
An Efficient Synthesis of 2-CF3-3-Benzylindoles



Abstract
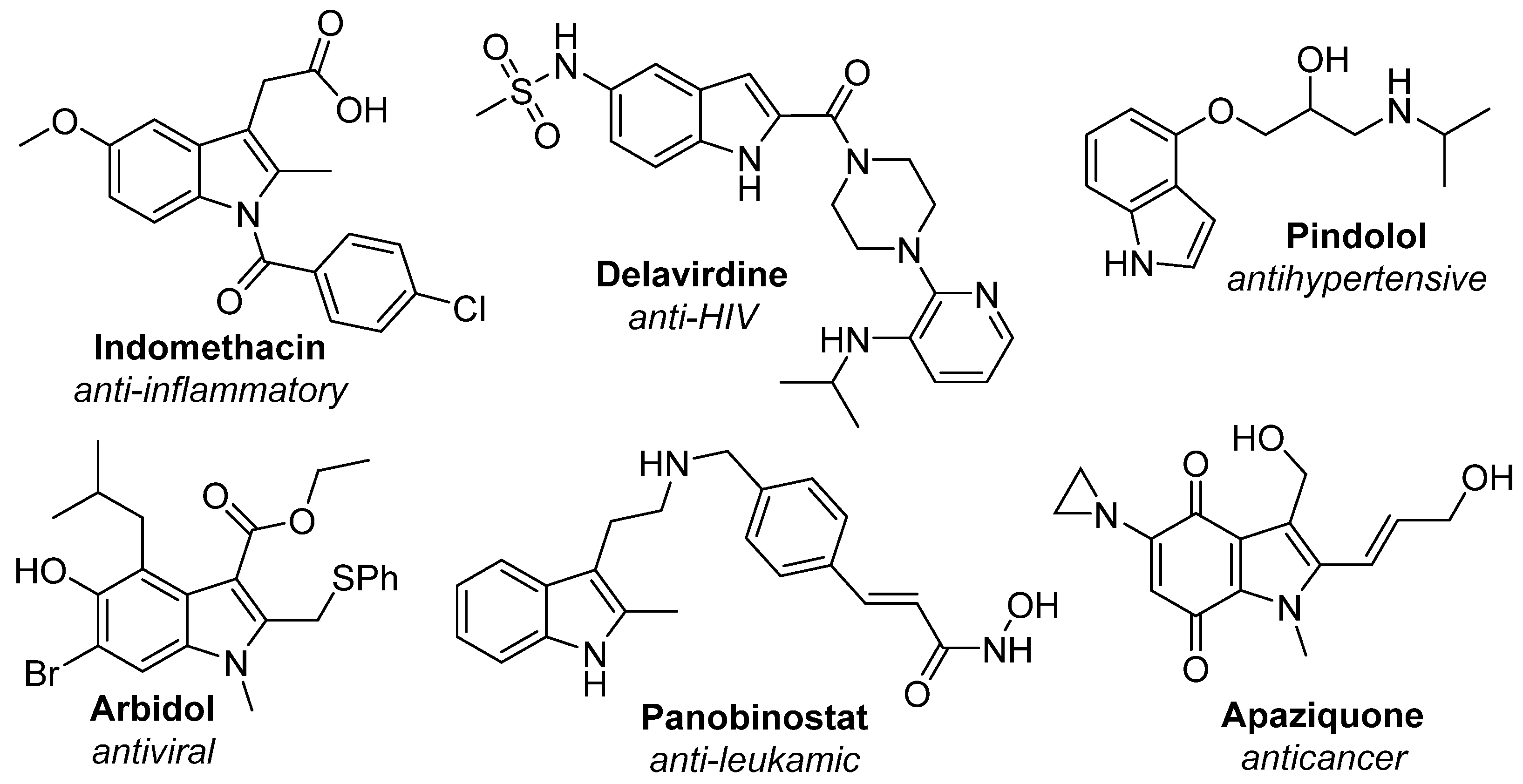
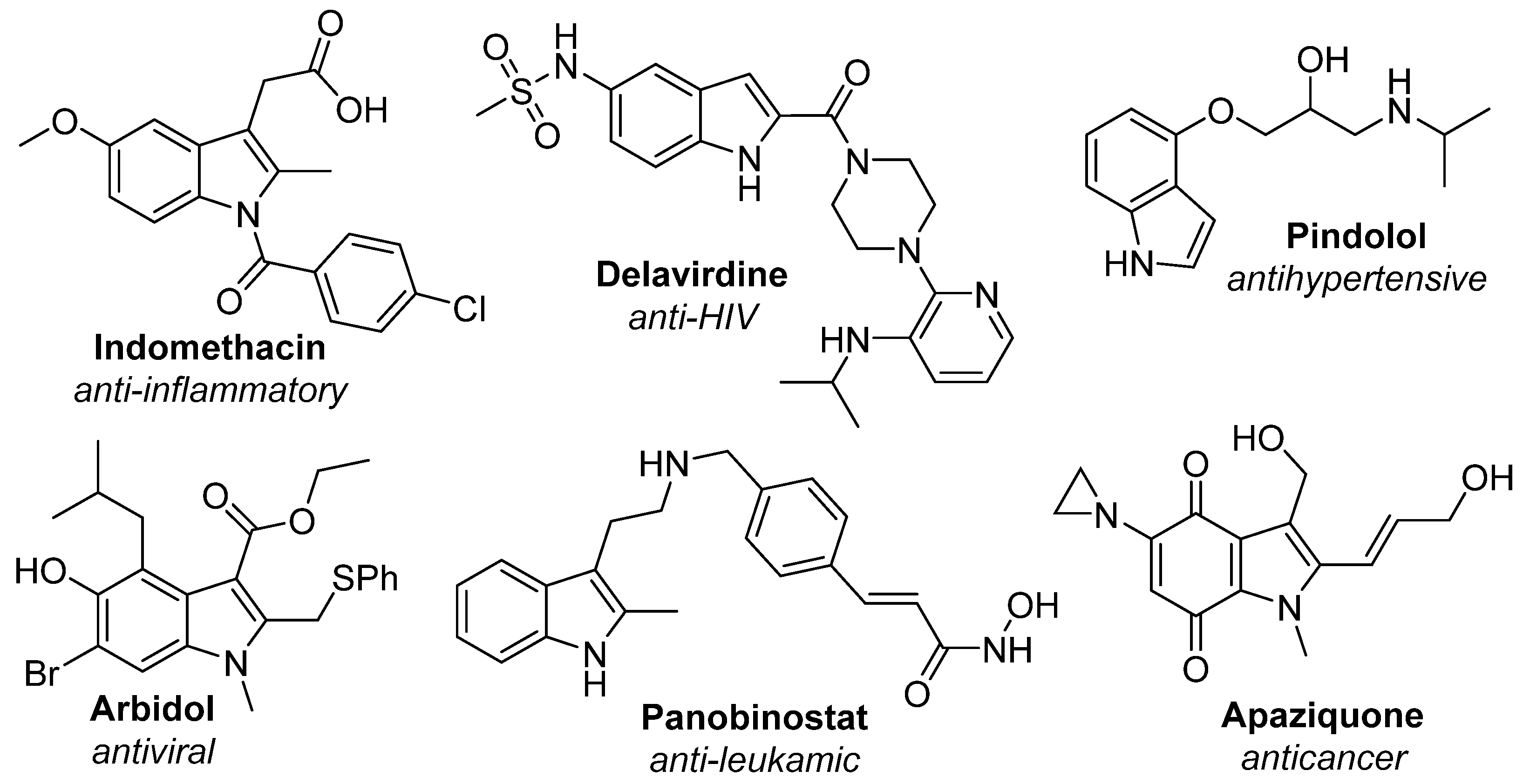
:1. Introduction
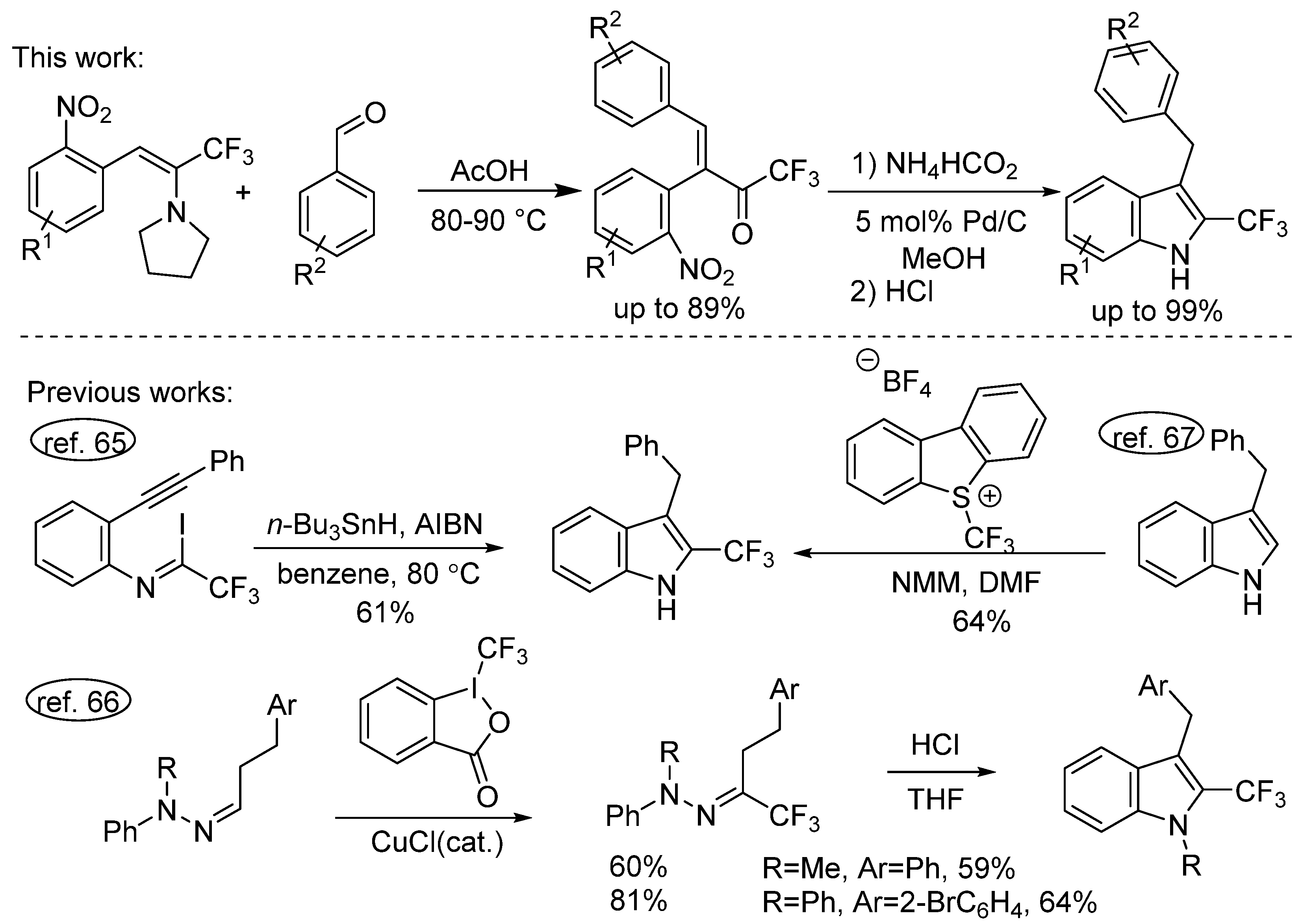
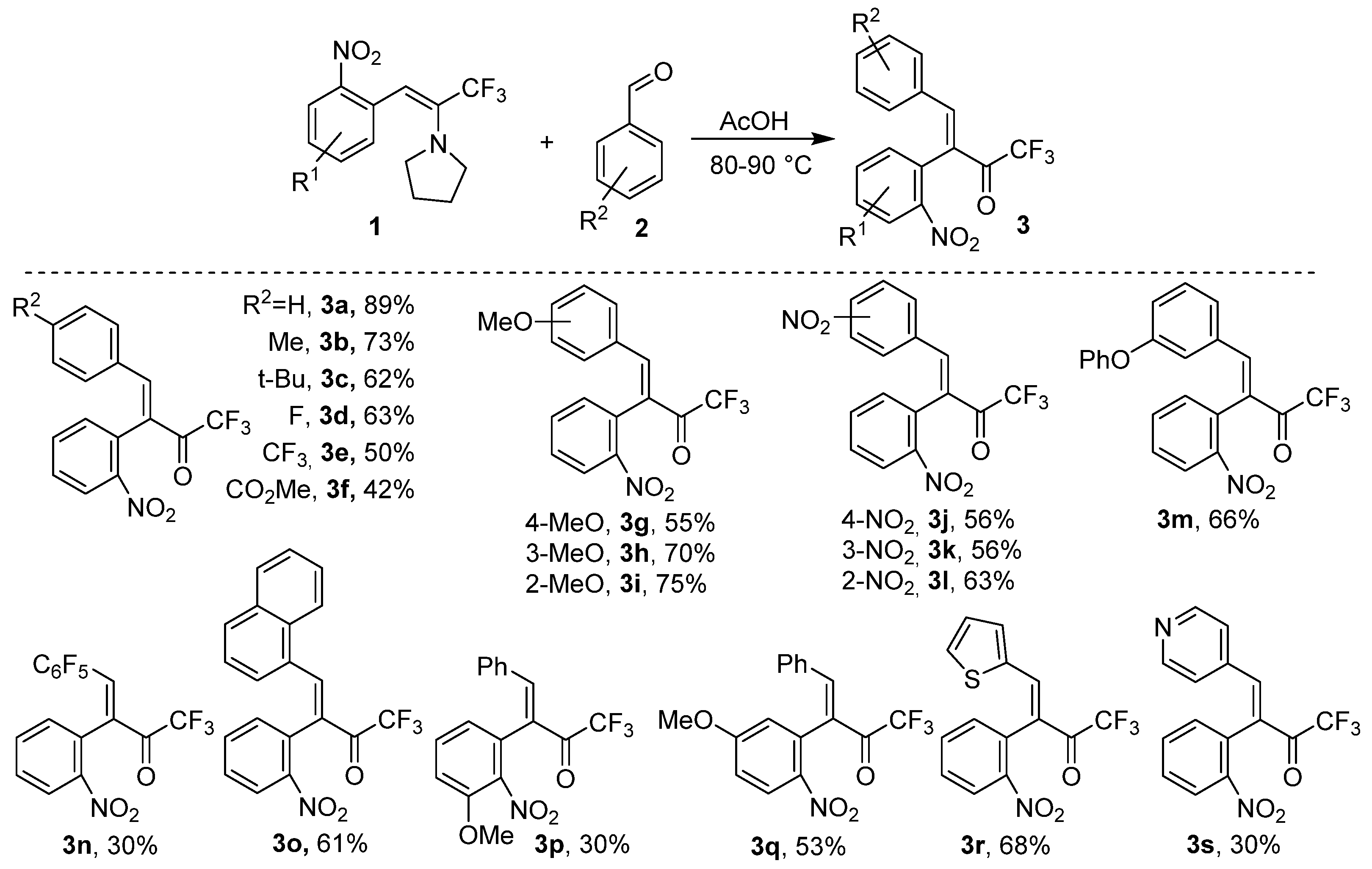
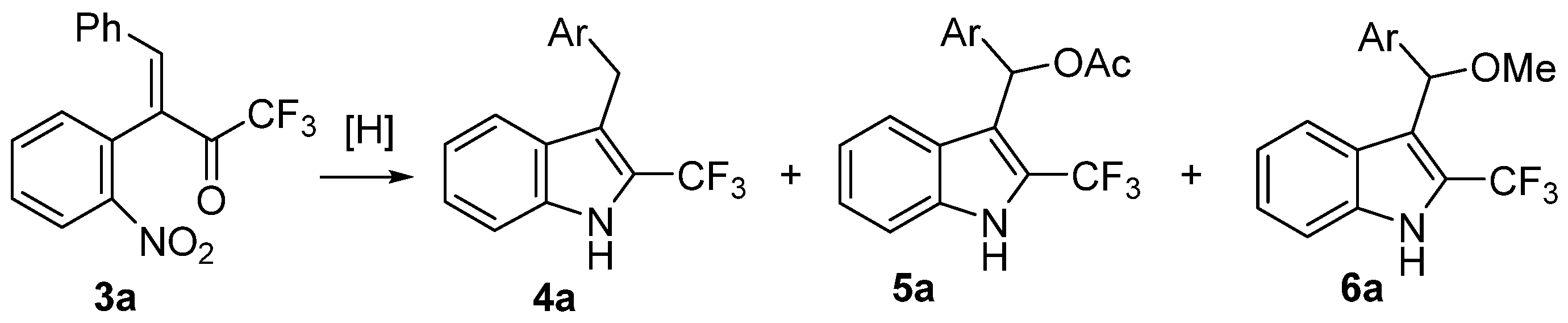
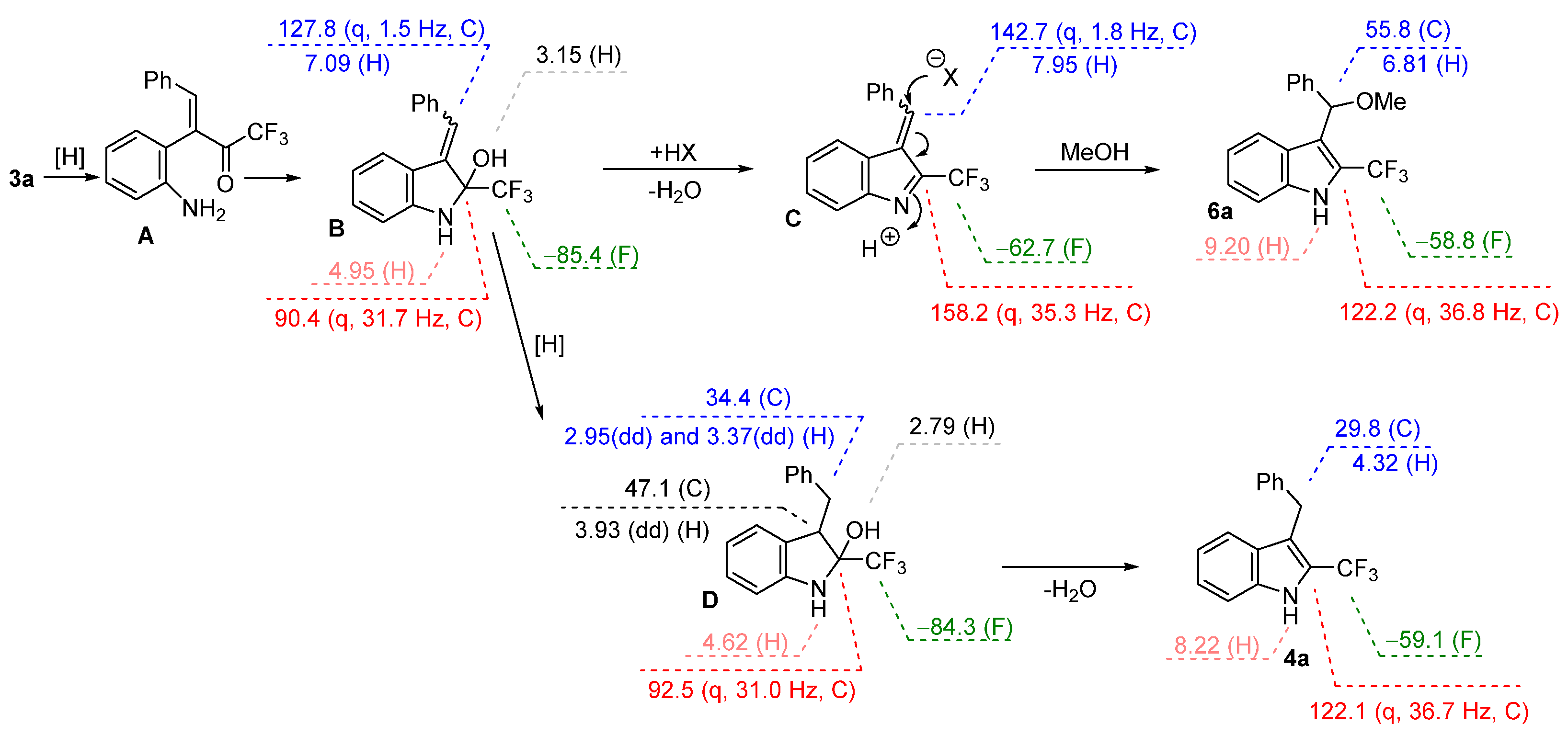
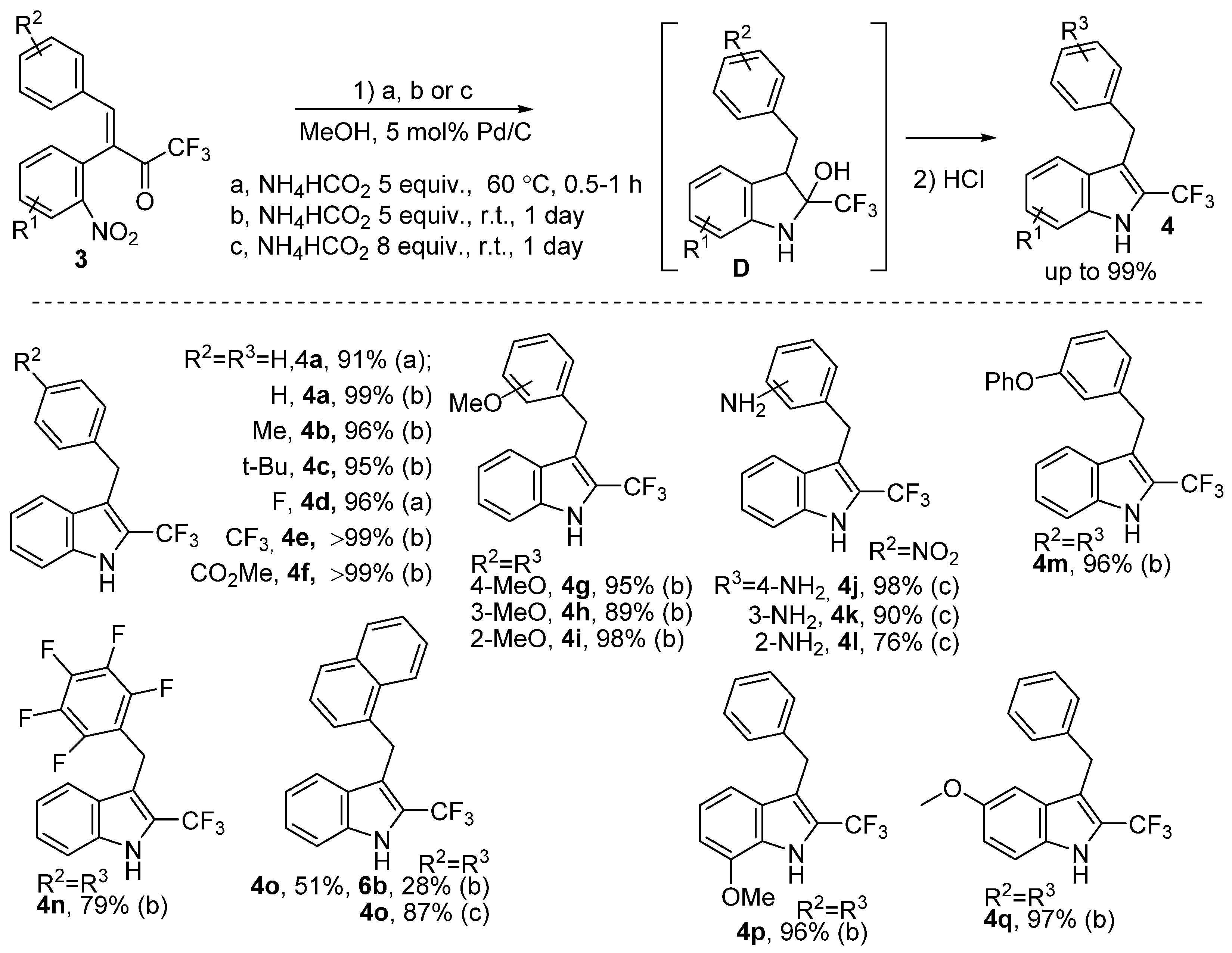
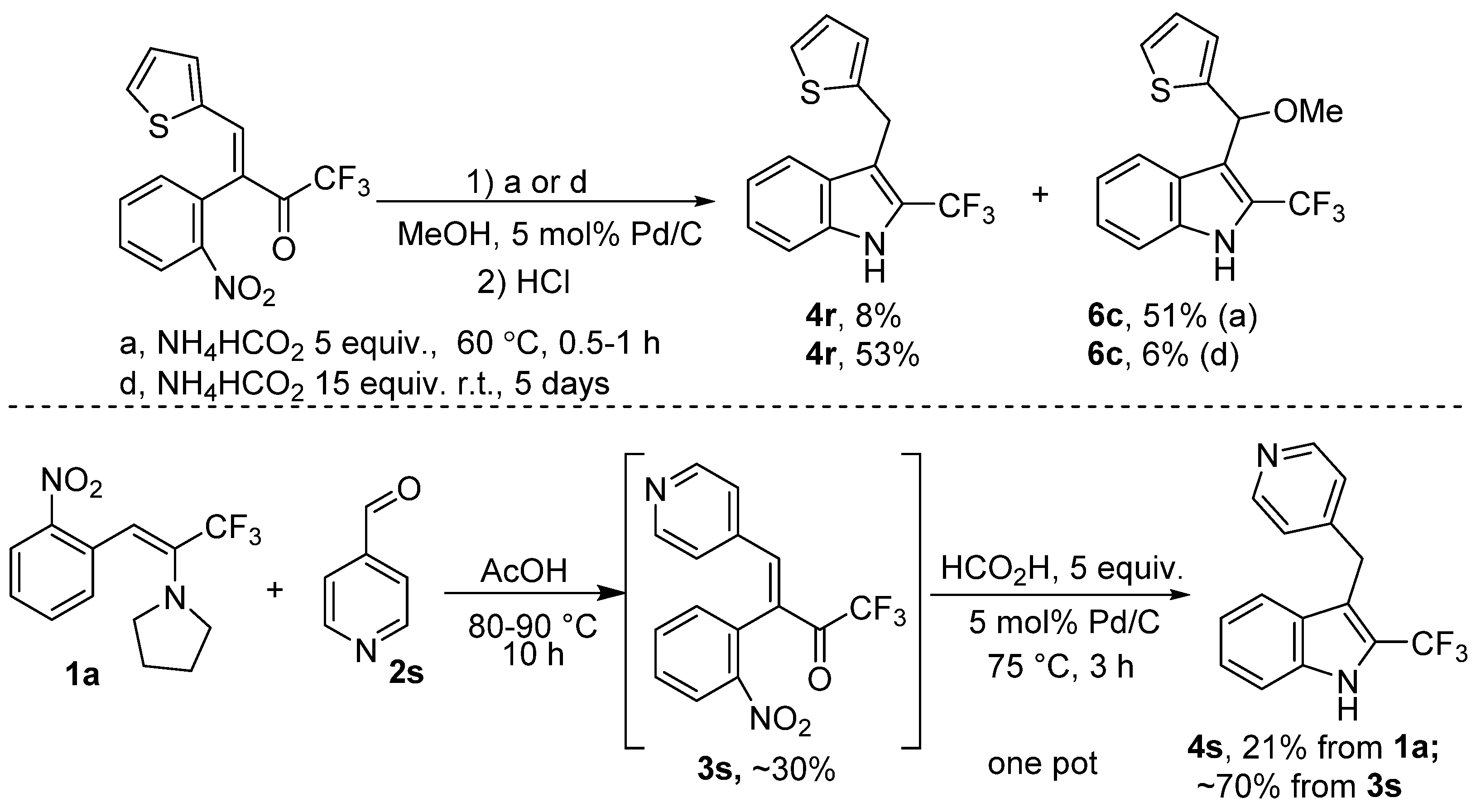
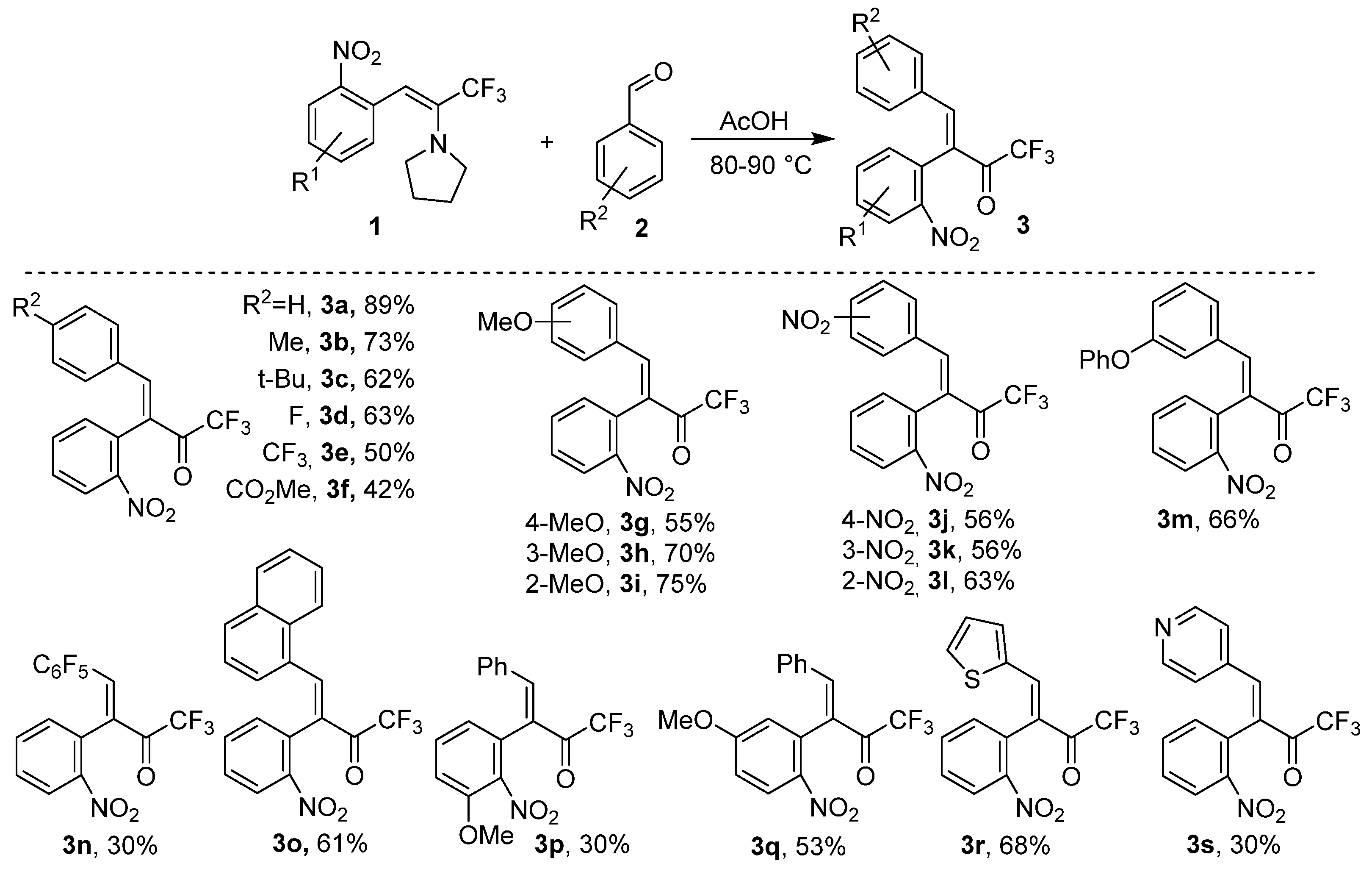
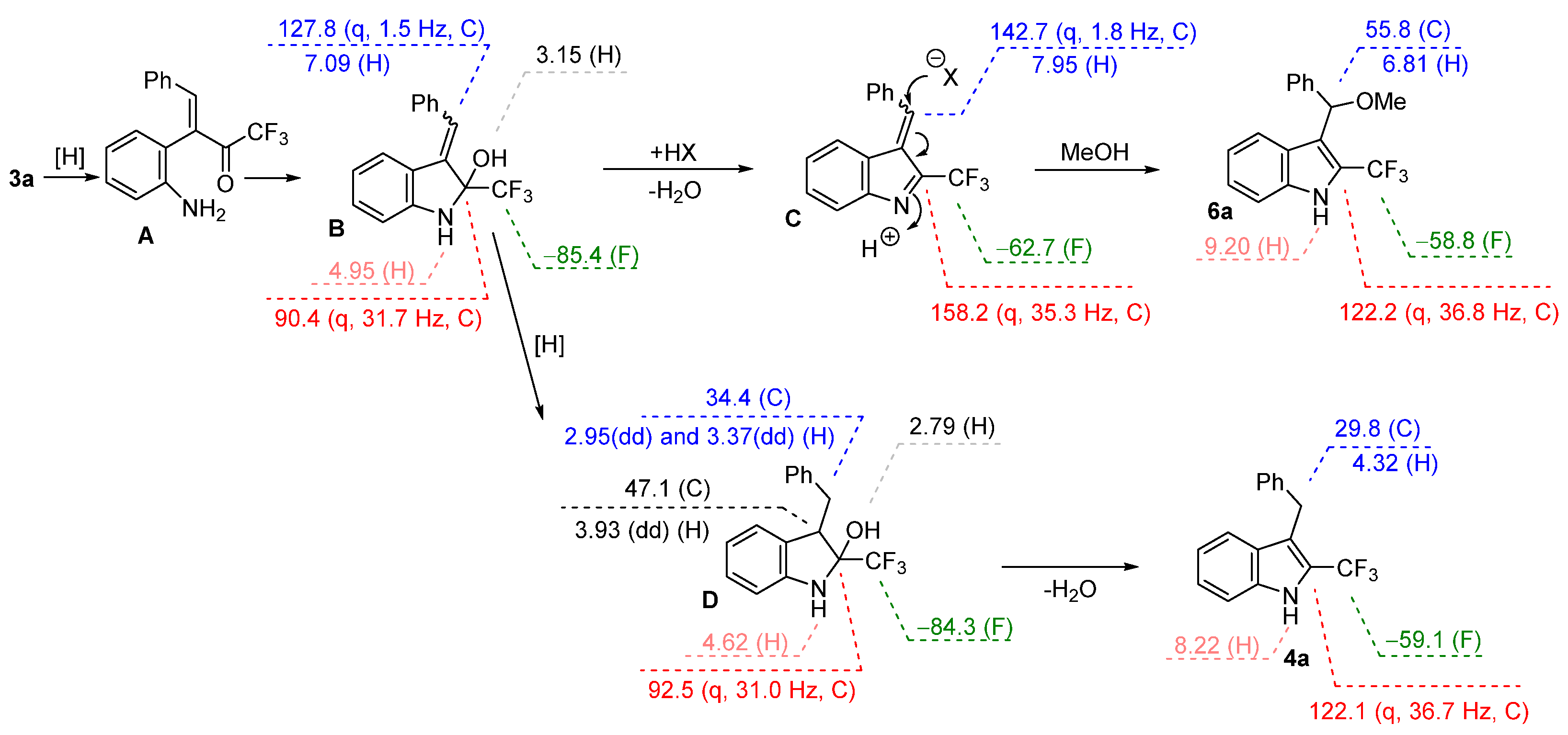
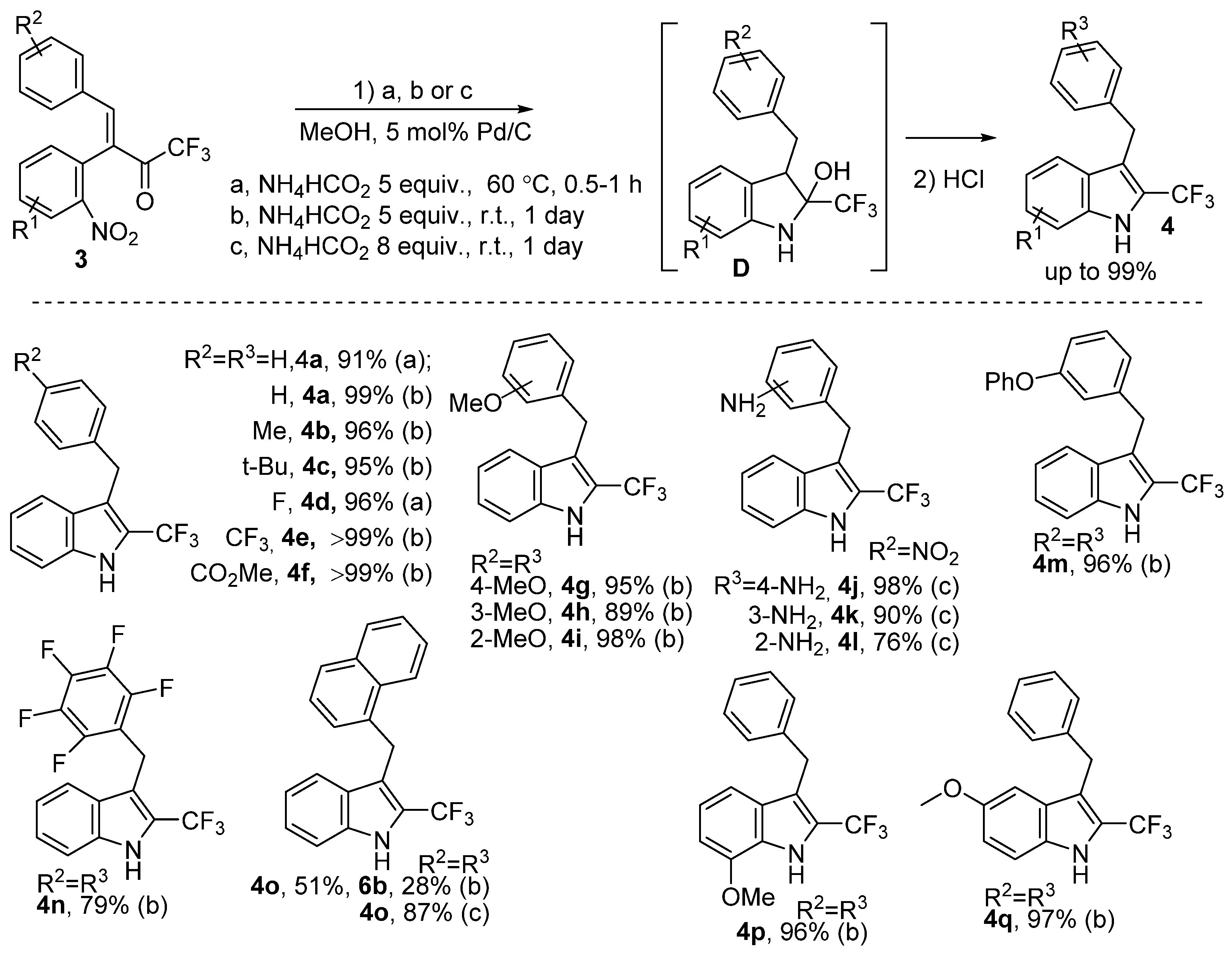
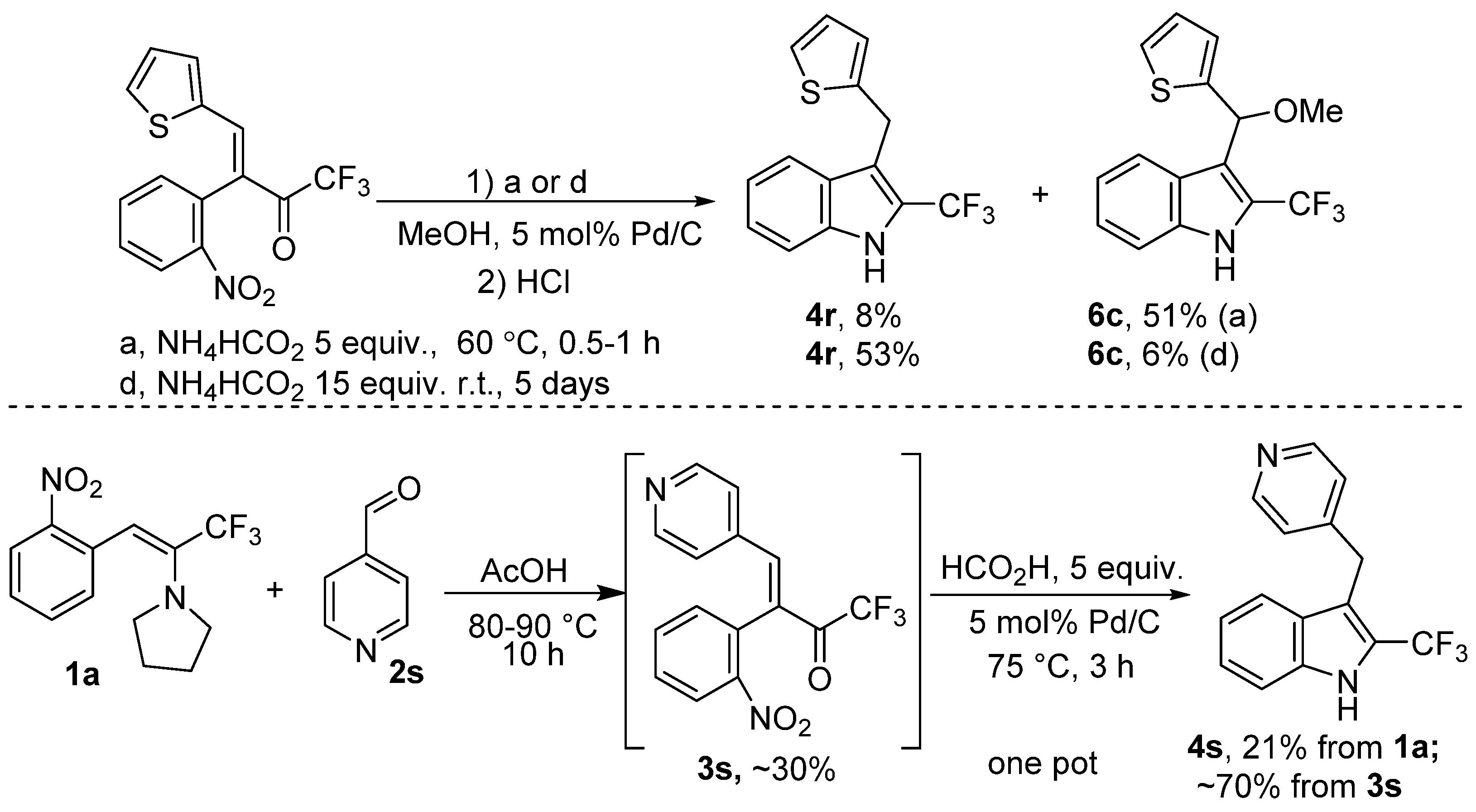
2. Results
3. Materials and Methods
General Remarks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wu, T.; Phipps, R.J.; Toste, F.D. Advances in catalytic enantioselective fluorination, mono-, di-, and trifluoromethylation, and trifluoromethylthiolation reactions. Chem. Rev. 2015, 115, 826–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahrens, T.; Kohlmann, J.; Ahrens, M.; Braun, T. Functionalization of fluorinated molecules by transition metal mediated C−F bond activation to access fluorinated building blocks. Chem. Rev. 2015, 115, 931–972. [Google Scholar] [CrossRef]
- Nenajdenko, V.G.; Muzalevskiy, V.M.; Shastin, A.V. Polyfluorinated ethanes as versatile fluorinated C2-building blocks for organic synthesis. Chem. Rev. 2015, 115, 973–1050. [Google Scholar] [CrossRef] [PubMed]
- Yerien, D.E.; Barata-Vallejo, S.; Postigo, A. Difluoromethylation reactions of organic compounds. Chem. Eur. J. 2017, 23, 14676–14701. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P. The unique role of fluorine in the design of active ingredients for modern crop protection. Chem. Bio. Chem. 2004, 5, 570–589. [Google Scholar] [CrossRef]
