
RNA Dynamics in Alzheimer’s Disease

Abstract
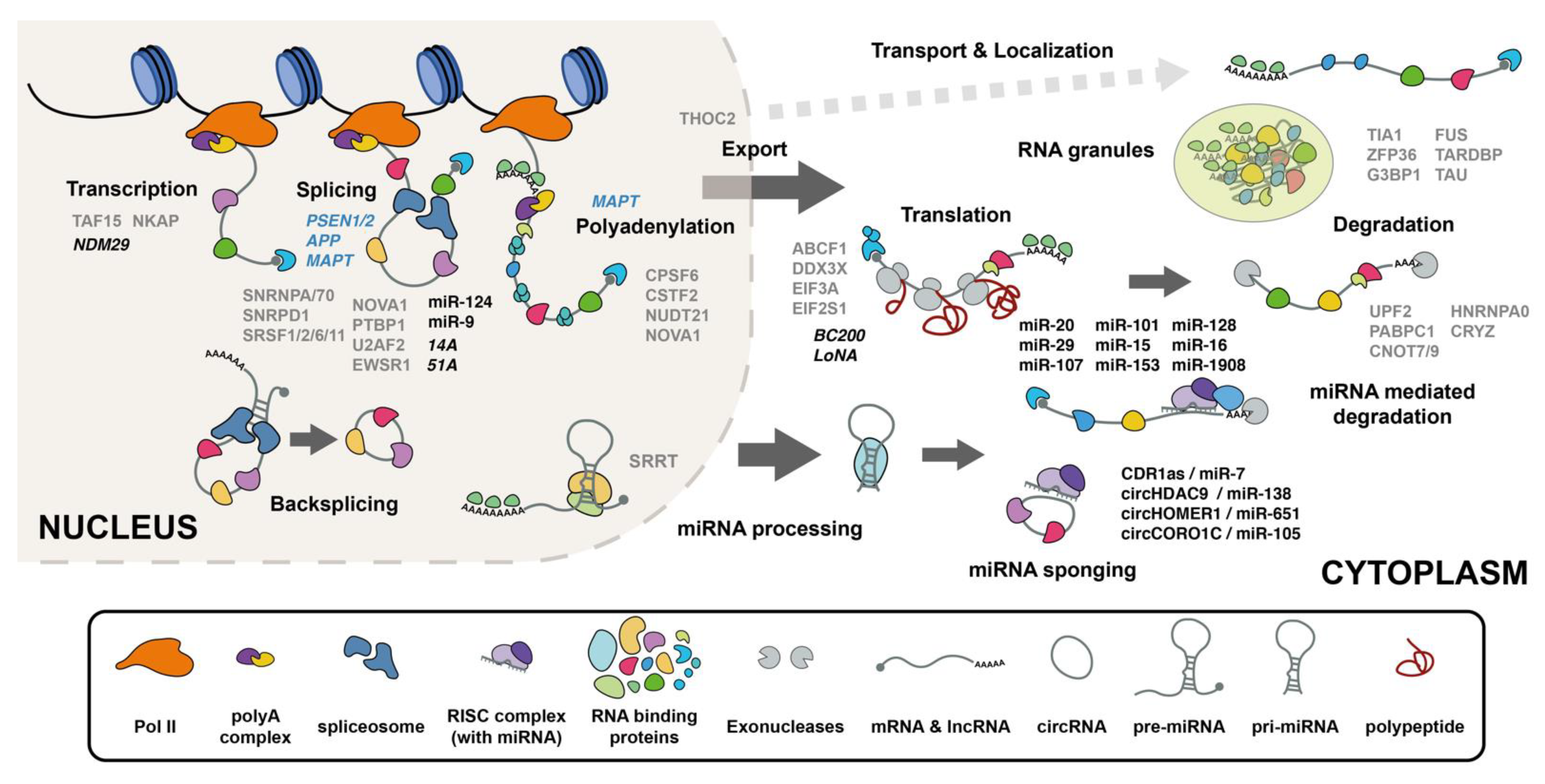
1. Introduction
2. RNA Processing Governs the RNA Makeup of Cells
3. RNA Binding Proteins Have Altered Functions in AD
3.1. Proteomics Studies Identify RNA Processing Modules Altered in AD
3.2. Transcriptomic Studies Identify Altered Expression of Splicing Factors in AD
3.3. Single-Cell Transcriptomics Confirm Cell-Type Independent RBP Alterations
3.4. RNA Binding Proteins Aggregate in AD
4. mRNA Changes in AD Are Due to Specific SNPs and Alterations in the Function of the RNA Processing Machinery
mRNA Isoform Changes Are Common in Known AD-Risk Genes
5. miRNAs in AD
5.1. miRNAs Regulate APP Expression
5.2. miRNA Function in Aβ Clearance
5.3. miRNAs and Tau-Related Pathologies
6. Circular RNA
circRNAs’ Function in AD
7. LncRNAs
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kawas, C.; Gray, S.; Brookmeyer, R.; Fozard, J.; Zonderman, A. Age-specific incidence rates of Alzheimer’s disease. Neurology 2000, 54, 2072–2077. [Google Scholar] [CrossRef]
- Campion, D.; Dumanchin, C.; Hannequin, D.; Dubois, B.; Belliard, S.; Puel, M.; Thomas-Anterion, C.; Michon, A.; Martin, C.; Charbonnier, F.; et al. Early-Onset Autosomal Dominant Alzheimer Disease: Prevalence, Genetic Heterogeneity, and Mutation Spectrum. Am. J. Hum. Genet. 1999, 65, 664–670. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2010, 121, 171–181. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the Pathologic Process in Alzheimer Disease: Age Categories From 1 to 100 Years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blen-now, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s Disease; Nature Publishing Group: Berlin, Germany, 2015; Volume 1, ISBN 0140-6736. [Google Scholar]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Oxford, A.E.; Stewart, E.S.; Rohn, T.T. Clinical Trials in Alzheimer’s Disease: A Hurdle in the Path of Remedy. Int. J. Alzheimer’s Dis. 2020, 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C. Defining cell types and states with single-cell genomics. Genome Res. 2015, 25, 1491–1498. [Google Scholar] [CrossRef]
- Plass, M.; Solana, J.; Wolf, F.A.; Ayoub, S.; Misios, A.; Glažar, P.; Obermayer, B.; Theis, F.J.; Kocks, C.; Rajewsky, N. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science 2018, 360, eaaq1723. [Google Scholar] [CrossRef]
- Han, X.; Zhou, Z.; Fei, L.; Sun, H.; Wang, R.; Chen, Y.; Chen, H.; Wang, J.; Tang, H.; Ge, W.; et al. Construction of a human cell landscape at single-cell level. Nature 2020, 581, 303–309. [Google Scholar] [CrossRef]
- Keene, J.D. RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef]
- Schieweck, R.; Ninkovic, J.; A Kiebler, M. RNA-binding proteins balance brain function in health and disease. Physiol. Rev. 2020, 101, 1309–1370. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, M.; Taylor, J.P.; Parker, R. Altered Ribostasis: RNA-Protein Granules in Degenerative Disorders. Cell 2013, 154, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Nussbacher, J.; Batra, R.; Lagier-Tourenne, C.; Yeo, G.W. RNA-binding proteins in neurodegeneration: Seq and you shall receive. Trends Neurosci. 2015, 38, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Apicco, D. RNA binding proteins and the genesis of neurodegenerative diseases. Single Mol. Single Cell Seq. 2014, 822, 11–15. [Google Scholar] [CrossRef]
- Cookson, M.R. RNA-binding proteins implicated in neurodegenerative diseases. Wiley Interdiscip. Rev. RNA 2016, 8, e1397. [Google Scholar] [CrossRef]
- Conlon, E.G.; Manley, J.L. RNA-binding proteins in neurodegeneration: Mechanisms in aggregate. Genes Dev. 2017, 31, 1509–1528. [Google Scholar] [CrossRef] [PubMed]
- Maziuk, B.; Ballance, H.I.; Wolozin, B. Dysregulation of RNA Binding Protein Aggregation in Neurodegenerative Disorders. Front. Mol. Neurosci. 2017, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Maurya, B.; Surabhi, S.; Pandey, P.; Mukherjee, A.; Mutsuddi, M. The expanding role of RNA-binding proteins in neurode-generation. In Insights into Human Neurodegeneration: Lessons Learnt from Drosophila; Springer: Singapore, 2019; pp. 373–403. ISBN 9789811322181. [Google Scholar]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, F.; Schwarzl, T.; Valcárcel, J.; Hentze, M.W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2020, 22, 185–198. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Bentmann, E.; Dormann, D.; Jawaid, A.; DeJesus-Hernandez, M.; Ansorge, O.; Roeber, S.; Kretzschmar, H.A.; Munoz, D.; Kusaka, H.; et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain 2011, 134, 2595–2609. [Google Scholar] [CrossRef] [PubMed]
- Jr., T.J.K.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.; Nishimura, A.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA Processing Protein, Cause Familial Amyotrophic Lateral Sclerosis Type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.C.; Ng, C.S.; Xiang, P.; Liu, H.; Zhang, K.; Mohamud, Y.; Luo, H. Dysregulation of RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2020, 13, 78. [Google Scholar] [CrossRef] [PubMed]
- Imbert, G.; Saudou, F.; Yvert, G.; Devys, D.; Trottier, Y.; Garnier, J.-M.; Weber, C.; Mandel, J.-L.; Cancel-Tassin, G.; Abbas, N.; et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat. Genet. 1996, 14, 285–291. [Google Scholar] [CrossRef]
- Pulst, S.-M.; Nechiporuk, A.; Nechiporuk, T.; Gispert, S.; Chen, X.-N.; Lopes-Cendes, I.; Pearlman, S.; Starkman, S.; Orozco-Diaz, G.; Lunkes, A.; et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 1996, 14, 269–276. [Google Scholar] [CrossRef]
- Sanpei, K.; Takano, H.; Igarashi, S.; Sato, T.; Oyake, M.; Sasaki, H.; Wakisaka, A.; Tashiro, K.; Ishida, Y.; Ikeuchi, T.; et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat. Genet. 1996, 14, 277–284. [Google Scholar] [CrossRef]
- Cairns, N.J.; Bigio, E.H.; MacKenzie, I.R.A.; Neumann, M.; Lee, V.M.-Y.; Hatanpaa, K.J.; White, C.; Schneider, J.A.; Grinberg, L.; Halliday, G.; et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007, 114, 5–22. [Google Scholar] [CrossRef]
- Davidson, Y.; Kelley, T.; MacKenzie, I.R.A.; Pickering-Brown, S.; Du Plessis, D.; Neary, D.; Snowden, J.S.; Mann, D.M.A. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 2007, 113, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, N.T.; Dammer, E.; Swarup, V.; Nandakumar, D.; Duong, D.; Yin, L.; Deng, Q.; Nguyen, T.; Hales, C.M.; Wingo, T.; et al. A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst. 2016, 4, 60–72.e4. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Yin, L.; Thambisetty, M.; Troncoso, J.C.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol. Neurodegener. 2018, 13, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Lanke, V.; Moolamalla, S.T.R.; Roy, D.; Vinod, P.K. Integrative Analysis of Hippocampus Gene Expression Profiles Identifies Network Alterations in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 153. [Google Scholar] [CrossRef]
- Van Rooij, J.G.; Meeter, L.H.; Melhem, S.; Nijholt, D.; Wong, T.H.; Rozemuller, A.; Uitterlinden, A.G.; van Meurs, J.G.; van Swieten, J.C. Hippocampal transcriptome profiling combined with protein-protein interaction analysis elucidates Alzheimer’s disease pathways and genes. Neurobiol. Aging 2018, 74, 225–233. [Google Scholar] [CrossRef]
