; Simons, J. Strategy for creating rational fraction fits to stabilization graph data on metastable electronic states. Chem. Phys. 2018, 515, 342–349. [Google Scholar] [CrossRef]
- Kairalapova, A.; Jordan, K.D.; Maienshein, D.N.; Fair, M.C.; Falcetta, M.F. Prediction of a nonvalence temporary anion shape resonance for a model (H2O) 4 system. J. Phys. Chem. A 2019, 123, 2719–2726. [Google Scholar] [CrossRef] [PubMed]
- Thodika, M.; Fennimore, M.; Karsili, T.N.; Matsika, S. Comparative study of methodologies for calculating metastable states of small to medium-sized molecules. J. Chem. Phys. 2019, 151, 244104. [Google Scholar] [CrossRef] [PubMed]
- Landau, A. Shaping and controlling stabilisation graphs for calculating stable complex resonance energies. Mol. Phys. 2019, 117, 2029–2042. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Pawlak, M.; Ben-Asher, A.; Landau, A.; Haritan, I.; Narevicius, E.; Moiseyev, N. Quantum effects in cold molecular collisions from spatial polarization of electronic wave function. J. Phys. Chem. Lett. 2019, 10, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Landau, A.; Haritan, I. The clusterization technique: A systematic search for the resonance energies obtained via Padé. J. Phys. Chem. A 2019, 123, 5091–5105. [Google Scholar] [CrossRef]
- Thodika, M.; Mackouse, N.; Matsika, S. Description of Two-Particle One-Hole Electronic Resonances Using Orbital Stabilization Methods. J. Phys. Chem. A 2020, 124, 9011–9020. [Google Scholar] [CrossRef]
- Chao, J.Y.; Falcetta, M.; Jordan, K. Application of the stabilization method to the N-2 (1 2Π g) and Mg-(1 2 P) temporary anion states. J. Chem. Phys. 1990, 93, 1125–1135. [Google Scholar] [CrossRef]
- Lee, W.; Matsika, S. Stabilization of the Triplet Biradical Intermediate of 5-Methylcytosine Enhances Cyclobutane Pyrimidine Dimer (CPD) Formation in DNA. Chem.-Eur. J. 2020, 26, 14181–14186. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Landau, A.; Moiseyev, N. Ab initio complex transition dipoles between autoionizing resonance states from real stabilization graphs. J. Phys. Chem. Lett. 2020, 11, 5601–5609. [Google Scholar] [CrossRef]
- Landau, A.; Ben-Asher, A.; Gokhberg, K.; Cederbaum, L.S.; Moiseyev, N. Ab initio complex potential energy curves of the He*(1 s 2 p 1P)–Li dimer. J. Chem. Phys. 2020, 152, 184303. [Google Scholar] [CrossRef]
- Carlson, B.J.; Falcetta, M.F.; Slimak, S.R.; Jordan, K.D. A Fresh Look at the Role of the Coupling of a Discrete State with a Pseudocontinuum State in the Stabilization Method for Characterizing Metastable States. J. Phys. Chem. Lett. 2021, 12, 1202–1206. [Google Scholar] [CrossRef]
- Slimak, S.R.; Jordan, K.D.; Falcetta, M.F. Role of Overlap between the Discrete State and Pseudocontinuum States in Stabilization Calculations of Metastable States. J. Phys. Chem. A 2021, 125, 4401–4408. [Google Scholar] [CrossRef] [PubMed]
- Ben-Asher, A.; Landau, A.; Moiseyev, N. Uniform vs Partial Scaling within Resonances via Padé Based on the Similarities to Other Non-Hermitian Methods: Illustration for the Beryllium 1 s2 2 p3 s State. J. Chem. Theory Comput. 2021, 17, 3435–3444. [Google Scholar] [CrossRef]
- Moiseyev, N. Non-Hermitian Quantum Mechanics; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar]
- Aguilar, J.; Combes, J.M. A class of analytic perturbations for one-body Schrödinger Hamiltonians. Commun. Math. Phys. 1971, 22, 269–279. [Google Scholar] [CrossRef]
- Balslev, E.; Combes, J.M. Spectral properties of many-body Schrödinger operators with dilatation-analytic interactions. Commun. Math. Phys. 1971, 22, 280–294. [Google Scholar] [CrossRef]
- Simon, B. Quadratic form techniques and the Balslev-Combes theorem. Commun. Math. Phys. 1972, 27, 1–9. [Google Scholar] [CrossRef]
- Simon, B. Resonances in n-body quantum systems with dilatation analytic potentials and the foundations of time-dependent perturbation theory. Ann. Math. 1973, 97, 247–274. [Google Scholar] [CrossRef]
- Moiseyev, N.; Weinhold, F. Criteria of accuracy of resonance eigenvalues. Int. J. Quantum Chem. 1980, 17, 1201–1211. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dann, R.; Elbaz, G.; Berkheim, J.; Muhafra, A.; Nitecki, O.; Wilczynski, D.; Moiseyev, N. Variational Solutions for Resonances by a Finite-Difference Grid Method. Molecules 2021, 26, 5248. https://doi.org/10.3390/molecules26175248
Dann R, Elbaz G, Berkheim J, Muhafra A, Nitecki O, Wilczynski D, Moiseyev N. Variational Solutions for Resonances by a Finite-Difference Grid Method. Molecules. 2021; 26(17):5248. https://doi.org/10.3390/molecules26175248
Chicago/Turabian StyleDann, Roie, Guy Elbaz, Jonathan Berkheim, Alan Muhafra, Omri Nitecki, Daniel Wilczynski, and Nimrod Moiseyev. 2021. "Variational Solutions for Resonances by a Finite-Difference Grid Method" Molecules 26, no. 17: 5248. https://doi.org/10.3390/molecules26175248
APA StyleDann, R., Elbaz, G., Berkheim, J., Muhafra, A., Nitecki, O., Wilczynski, D., & Moiseyev, N. (2021). Variational Solutions for Resonances by a Finite-Difference Grid Method. Molecules, 26(17), 5248. https://doi.org/10.3390/molecules26175248