- Jeschke, P. The unique role of halogen substituents in the design of modern agrochemicals. Pest Manage. Sci. 2010, 66, 10–27. [Google Scholar] [CrossRef]
- Fujiwara, T.; O’Hagan, D. Successful fluorine-containing herbicide agrochemicals. J. Fluorine Chem. 2014, 167, 16–29. [Google Scholar] [CrossRef]
- Jeschke, P. Latest generation of halogen-containing pesticides. Pest Manage. Sci. 2017, 73, 1053–1056. [Google Scholar] [CrossRef] [PubMed]
- Bégué, J.P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Fluorine and Health. Molecular Imaging, Biomedical Materials and Pharmaceuticals; Tressaud, A., Haufe, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 553–778. [Google Scholar]
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001−2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef] [PubMed]
- Ilardi, E.A.; Vitaku, E.; Njardarson, J.T. Data-mining for sulfur and fluorine: An evaluation of pharmaceuticals to reveal opportunities for drug design and discovery. J. Med. Chem. 2014, 57, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J.L.; Izawa, K.; Liu, H.; Soloshonok, V.A. Recent advances in the trifluoromethylation methodology and new CF3-containing drugs. J. Fluorine Chem. 2014, 167, 37–54. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next generation of fluorine containing pharmaceuticals, compounds currently in phase II−III clinical trials of major pharmaceutical companies: New structural trends and therapeutic areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2018. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2019, 24, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2019. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2020, 25, 745. [Google Scholar] [CrossRef] [Green Version]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Nenajdenko, V.G. (Ed.) Fluorine in Heterocyclic Chemistry; Springer: Heidelberg, Germany, 2014; Volume 1, p. 681; Volume 2, p. 760. [Google Scholar]