- Hsieh, Y.-C.; Guo, C.; Yalamanchili, H.K.; Abreha, M.; Al-Ouran, R.; Li, Y.; Dammer, E.B.; Lah, J.J.; Levey, A.I.; Bennett, D.A.; et al. Tau-Mediated Disruption of the Spliceosome Triggers Cryptic RNA Splicing and Neurodegeneration in Alzheimer’s Disease. Cell Rep. 2019, 29, 301–316.e10. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef]
- Twine, N.A.; Janitz, K.; Wilkins, M.R.; Janitz, M. Whole Transcriptome Sequencing Reveals Gene Expression and Splicing Differences in Brain Regions Affected by Alzheimer’s Disease. PLoS ONE 2011, 6, e16266. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Wang, Z.; Hortobágyi, T.; Witten, J.T.; Zarnack, K.; Kayikci, M.; Clark, T.; Schweitzer, A.C.; Rot, G.; Curk, T.; et al. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res. 2011, 21, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Wong, J. Altered Expression of RNA Splicing Proteins in Alzheimer’s Disease Patients: Evidence from Two Microarray Studies. Dement. Geriatr. Cogn. Disord. Extra 2013, 3, 74–85. [Google Scholar] [CrossRef]
- Barbash, S.; Garfinkel, B.; Maoz, R.; Simchovitz, A.; Nadorp, B.; Guffanti, A.; Bennett, E.R.; Nadeau, C.; Türk, A.; Paul, L.; et al. Alzheimer’s brains show inter-related changes in RNA and lipid metabolism. Neurobiol. Dis. 2017, 106, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, S.; Gaiteri, C.; Sullivan, S.E.; White, C.C.; Tasaki, S.; Xu, J.; Taga, M.; Klein, H.-U.; Patrick, E.; Komashko, V.; et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 2018, 21, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Raj, T.; Li, Y.I.; Wong, G.; Humphrey, J.; Wang, M.; Ramdhani, S.; Wang, Y.-C.; Ng, B.; Gupta, I.; Haroutunian, V.; et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet. 2018, 50, 1584–1592. [Google Scholar] [CrossRef]
- Abreu, R.D.S.; Penalva, L.; Marcotte, E.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. BioSyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Younas, N.; Zafar, S.; Shafiq, M.; Noor, A.; Siegert, A.; Arora, A.S.; Galkin, A.; Zafar, A.; Schmitz, M.; Stadelmann, C.; et al. SFPQ and Tau: Critical factors contributing to rapid progression of Alzheimer’s disease. Acta Neuropathol. 2020, 140, 317–339. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free Formation of RNA Granules: Low Complexity Sequence Domains Form Dynamic Fibers within Hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef]
- Bishof, I.; Dammer, E.B.; Duong, D.M.; Kundinger, S.R.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. RNA-binding proteins with basic-acidic dipeptide (BAD) domains self-assemble and aggregate in Alzheimer’s disease. J. Biol. Chem. 2018, 293, 11047–11066. [Google Scholar] [CrossRef]
- Canchi, S.; Raao, B.; Masliah, D.; Rosenthal, S.B.; Sasik, R.; Fisch, K.M.; De Jager, P.L.; Bennett, D.A.; Rissman, R.A. Integrating Gene and Protein Expression Reveals Perturbed Functional Networks in Alzheimer’s Disease. Cell Rep. 2019, 28, 1103–1116.e4. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Zhang, Y.; Enge, M.; Caneda, C.; Shuer, L.M.; Gephart, M.G.H.; Barres, B.A.; Quake, S.R. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA 2015, 112, 7285–7290. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.; Ai, R.; Kaeser, G.E.; Salathia, N.; Yung, Y.C.; Liu, R.; Wildberg, A.; Gao, D.; Fung, H.-L.; Chen, S.; et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 2016, 352, 1586–1590. [Google Scholar] [CrossRef]
- Lake, B.B.; Chen, S.; Sos, B.C.; Fan, J.; Kaeser, G.E.; Yung, Y.C.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.V.; et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2017, 36, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Zhang, S.; Fan, X.; Wu, Q.; Yan, L.; Dong, J.; Zhang, H.; Li, L.; Sun, L.; Pan, N.; et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 2018, 555, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Fu, Y.; Zhou, X.; Sun, L.; Yang, M.; Wang, M.; Chen, R.; Wu, Q.; Yong, J.; Dong, J.; et al. Single-cell transcriptome analysis reveals cell lineage specification in temporal-spatial patterns in human cortical development. Sci. Adv. 2020, 6, eaaz2978. [Google Scholar] [CrossRef] [PubMed]
- Eze, U.C.; Bhaduri, A.; Haeussler, M.; Nowakowski, T.J.; Kriegstein, A.R. Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat. Neurosci. 2021, 24, 584–594. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef]
- Del-Aguila, J.L.; Li, Z.; Dube, U.; Mihindukulasuriya, K.A.; Budde, J.P.; Fernandez, M.V.; Ibanez, L.; Bradley, J.; Wang, F.; Bergmann, K.; et al. A single-nuclei RNA sequencing study of Mendelian and sporadic AD in the human brain. Alzheimer’s Res. Ther. 2019, 11, 1–16. [Google Scholar] [CrossRef]
- Lau, S.-F.; Cao, H.; Fu, A.K.Y.; Ip, N.Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 25800–25809. [Google Scholar] [CrossRef]
- Alsema, A.M.; Jiang, Q.; Kracht, L.; Gerrits, E.; Dubbelaar, M.L.; Miedema, A.; Brouwer, N.; Hol, E.M.; Middeldorp, J.; Van Dijk, R.; et al. Profiling Microglia from Alzheimer’s Disease Donors and Non-demented Elderly in Acute Human Postmortem Cortical Tissue. Front. Mol. Neurosci. 2020, 13, 134. [Google Scholar] [CrossRef]
- Otero-Garcia, M.; Xue, Y.-Q.; Shakouri, T.; Deng, Y.; Morabito, S.; Allison, T.; Lowry, W.; Kawaguchi, R.; Swarup, V.; Cobos, I. Single-soma transcriptomics of tangle-bearing neurons in Alzheimer’s disease reveals the signatures of tau-associated sy-naptic dysfunction. bioRxiv 2020. [Google Scholar] [CrossRef]
- Leng, K.; Li, E.; Eser, R.; Piergies, A.; Sit, R.; Tan, M.; Neff, N.; Li, S.H.; Rodriguez, R.D.; Suemoto, C.K.; et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 2021, 24, 276–287. [Google Scholar] [CrossRef]
- Han, T.W.; Kato, M.; Xie, S.; Wu, L.C.; Mirzaei, H.; Pei, J.; Chen, M.; Xie, Y.; Allen, J.; Xiao, G.; et al. Cell-free Formation of RNA Granules: Bound RNAs Identify Features and Components of Cellular Assemblies. Cell 2012, 149, 768–779. [Google Scholar] [CrossRef]
- Protter, D.S.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Drechsel, D.N.; Hyman, A.; Cobb, M.; Kirschner, M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- Brandt, R.; Lee, G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. J. Biol. Chem. 1993, 268, 3414–3419. [Google Scholar] [CrossRef]
- Chang, C.-W.; Shao, E.; Mucke, L. Tau: Enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science 2021, 371, eabb8255. [Google Scholar] [CrossRef]
- Schröder, H.C.; Bernd, A.; Zahn, R.K.; Müller, W.E. Binding of polyribonucleotides and polydeoxyribonucleotides to bovine brain microtubule protein: Age-dependent modulation via phosphorylation of high-molecular-weight microtubule-associated proteins and tau proteins. Mech. Ageing Dev. 1984, 24, 101–117. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, Y.; Eschmann, N.A.; Zhou, H.; Rauch, J.N.; Hernandez, I.; Guzman, E.; Kosik, K.S.; Han, S. RNA stores tau reversibly in complex coacervates. PLoS Biol. 2017, 15, e2002183. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Ferdosh, S.; Ghosh, A.N.; Barat, C. Tau protein- induced sequestration of the eukaryotic ribosome: Implications in neurodegenerative disease. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Apicco, D.; Zhang, C.; Maziuk, B.; Jiang, L.; Ballance, H.I.; Boudeau, S.; Ung, C.; Li, H.; Wolozin, B. Dysregulation of RNA Splicing in Tauopathies. Cell Rep. 2019, 29, 4377–4388.e4. [Google Scholar] [CrossRef]
