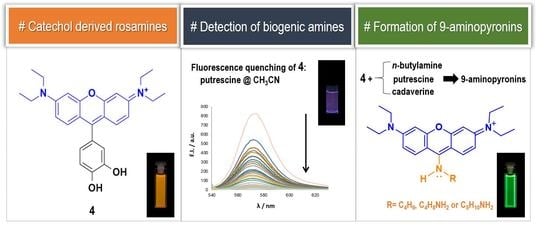
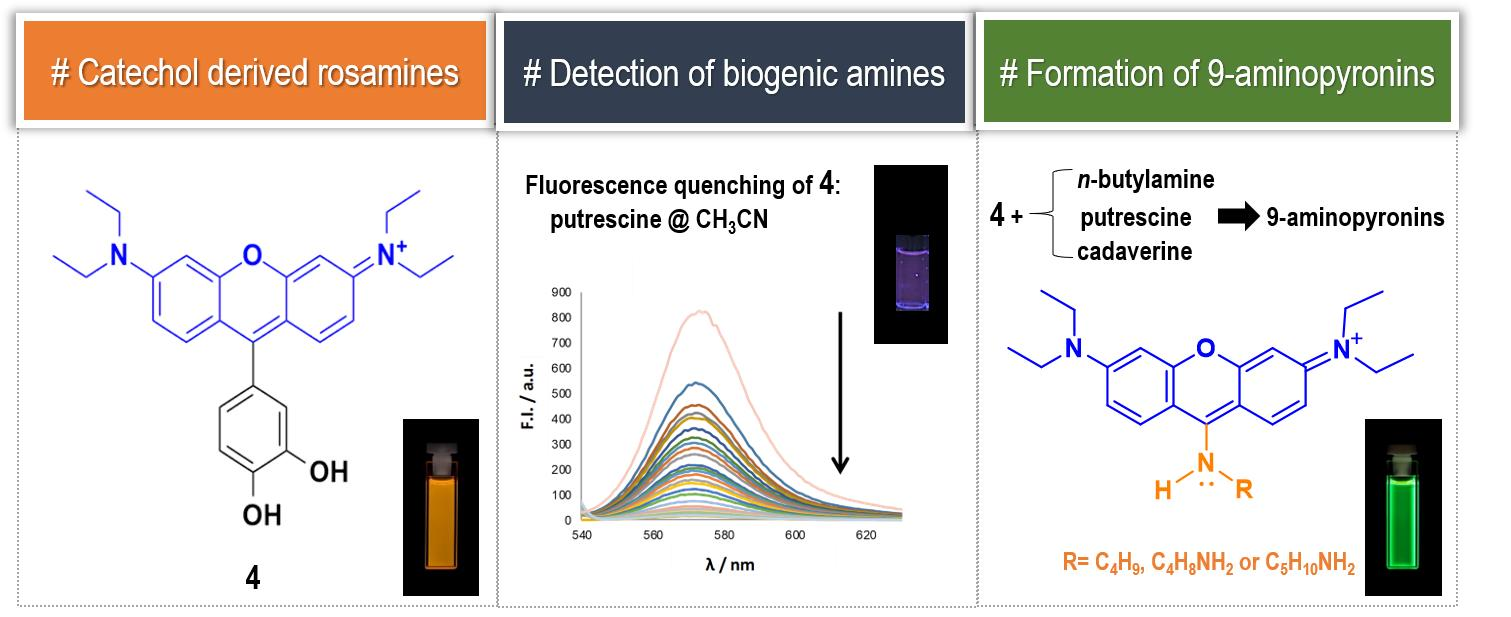
2. Results and Discussion
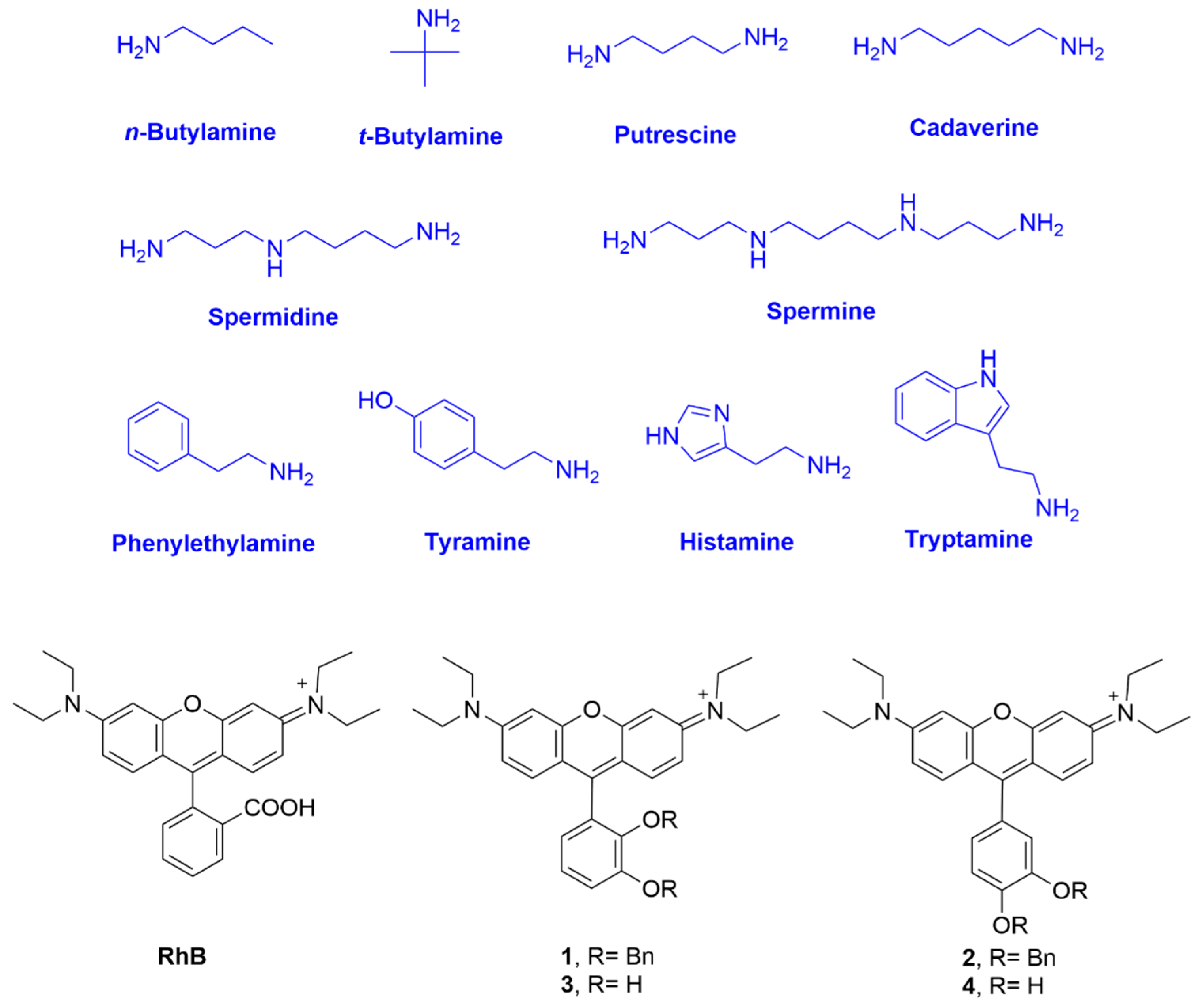
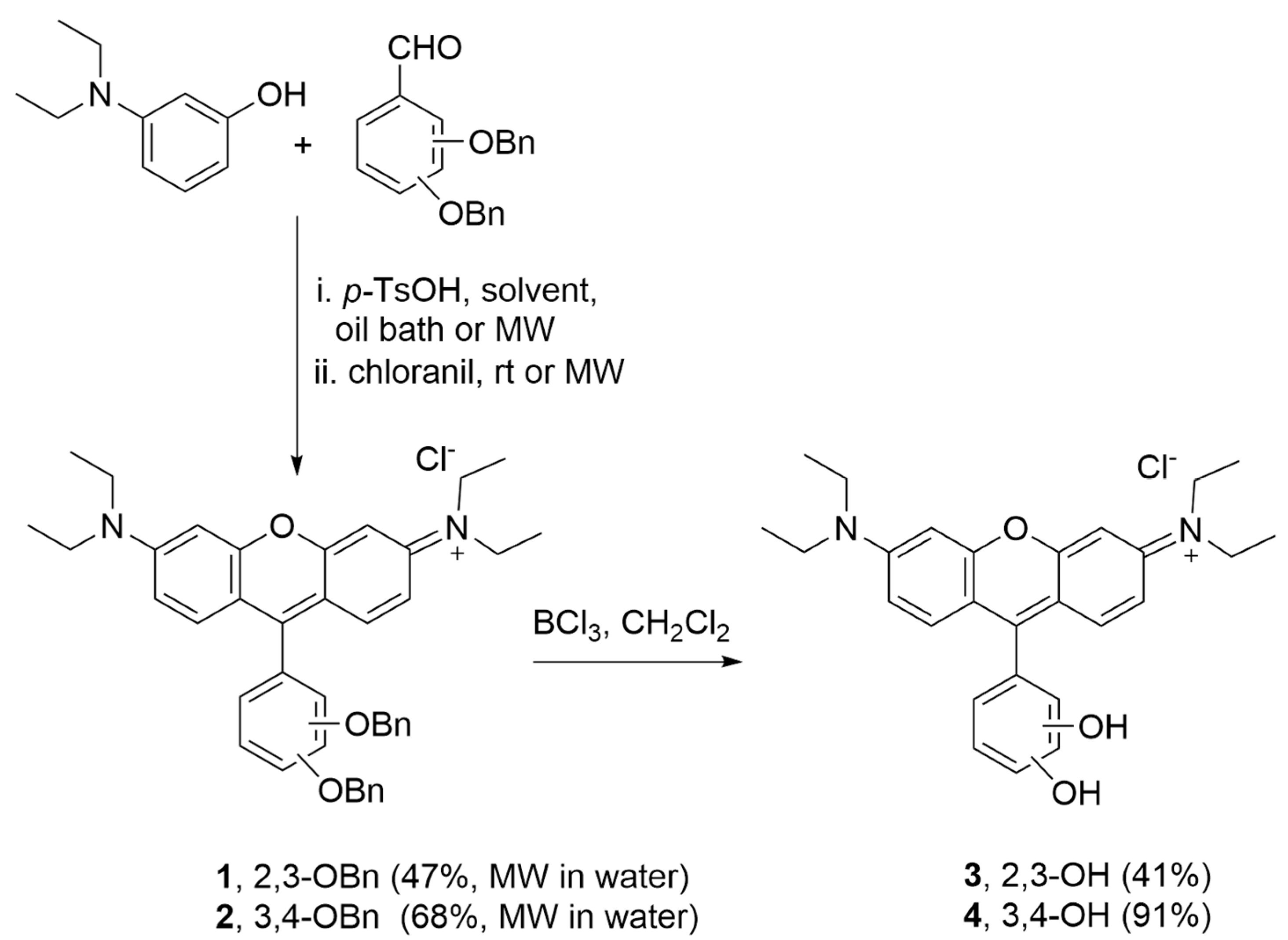
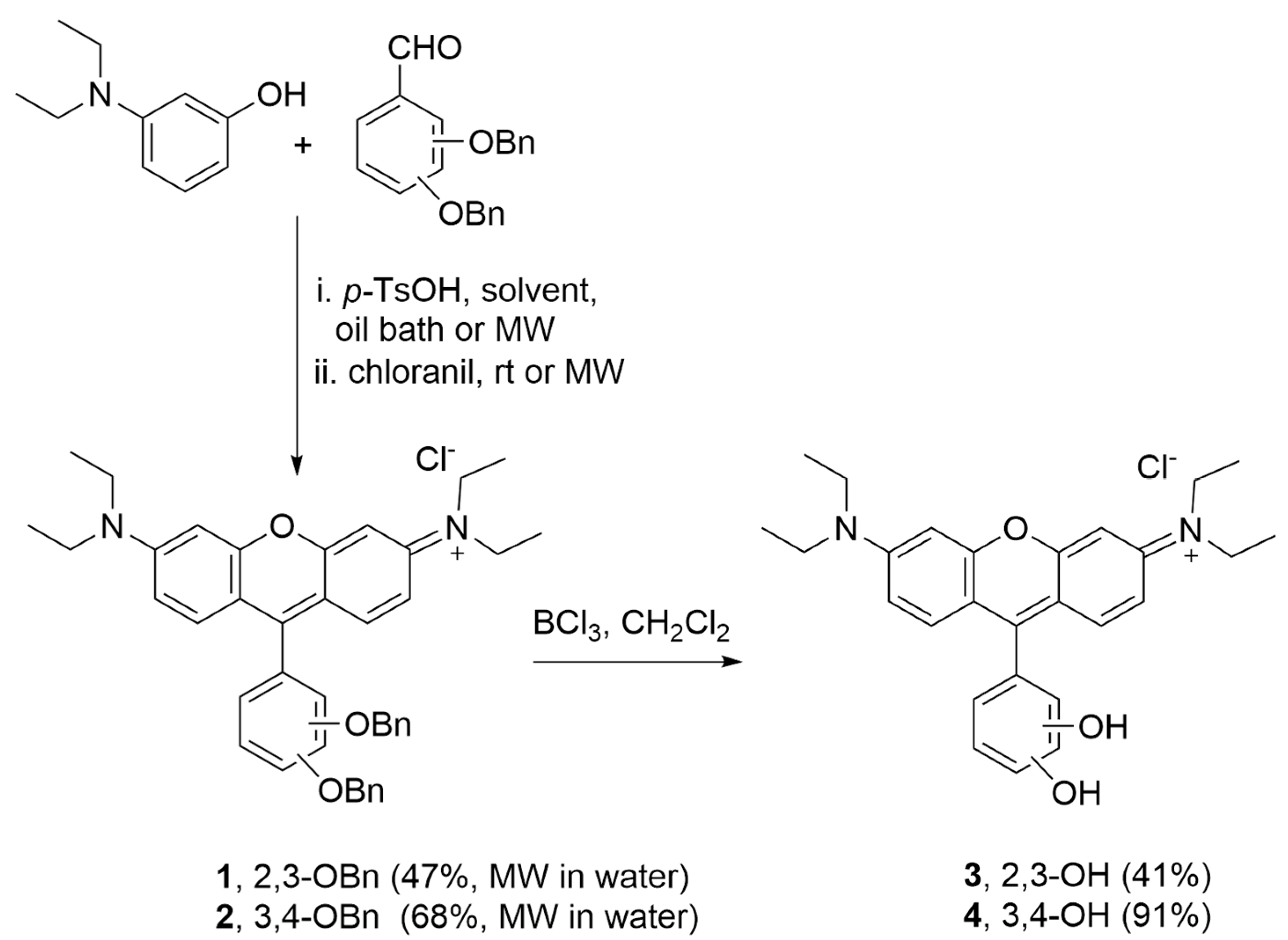
The synthetic route for rosamines
1–
4 is simple and effective (
Scheme 1). It consists of the condensation of the appropriate dibenzyloxybenzaldehyde and 3-(diethylamino)phenol in the presence of a catalytic amount of
p-toluenesulfonic acid (
p-TsOH), with subsequent oxidation with chloranil to give
1 and
2. The removal of the benzyl protecting groups was performed using boron trichloride in dichloromethane to afford
3 and
4. After screening different experimental conditions for condensation including (i) the heating method (oil bath or microwave irradiation, MW) and (ii) the reaction solvent (propionic acid or water), we verified that the combination of MW and water provided higher yields for compounds
1 and
2 (47% and 68%, respectively) in a remarkable shorter period of time (10 min) (please refer to the Materials and Methods section for more details).
Rosamines (
1–
4) were characterized by
1H NMR,
13C NMR, HRMS, UV–Vis, and fluorescence spectroscopy. These were obtained as a dark pink powder soluble in organic solvents, but were only sparingly soluble in an aqueous solution. Their photophysical properties including absorption extinction coefficients (
ε) and fluorescence quantum yield (
ΦF) were studied in aprotic and protic solvents including chloroform, acetonitrile, and methanol and compared with
RhB. The results are listed in
Table 1.
The data showed that the molecular rigidity and the electron density of the substituent groups have remarkable effects on the photophysical properties of the compounds, namely: (i) in chloroform, the absorption bands of the catecholated compounds were in the shorter wavelength region than those of the benzylated ones; (ii) a marginally higher
ΦF value (0.60) was obtained for
1 in chloroform, where the orthogonal OBn substituent constrains the rotation of the 9-phenyl ring and thus enhances its fluorescence properties [
27]; and (iii) the presence of the catechol substituent (