- Petrov, V.A. (Ed.) Fluorinated Heterocyclic Compounds: Synthesis, Chemistry, and Applications; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Gakh, A.; Kirk, K.L. (Eds.) Fluorinated Heterocycles; Oxford University Press: Oxford, UK, 2008. [Google Scholar]
- Muzalevskiy, V.M.; Nenajdenko, V.G.; Shastin, A.V.; Balenkova, E.S.; Haufe, G. Synthesis of Trifluoromethyl Pyrroles and Their Benzo Analogues. Synthesis 2009, 23, 3905–3929. [Google Scholar]
- Serdyuk, O.V.; Abaev, V.T.; Butin, A.V.; Nenajdenko, V.G. Synthesis of Fluorinated Thiophenes and Their Analogues. Synthesis 2011, 16, 2505–2529. [Google Scholar] [CrossRef]
- Serdyuk, O.V.; Muzalevskiy, V.M.; Nenajdenko, V.G. Synthesis and Properties of Fluoropyrroles and Their Analogues. Synthesis 2012, 44, 2115–2137. [Google Scholar]
- Politanskaya, L.V.; Selivanova, G.A.; Panteleeva, E.V.; Tretyakov, E.V.; Platonov, V.E.; Nikul’shin, P.V.; Vinogradov, A.S.; Zonov, Y.V.; Karpov, V.M.; Mezhenkova, T.V.; et al. Organofluorine chemistry: Promising growth areas and challenges. Rus. Chem. Rev. 2019, 88, 425–569. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Metal-Free Synthesis of Fluorinated Indoles Enabled by Oxidative Dearomatization. Angew. Chem. 2016, 128, 2283–2287. [Google Scholar] [CrossRef]
- Pindur, U.; Adam, R. Synthetically attractive indolization processes and newer methods for the preparation of selectively substituted indole. J. Heterocycl. Chem. 1988, 25, 1–8. [Google Scholar] [CrossRef]
- Cacchi, S.; Fabrizi, G. Synthesis and Functionalization of Indoles Through Palladium-catalyzed Reactions. Chem. Rev. 2005, 105, 2873–2920. [Google Scholar] [CrossRef]