- Montalbano, M.; Jaworski, E.; Garcia, S.; Ellsworth, A.; McAllen, S.; Routh, A.; Kayed, R. Tau modulates mRNA transcrip-tion, alternative polyadenylation profiles of hnRNPs, chromatin remodeling and spliceosome complexes. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lester, E.; Ooi, F.K.; Bakkar, N.; Ayers, J.; Woerman, A.L.; Wheeler, J.; Bowser, R.; Carlson, G.A.; Prusiner, S.B.; Parker, R. Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron 2021, 109, 1675–1691.e9. [Google Scholar] [CrossRef]
- Vanderweyde, T.; Yu, W.H.; Varnum, M.; Liu-Yesucevitz, L.; Citro, A.; Ikezu, T.; Duff, K.; Wolozin, B. Contrasting Pathology of the Stress Granule Proteins TIA-1 and G3BP in Tauopathies. J. Neurosci. 2012, 32, 8270–8283. [Google Scholar] [CrossRef]
- Meyer, C.; Garzia, A.; Mazzola, M.; Gerstberger, S.; Molina, H.; Tuschl, T. The TIA1 RNA-Binding Protein Family Regulates EIF2AK2-Mediated Stress Response and Cell Cycle Progression. Mol. Cell 2018, 69, 622–635.e6. [Google Scholar] [CrossRef]
- Mukherjee, N.; Jacobs, N.C.; Hafner, M.; A Kennington, E.; Nusbaum, J.D.; Tuschl, T.; Blackshear, P.J.; Ohler, U. Global target mRNA specification and regulation by the RNA-binding protein ZFP36. Genome Biol. 2014, 15, R12. [Google Scholar] [CrossRef]
- Sahoo, P.K.; Lee, S.J.; Jaiswal, P.B.; Alber, S.; Kar, A.; Miller-Randolph, S.; Taylor, E.E.; Smith, T.; Singh, B.; Ho, T.S.-Y.; et al. Axonal G3BP1 stress granule protein limits axonal mRNA translation and nerve regeneration. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Vanderweyde, T.; Apicco, D.; Youmans-Kidder, K.; Ash, P.E.; Cook, C.; da Rocha, E.L.; Jansen-West, K.; Frame, A.A.; Citro, A.; Leszyk, J.D.; et al. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep. 2016, 15, 1455–1466. [Google Scholar] [CrossRef]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef]
- Sengupta, U.; Montalbano, M.; McAllen, S.; Minuesa, G.; Kharas, M.; Kayed, R. Formation of Toxic Oligomeric Assemblies of RNA-binding Protein: Musashi in Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 1–14. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Sengupta, U.; Puangmalai, N.; Bhatt, N.; Ellsworth, A.; Kayed, R. Tau oligomers mediate aggregation of RNA-binding proteins Musashi1 and Musashi2 inducing Lamin alteration. Aging Cell 2019, 18, e13035. [Google Scholar] [CrossRef]
- Bai, B.; Hales, C.M.; Chen, P.-C.; Gozal, Y.; Dammer, E.; Fritz, J.J.; Wang, X.; Xia, Q.; Duong, D.; Street, C.; et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16562–16567. [Google Scholar] [CrossRef]
- Hales, C.M.; Dammer, E.B.; Deng, Q.; Duong, D.; Gearing, M.; Troncoso, J.C.; Thambisetty, M.; Lah, J.J.; Shulman, J.M.; Levey, A.I.; et al. Changes in the detergent-insoluble brain proteome linked to amyloid and tau in Alzheimer’s Disease progression. Proteomics 2016, 16, 3042–3053. [Google Scholar] [CrossRef]
- Balasubramaniam, M.; Ayyadevara, S.; Ganne, A.; Kakraba, S.; Penthala, N.R.; Du, X.; Crooks, P.A.; Griffin, S.T.; Reis, R.J.S. Aggregate Interactome Based on Protein Cross-linking Interfaces Predicts Drug Targets to Limit Aggregation in Neurodegenerative Diseases. iScience 2019, 20, 248–264. [Google Scholar] [CrossRef]
- Gunawardana, C.G.; Mehrabian, M.; Wang, X.; Mueller, I.; Lubambo, I.B.; Jonkman, J.; Wang, H.; Schmitt-Ulms, G. The Human Tau Interactome: Binding to the Ribonucleoproteome, and Impaired Binding of the Proline-to-Leucine Mutant at Position 301 (P301L) to Chaperones and the Proteasome. Mol. Cell. Proteom. 2015, 14, 3000–3014. [Google Scholar] [CrossRef]
- Ando, K.; Ndjim, M.; Turbant, S.; Fontaine, G.; Pregoni, G.; Dauphinot, L.; Yilmaz, Z.; Suain, V.; Mansour, S.; Authelet, M.; et al. The lipid phosphatase Synaptojanin 1 undergoes a significant alteration in expression and solubility and is associated with brain lesions in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 1–15. [Google Scholar] [CrossRef]
- Higashi, S.; Iseki, E.; Yamamoto, R.; Minegishi, M.; Hino, H.; Fujisawa, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Concurrence of TDP-43, tau and α-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007, 1184, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Amador, C.; Lin, W.-L.; Ahmed, Z.; Ms, D.P.; Davies, P.; Duara, R.; Graff-Radford, N.R.; Hutton, M.L.; Dickson, D.W. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 2007, 61, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Tomé, S.O.; Vandenberghe, R.; Ospitalieri, S.; Van Schoor, E.; Tousseyn, T.; Otto, M.; Von Arnim, C.A.F.; Thal, D.R. Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: Relationship with clinical phenotypes. Acta Neuropathol. Commun. 2020, 8, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.-H.; Tu, L.-H.; Chang, T.-Y.; Ganesan, K.; Chang, W.-W.; Chang, P.-S.; Fang, Y.-S.; Lin, Y.-T.; Jin, L.-W.; Chen, Y.-R. TDP-43 interacts with amyloid-β, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Rees, H.D.; Weintraub, S.T.; Levey, A.I.; Chin, L.-S.; Li, L. Oxidative Modifications and Aggregation of Cu,Zn-Superoxide Dismutase Associated with Alzheimer and Parkinson Diseases. J. Biol. Chem. 2005, 280, 11648–11655. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.J.; Park, H.-J.; Kim, G.-Y.; Cho, H.; Choi, J.-H.; Park, H.-Y.; Jang, J.-Y.; Rhim, H.; Kang, S. Intracellular amyloid beta interacts with SOD1 and impairs the enzymatic activity of SOD1: Implications for the pathogenesis of amyotrophic lateral sclerosis. Exp. Mol. Med. 2009, 41, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.; Riascos, D.; Kovalik, T.; An, J.; Krupa, K.; Krupa, K.; Hood, B.L.; Conrads, T.P.; Renton, A.E.; Traynor, B.J.; et al. The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 2012, 124, 717–732. [Google Scholar] [CrossRef]
- Hosp, F.; Vossfeldt, H.; Heinig, M.; Vasiljevic, D.; Arumughan, A.; Wyler, E.; Landthaler, M.; Hubner, N.; Wanker, E.E.; Lannfelt, L.; et al. Quantitative Interaction Proteomics of Neurodegenerative Disease Proteins. Cell Rep. 2015, 11, 1134–1146. [Google Scholar] [CrossRef]
- Karikari, T.; Nagel, D.A.; Grainger, A.; Clarke-Bland, C.; Crowe, J.; Hill, E.J.; Moffat, K.G. Distinct Conformations, Aggregation and Cellular Internalization of Different Tau Strains. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Maziuk, B.F.; Apicco, D.; Cruz, A.L.; Jiang, L.; Ash, P.E.A.; da Rocha, E.L.; Zhang, C.; Yu, W.H.; Leszyk, J.; Abisambra, J.F.; et al. RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol. Commun. 2018, 6, 1–14. [Google Scholar] [CrossRef]
- Ke, Y.; Dramiga, J.; Schütz, U.; Kril, J.J.; Ittner, L.M.; Schröder, H.; Götz, J. Tau-Mediated Nuclear Depletion and Cytoplasmic Accumulation of SFPQ in Alzheimer’s and Pick’s Disease. PLoS ONE 2012, 7, e35678. [Google Scholar] [CrossRef]
- Hales, C.M.; Dammer, E.B.; Diner, I.; Yi, H.; Seyfried, N.T.; Gearing, M.; Glass, J.D.; Montine, T.J.; Levey, A.I.; Lah, J.J. Aggregates of small nuclear ribonucleic acids (snRNAs) in Alzheimer’s disease. Brain Pathol. 2014, 24, 344–351. [Google Scholar] [CrossRef]