3 and
4) remarkably decreased the
ΦF value of the molecules due to the intramolecular photoinduced electron transfer (PET), which occurs from the hydroxyl electron donating groups (D) to the xanthene electron acceptor fragment (A), producing the donor–acceptor (D–A) pair [
28]. Although the catecholate derivatives are always less emissive than the respective benzylated derivatives, the emission of
3 and
4 (especially in terms of
ΦF values) was significantly higher in acetonitrile than in chloroform and methanol. Lower Stokes shifts were observed for rosamines
1 and
2 in chloroform, the same being true for
RhB. On the other hand, for rosamines
3 and
4, the solvent did not influence the Stokes shift values as significantly.
Taking into account the yields obtained in the synthesis and the peculiar spectral properties of the catechol derived compounds, we considered rosamine
4 as the most interesting dye in this series. Accordingly, we started by studying the influence of pH variation (2 < pH < 12) in the fluorescence intensity of rosamine
4. The graphical representation of the fluorescence intensity as a function of the pH value showed a decrease in intensity from 2 to approximately 10 (please refer to
Figure S27 in the Supplementary Materials). In this range, two deprotonations can be observed both related to the deprotonation of the two hydroxyl groups, the first occurs for pH values from pH 2 to 6.5 (zwitterionic form) and the second deprotonation occurs for pH values above 6.5 until pH 9.90 (anionic form).
Subsequently, we investigated the color and fluorescence qualitative changes of acetonitrile solutions of
4 in the presence of the most common BAs diluted in water at room temperature using 2, 4, and 8 equiv. of amine. As shown in
Figure S28 of the Supplementary Materials, by adding increasing amounts of amines, there was a change (quench of fluorescence) observed under ultraviolet radiation, indicating that rosamine
4 is sensitive to 2 equiv. of cadaverine, putrescine, spermidine, and spermine. Furthermore, upon addition of 4 and 8 equiv., a quench in fluorescence was also observed for
n-butylamine and histamine, respectively.