- Humphrey, G.R.; Kuethe, J.T. Practical methodologies for the synthesis of indoles. Chem. Rev. 2006, 106, 2875–2911. [Google Scholar] [CrossRef]
- Taber, D.F.; Tirunahari, P.K. Indole synthesis: A review and proposed classification. Tetrahedron 2011, 67, 7195–7210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacchi, S.; Fabrizi, G. Update 1 of: Synthesis and Functionalization of Indoles Through Palladium-Catalyzed Reactions. Chem. Rev. 2011, 111, PR215–PR283. [Google Scholar] [CrossRef] [PubMed]
- Platon, M.; Amardeil, R.; Djakovitch, L.; Hierso, J.C. Progress in palladium-based catalytic systems for the sustainable synthesis of annulated heterocycles: A focus on indole backbones. Chem. Soc. Rev. 2012, 41, 3929–3968. [Google Scholar] [CrossRef] [PubMed]
- De Sa Alves, F.R.; Barreiro, E.J.; Fraga, C.A.M. From nature to drug discovery: The indole scaffold as a “privileged structure”. Mini-Rev. Med. Chem. 2009, 9, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Sravanthi, T.V.; Manju, S.L. Indoles—A promising scaffold for drug development. Eur. J. Pharm. Sciences 2016, 91, 1–10. [Google Scholar] [CrossRef]
- Bugaenko, D.I.; Karchava, A.V.; Yurovskaya, M.A. Synthesis of indoles: Recent advances. Russ. Chem. Rev. 2019, 88, 99–159. [Google Scholar] [CrossRef]
- Hart, F.D.; Boardman, P.L. Indomethacin: A new non-steroid anti-inflammatory agent. Br. Med. J. 1963, 2, 965–970. [Google Scholar] [CrossRef] [Green Version]
- Lucas, S. The Pharmacology of Indomethacin. Headache. 2016, 56, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Geitmann, M.; Unge, T.; Danielson, U.H. Biosensor-based kinetic characterization of the interaction between HIV-1 reverse transcriptase and non-nucleoside inhibitors. J. Med. Chem. 2006, 49, 2367–2374. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Radzio, J.; Anderson, K.S.; Sluis-Cremer, N. Probing nonnucleoside inhibitor-induced active-site distortion in HIV-1 reverse transcriptase by transient kinetic analyses. Protein Sci 2007, 16, 1728–1737. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ji, Z.L.; Chen, Y.Z. TTD: Therapeutic Target Database. Nucleic Acids Res. 2002, 30, 412–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, S.S.; Lynham, J.A.; Molenaar, P.; Grace, A.A.; Colledge, W.H.; Kaumann, A.J. Intrinsic sympathetic activity of (−)-pindolol mediated through a (−)-propranolol-resistant site of the β1-adrenoceptor in human atrium and recombinant receptors. Naunyn Schmiedebergs Arch. Pharmacol. 2003, 368, 496–503. [Google Scholar] [CrossRef]
- Qian, D.Z.; Kato, Y.; Shabbeer, S.; Wei, Y.; Verheul, H.M.; Salumbides, B.; Sanni, T.; Atadja, P.; Pili, R. Targeting tumor angiogenesis with histone deacetylase inhibitors: The hydroxamic acid derivative LBH589. Clin. Cancer Res. 2006, 12, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, K.; Webster, R. The bladder cancer drug market. Nat. Rev. Drug Discov. 2016, 15, 599–600. [Google Scholar] [CrossRef]
- Phillips, R.M.; Hendriks, H.R.; Sweeney, J.B.; Reddy, G.; Peters, G.J. Efficacy, pharmacokinetic and pharmacodynamic evaluation of apaziquone in the treatment of nonmuscle invasive bladder cancer. Expert Opin. Drug Metab. Toxicol. 2017, 13, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Nenaidenko, V.G.; Balenkova, E.S. Perfluoroacylation of alkenes. Zh. Org. Khim. 1992, 28, 600–602. [Google Scholar]
- Nenajdenko, V.G.; Leshcheva, I.F.; Balenkova, E.S. The perfluoroacylation of cyclopropyl-containing alkenes. Tetrahedron 1994, 50, 775–782. [Google Scholar] [CrossRef]