- Ash, P.E.; Vanderweyde, T.E.; Youmans, K.L.; Apicco, D.; Wolozin, B. Pathological stress granules in Alzheimer’s disease. Brain Res. 2014, 1584, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Brophy, C.; Hickling, M.; Neve, J.; Furger, A. Alternative cleavage and polyadenylation of genes associated with protein turnover and mitochondrial function are deregulated in Parkinson’s, Alzheimer’s and ALS disease. BMC Med. Genom. 2019, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Fu, Y.; Yamazaki, Y.; Ren, Y.; Davis, M.D.; Liu, C.-C.; Lu, W.; Wang, X.; Chen, K.; Cherukuri, Y.; et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and regulation of alternative Pre-mRNA splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2016, 18, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Łabno, A.; Tomecki, R.; Dziembowski, A. Cytoplasmic RNA decay pathways-Enzymes and mechanisms. Biochim. Biophys. Acta (BBA) 2016, 1863, 3125–3147. [Google Scholar] [CrossRef]
- Dharshini, S.A.P.; Taguchi, Y.-H.; Gromiha, M.M. Investigating the energy crisis in Alzheimer disease using transcriptome study. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Morabito, S.; Miyoshi, E.; Michael, N.; Swarup, V. Integrative genomics approach identifies conserved transcriptomic networks in Alzheimer’s disease. Hum. Mol. Genet. 2020, 29, 2899–2919. [Google Scholar] [CrossRef]
- Lai, M.K.; Esiri, M.M.; Tan, M.G. Genome-wide profiling of alternative splicing in Alzheimer’s disease. Genom. Data 2014, 2, 290–292. [Google Scholar] [CrossRef]
- Martiskainen, H.; Viswanathan, J.; Nykänen, N.-P.; Kurki, M.; Helisalmi, S.; Natunen, T.; Sarajärvi, T.; Kurkinen, K.M.; Pursiheimo, J.-P.; Rauramaa, T.; et al. Transcriptomics and mechanistic elucidation of Alzheimer’s disease risk genes in the brain and in vitro models. Neurobiol. Aging 2015, 36, 1221.e5–1221.e8. [Google Scholar] [CrossRef]
- Dharshini, S.A.P.; Taguchi, Y.-H.; Gromiha, M.M. Exploring the selective vulnerability in Alzheimer disease using tissue specific variant analysis. Genomics 2019, 111, 936–949. [Google Scholar] [CrossRef]
- Han, S.; Initiative, F.A.D.N.; Miller, J.E.; Byun, S.; Kim, D.; Risacher, S.L.; Saykin, A.J.; Lee, Y.; Nho, K. Identification of exon skipping events associated with Alzheimer’s disease in the human hippocampus. BMC Med. Genom. 2019, 12, \linebreak . [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.; Steinberg, S.; Sealock, J.; Karlsson, I.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; Van Der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.; Hill, M.; Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 2020, 23, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Cooper, S.; Liu, J.Z.; Barrio-Hernandez, I.; Bello, E.; Kumasaka, N.; Young, A.M.H.; Franklin, R.J.M.; Johnson, T.; Estrada, K.; et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat. Genet. 2021, 53, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xu, J.; Liu, H.; Zhang, S.; Li, M.; Zhou, Y.; Qin, W.; Li, M.J.; Yu, C.; Initiative, F.T.A.D.N. Hippocampal transcriptome-wide association study and neurobiological pathway analysis for Alzheimer’s disease. PLoS Genet. 2021, 17, e1009363. [Google Scholar] [CrossRef] [PubMed]
- Courtney, E.; Kornfeld, S.; Janitz, K.; Janitz, M. Transcriptome profiling in neurodegenerative disease. J. Neurosci. Methods 2010, 193, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Biamonti, G.; Amato, A.; Belloni, E.; Di Matteo, A.; Infantino, L.; Pradella, D.; Ghigna, C. Alternative splicing in Alzheimer’s disease. Aging Clin. Exp. Res. 2019, 33, 747–758. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and -Secretase: Structure, Function, and Role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 2, a006304. [Google Scholar] [CrossRef]
- Rogaev, E.; Sherrington, R.; Wu, C.; Levesque, G.; Liang, Y.; Rogaeva, E.; Ikeda, M.; Holman, K.; Lin, C.; Lukiw, W.; et al. Analysis of the 5′ Sequence, Genomic Structure, and Alternative Splicing of thepresenilin-1Gene (PSEN1) Associated with Early Onset Alzheimer Disease. Genomics 1997, 40, 415–424. [Google Scholar] [CrossRef] [PubMed]
- De Jonghe, C.; Cruts, M.; Rogaeva, E.A.; Tysoe, C.; Singleton, A.; Vanderstichele, H.; Meschino, W.; Dermaut, B.; Vanderhoeven, I.; Backhovens, H.; et al. Aberrant Splicing in the Presenilin-1 Intron 4 Mutation Causes Presenile Alzheimer’s Disease by Increased A 42 Secretion. Hum. Mol. Genet. 1999, 8, 1529–1540. [Google Scholar] [CrossRef] [PubMed]
- Kwok, J.B.J.; Halliday, G.M.; Brooks, W.S.; Dolios, G.; Laudon, H.; Murayama, O.; Hallupp, M.; Badenhop, R.F.; Vickers, J.; Wang, R.; et al. Presenilin-1 Mutation L271V Results in Altered Exon 8 Splicing and Alzheimer’s Disease with Non-cored Plaques and No Neuritic Dystrophy. J. Biol. Chem. 2003, 278, 6748–6754. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Zwart, R.; Baas, F. Alternative splicing in the N-terminus of Alzheimer?s presenilin 1. Neurogenetics 2004, 5, 223–227. [Google Scholar] [CrossRef]
- Braggin, J.E.; Bucks, S.A.; Course, M.M.; Smith, C.L.; Sopher, B.; Osnis, L.; Shuey, K.D.; Domoto-Reilly, K.; Caso, C.; Kinoshita, C.; et al. Alternative splicing in a presenilin 2 variant associated with Alzheimer disease. Ann. Clin. Transl. Neurol. 2019, 6, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Guo, Z. Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J. Neurochem. 2013, 126, 305–311. [Google Scholar] [CrossRef]
- Sandbrink, R.; Masters, C.L.; Beyreuther, K. APP Gene Family Alternative Splicing Generates Functionally Related Isoformsa. Ann. N. Y. Acad. Sci. 1996, 777, 281–287. [Google Scholar] [CrossRef]
- Menéndez-González, M.; Pérez-Pinera, P.; Martínez-Rivera, M.; Calatayud, M.; Menes, B.B. APP Processing and the APP-KPI Domain Involvement in the Amyloid Cascade. Neurodegener. Dis. 2005, 2, 277–283. [Google Scholar] [CrossRef]
- Smith, P.; Al Hashimi, A.; Girard, J.; DeLay, C.; Hebert, S. In Vivo regulation of amyloid precursor protein neuronal splicing by microRNAs. J. Neurochem. 2010, 116, 240–247. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2015, 17, 22–35. [Google Scholar] [CrossRef]
- Gu, J.; Chen, F.; Iqbal, K.; Gong, C.-X.; Wang, X.; Liu, F. Transactive response DNA-binding protein 43 (TDP-43) regulates alternative splicing of tau exon 10: Implications for the pathogenesis of tauopathies. J. Biol. Chem. 2017, 292, 10600–10612. [Google Scholar] [CrossRef]
- Yin, X.; Jiang, X.; Wang, J.; Qian, S.; Liu, F.; Qian, W. SIRT1 Deacetylates SC35 and Suppresses Its Function in Tau Exon 10 Inclusion. J. Alzheimer’s Dis. 2017, 61, 561–570. [Google Scholar] [CrossRef] [PubMed]
- García-Escudero, V.; Ruiz-Gabarre, D.; Gargini, R.; Pérez, M.; García, E.; Cuadros, R.; Hernández, I.H.; Cabrera, J.R.; García-Escudero, R.; Lucas, J.J.; et al. A new non-aggregative splicing isoform of human Tau is decreased in Alzheimer’s disease. Acta Neuropathol. 2021, 142, 159–177. [Google Scholar] [CrossRef]
- Jovanovic, M.; Hengartner, M. miRNAs and apoptosis: RNAs to die for. Oncogene 2006, 25, 6176–6187. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Githinji, J.; McLaughlin, B.; Wilczek, K.; Nolta, J. Role of miRNAs in Neuronal Differentiation from Human Embryonic Stem Cell—Derived Neural Stem Cells. Stem Cell Rev. Rep. 2012, 8, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Bu, H.; Wedel, S.; Cavinato, M.; Jansen-Dürr, P. MicroRNA Regulation of Oxidative Stress-Induced Cellular Senescence. Oxidative Med. Cell. Longev. 2017, 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.I.; Yan, K.-P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Güttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear Export of MicroRNA Precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef]