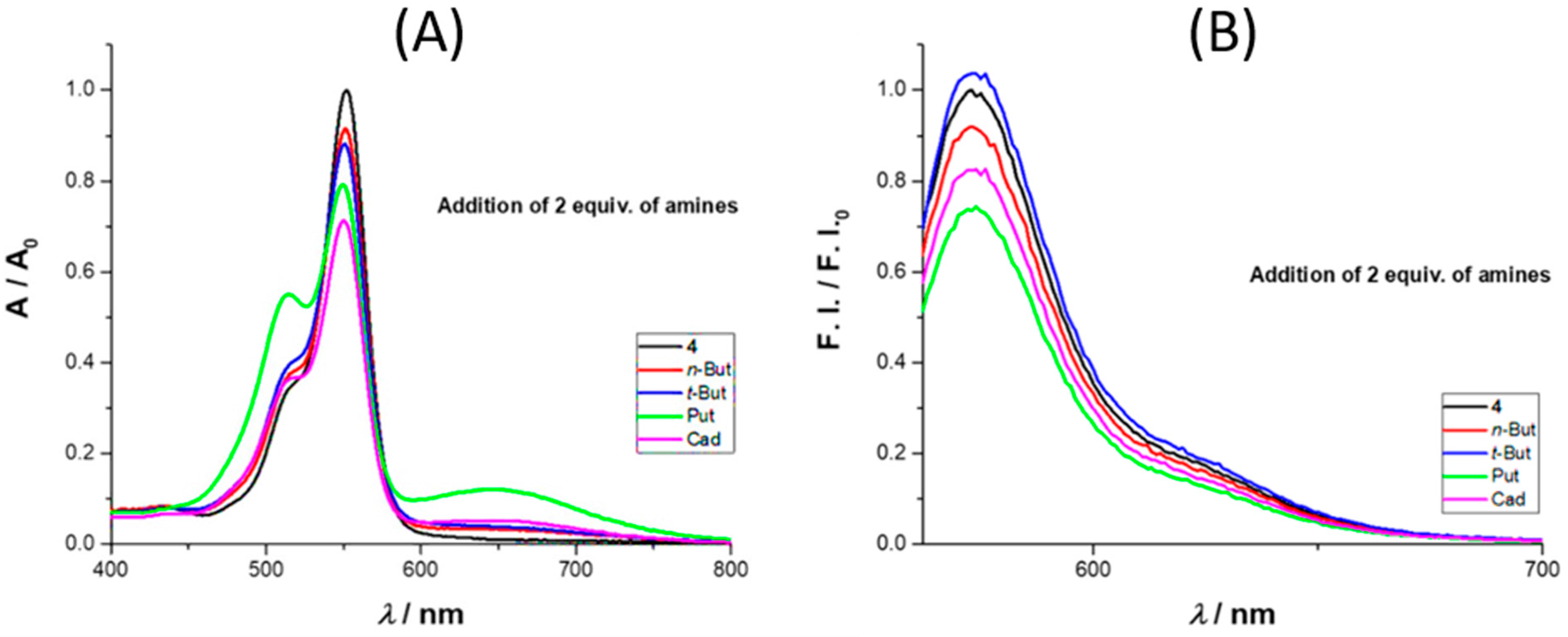
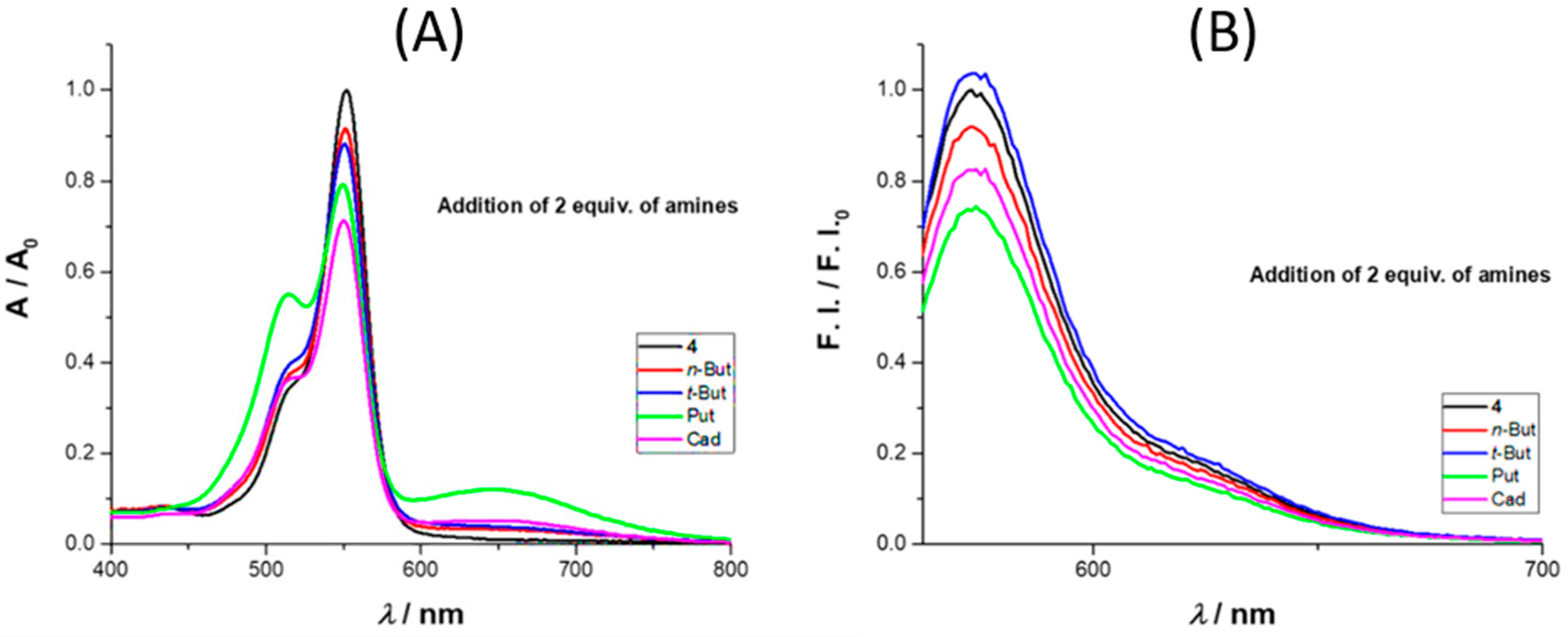
We also tested the influence of selected amines, namely
n- and
t-butylamine, cadaverine, and putrescine on the spectroscopic behavior of rosamine
4 in acetonitrile, using UV–Vis and fluorescence (
Figure 2). With the addition of 2 equiv. of amine, there was an absorbance decrease at 552 nm, more expressive for cadaverine. On the emission spectra, there was an intensity quench at 573 nm with the addition of 2 equiv. of amine, with the exception of
t-butylamine, where a small increase was observed. In terms of percentage values, for the maximum absorbance, the decrease was in the order of 8%, 12%, 21%, and 29% for
n-butylamine,
t-butylamine, putrescine, and cadaverine, respectively, while for the emission intensity, the percentage values were 8%, 4% (increase), 27%, and 18%, respectively.
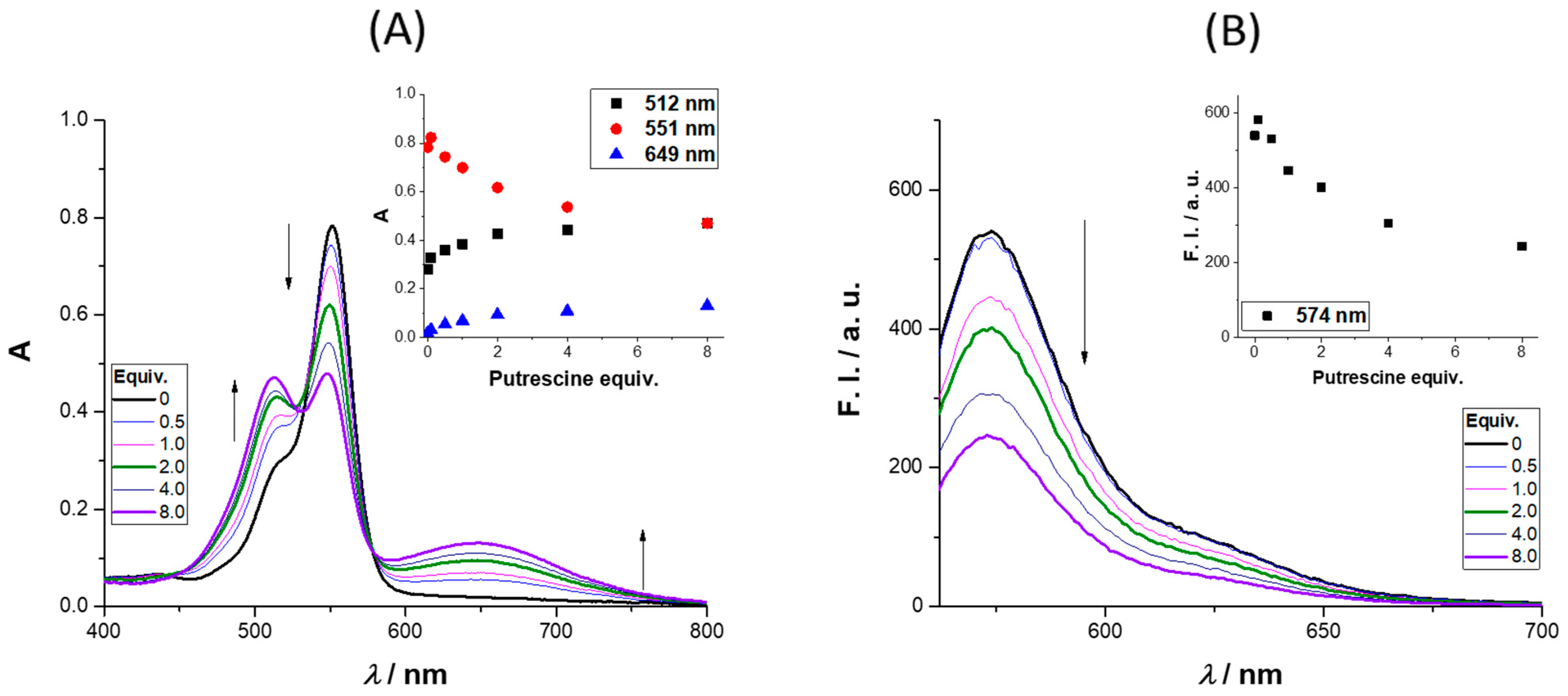
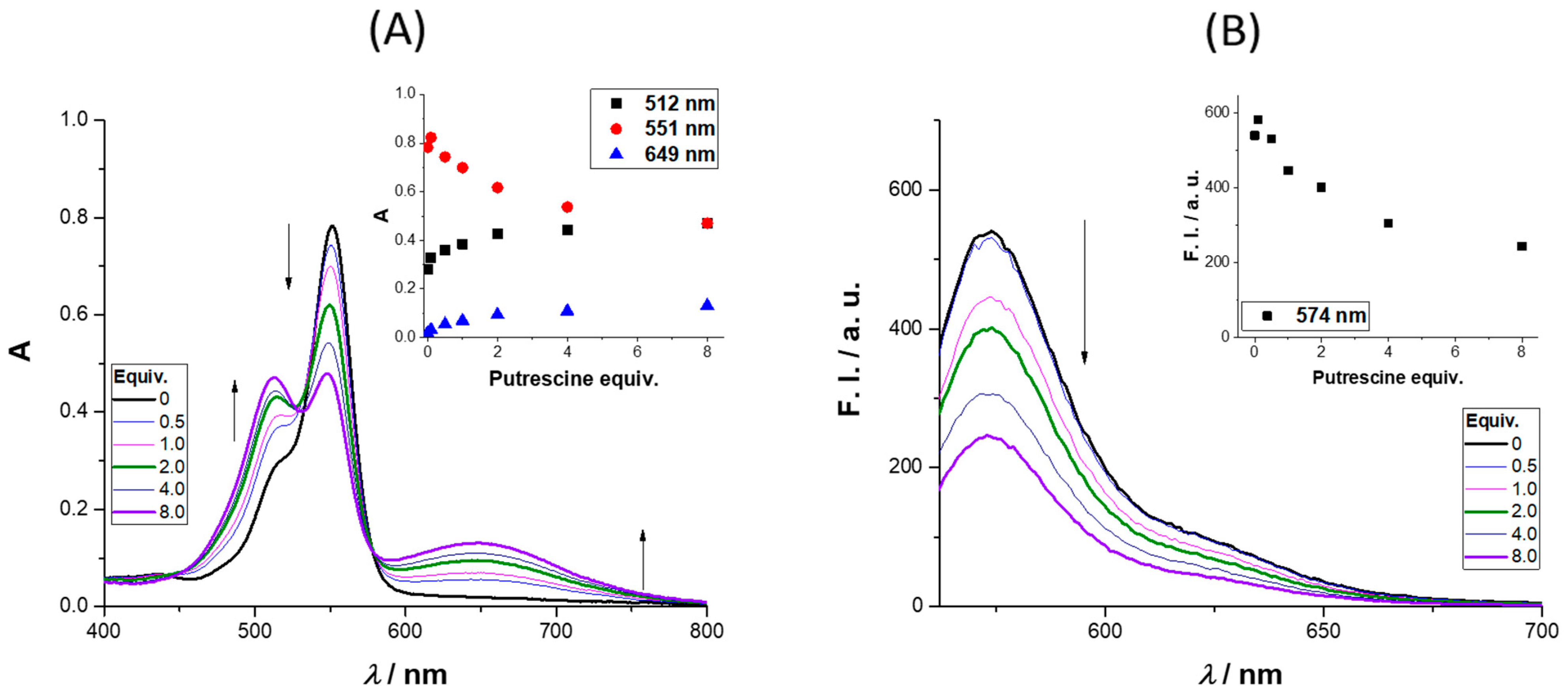
The absorbance and emission spectra of
4 after the addition of up to 8 equiv. of putrescine is presented in
Figure 3 (further amine results are available in the
Supplementary Materials). The absorbance spectra revealed three regions of interest, two with an intensity increase (512 and 649 nm) and the third (at 551 nm) where a quench was observed as the molar equivalents of putrescine increased. The intensity enhancement regions can be used to monitor the presence of putrescine in the concentration range (0 to 122 μM). The region between 649 and 657 nm is particularly interesting due to the enhancement of the absorbance; the same behavior was observed for cadaverine (please refer to the
Supplementary Materials, Figure S29, and for different concentrations of cadaverine (0.1–40 equiv.) please refer to
Figures S30 and S31). The excitation of
4 in the presence of 8 equiv. of putrescine at 649 nm did not lead to a new emission band, the same being true for the other BAs. Analyzing the emission spectra, a quench percentage of 55% was obtained for an addition of 8 equiv. of putrescine, much higher values than those observed for
n-butylamine and cadaverine (12% and 32%, respectively), whereas for
t-butylamine, a slight increase was obtained (6%).