- Nenajdenko, V.G.; Krasovsky, A.L.; Lebedev, M.V.; Balenkova, E.S. A novel efficient synthesis of heteroaryl substituted alpha, beta-unsaturated trifluoromethyl ketones. Synlett 1997, 12, 1349–1350. [Google Scholar] [CrossRef]
- Krasovsky, A.L.; Nenajdenko, V.G.; Balenkova, E.S. Diels-Alder reactions of beta-trifluoroacetylvinylsulfones. Tetrahedron 2001, 57, 201–209. [Google Scholar] [CrossRef]
- Nenajdenko, V.G.; Krasovsky, A.L.; Balenkova, E.S. The chemistry of sulfinyl and sulfonyl enones. Tetrahedron 2007, 63, 12481–12539. [Google Scholar] [CrossRef]
- Nenajdenko, V.G.; Sanin, A.V.; Balenkova, E.S. Preparation of α,β-Unsaturated Ketones Bearing a Trifluoromethyl Group and Their Application in Organic Synthesis. Molecules 1997, 2, 186–232. [Google Scholar] [CrossRef] [Green Version]
- Nenajdenko, V.G.; Sanin, A.V.; Balenkova, E.S. Methods for the synthesis of α,β-unsaturated trifluoromethyl ketones and their use in organic synthesis. Russ. Chem. Rev. 1999, 68, 483–505. [Google Scholar] [CrossRef]
- Druzhinin, S.V.; Balenkova, E.S.; Nenajdenko, V.G. Recent advances in the chemistry of α,β-unsaturated trifluoromethylketones. Tetrahedron 2007, 63, 7753–7808. [Google Scholar] [CrossRef]
- Nenajdenko, V.G.; Balenkova, E.S. Preparation of α,β-unsaturated trifluoromethylketones and their application in the synthesis of heterocycles. ARKIVOC 2011, 1, 246–328. [Google Scholar] [CrossRef] [Green Version]
- Rulev, A.Y.; Muzalevskiy, V.M.; Kondrashov, E.V.; Ushakov, I.A.; Romanov, A.R.; Khrustalev, V.N.; Nenajdenko, V.G. Reaction of α-Bromo Enones with 1,2-Diamines. Cascade Assembly of 3-(Trifluoromethyl)piperazin-2-ones via Rearrangement. Org. Lett. 2013, 15, 2726–2729. [Google Scholar] [CrossRef]
- Muzalevskiy, V.M.; Sizova, Z.A.; Panyushkin, V.V.; Chertkov, V.A.; Khrustalev, V.N.; Nenajdenko, V.G. α,β-Disubstituted CF3-Enones as a Trifluoromethyl Building Block: Regioselective Preparation of Totally Substituted 3-CF3-Pyrazoles. J. Org. Chem. 2021, 86, 2385–2405. [Google Scholar] [CrossRef] [PubMed]