- Hammond, S.M. Dicing and slicing: The core machinery of the RNA interference pathway. FEBS Lett. 2005, 579, 5822–5829. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Hutvagner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P.D. Asymmetry in the Assembly of the RNAi Enzyme Complex. Cell 2003, 115, 199–208. [Google Scholar] [CrossRef]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs Exhibit Strand Bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef]
- Tijsterman, M.; Plasterk, R.H.A. Dicers at RISC: The mechanism of RNAi. Cell 2004, 117, 1–3. [Google Scholar] [CrossRef]
- Long, J.M.; Maloney, B.; Rogers, J.; Lahiri, D.K. Novel upregulation of amyloid-β precursor protein (APP) by microRNA-346 via targeting of APP mRNA 5′-untranslated region: Implications in Alzheimer’s disease. Mol. Psychiatry 2018, 24, 345–363. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.M.; Krüger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; Kaul, D.; et al. An unconventional role for miRNA: Let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Lauressergues, D.; Couzigou, J.-M.; Clemente, H.S.; Martinez, Y.; Dunand, C.; Bécard, G.; Combier, J.-P. Primary transcripts of microRNAs encode regulatory peptides. Nature 2015, 520, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Dragomir, M.P.; Knutsen, E.; Calin, G. SnapShot: Unconventional miRNA Functions. Cell 2018, 174, 1038–1038.e1. [Google Scholar] [CrossRef] [PubMed]
- Hébert, S.S.; Papadopoulou, A.S.; Smith, P.; Galas, M.-C.; Planel, E.; Silahtaroglu, A.N.; Sergeant, N.; Buée, L.; De Strooper, B. Genetic ablation of Dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum. Mol. Genet. 2010, 19, 3959–3969. [Google Scholar] [CrossRef]
- Patel, N.; Hoang, D.; Miller, N.; Ansaloni, S.; Huang, Q.; Rogers, J.T.; Lee, J.C.; Saunders, A.J. MicroRNAs can regulate hu-man APP levels. Mol. Neurodegener. 2008, 3, 1–6. [Google Scholar] [CrossRef]
- Hebert, S.; Horré, K.; Nicolaï, L.; Bergmans, B.; Papadopoulou, A.S.; Delacourte, A.; De Strooper, B. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol. Dis. 2009, 33, 422–428. [Google Scholar] [CrossRef]
- Vilardo, E.; Barbato, C.; Ciotti, M.; Cogoni, C.; Ruberti, F. MicroRNA-101 Regulates Amyloid Precursor Protein Expression in Hippocampal Neurons. J. Biol. Chem. 2010, 285, 18344–18351. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, C.; Zhu, J.; Shu, P.; Yin, B.; Gong, Y.; Qiang, B.; Yuan, J.; Peng, X. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol. Aging 2012, 33, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Barbato, C.; Giacovazzo, G.; Albiero, F.; Scardigli, R.; Scopa, C.; Ciotti, M.T.; Strimpakos, G.; Coccurello, R.; Ruberti, F. Cognitive Decline and Modulation of Alzheimer’s Disease-Related Genes After Inhibition of MicroRNA-101 in Mouse Hippocampal Neurons. Mol. Neurobiol. 2020, 57, 3183–3194. [Google Scholar] [CrossRef]
- Long, J.; Ray, B.; Lahiri, D.K. MicroRNA-153 Physiologically Inhibits Expression of Amyloid-β Precursor Protein in Cultured Human Fetal Brain Cells and Is Dysregulated in a Subset of Alzheimer Disease Patients. J. Biol. Chem. 2012, 287, 31298–31310. [Google Scholar] [CrossRef]
- Hébert, S.; Horré, K.; Nicolaï, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/-secretase expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef]
- Lei, X.; Lei, L.; Zhang, Z.; Zhang, Z.; Cheng, Y. Downregulated miR-29c correlates with increased BACE1 expression in spo-radic Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2015, 8, 1565–1574. [Google Scholar] [PubMed]
- Wang, W.-X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The Expression of MicroRNA miR-107 Decreases Early in Alzheimer’s Disease and May Accelerate Disease Progression through Regulation of -Site Amyloid Precursor Protein-Cleaving Enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef]
- Nelson, P.T.; Wang, W.-X. MiR-107 is Reduced in Alzheimer’s Disease Brain Neocortex: Validation Study. J. Alzheimer’s Dis. 2010, 21, 75–79. [Google Scholar] [CrossRef]
- Liu, W.; Cai, H.; Lin, M.; Zhu, L.; Gao, L.; Zhong, R.; Bi, S.; Xue, Y.; Shang, X. MicroRNA-107 prevents amyloid-beta induced blood-brain barrier disruption and endothelial cell dysfunction by targeting Endophilin-1. Exp. Cell Res. 2016, 343, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Shu, B.; Zhang, X.; Du, G.; Fu, Q.; Huang, L. MicroRNA-107 prevents amyloid-β-induced neurotoxicity and memory impairment in mice. Int. J. Mol. Med. 2017, 41, 1665–1672. [Google Scholar] [CrossRef]
- Boissonneault, V.; Plante, I.; Rivest, S.; Provost, P. MicroRNA-298 and MicroRNA-328 Regulate Expression of Mouse β-Amyloid Precursor Protein-converting Enzyme 1. J. Biol. Chem. 2009, 284, 1971–1981. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.-C.; Wang, L.-M.; Wang, M.; Song, B.; Tan, S.; Teng, J.-F.; Duan, D.-X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-β production by targeting BACE1. Brain Res. Bull. 2012, 88, 596–601. [Google Scholar] [CrossRef]
- Liu, T.; Huang, Y.; Chen, J.; Chi, H.; Yu, Z.; Wang, J.; Chen, C. Attenuated ability of BACE1 to cleave the amyloid precursor protein via silencing long noncoding RNA BACE1-AS expression. Mol. Med. Rep. 2014, 10, 1275–1281. [Google Scholar] [CrossRef]
- Zhang, Y.; Xing, H.; Guo, S.; Zheng, Z.; Wang, H.; Xu, D. MicroRNA-135b has a neuroprotective role via targeting of β-site APP-cleaving enzyme 1. Exp. Ther. Med. 2016, 12, 809–814. [Google Scholar] [CrossRef]
- Xie, H.; Zhao, Y.; Zhou, Y.; Liu, L.; Liu, Y.; Wang, D.; Zhang, S.; Yang, M. MiR-9 Regulates the Expression of BACE1 in Dementia Induced by Chronic Brain Hypoperfusion in Rats. Cell. Physiol. Biochem. 2017, 42, 1213–1226. [Google Scholar] [CrossRef]
- Chopra, N.; Wang, R.; Maloney, B.; Nho, K.; Beck, J.S.; Pourshafie, N.; Niculescu, A.; Saykin, A.J.; Rinaldi, C.; Counts, S.E.; et al. MicroRNA-298 reduces levels of human amyloid-β precursor protein (APP), β-site APP-converting enzyme 1 (BACE1) and specific tau protein moieties. Mol. Psychiatry 2020, 1–22. [Google Scholar] [CrossRef]
- Tiribuzi, R.; Crispoltoni, L.; Porcellati, S.; Di Lullo, M.; Florenzano, F.; Pirro, M.; Bagaglia, F.; Kawarai, T.; Zampolini, M.; Orlacchio, A.; et al. miR128 up-regulation correlates with impaired amyloid β(1–42) degradation in monocytes from patients with sporadic Alzheimer’s disease. Neurobiol. Aging 2014, 35, 345–356. [Google Scholar] [CrossRef]
- Zhao, Y.; Jaber, V.; Lukiw, W.J. Over-Expressed Pathogenic miRNAs in Alzheimer’s Disease (AD) and Prion Disease (PrD) Drive Deficits in TREM2-Mediated Aβ42 Peptide Clearance. Front. Aging Neurosci. 2016, 8, 140. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Zhao, Y.; Dua, P.; Rogaev, E.I.; Lukiw, W.J. microRNA-34a-Mediated Down-Regulation of the Microglial-Enriched Triggering Receptor and Phagocytosis-Sensor TREM2 in Age-Related Macular Degeneration. PLoS ONE 2016, 11, e0150211. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.; et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef]
- Wang, Z.; Qin, W.; Wei, C.; Tang, Y.; Zhao, L.; Jin, H.; Li, Y.; Wang, Q.; Luan, X.; He, J.; et al. The microRNA-1908 up-regulation in the peripheral blood cells impairs amyloid clearance by targeting ApoE. Int. J. Geriatr. Psychiatry 2018, 33, 980–986. [Google Scholar] [CrossRef]