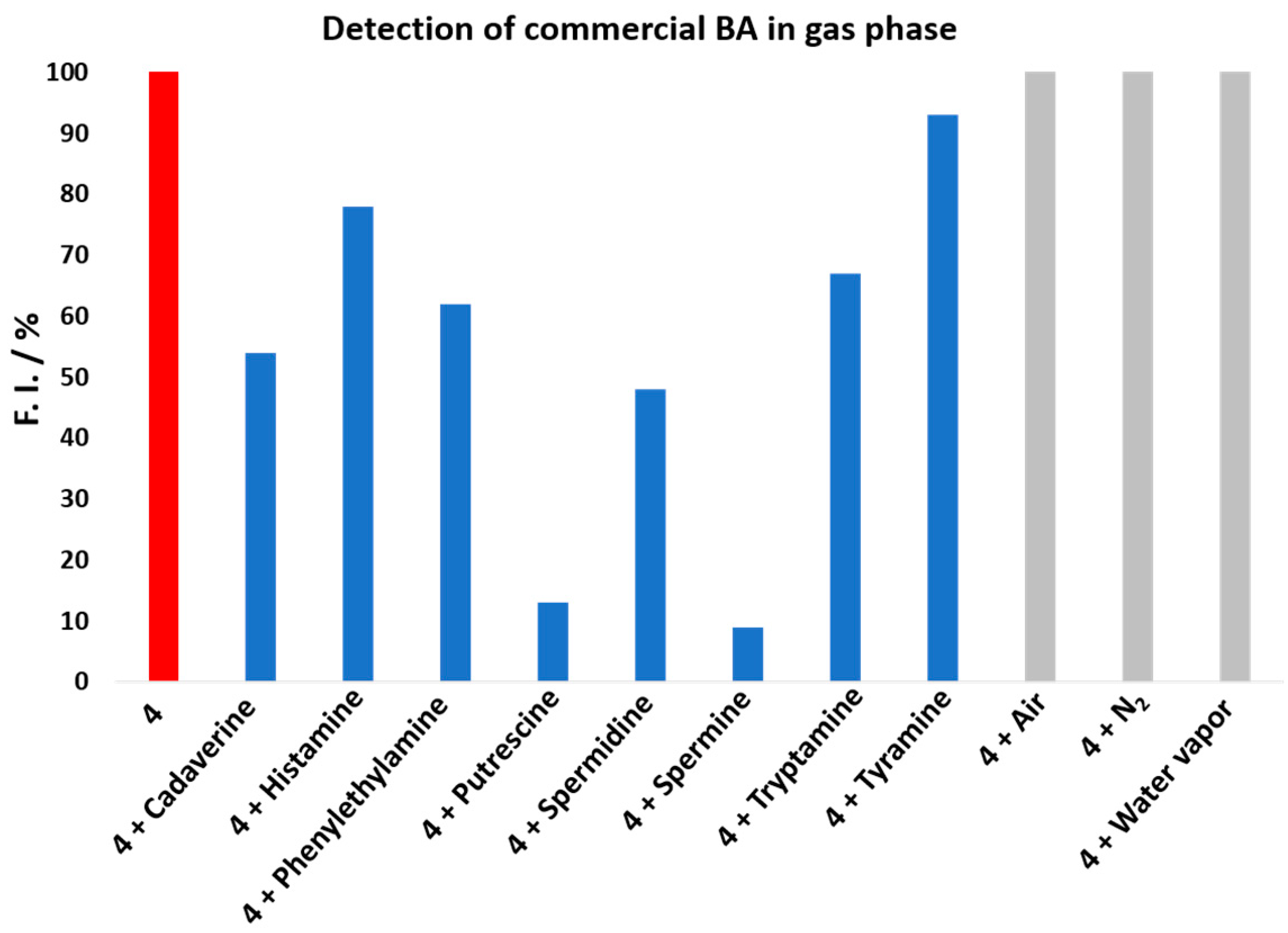
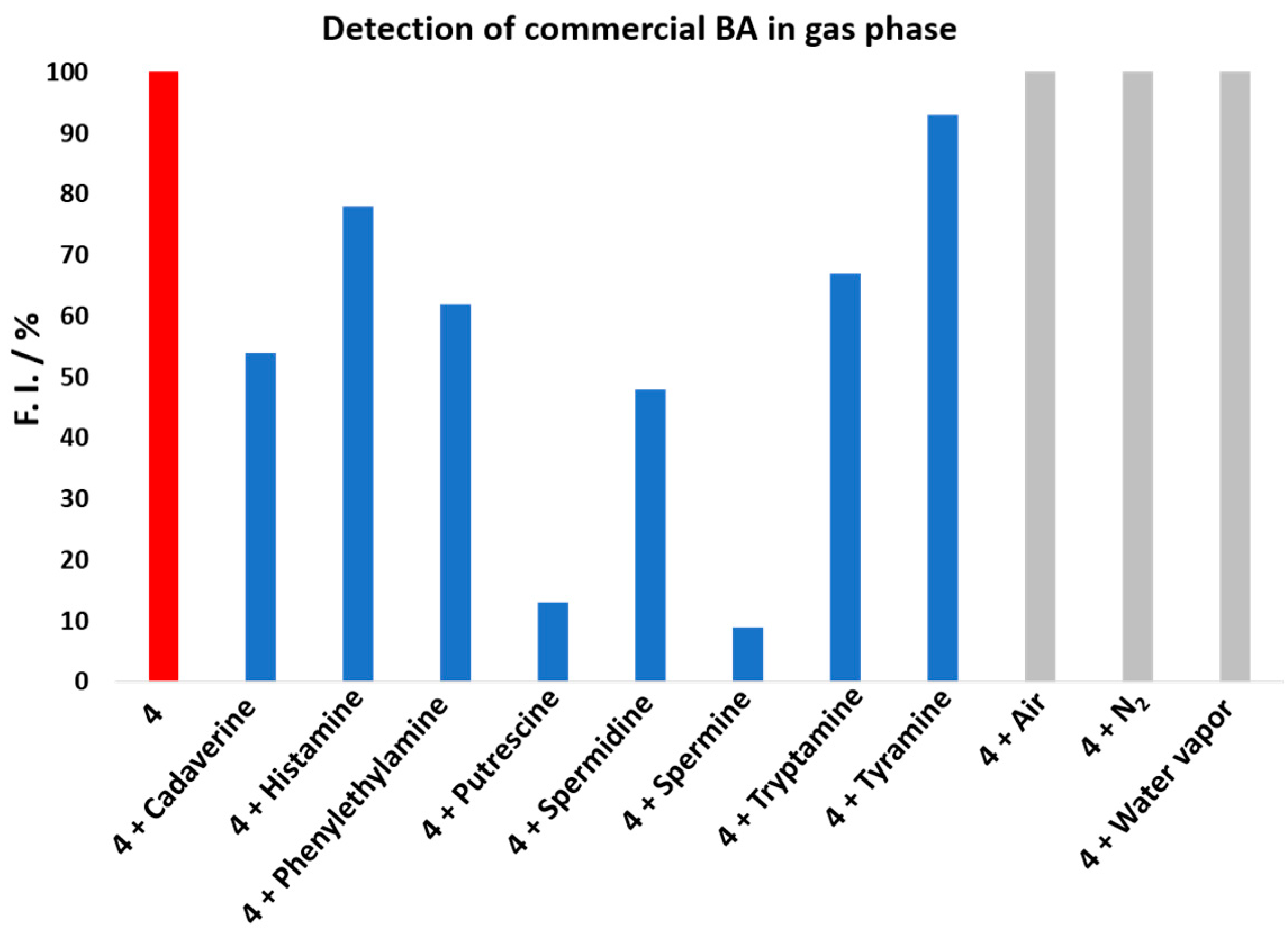
To demonstrate that rosamine
4 can be used in the detection of volatile amine vapors, which are highly toxic, irritant, and corrosive, being important indicators of food quality, for example, in the assessment for fish freshness [
29], we investigated its reactivity toward BAs in the gas phase. The study was carried out by injecting vapors of different commercial BAs in solutions of
4 dissolved in acetonitrile (see the Materials and Methods section for more details and the apparatus in
Figure S32 in the Supplementary Materials). The spectrophotometrical measurements were carried at 533 nm. As shown in
Figure 4, nitrogen, air, and water vapor did not affect rosamine
4, since the value of fluorescence intensity did not change when they were injected. However, when the commercial BA vapor was introduced, a decrease in the fluorescence intensity was observed, which was more expressive upon spermine addition, resulting in decreases greater than 90%, 87% for putrescine, 52% for spermidine, 47% for cadaverine, 32% for tryptamine, 21% for histamine, and 8% for tyramine. The results for putrescine and cadaverine confirmed rosamine
4 sensitivity toward these BAs with higher quenching percentages in the gas phase comparatively to those reported in solution for 8 equiv.