- Muzalevskiy, V.M.; Sizova, Z.A.; Abaev, V.T.; Nenajdenko, V.G. Synthesis of 2-trifluoromethylated quinolines from CF3-alkenes. Org. Biomol. Chem. 2021, 19, 4303–4319. [Google Scholar] [CrossRef]
- Muzalevskiy, V.M.; Sizova, Z.A.; Nenajdenko, V.G. Modular Construction of Functionalized 2-CF3-Indoles. Org. Lett. 2021, 23, 5973–5977. [Google Scholar] [CrossRef] [PubMed]
- Dan-oh, Y.; Matta, H.; Uemura, J.; Watanabe, H.; Uneyama, K. Generation and Reactions of Trifluoroacetimidoyl Radicals. Bull. Chem. Soc. Jpn. 1995, 68, 1497–1507. [Google Scholar] [CrossRef]
- Prieto, A.; Landart, M.; Baudoin, O.; Monteiro, N.; Bouyssi, D. Copper-Catalyzed Trifluoromethylation of Aliphatic N -Arylhydrazones: A Concise Synthetic Entry to 2-Trifluoromethylindoles from Simple Aldehydes. Adv. Synth. Catal. 2015, 357, 2939–2943. [Google Scholar] [CrossRef]
- Cheng, Y.; Yuan, X.; Ma, J.; Yu, S. Direct Aromatic C-H Trifluoromethylation via an Electron-Donor–Acceptor Complex. Chem. Eur. J. 2015, 21, 8355–8359. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. (Ed.) Leimgruber–Batcho Indole Synthesis. In Indole Ring Synthesis: From Natural Products to Drug Discovery; JWiley-VCH: Chichester, West Sussex, UK, 2016. [Google Scholar]
- Gribble, G.W. (Ed.) Reissert Indole Synthesis. In Indole Ring Synthesis: From Natural Products to Drug Discovery; JWiley-VCH: Chichester, West Sussex, UK, 2016. [Google Scholar]
- Seoane, X.L.; L’Argentiere, P.C.; Fígoli, N.S.; Arcoya, A. On the deactivation of supported palladium hydrogenation catalysts by thiophene poisoning. Catal. Lett. 1992, 16, 137–148. [Google Scholar] [CrossRef]