- Zhao, Y.; Alexandrov, P.N.; Jaber, V.; Lukiw, W.J. Deficiency in the Ubiquitin Conjugating Enzyme UBE2A in Alzheimer’s Disease (AD) is Linked to Deficits in a Natural Circular miRNA-7 Sponge (circRNA; ciRS-7). Genes 2016, 7, 116. [Google Scholar] [CrossRef]
- Subramanian, M.; Hyeon, S.J.; Das, T.; Suh, Y.S.; Kim, Y.K.; Lee, J.-S.; Song, E.J.; Ryu, H.; Yu, K. UBE4B, a microRNA-9 target gene, promotes autophagy-mediated Tau degradation. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Maqbool, M.; Mobashir, M.; Hoda, N. Pivotal role of glycogen synthase kinase-3: A therapeutic target for Alzheimer’s disease. Eur. J. Med. Chem. 2016, 107, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Deng, J.; Chu, X.; Zhao, Y.; Guo, Y. Role of Post-Transcriptional Control of Calpain by miR-124-3p in the Development of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 67, 571–581. [Google Scholar] [CrossRef]
- Smith, P.Y.; Delay, C.; Girard, J.; Papon, M.-A.; Planel, E.; Sergeant, N.; Buée, L.; Hébert, S.S. MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Hum. Mol. Genet. 2011, 20, 4016–4024. [Google Scholar] [CrossRef] [PubMed]
- Bazrgar, M.; Khodabakhsh, P.; Mohagheghi, F.; Prudencio, M.; Ahmadiani, A. Brain microRNAs dysregulation: Implication for missplicing and abnormal post-translational modifications of tau protein in Alzheimer’s disease and related tauopathies. Pharmacol. Res. 2020, 155, 104729. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Mucke, L. Paths of Convergence: Sirtuins in Aging and Neurodegeneration. Neuron 2008, 58, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Fan, G.; Zhang, J.; Wu, C.; Du, Y.; Ye, H.; Li, Z.; Wang, L.; Zhang, Z.; Zhang, L.; et al. A 9-microRNA Signature in Serum Serves as a Noninvasive Biomarker in Early Diagnosis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 1365–1377. [Google Scholar] [CrossRef]
- Sheinerman, K.S.; Tsivinsky, V.G.; Abdullah, L.; Crawford, F.; Umansky, S.R. Plasma microRNA biomarkers for detection of mild cognitive impairment: Biomarker Validation Study. Aging 2013, 5, 925–938. [Google Scholar] [CrossRef]
- Galimberti, D.; Villa, C.; Fenoglio, C.; Serpente, M.; Ghezzi, L.; Cioffi, S.M.; Arighi, A.; Fumagalli, G.; Scarpini, E. Circulating miRNAs as Potential Biomarkers in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 42, 1261–1267. [Google Scholar] [CrossRef]
- Wiedrick, J.T.; Phillips, J.I.; Lusardi, T.A.; McFarland, T.J.; Lind, B.; Sandau, U.S.; Harrington, C.A.; Lapidus, J.A.; Galasko, D.R.; Quinn, J.F.; et al. Validation of MicroRNA Biomarkers for Alzheimer’s Disease in Human Cerebrospinal Fluid. J. Alzheimer’s Dis. 2019, 67, 875–891. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, C.; Mostile, G.; Baglieri, G.; Giunta, F.; Luca, A.; Raciti, L.; Zappia, M.; Purrello, M.; Ragusa, M.; Nicoletti, A. Specific Signatures of Serum miRNAs as Potential Biomarkers to Discriminate Clinically Similar Neurodegenerative and Vascular-Related Diseases. Cell. Mol. Neurobiol. 2019, 40, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Danborg, P.B.; Simonsen, A.H.; Waldemar, G.; Heegaard, N.H.H. The potential of microRNAs as biofluid markers of neuro-degenerative diseases-a systematic review. Biomarkers 2014, 19, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, S.; Wragg, N.; Ghosh, S.; Stolzing, A. Systematic Review of miRNA as Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 6156–6167. [Google Scholar] [CrossRef]
- Denk, J.; Oberhauser, F.; Kornhuber, J.; Wiltfang, J.; Fassbender, K.; Schroeter, M.L.; Volk, A.E.; Diehl-Schmid, J.; Prudlo, J.; Danek, A.; et al. Specific serum and CSF microRNA profiles distinguish sporadic behavioural variant of frontotemporal dementia compared with Alzheimer patients and cognitively healthy controls. PLoS ONE 2018, 13, e0197329. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak-Wolf, A.; Maier, L.; Mackowiak, S.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-Type Specific Features of Circular RNA Expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Jeck, W.; Sharpless, N. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Lasda, E.; Parker, R. Circular RNAs: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA Biogenesis Competes with Pre-mRNA Splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef]
- Zhang, X.-O.; Wang, H.-B.; Zhang, Y.; Lu, X.; Chen, L.-L.; Yang, L. Complementary Sequence-Mediated Exon Circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef]
- Mo, D.; Li, X.; Raabe, C.; Rozhdestvensky, T.; Skryabin, B.; Brosius, J. Circular RNA Encoded Amyloid Beta Peptides—A Novel Putative Player in Alzheimer’s Disease. Cells 2020, 9, 2196. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. Circular RNA (circRNA) in Alzheimer’s disease (AD). Front. Genet. 2013, 4, 307. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Shang, H.; Chen, X.; Yang, S.; Qu, Y.; Ding, J.; Li, X. Circular RNA circ_0000950 promotes neuron apoptosis, suppresses neurite outgrowth and elevates inflammatory cytokines levels via directly sponging miR-103 in Alzheimer’s disease. Cell Cycle 2019, 18, 2197–2214. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Tan, L.; Wang, X. Circular HDAC9/microRNA-138/Sirtuin-1 Pathway Mediates Synaptic and Amyloid Precursor Protein Processing Deficits in Alzheimer’s Disease. Neurosci. Bull. 2019, 35, 877–888. [Google Scholar] [CrossRef]
- Ma, N.; Pan, J.; Wen, Y.; Wu, Q.; Yu, B.; Chen, X.; Wan, J.; Zhang, W. circTulp4 functions in Alzheimer’s disease pathogenesis by regulating its parental gene, Tulp4. Mol. Ther. 2021, 29, 2167–2181. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, Y.; Zhang, L.; Dong, Y.; Ji, H.; Shen, L. The Potential Markers of Circulating microRNAs and long non-coding RNAs in Alzheimer’s Disease. Aging Dis. 2019, 10, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Dube, U.; (Dian), T.D.I.A.N.; Del Aguila, J.L.; Li, Z.; Budde, J.P.; Jiang, S.; Hsu, S.; Ibanez, L.; Fernandez, M.V.; Farias, F.; et al. An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat. Neurosci. 2019, 22, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.; Preiss, T. Circular RNAs: Splicing’s enigma variations. EMBO J. 2013, 32, 923–925. [Google Scholar] [CrossRef]
- Hansen, T.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Chen, T.; Yao, Q.; Zheng, L.; Zhang, Z.; Wang, J.; Hu, Z.; Cui, H.; Han, Y.; Han, X.; et al. The circular RNA ci RS -7 promotes APP and BACE 1 degradation in an NF -κB-dependent manner. FEBS J. 2017, 284, 1096–1109. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, F.; Bao, S.; Sun, J. Systematic Characterization of Circular RNA-Associated CeRNA Network Identified Novel circRNA Biomarkers in Alzheimer’s Disease. Front. Bioeng. Biotechnol. 2019, 7, 222. [Google Scholar] [CrossRef]
- Long, J.; Lahiri, D.K. MicroRNA-101 downregulates Alzheimer’s amyloid-β precursor protein levels in human cell cultures and is differentially expressed. Biochem. Biophys. Res. Commun. 2011, 404, 889–895. [Google Scholar] [CrossRef]
- Pereira, P.; Tomás, J.F.M.; Queiroz, J.; Figueiras, A.; Sousa, F. Recombinant pre-miR-29b for Alzheimer’s disease therapeutics. Sci. Rep. 2016, 6, 19946. [Google Scholar] [CrossRef]