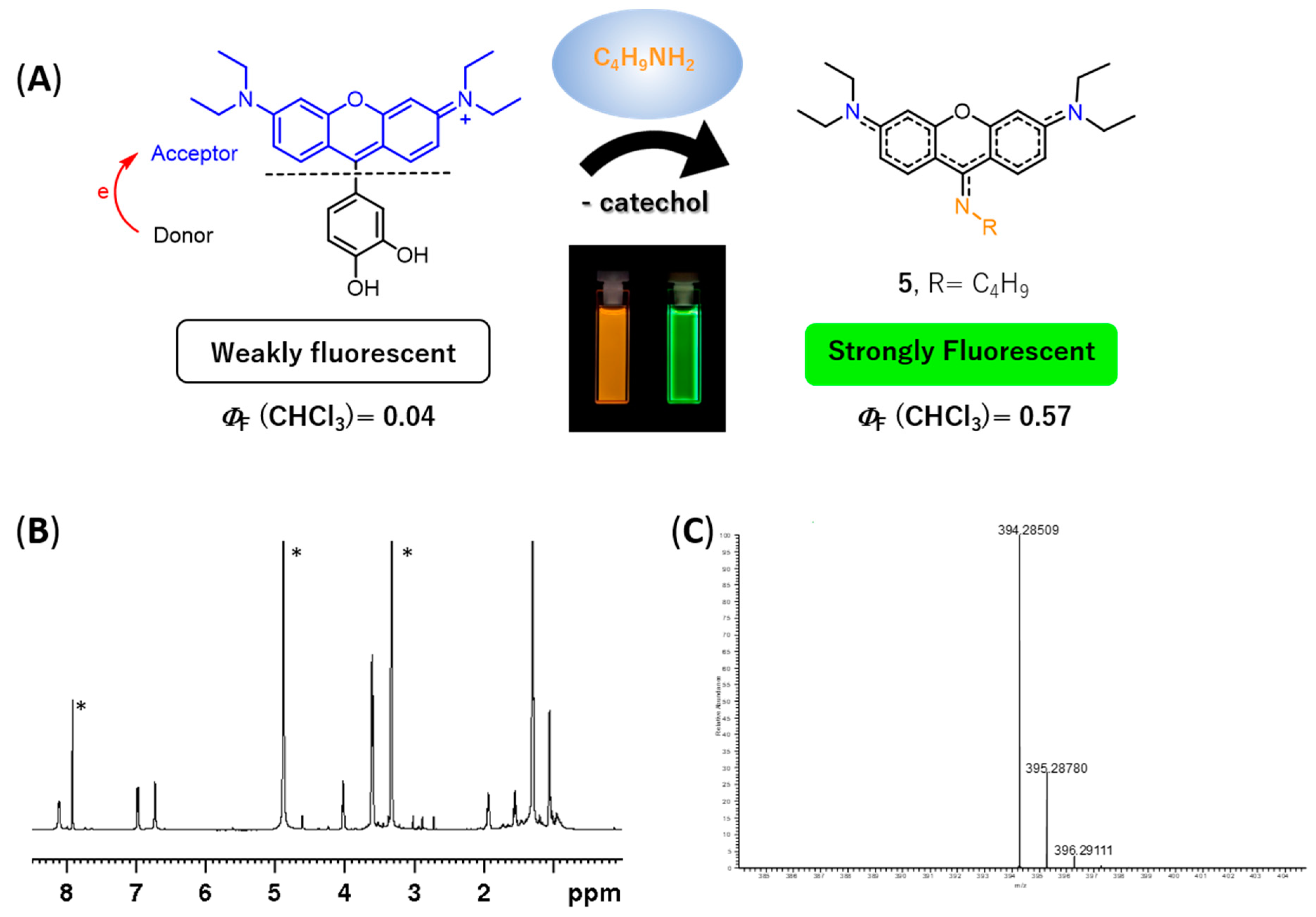
During the experimental assays, when compound
4 was brought to the presence of amines, we observed that—particularly for methanolic solutions of
4 with
n-butylamine—a color change from pink to yellow was observed over time, at room temperature and at 4 °C (refrigerator), which appeared to be due to the formation of a new compound. To verify the aforementioned hypothesis, a solution of rosamine
4 and
n-butylamine (15 equiv.) in methanol was prepared, which was kept at 4 °C for 120 h. The TLC analysis of the resulting solution revealed the presence of a new compound with a bright yellow color, which was subsequently isolated by preparative thin layer chromatography and characterized by NMR and ESI-MS. The corresponding
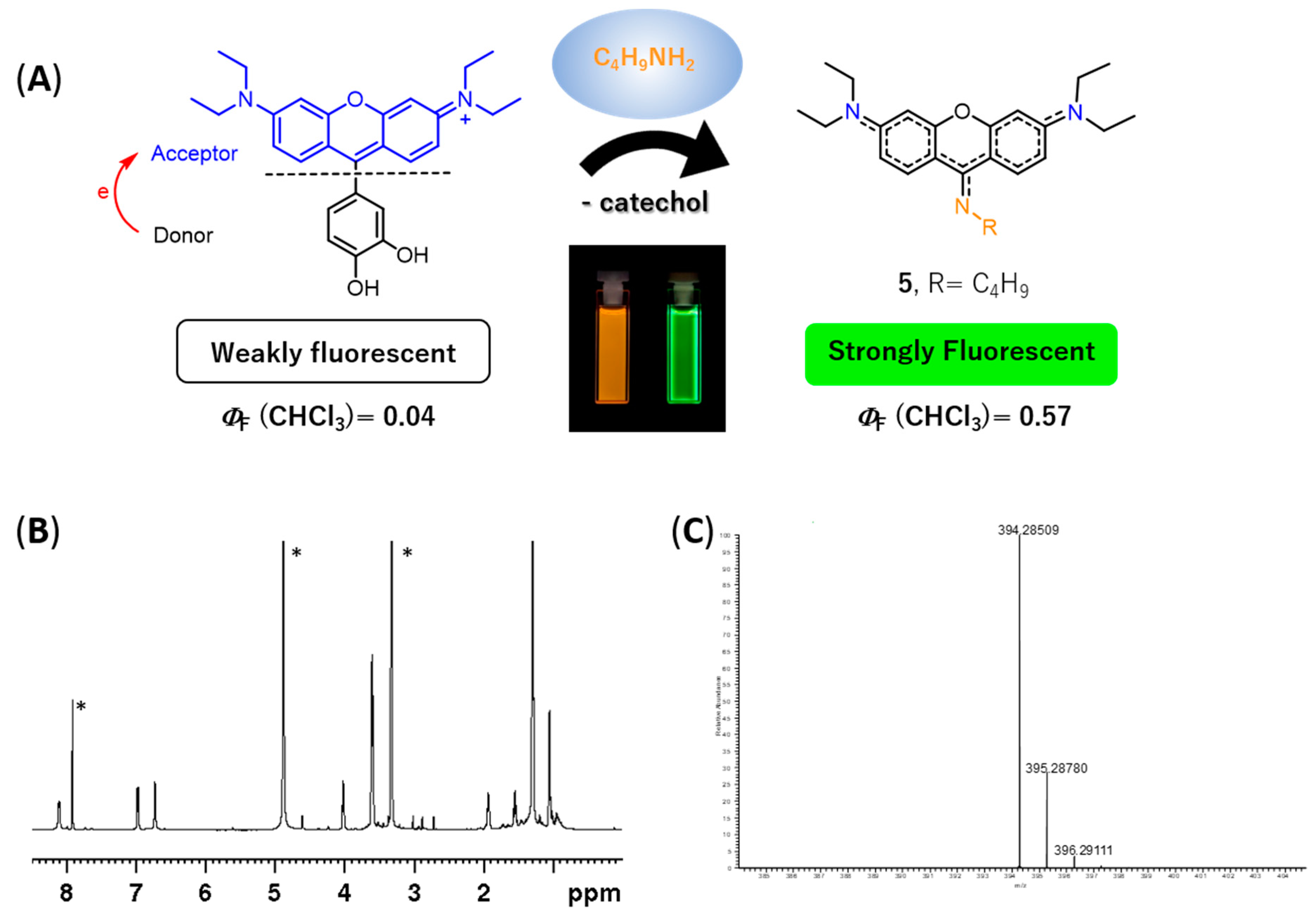
1H NMR spectrum (please refer to
Figure 5B and
Figure S33 of the Supplementary Materials) exhibited: (i) three set of signals from 6.73 to 8.12 ppm due to the resonance of the xanthene ring (no signals due to the catechol unit were detected indicating the absence of the catechol substituent in the molecule) and (ii) a new set of signals appeared at 1.05, 1.55, 1.93, and 4.02 ppm due to the entry of a butylamine moiety, suggesting the formation of 9-aminopyronin
5 (
Figure 5A). A comparison of the
1H NMR spectra of compounds
4 and
5 is provided in the
Supplementary Materials (please refer to
Figure S35). Further evidences of
5 were obtained by ESI-MS, through the molecular ion peak [M + H]
+ at
m/z 394.2851 (
Figure 5C and
Figure S39 in the Supplementary Materials).
In fact, 9-aminopyronin dyes are described in the literature as chemically stable dyes having high molar absorption coefficients and high fluorescence quantum yields. Being initially introduced by K. Burgess et al. [
30], their synthesis typically involves the reaction of xanthenone derivatives with amines in the presence of trifluoromethanesulfonic anhydride or oxalyl chloride [
31,
32,
33,
34]. Alternatively, 9-aminopyronins can be prepared by reacting 9H-xanthene-9-thione with electron-withdrawing groups such as 4-aminopyridine and 2-aminoquinoline [
35]. However, to the best of our knowledge, there are no preceding reports describing the synthesis of 9-aminopyronine dyes from catecholate xanthenes with amines.