- Muzalevskiy, V.M.; Nenajdenko, V.G.; Rulev, A.Y.; Ushakov, I.A.; Romanenko, G.V.; Shastin, A.V.; Balenkova, E.S.; Haufe, G. Selective synthesis of α-trifluoromethyl-β-arylenamines or vinylogous guanidinium salts by treatment of β-halo-β-trifluoromethylstyrenes with secondary amines under different conditions. Tetrahedron 2009, 65, 6991–7000. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title 1 | Reaction Conditions | Yield of 4a, % | Yield of 5a, % | Yield of 6a, % |
|---|---|---|---|---|
| entry 1 | 6 eq. Zn, AcOH, 80 °C, 4h | 20 | 47 | - |
| entry 2 | 12 eq. Zn, AcOH, 80 °C, 14h | 43 | 3 | - |
| entry 3 | 6 eq. Zn, AcOH-MeOH, 65 °C, 8h | 8 | 2 | 77 |
| entry 4 | H2, MeOH, 5 mol% Pd/C, r.t., 1 day | 89 | - | <1 |
| entry 5 | 5 eq. NH4HCO2, MeOH, 5 mol% Pd/C, r.t., 60 °C, 1 h | 91 | - | <1 |
| entry 6 | 5 eq. NH4HCO2, MeOH, 5 mol% Pd/C, r.t., 1 day | 99 | - | - |
| entry 7 | 3.3 equiv. NH4HCO2, MeOH, 5 mol% Pd/C, 60 °C, 1 h | <1 | - | 86 |
| entry 8 | 3.3 equiv. NH4HCO2, THF, 5 mol% Pd/C, r.t., 1 day; then pTSA, MeOH | traces | - | 81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muzalevskiy, V.M.; Sizova, Z.A.; Nenajdenko, V.G. An Efficient Synthesis of 2-CF3-3-Benzylindoles. Molecules 2021, 26, 5084. https://doi.org/10.3390/molecules26165084
Muzalevskiy VM, Sizova ZA, Nenajdenko VG. An Efficient Synthesis of 2-CF3-3-Benzylindoles. Molecules. 2021; 26(16):5084. https://doi.org/10.3390/molecules26165084
Chicago/Turabian StyleMuzalevskiy, Vasiliy M., Zoia A. Sizova, and Valentine G. Nenajdenko. 2021. "An Efficient Synthesis of 2-CF3-3-Benzylindoles" Molecules 26, no. 16: 5084. https://doi.org/10.3390/molecules26165084
APA StyleMuzalevskiy, V. M., Sizova, Z. A., & Nenajdenko, V. G. (2021). An Efficient Synthesis of 2-CF3-3-Benzylindoles. Molecules, 26(16), 5084. https://doi.org/10.3390/molecules26165084