- Diling, C.; Yinrui, G.; Longkai, Q.; Xiaocui, T.; Yadi, L.; Xin, Y.; Guoyan, H.; Ou, S.; Tianqiao, Y.; Dongdong, W.; et al. Circular RNA NF1-419 enhances autophagy to ameliorate senile dementia by binding Dynamin-1 and Adaptor protein 2 B1 in AD-like mice. Aging 2019, 11, 12002–12031. [Google Scholar] [CrossRef]
- Huang, J.-L.; Qin, M.-C.; Zhou, Y.; Xu, Z.-H.; Yang, S.-M.; Zhang, F.; Zhong, J.; Liang, M.-K.; Chen, B.; Zhang, W.-Y.; et al. Comprehensive analysis of differentially expressed profiles of Alzheimer’s disease associated circular RNAs in an Alzheimer’s disease mouse model. Aging 2018, 10, 253–265. [Google Scholar] [CrossRef]
- Huang, J.-L.; Xu, Z.-H.; Yang, S.-M.; Yu, C.; Zhang, F.; Qin, M.-C.; Zhou, Y.; Zhong, Z.-G.; Wu, D.-P. Identification of Differentially Expressed Profiles of Alzheimer’s Disease Associated Circular RNAs in a Panax Notoginseng Saponins-Treated Alzheimer’s Disease Mouse Model. Comput. Struct. Biotechnol. J. 2018, 16, 523–531. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2020, 22, 96–118. [Google Scholar] [CrossRef]
- Lee, D.Y.; Moon, J.; Lee, S.-T.; Jung, K.-H.; Park, D.-K.; Yoo, J.-S.; Sunwoo, J.-S.; Byun, J.-I.; Shin, J.-W.; Jeon, D.; et al. Distinct Expression of Long Non-Coding RNAs in an Alzheimer’s Disease Model. J. Alzheimer’s Dis. 2015, 45, 837–849. [Google Scholar] [CrossRef]
- Fang, M.; Zhang, P.; Zhao, Y.; Liu, X. Bioinformatics and co-expression network analysis of differentially expressed lncRNAs and mRNAs in hippocampus of APP/PS1 transgenic mice with Alzheimer disease. Am. J. Transl. Res. 2017, 9, 1381–1391. [Google Scholar]
- Zhou, X.; Xu, J. Identification of Alzheimer’s disease–associated long noncoding RNAs. Neurobiol. Aging 2015, 36, 2925–2931. [Google Scholar] [CrossRef] [PubMed]
- Annese, A.; Manzari, C.; Lionetti, C.; Picardi, E.; Horner, D.S.; Chiara, M.; Caratozzolo, M.F.; Tullo, A.; Fosso, B.; Pesole, G.; et al. Whole transcriptome profiling of Late-Onset Alzheimer’s Disease patients provides insights into the molecular changes involved in the disease. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Li, H.; Zhao, J.; Cui, J.; Hu, G. Identification of age- and gender-associated long noncoding RNAs in the human brain with Alzheimer’s disease. Neurobiol. Aging 2019, 81, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, Y.; Faghihi, M.A.; Magistri, M.; Alvarez-Garcia, O.; Lotz, M.; Wahlestedt, C. Antisense RNA Controls LRP1 Sense Transcript Expression through Interaction with a Chromatin-Associated Protein, HMGB2. Cell Rep. 2015, 11, 967–976. [Google Scholar] [CrossRef]
- Massone, S.; Ciarlo, E.; Vella, S.; Nizzari, M.; Florio, T.; Russo, C.; Cancedda, R.; Pagano, A. NDM29, a RNA polymerase III-dependent non coding RNA, promotes amyloidogenic processing of APP and amyloid β secretion. Biochim. Biophys. Acta (BBA) 2012, 1823, 1170–1177. [Google Scholar] [CrossRef]
- Massone, S.; Vassallo, I.; Fiorino, G.; Castelnuovo, M.; Barbieri, F.; Borghi, R.; Tabaton, M.; Robello, M.; Gatta, E.; Russo, C.; et al. 17A, a novel non-coding RNA, regulates GABA B alternative splicing and signaling in response to inflammatory stimuli and in Alzheimer disease. Neurobiol. Dis. 2011, 41, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Gavazzo, P.; Vassalli, M.; Costa, D.; Pagano, A. Novel ncRNAs transcribed by Pol III and elucidation of their functional relevance by biophysical approaches. Front. Cell. Neurosci. 2013, 7, 203. [Google Scholar] [CrossRef]
- Ciarlo, E.; Massone, S.; Penna, I.; Nizzari, M.; Gigoni, A.; Dieci, G.; Russo, C.; Florio, T.; Cancedda, R.; Pagano, A. An intronic ncRNA-dependent regulation of SORL1 expression affecting Aβ formation is upregulated in post-mortem Alzheimer’s disease brain samples. Dis. Model. Mech. 2012, 6, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Chen, C.; Wu, R.; Dong, T.; Hu, X.; Yao, Y.; Zhang, Y. Long Noncoding RNA EBF3-AS Promotes Neuron Apoptosis in Alzheimer’s Disease. DNA Cell Biol. 2018, 37, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; E Wood, D.; Sahagan, B.G.; E Morgan, T.; E Finch, C.; Iii, G.S.L.; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of β-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef]
- Kang, M.-J.; Abdelmohsen, K.; Hutchison, E.R.; Mitchell, S.J.; Grammatikakis, I.; Guo, R.; Noh, J.H.; Martindale, J.L.; Yang, X.; Lee, E.K.; et al. HuD Regulates Coding and Noncoding RNA to Induce APP→Aβ Processing. Cell Rep. 2014, 7, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Ni, H.; Yu, Y.; Zhang, M.; Wu, M.; Wang, Q.; Wang, L.; Xu, S.; Xu, Z.; Xu, C.; et al. BACE1-AS prevents BACE1 mRNA degradation through the sequestration of BACE1-targeting miRNAs. J. Chem. Neuroanat. 2019, 98, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.P.; Neyt, C.; Wilkins, S.J.; Askarian-Amiri, M.E.; Sunkin, S.M.; Perkins, A.C.; Mattick, J.S. Complex architecture and regulated expression of the Sox2ot locus during vertebrate development. RNA 2009, 15, 2013–2027. [Google Scholar] [CrossRef] [PubMed]
- Askarian-Amiri, M.E.; Seyfoddin, V.; Smart, C.E.; Wang, J.; Kim, J.E.; Hansji, H.; Baguley, B.C.; Finlay, G.J.; Leung, E.Y. Emerging Role of Long Non-Coding RNA SOX2OT in SOX2 Regulation in Breast Cancer. PLoS ONE 2014, 9, e102140. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-Y.; Wang, G.-Q.; Wang, N.-N.; Yu, Q.-Y.; Liu, R.-L.; Shi, W.-Q. The long-non-coding RNA NEAT1 is a novel target for Alzheimer’s disease progression via miR-124/BACE1 axis. Neurol. Res. 2019, 41, 489–497. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Y.; Xu, N.; Zhang, S.; Wang, S.; Mao, Y.; Zhu, Y.; Li, B.; Jiang, Y.; Tan, Y.; et al. NEAT1 regulates neuroglial cell mediating Aβ clearance via the epigenetic regulation of endocytosis-related genes expression. Experientia 2019, 76, 3005–3018. [Google Scholar] [CrossRef]
- Ke, S.; Yang, Z.; Yang, F.; Wang, X.; Tan, J.; Liao, B. Long Noncoding RNA NEAT1 Aggravates Aβ-Induced Neuronal Damage by Targeting miR-107 in Alzheimer’s Disease. Yonsei Med. J. 2019, 60, 640–650. [Google Scholar] [CrossRef]
- Parenti, R.; Paratore, S.; Torrisi, A.; Cavallaro, S. A natural antisense transcript against Rad18, specifically expressed in neurons and upregulated during β-amyloid-induced apoptosis. Eur. J. Neurosci. 2007, 26, 2444–2457. [Google Scholar] [CrossRef]
- Guo, C.-C.; Jiao, C.-H.; Gao, Z.-M. Silencing of LncRNA BDNF-AS attenuates Aβ25-35-induced neurotoxicity in PC12 cells by suppressing cell apoptosis and oxidative stress. Neurol. Res. 2018, 40, 795–804. [Google Scholar] [CrossRef]
- Mus, E.; Hof, P.R.; Tiedge, H. Dendritic BC200 RNA in aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 10679–10684. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, J.; Wang, M.; Li, X.; Gong, H.; Tang, H.; Chen, L.; Wan, L.; Liu, Q. Activity dependent LoNA regulates translation by coordinating rRNA transcription and methylation. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Handley, P.; Wong, L.; McLachlan, D.R.C. BC200 RNA in normal human neocortex, non-Alzheimer dementia (NAD), and senile dementia of the Alzheimer type (AD). Neurochem. Res. 1992, 17, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Bujo, H.; Saito, Y. A Novel Member of the LDL Receptor Gene Family with Eleven Binding Repeats is Structurally Related to Neural Adhesion Molecules and a Yeast Vacuolar Protein Sorting Receptor. J. Atheroscler. Thromb. 1997, 4, 20–26. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rogaeva, E.; Meng, Y.; Lee, J.H.; Gu, Y.; Kawarai, T.; Zou, F.; Katayama, T.; Baldwin, C.T.; Cheng, R.; Hasegawa, H.; et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 2007, 39, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Barral, S.; Reitz, C. The neuronal sortilin-related receptor gene SORL1 and late-onset Alzheimer’s disease. Curr. Neurol. Neurosci. Rep. 2008, 8, 384–391. [Google Scholar] [CrossRef]