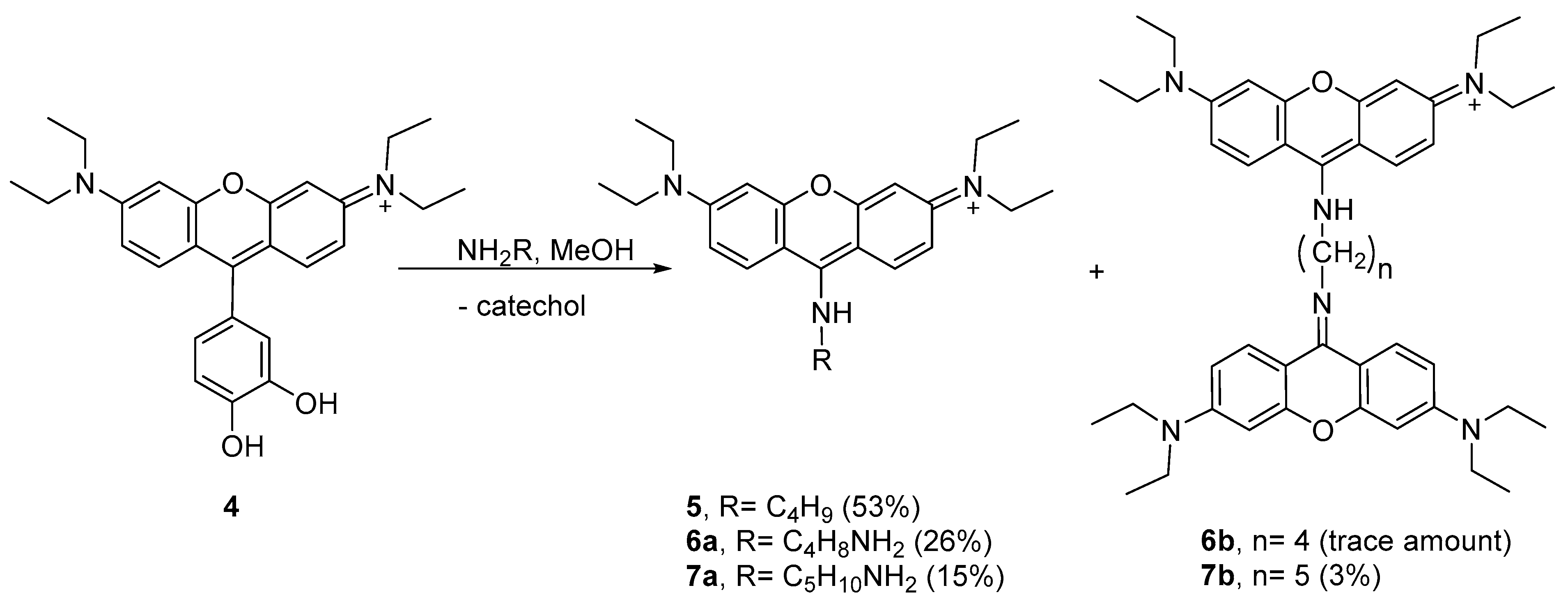
Several attempts have been conducted in order to improve the yield of aminopyronin 5, which included reactions at 4 °C using chloroform or methanol and the use of MW heating (N-methylpyrrolidine as solvent, 200 °C, 10 min; detailed procedures in the Materials and Methods section). The best result was obtained when methanol was used as a solvent (4 °C, for ca. 144 h) with a 53% yield.
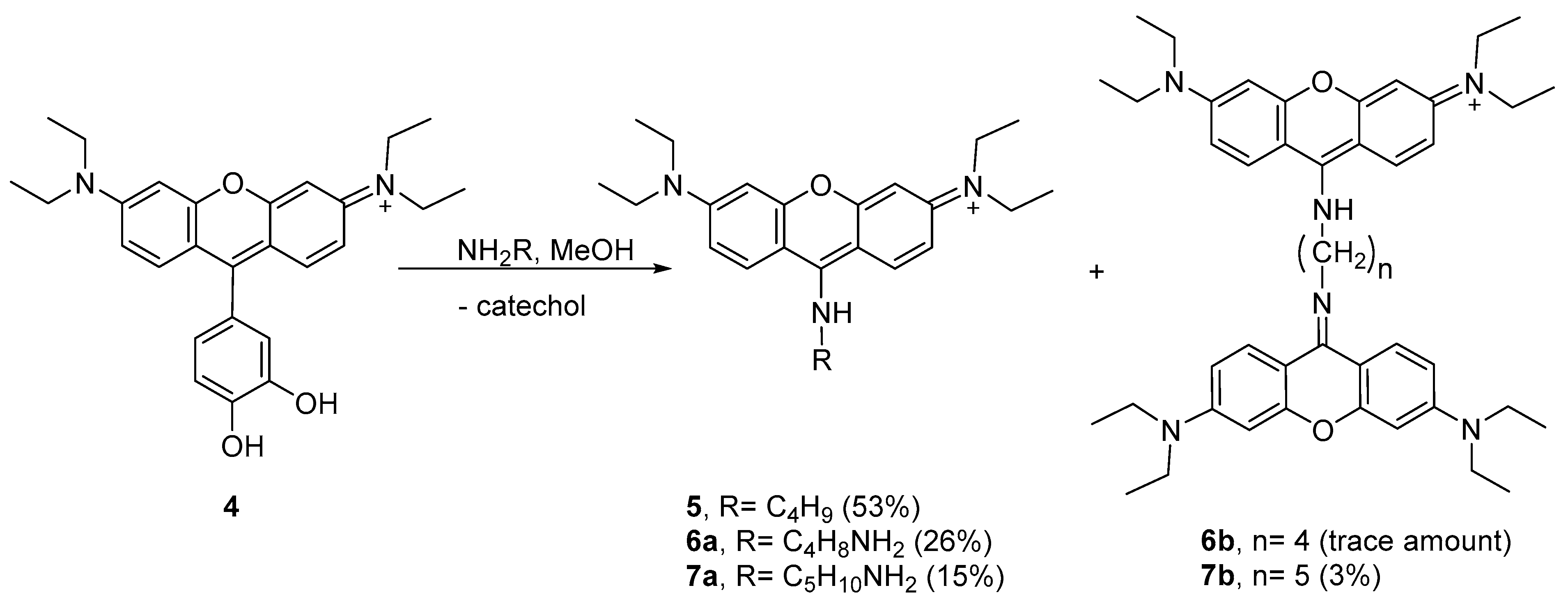
Considering the results obtained with
n-butylamine, we decided to study the peculiar reactivity of
4 using two other nucleophilic amines—putrescine and cadaverine. Using methanol as the solvent at 4 °C, the reactions took place in different extensions, leading to aminopyronins
6a,b and
7a,b (
Scheme 2, see
1H NMR spectra in
Figures S36–S38 in the Supplementary Materials).
The reaction of 4 with putrescine mainly afforded the monomer 6a, as supported by the NMR spectral analysis and MALDI-TOF mass spectrometry, showing [M + H]+ at m/z 409.409 (theoretical [M + H]+ = 409.296), while only a trace amount of dimer 6b was obtained. Higher reaction extension was achieved when cadaverine was used, affording monomer 7a together with the dimer 7b. The 1H NMR profile of 7a follows the same profile as compound 5 while the MALDI-TOF spectrum showed the molecular ion [M + H]+ at m/z 423.450 (theoretical [M + H]+ = 423.312) supporting the structure 7a. In the 1H NMR spectrum of dimer 7b, the presence of two equivalent xanthene rings per one cadaverine frame was confirmed by the 12 aromatic protons and the 10 aliphatic protons, which is consistent with the MALDI-TOF spectrum that showed [M + H]+ at m/z 743.969 (theoretical [M + H]+ = 743.501).
For comparison purposes, the photophysical properties of all isolated aminopyronins (
5,
6a,
7a,
7b) were accessed in chloroform, methanol, and acetonitrile (
Table 2). All compounds presented an intense yellow color (naked eye), exhibiting absorption in the range of 430–440 nm with high extinction coefficients (10.6 × 10
4–4.35 × 10
4). Their emissions were in the range of 507–526 nm with large Stokes shifts (72–88 nm; 3265–3820 cm
−1), values around 5.4–6.6 times higher than those found for rosamines
1–
4. High fluorescence quantum yields were obtained, especially in chloroform (
ΦF = 0.57, 0.36, 0.58, and 0.53, respectively, for
5,
6a,
7a, and
7b), a fact in clear contrast with the starting rosamines
3 and
4 for which low
ΦF values were obtained in chloroform (
Table 1).
Considering the possible similar reactivity of 2,3-dihydroxylphenyl substituted rosamine
3, a similar study was performed from a solution of
3 and
n-butylamine (15 equiv.) in methanol at 4 °C for 120 h. In this case, TLC analysis of the resulting solution showed a complex mixture of products very difficult to purify by chromatographic methods. It is very probable that, according to the generally accepted crosslinking chemistry of catechols and amines [
11], the catechol unit would suffer different nucleophilic attacks of
n-butylamine by either Michael-type addition or via the formation of a Schiff base, thus supporting the formation of many different products. However, in the case of the reaction of rosamine
4 with
n-butylamine, 9-aminopyronin
5 was obtained as the major product of the reaction. The data suggest that the nucleophilic attack of the amine to the electrophilic 9-position of the xanthene takes place with the formation of the addition adduct, and that the loss of the catechol moiety occurs, potentially, during the isolation process.
3. Materials and Methods
Reagents and solvents were purchased as reagent-grade and used without further purification unless otherwise stated. NMR spectra were recorded on a Bruker Avance III 400 spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany; 400.15 MHz for
1H and 100.63 MHz for
13C) or on a Bruker Avance III HD 600 spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany; 600.13 MHz for
1H and 150.92 MHz for
13C). Chemical shifts (δ) were reported in ppm and coupling constants (
J) in Hz; internal standard was TMS. Two-dimensional
1H/
1H correlation spectra (COSY), gradient selected
1H/
13C heteronuclear single quantum coherence (HSQC), and
1H/
13C heteronuclear multiple bond coherence (HMBC) spectra were acquired using the standard Bruker software. Flash chromatography was carried out using silica gel (230–400 mesh,Merck, Kenilworth, NJ, USA). Microwave-assisted reactions were carried out in a CEM Discovery Labmate circular single-mode cavity instrument (300 W max magnetron power output) from CEM Corporation (CEM Microwave Technology Ltd., Buckingham, UK). Reactions were performed under closed-vessel conditions. MS analysis of compounds
1,
2, and
5 were carried out by electrospray ionization (ESI) in a LTQ-Orbitrap-XL instrument (Thermo Fischer Scientific, Winsford, UK) with the following ESI source parameters: electrospray needle voltage 3.1 kV, sheath gas nitrogen 6, capillary temperature 275 °C, capillary voltage 21 V, and tube lens voltage 55 V. Ionization polarity was adjusted according to sample. Mass spectra of rosamines
3 and
4 were acquired by Unidade De Espectrometria De Masas of Santiago de Compostela and microanalyses were acquired by Unidad De Análisis Elemental of Santiago de Compostela. Mass spectra of compounds
6a and
7a,b were acquired on a Bruker UltrafleXtreme MALDI-TOF/TOF equipped with a 200 Hz smartbeam laser (Bruker Daltonik GmbH, Bremen, Germany). Electronic absorption spectra were recorded with a Varian Cary bio50 spectrophotometer (Agilent Technologies, Santa Clara, CA, USA), equipped with a Varian Cary single-cell Peltier accessory, using 1 cm path-length quartz cells. Steady-state fluorescence measurements were carried out in a Varian spectrofluorometer, model Cary Eclipse (Agilent Technologies, Santa Clara, CA, USA), equipped with a constant-temperature cell holder (Peltier single-cell holder) with 5 nm slit width for excitation and emission. All photophysical assays were performed under controlled temperature conditions (25 °C), using the maximum
λexc and the appropriate
λem range, for each rosamine and considering the different solvents used. Rosamine stock solutions were prepared in anhydrous dimethylsulfoxide (DMSO) and DMSO percentage was kept under 1% regarding the reported studies with different solvents. Rhodamine B (
RhB) was used as standard in the photophysical assays and the respective solutions were prepared in anhydrous ethanol. Solutions of the compounds, in appropriate concentration ranges, were prepared previously to molar absorptivity coefficients (
ε) and fluorescence quantum yield (
ΦF) determination assays. Generally, fluorescence quantum yield determination was done combining the literature-described [
36,
37], pH variation study was carried out from a stock solution of rosamine
4 in water, and dilutions were performed with ionic force adjustment (0.1 mol/L NaCl). Each pH adjustment from 2–12 was obtained by the addition of small amounts of concentrated HCl and NaOH, followed by UV–Vis and fluorescence measurements between 200–800 nm or 555–700 nm for UV–Vis and fluorescence, respectively.