- Matsui, T.; Ingelsson, M.; Fukumoto, H.; Ramasamy, K.; Kowa, H.; Frosch, M.P.; Irizarry, M.C.; Hyman, B.T. Expression of APP pathway mRNAs and proteins in Alzheimer’s disease. Brain Res. 2007, 1161, 116–123. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| GENE NAME | Description | Tau Association | Aβ Association | References |
|---|---|---|---|---|
| MAPT | Microtubule-associated protein tau | YES | YES | [84] |
| MSI1 | Musashi 1 | YES | NO | [85,86] |
| MSI2 | Musashi 2 | YES | NO | [85,86] |
| SNRNP70 | U1 small nuclear ribonucleoprotein 70 kDa | YES | NO | [51,87,88] |
| LUC7L3 | LUC7 like 3 pre-mRNA splicing factor | YES | NO | [38,51] |
| SRRM2 | serine/arginine repetitive matrix 2 | YES | YES | [51,87,88] |
| PRPF40A | pre-mRNA processing factor 40 homolog A | NO | NO | [51] |
| ADP ribosylation factor like GTPase 6 interacting protein 4 | NO | NO | [51] | |
| THOC2 | THO complex 2 | NO | NO | [51] |
| PRPF4B | pre-mRNA processing factor 4B | NO | NO | [51] |
| RNF20 | ring finger protein 20 | NO | NO | [51] |
| RBM15 | RNA binding motif protein 15 | NO | NO | [51] |
| NKAP | NFKB-activating protein | NO | NO | [51] |
| RNPS1 | RNA binding protein with serine-rich domain 1 | NO | NO | [51] |
| ACIN1 | apoptotic chromatin condensation inducer 1 | NO | NO | [51] |
| GPATCH8 | G-patch domain containing 8 | NO | NO | [51] |
| EIF3A | eukaryotic translation initiation factor 3 subunit A | YES | NO | [51,89] |
| YTHDC1 | YTH domain containing 1 | NO | NO | [51] |
| SRRT | Serrate RNA effector molecule | YES | YES | [51,89] |
| SRSF11 | Serine- and arginine-rich splicing factor 11 | NO | NO | [51] |
| PLEC | Plectin | YES | NO | [51,90] |
| TCERG1 | transcription elongation regulator 1 | YES | NO | [51,90] |
| PRPF38B | pre-mRNA processing factor 38B | NO | NO | [51] |
| Zinc finger protein 638 | NO | NO | [51] | |
| CPSF6 | Cleavage- and polyadenylation-specific factor 6 | NO | NO | [51] |
| SNRPA | U1 small nuclear RNP-specific A | YES | NO | [38,87,88] |
| SYNJ1 | Synaptojanin 1 | YES | NO | [87,91] |
| RIMS1 | Regulating synaptic membrane exocytosis 1 | NO | NO | [87] |
| DDX46 | DEAD-Box Helicase 46 | NO | NO | [87] |
| TARDBP | TAR DNA-binding protein 43 | YES | YES | [79,92,93,94,95] |
| TIA1 | T-cell intracellular antigen 1 | YES | NO | [79] |
| G3BP1 | Ras GTPase-activating protein-binding protein 1 | NO | NO | [79] |
| ZFP36 | ZFP36 Ring Finger Protein | YES | NO | [79] |
| FUS | Fused in sarcoma | NO | NO | [79] |
| SOD1 | Superoxide dismutase | YES | YES | [96,97] |
| RBM45 | RNA binding motif protein 45 | NO | NO | [98] |
| SRSF6 | Serine- and arginine-rich splicing factor 6 | YES | YES | [89] |
| SRRM1 | serine/arginine repetitive matrix 1 | YES | NO | [89] |
| MAK16 | MAK16 homolog | NO | YES | [89] |
| ABCF1 | ATP binding cassette subfamily F member 1 | YES | NO | [89] |
| SRSF1 | Serine- and arginine-rich splicing factor 1 | YES | NO | [89] |
| DDX3X | DEAD-Box helicase 3 X-linked | YES | NO | [89] |
| UTP20 | UTP20 small subunit processome component | YES | NO | [89] |
| SNRPD1 | Small nuclear ribonucleoprotein Sm D1 | YES | NO | [88] |
| SLIRP | SRA stem-loop-interacting RNA-binding protein, mitochondrial | YES | NO | [88,99] |
| U2AF2 | Splicing factor U2AF 65 kDa subunit | NO | NO | [88] |
| UPF2 | Regulator of nonsense transcripts 2 | NO | NO | [88] |
| NCL | Nucleolin | YES | NO | [88,90,100] |
| NUDT21 | Cleavage and polyadenylation specificity factor subunit 5 | YES | YES | [88] |
| CRYZ | Quinone oxidoreductase | NO | NO | [88] |
| DARS2 | Aspartate tRNA ligase, mitochondrial | NO | NO | [88] |
| EWSR1 | EWS RNA binding protein 1 | YES | NO | [101] |
| TAF15 | TATA-Box binding protein-associated factor 15 | YES | NO | [101] |
| RPL7 | Ribosomal protein L7 | YES | NO | [101] |
| DDX5 | DEAD-Box helicase 5 | YES | NO | [101] |
| HNRNPA0 | Heterogeneous nuclear ribonucleoprotein A0 | YES | NO | [101] |
| PABPC1 | Poly(A) binding protein cytoplasmic 1 | YES | NO | [101] |
| DDX6 | DEAD-Box helicase 6 | YES | NO | [101] |
| EIF2S1 | Eukaryotic Translation Initiation Factor 2 Subunit Alpha | YES | NO | [101] |
| SFPQ | Splicing factor proline, glutamine rich | YES | NO | [48,102] |
| circRNA | Function in AD | References |
|---|---|---|
| ciRS-7/ciRS-7/CDR1as | regulating protein levels of APP and β-secretase (BACE1) | [177,199] |
| CircRNA_0000950 | miR-103, function in apoptosis, neurite outgrowth, and neuroinflammation in AD | [200] |
| circHDAC9 | miR-138 sponge regulation of ADAM10 and Aβ production | [201] |
| circTulp4 | interacts with U1 snRNP and RNA polymerase II to modulate the transcription of its parental gene, Tulp4 | [202] |
| circHOMER1 | binding sites for mir miR-651 | [203] |
| circCORO1C | bind to miR-105, which is predicted to target both APP and SNCA42 | [204] |
| circRNA KIAA1586 | sponge for several miRNAs including miR-29b, miR-101, and miR-15a | [203] |
| Process Regulated | circRNA | Function in AD | References |
|---|---|---|---|
| Transcription | LRP1-AS | Inhibits the activity of Hmgb2 to enhance Srebp1a-dependent transcription of Lrp1 | [220] |
| NDM29 | Upregulated in AD. Enhances ratio of Aβ42/Aβ40 and accumulation of Aβ | [221] | |
| Splicing | 17A | Synthesis of an alternative splicing isoform that interferes with GABA B2 signaling and enhances the secretion of amyloid β peptide (Aβ) | [222,223] |
| 51A | Regulates SORL1 gene splicing | [224] | |
| RNA stability | EBF3-AS | Upregulated in AD mouse models Regulates EBF3 stability and promotes neuronal apoptosis | [225] |
| BACE1-AS | Upregulated in AD. Regulates BACE1 stability and enhances Aβ production | [226,227,228] | |
| SOX2OT | Dysregulated in mouse AD models. Regulates SOX2 expression | [229,230] | |
| NEAT1 | Reduced in AD and in mouse AD models. Inhibits the expression of endocytosis-related genes and decreases Aβ clearance Regulates miR-124/BACE1 axis. Target of miR-107 | [231,232,233] | |
| NAT-RAD18 | Upregulated in AD. Regulates RAD18 | [234] | |
| BDNF-AS | Negatively regulates BDNF expression | [235] | |
| Translation | BC200 | Dysregulated in AD Modulates local protein synthesis by targeting EIF4A | [236] |
| LoNA | Upregulated in hippocampus of AD mouse. Regulates synaptic plasticity by regulating ribosomal assembly and protein translation | [237] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rybak-Wolf, A.; Plass, M. RNA Dynamics in Alzheimer’s Disease. Molecules 2021, 26, 5113. https://doi.org/10.3390/molecules26175113
Rybak-Wolf A, Plass M. RNA Dynamics in Alzheimer’s Disease. Molecules. 2021; 26(17):5113. https://doi.org/10.3390/molecules26175113
Chicago/Turabian StyleRybak-Wolf, Agnieszka, and Mireya Plass. 2021. "RNA Dynamics in Alzheimer’s Disease" Molecules 26, no. 17: 5113. https://doi.org/10.3390/molecules26175113
APA StyleRybak-Wolf, A., & Plass, M. (2021). RNA Dynamics in Alzheimer’s Disease. Molecules, 26(17), 5113. https://doi.org/10.3390/molecules26175113