Synthesis of benzylated precursors: To a solution of 2,3-dihydroxybenzaldehyde or 3,4-dihydroxybenzaldehyde (1.00 g, 7.24 mmol) and K2CO3 (2.20 g, 15.9 mmol) in DMF (20 mL) at 0 °C, benzyl bromide (1.89 mL, 15.9 mmol) was added drop-by-drop, under N2 atmosphere. After stirring at 0 °C for 15 min, the reaction mixture was allowed to warm up to room temperature and the stirring was maintained for 4 h. After that time, the reaction mixture was precipitated into an ice/water mixture and neutralized using citric acid. The white solid obtained was filtered, washed with water, dissolved in chloroform, and crystallized in n-hexane to afford the respective products.
2,3-Dibenzyloxybenzaldehyde: Yield 75% (1.73 g). 1H NMR (400 MHz, CDCl3) δ: 5.19 and 5.20 (4H, 2s, 2xCH2C6H5), 7.12 (1H, dt, J 8.0 Hz and J 0.6 Hz, H-Ar), 7.23–7.26, 7.32–7.43 and 7.47–7.49 (12H, 3m, H-Ar), 10.26 (1H, s, CHO). 13C NMR (CDCl3, 100.62 MHz) δ: 71.3 (CH2), 76.5 (CH2), 119.6, 119.9, 124.2, 127.6, 128.3, 128.5, 128.6, 128.7, 128.8, 130.5, 136.3, 151.5, 152.1, 190.2 (CHO). MS (EI) m/z: 318 M+. C21H18O3 ¼ H2O calcd. C 78.12, H 5.78; found C 78.46, H 5.34.
3,4-Dibenzyloxybenzaldehyde: Yield 84% (1.94 g). 1H NMR (400 MHz, CDCl3) δ: 5.22 and 5.26 (4H, 2s, 2×CH2C6H5), 7.02 (1H, d, J 8.0 Hz, H-5), 7.32–7.49 (12H, m, H-Ar), 9.81 (1H, s, CHO) ppm. HRMS (ESI) m/z: calcd. for C21H19O3+, 319.129; found, 319.133.
3.1. Synthesis of Rosamines 1 and 2
(a) Via conventional heating using propionic acid as solvent: a solution of 3-(diethylamino)phenol (0.28 g, 1.72 mmol) with the appropriate benzaldehyde (0.27 g, 0.86 mmol) and p-TsOH (17.2 mg, 0.10 mmol) in propionic acid (10 mL) was placed in a reaction flask and heated to 65 °C for 16 h. After cooling to room temperature, the mixture was neutralized with NaOAc (3 mol/L, 100 mL). The resulting suspension was extracted with chloroform. The combined organic extracts were dried (Na2SO4 anhydrous) and the solvent evaporated. The resulting residue was dissolved in 40 mL of a mixture of CHCl3/MeOH (1:1), to which chloranil (0.10 g, 0.43 mmol) was added. The mixture was vigorously stirred for 2 h and concentrated in vacuum. The residue was purified by flash chromatography using a mixture of CHCl3/MeOH (9:1) and vacuum dried.
(b) Via MW using propionic acid or water as solvent: a solution of 3-(diethylamino)phenol (0.14 g, 0.86 mmol) with the appropriate benzaldehyde (0.13, 0.43 mmol) and p-TsOH (10.0 mg, 0.06 mmol) in water or propionic acid (5 mL) was placed in a 10 mL reaction vial, which was then sealed and placed in the cavity of a CEM microwave reactor. The reaction was irradiated at 80 °C (1 min ramp to 80 °C and 10 min hold at 80 °C, using 100 W maximum power). The solvent was decanted and the resulting solid was dissolved in 10 mL of a mixture of CHCl3/MeOH (1:1), to which chloranil (0.10 g, 0.43 mmol) was added. The mixture was placed into the cavity of the CEM microwave using 1 min ramp to 60 °C and 10 min hold at 60 °C, using 50 W maximum power. The residue was purified by flash chromatography using a mixture of CHCl3/MeOH (9:1) and vacuum dried.
Rosamine 1: (a) Yield 36% (95 mg); (b) Yields 43% (113 mg) and 47% (124 mg) in propionic acid and water, respectively. 1H NMR (400 MHz, CDCl3) δ: 1.33 (12H, t, J 7.2 Hz, 4×CH3), 3.64 (8H, q, J 7.2 Hz, 4×CH2), 4.88 (2H, s, 2′-CH2C6H5), 5.27 (2H, s, 3′-CH2C6H5), 6.74–6.76 (3H, m, H-Ar), 6.77 (2H, d, J 2.4 Hz, H-4 and H-5), 6.82 (2H, dd, J 9.4 and J 2.4 Hz, H-2 and H-7), 6.93–7.03 (3H, m, H-Ar), 7.19 (2H, d, J 9.4 Hz, H-1 and H-8), 7.23–7.27 and 7.36–7.52 (7H, 2m, H-Ar) ppm. 13C NMR (100 MHz, CDCl3) δ: 12.7 (CH3), 46.2 (CH2), 71.2 (3′-CH2C6H5), 75.2 (2′-CH2C6H5), 96.2 (C-4 and C-5), 113.7, 114.0, 116.0 (C-2 and C-7), 122.4, 124.8, 126.7, 127.6, 127.8, 128.0, 128.4, 128.7, 132.2 (C-1 and C-8), 136.2, 136.6, 145.5 (C-2′), 152.3 (C-3′), 155.1 (C-9), 155.4 (C-3 and C-6), 157.7 (C-4a and C-5a) ppm. HRMS (ESI) m/z: calcd. for C41H43N2O3+, 611.327; found 611.326.
Rosamine 2: (a) Yield 12% (32 mg); (b) Yields 61% (160 mg) and 68% (179 mg) in propionic acid and water, respectively. 1H NMR (400 MHz, CDCl3) δ: 1.32 (12H, t, J 7.2 Hz, 4×CH3), 3.63 (8H, q, J 7.2 Hz, 4×CH2), 5.24 (2H, s, 3′-CH2C6H5), 5.31 (2H, s, 4′-CH2C6H5), 6.77 (2H, d, J 2.4 Hz, H-4 and H-5), 6.80 (2H, dd, J 9.6 and J 2.4 Hz, H-2 and H-7), 6.89 (1H, d, J 2.0 Hz, H-2′), 6.92 (1H, dd, J 8.0 and J 2.0 Hz, H-6′), 7.17 (1H, d, J 8.0 Hz, H-5′), 7.27 (2H, d, J 9.6 Hz, H-1 and H-8), 7.31–7.45 (8H, m, H-Ar), 7.53 (2H, d, J 7.2 Hz, H-Ar) ppm. 13C NMR (100 MHz, CDCl3) δ: 12.8 (CH3), 46.2 (CH2), 71.2 and 71.3 (CH2C6H5), 96.5 (C-4 and C-5), 113.2 (C-1a and C-8a), 114.1 (C-2 and C-7), 114.4 (C-5′), 116.9 (C-2′), 123.8 (C-6′), 124.2 (C-1′), 127.37, 127.41, 128.0, 128.2, 128.76, 128.79, 132.2 (C-1 and C-8), 136.6, 136.7, 148.4 (C-3′), 151.1 (C-4′), 155.4 (C-3 and C-6), 157.1 (C-9), 158.0 (C-4a and C-5a) ppm. HRMS (ESI) m/z: calcd. for C41H43N2O3+, 611.327; found, 611.326.
3.2. Synthesis of Rosamines 3 and 4
A 1 mol/L solution of boron trichloride in dichloromethane (2 mL) was dropped slowly into an ice-bath cooled suspension of rosamine 1 or 2 (85.6 mg, 0.14 mmol) in dry dichloromethane (8 mL) under a N2 atmosphere. The mixture was stirred at room temperature for 18 h. Methanol (20 mL) was added to stop the reaction, followed by several washings with acetone and methanol. After removal of the solvent in vacuum, the residue was precipitated with methanol/acetone.
Rosamine 3: Yield 41% (25 mg). 1H NMR (400 MHz, DMSO-d6) δ: 1.21 (12H, t, J 7.2 Hz, 4×CH3), 3.65 (8H, q, J 6.8 Hz, 4×CH2), 6.66 (1H, d, J 7.6 Hz, H-6′), 6.89 (1H, dd, J 8.0 and J 7.6 Hz, H-5′), 6.96 (2H, d, J 2.2 Hz, H-4 and H-5), 7.07 (1H, d, J 8.0 Hz, H-4′), 7.14 (2H, dd, J 9.4 and J 2.2 Hz, H-2 and H-7), 7.26 (2H, d, J 9.4 Hz, H-1 and H-8), 9.00 (1H, s, OH), 9.96 (1H, s, OH) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 12.5 (CH3), 45.3 (CH2), 95.8 (C-4 and C-5), 113.0 (C-1a and C-8a), 114.3 (C-2 and C-7), 117.2 (C-4′), 119.3 (C-1′), 119.5 (C-5′), 120.4 (C-6′), 131.9 (C-1 and C-8), 143.0 (C-2′), 145.8 (C-3′), 155.1 (C-3 and C-6), 155.6 (C-9), 157.4 (C-4a and C-5a) ppm. MS (ESI) m/z: 431.2 M+. C27H31ClN2O3.3/2CH2Cl2 calcd. C, 57.59, H, 5.77, N, 4.71; found C, 58.07, H, 5.46, N, 4.75.
Rosamine 4: Yield 91% (55 mg). 1H NMR (400 MHz, DMSO-d6) δ: 1.22 (12H, t, J 7.2 Hz, 4×CH3), 3.65 (8H, q, J 6.8 Hz, 4×CH2), 6.79 (1H, dd, J 8.0 and J 2.1 Hz, H-6′), 6.94 (3H, d, J 2.1 Hz, H-4, H-5 and H-2′), 7.05 (1H, d, J 8.0 Hz, H-5′), 7.16 (2H, dd, J 9.8 and J 2.1 Hz, H-2 and H-7), 7.47 (2H, d, J 9.8 Hz, H-1 and H-8), 9.77 (2H, s broad, 2×OH) ppm. 13C NMR (100 MHz, DMSO-d6) δ: 12.5 (CH3), 45.3 (CH2), 95.9 (C-4 and C-5), 112.5 (C-1a and C-8a), 114.2 (C-2 and C-7), 116.0 (C-5′), 117.3 (C-2′), 121.7 (C-6′), 122.3 (C-1′), 132.1 (C-1 and C-8), 145.6 (C-3′), 147.8 (C-4′), 154.9 (C-3 and C-6), 157.3 (C-9), 157.4 (C-4a and C-5a) ppm. MS (ESI) m/z: 431.2 M+. C27H31ClN2O3.½CH2Cl2 calcd. C, 64.83, H, 6.33, N, 5.50; found C, 64.41, H, 6.28, N, 5.42.
3.3. Reaction of Rosamine 4 with Amines
(a) Protocol at 4 °C with n-butylamine: To an ice-bath cooled solution of rosamine 4 (9.9 mg, 23 mmol) in chloroform (500 μL), n-butylamine (34.3 μL, 0.35 mol, 15 equiv.) was added dropwise and the mixture was stirred at 0 °C for 2 h. Afterward, the reactional mixture was placed in the refrigerator for 22 h. After that time, the reaction mixture was washed with diethyl ether to remove the n-butylamine excess and purified by preparative TLC using a chloroform/methanol mixture (9:1). Amino-pyronin 5 was obtained as a yellow solid (24% yield, 2.2 mg), whereas initial rosamine 4 (22%, 2.4 mg) was recovered. A similar procedure was performed using methanol as the solvent, with an extended reaction time to six days. Amino-pyronin 5 was obtained as a yellow solid (53% yield, 4.8 mg), whereas no rosamine 4 was recovered. A similar experiment using rosamine 3 in chloroform at 4 °C was performed but, in this case, a complex mixture of products was obtained.
(b) Protocol under MW with n-butylamine: A solution of rosamine 4 (9.9 mg, 23 mmol) in NMP (N-methylpyrrolidine, 0.1 mL) was placed in a 10 mL reaction vial, to which n-butylamine (34.0 μL, 0.35 mol, 15 equiv.) was added. The vial was then sealed and placed in the cavity of a CEM microwave reactor. The reaction was irradiated at 200 °C (1 min ramp to 200 °C and 35 min hold at 200 °C, using 150 W maximum power). The reaction was controlled by TLC at 5, 20, and 35 min. The resulting mixture was purified by preparative TLC using a mixture of chloroform/methanol (8:2) and vacuum dried, affording amino-pyronin 5 (10% yield, 0.9 mg), whereas rosamine 4 (7% yield, 0.7 mg) was recovered.
Amino-pyronin 5: (a) Yield 53% (4.8 mg) and (b) Yield 10% (0.9 mg) 1H NMR (600 MHz, MeOD-d4) δ: 1.05 (3H, t, J 7.4 Hz, NHCH2CH2CH2CH3), 1.29 (12H, t, J 6.8 Hz, 4×CH2CH3), 1.55 (2H, q, J 7.4 Hz, NHCH2CH2CH2CH3), 1.93 (2H, t, J 7.4 Hz, NHCH2CH2CH2CH3), 3.60 (8H, q, J 6.8 Hz, 4×CH2CH3), 4.02 (2H, t, J 7.4 Hz, NHCH2CH2CH2CH3), 6.73 (2H, s broad, H-4 and H-5), 6.98 (2H, dd, J 9.0 and J 1.8 Hz, H-2 and H-7), 8.12 (2H, d, J 8.4 Hz, H-1 and H-8) ppm. 13C NMR (150 MHz, MeOD-d4) δ: 11.3 (NHCH2CH2CH2CH3), 12.6 (CH2CH3), 19.7 and 31.2 and 47.2–48.2 (CH2, where one of the signals is under the signal of MeOD-d4), 44.5 (CH2CH3), 67.7, 81.7, 96.2 (C-4 and C-5), 110.7 (C-2 and C-7), 128.5, 153.4, 154.6. HRMS (ESI) m/z: calcd. for C25H36N3O+, 394.3285; found, 394.285.
(c) Protocol at 4 °C with putrescine: To an ice-bath solution of rosamine 4 (10.2 mg, 2.4 × 10−2 mol) in chloroform (500 μL), putrescine (17.8 μL, 0.18 mol, 7.5 equiv.) was added dropwise and the mixture was stirred at 0 °C for 2 h. Then, the reactional mixture was placed in the refrigerator for 22 h. After that time, the reaction mixture was washed with diethyl ether to remove the amine excess and purified by preparative TLC using a chloroform/methanol mixture (9:1), to give amino-pyronin 6a (26% yield, 2.5 mg) as a yellow solid, and only trace amount of 6b (dimer).
Amino-pyronin 6a: Yield 26% (2.5 mg) 1H NMR (600 MHz, MeOD-d4) δ: 1.28 (12H, t, J 6.8 Hz, 4×CH2CH3), 1.83 and 2.01 (4H, 2t, J 7.2 Hz, NHCH2CH2CH2CH2NH2), 3.02 (2H, t, J 7.2 Hz, NHCH2CH2CH2CH2NH2), 3.59 (8H, q, J 7.2 Hz, 4×CH2CH3), 4.08 (2H, t, J 7.2 Hz, NHCH2CH2CH2CH2NH2), 6.73 (2H, s broad, H-4 and H-5), 6.97 (2H, dd, J 9.3 and J 2.4 Hz, H-2 and H-7), 8.13 (2H, d, J 9.3 Hz, H-1 and H-8) ppm. MS (MALDI) m/z: calcd for C25H37N4O+, 409.296; found, 409.409.
(d) Protocol at 4 °C with cadaverine: To an ice-bath cooled solution of rosamine 4 (10.2 mg, 2.4 × 10−2 mol) in chloroform (500 μL), cadaverine (20.8 μL, 0.18 mol, 7.5 equiv.) was added dropwise and the mixture was stirred at 0 °C for 2 h. Afterward, the reactional mixture was placed in the refrigerator for 22 h. After that time, the reaction mixture was washed with diethyl ether to remove the amine excess and purified by preparative TLC using a chloroform/methanol mixture (9:1) to give amino-pyronin 7a (15% yield, 1.5 mg) and 7b (0.6 mg, 3%).
Amino-pyronin 7a: Yield 15% (1.5 mg) 1H NMR (600 MHz, MeOD-d4) δ: 1.29 (12H, t, J 6.8 Hz, 4×CH2CH3), 1.60, 1.78 and 2.01 (6H, 3t, J 7.3 Hz, NHCH2CH2CH2CH2CH2NH2), 2.99 (2H, t, J 7.3 Hz, NHCH2CH2CH2CH2CH2NH2), 3.61 (8H, q, J 6.8 Hz, 4×CH2CH3), 4.06 (2H, t, J 7.3 Hz, NHCH2(CH2)4NH2), 6.74 (2H, s broad, H-4 and H-5), 6.99 (2H, dd, J 9.0 and J 2.1 Hz, H-2 and H-7), 8.15 (2H, d, J 9.0 Hz, H-1 and H-8) ppm. MS (MALDI) m/z: calcd. for C26H39N4O+, 423.312; found, 423.450.
Amino-pyronin 7b: Yield 3% (0.6 mg) 1H NMR (400 MHz, MeOD-d4) δ: 1.26 (24H, t, J 7.2 Hz, 8×CH2CH3), 1.71 (2H, t, J 7.3 Hz, NHCH2CH2CH2CH2CH2NH), 2.04 (4H, t, J 7.3 Hz, NHCH2CH2CH2CH2CH2NH), 3.57 (16H, q, J 7.2 Hz, 8×CH2CH3), 4.05 (4H, t, J 7.3 Hz, NHCH2(CH2)3CH2NH2), 6.66 (4H, d, J 2.5 Hz, H-4 and H-5), 6.89 (4H, dd, J 9.6 and J 2.5 Hz, H-2 and H-7), 8.07 (4H, d, J 9.6 Hz, H-1 and H-8) ppm. MS (MALDI) m/z: calcd. for C47H63N6O2+, 743.501; found, 743.969.
3.4. Sensing Study of Rosamine 4 with Different Biogenic Amines
(a) Preliminary studies in solution. Nine samples were prepared from 500 μL of rosamine 4 dissolved in acetonitrile with a concentration of 10−5 M. Then, 2, 4, and up to 8 equiv. of 5 μL solutions of biogenic amines (1-histamine, 2-tyramine, 3-cadaverine, 4-putrescine, 5-phenylethylamine, 6-spermidine, 7-spermine, 8-n-butylamine, and 9-rosamine 4) dissolved in water were added. The changes were observed under visible and UV light and registered.
(b) UV-Vis and fluorescence studies in solution. A stock solution of rosamine 4 in DMSO was diluted to 35 μM in acetonitrile. Then 0.1, 0.5, 1.0, 2.0, 4.0, and 8.0 equiv. of amine solutions (n-butylamine, t-butylamine, cadaverine, and putrescine) in acetonitrile were added. UV–Vis and fluorescence spectra were recorded in the appropriate concentration and wavelength ranges between 225 and 800 nm (absorbance) and 561–800 nm (emission; λexc = 551 nm, 5 nm slit width for excitation and emission and 650 V) at 25 °C.
(c) Studies in gas phase. The study was carried out using 2.5 mL of rosamine
4 dissolved in acetonitrile and with a concentration of 5 μM. To dispose the amines in the gas phase, a small amount of each commercial biogenic amine (cadaverine, spermidine, spermine, phenylethylamine, histamine, tyramine, tryptamine, and putrescine) was placed in 10 mL vials, closed with a septum, and heated on a heating plate, as shown in
Figure S32, at different temperatures depending on the boiling point of each amine. The vapor produced was transferred with a syringe and bubbled into a cuvette containing 2.5 mL of
4. Afterward, the measurement was carried out in the spectrophotometer at 533 nm. The same procedure was carried out using nitrogen, air, and water vapor to check any putative effects on probe
4.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}